")
Back to Journals » Nature and Science of Sleep » Volume 8
Chronic sleep disturbance and neural injury: links to neurodegenerative disease
Authors Abbott S, Videnovic A
Received 20 September 2015
Accepted for publication 30 November 2015
Published 25 January 2016 Volume 2016:8 Pages 55—61
DOI https://doi.org/10.2147/NSS.S78947
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Steven A Shea
Sabra M Abbott,1 Aleksandar Videnovic2
1Department of Neurology, Northwestern Feinberg School of Medicine, Chicago, IL, USA; 2Department of Neurology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
Abstract: Sleep–wake disruption is frequently observed and often one of the earliest reported symptoms of many neurodegenerative disorders. This provides insight into the underlying pathophysiology of these disorders, as sleep–wake abnormalities are often accompanied by neurodegenerative or neurotransmitter changes. However, in addition to being a symptom of the underlying neurodegenerative condition, there is also emerging evidence that sleep disturbance itself may contribute to the development and facilitate the progression of several of these disorders. Due to its impact both as an early symptom and as a potential factor contributing to ongoing neurodegeneration, the sleep–wake cycle is an ideal target for further study for potential interventions not only to lessen the burden of these diseases but also to slow their progression. In this review, we will highlight the sleep phenotypes associated with some of the major neurodegenerative disorders, focusing on the circadian disruption associated with Alzheimer’s disease, the rapid eye movement behavior disorder and sleep fragmentation associated with Parkinson’s disease, and the insomnia and circadian dysregulation associated with Huntington’s disease.
Keywords: sleep, neurodegeneration, Alzheimer's disease, Parkinson's disease, Huntington's disease
Introduction
Sleep and neurodegenerative disorders are highly intertwined, and this link is bidirectional. As will be illustrated in this review, sleep disturbances are often early markers of ongoing neurodegeneration. However, it is unclear whether these sleep disturbances may be the cause of, or are simply a symptom of, the underlying disease. In this review, we will present evidence pointing toward both sides of this causal pathway. In addition, each of the major neurodegenerative disorders has its own sleep “phenotype” or characteristic aspects of the sleep–wake cycle that are disrupted in the disorder which we will review (Figure 1).
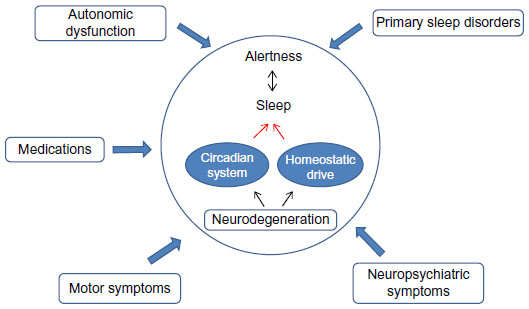 | Figure 1 A model of sleep–wake disruption across neurodegenerative disorders. |
Sleep disruption as a symptom of neurodegenerative disease
Sleep and wake are distinct phenomena regulated by specific brain regions and sets of neurotransmitters. Wakefulness is controlled by multiple brain regions, including cholinergic neurons in the basal forebrain and laterodorsal and pedunculopontine tegmental nuclei and norepinephrine-containing neurons in the locus coeruleus. The brain regions are innervated by neurons containing the peptide orexin, which promotes wakefulness. Sleep is initiated by GABA-containing neurons in the ventrolateral preoptic nucleus and can be divided into NREM and REM sleep. NREM sleep is initiated through ventrolateral preoptic nucleus-mediated inhibition of the ascending arousal pathways, while REM is mediated primarily by activation of cholinergic neurons in the laterodorsal and pedunculopontine tegmental nuclei.1
Sleep disturbances are a common early symptom of neurodegenerative disease, ranging from the increased sleep fragmentation and irregular sleep–wake patterns seen in Alzheimer’s disease (AD) to the increased risk for REM sleep behavior disorder (RBD) in the synucleinopathies, including Parkinson’s disease (PD), multiple system atrophy, and Lewy body dementia to the circadian dysregulation and insomnia seen in Huntington’s disease (HD).
Sleep disruption as an early symptom of AD
It has long been recognized that as AD progresses, sleep disturbances increase. AD has been associated with increased fragmentation of the overall sleep–wake pattern, increased sleep during the daytime, increased frequency of nocturnal awakenings, and a decrease in both slow wave and REM sleep.2 Individuals often develop irregular sleep–wake rhythm disorder, in which sleep is fragmented into at least three distinct bouts throughout the 24-hour period, and individuals also frequently develop “sundowning” whereby confusion and agitation worsen later in the day.3 These sleep disturbances do appear to be associated with adverse clinical outcomes, as both greater sleep disturbance and greater time spent in bed have been associated with greater functional impairment in individuals with AD.4
In addition, there does appear to be progressive circadian dysfunction in these individuals, which can in turn result in significant sleep disruption. In the Tg2576 mouse model of AD, which exhibits age-dependent amyloid-β deposition in the brain, animals demonstrate a significantly longer circadian period of wheel running rhythms when compared to control mice, and they also lacked the normally observed increase in delta power following sleep deprivation.5 Based on caregiver reports, individuals with AD have a decrease in the overall amplitude of the rest–activity rhythm that corresponds with abnormal patterns of the core body temperature rhythm.6 Use of objective actigraphy data to measure rest–activity patterns in AD has demonstrated increased fragmentation and decreased amplitude of activity that correlates with the severity of dementia7 and the degree of impairment in cognitive performance.8
Interestingly, there is increasing evidence that the sleep and circadian disturbances may actually develop prior to the clinical symptoms of AD being evident. In a large actigraphy study of over 1,200 healthy women, a decrease in the amplitude and robustness of the circadian rest–activity rhythm at baseline was associated with an increased risk for developing either mild cognitive impairment or dementia over the next 5 years, suggesting that these sleep disturbances are either an early symptom or contribute to the underlying pathophysiology.9
Furthermore, the use of hypnotics to treat sleep disturbances may further contribute to the development of dementia through unclear mechanisms. There is evidence that in individuals with traumatic brain injury, those with complaints of insomnia who are treated with hypnotics have a higher risk of developing dementia when compared to control individuals, either with or without insomnia.10 Neuroleptic medications are also frequently used to treat sleep disturbances in patients with dementia; however, these have also been associated with an increased risk of morbidity and mortality in these patients.11 This suggests that simply using medication to treat the underlying sleep disturbance will not necessarily improve risk, and may actually do more harm.
Sleep and circadian disruption in movement disorders
Impaired sleep and alertness are frequently encountered in movement disorders such as PD and HD. Sleep disturbances are one of the most common and disabling nonmotor manifestations of PD, affecting as many as 90% of patients.12 The most common sleep disturbances observed include sleep fragmentation, excessive daytime sleepiness, and RBD. Increasing evidence also suggests significant modifications of the circadian system, likely in turn affecting sleep–wake homeostasis in the PD population.13
In most cases, poor sleep is related to the underlying pathology of PD; however in some individuals, the dopamine agonists used to treat the motor symptoms may also contribute to the development of excessive sleepiness. Excessive sleepiness was reported in 13%–36% of PD participants in pivotal clinical trials of levodopa and dopamine agonists.14,15 Levodopa monotherapy confers the lowest risk for sleepiness, while the combination therapy with levodopa and dopamine agonist presents the highest risk.16 In these individuals, the addition of stimulating medications such as armodafinil, buproprion, or one of the more traditional stimulants may be beneficial.17
Poor sleep affects up to 90% of HD patients18 with symptoms including sleep initiation and maintenance insomnia, reduced sleep efficiency, and reduced total sleep time.19 Circadian rhythms also become progressively more disrupted as the HD-specific neurodegenerative process advances, resulting in further sleep–wake disruption.20 Interventions directed toward sleep consolidation, in this case by using hypnotics to impose a regular sleep–wake schedule do seem to be effective in improving circadian function and cognitive performance in HD.21
RBD is a parasomnia characterized by abnormal motor and vocal behaviors in the context of vivid dreams, associated with loss of muscle atonia during REM sleep. The disorder is strongly associated with synuclein-related disorders and is considered one of its earliest manifestations.22 This is likely reflective of early involvement of neuroanatomic networks responsible for the regulation of REM sleep by synuclein-related neurodegeneration. Idiopathic RBD confers a strong risk for the development of Parkinsonism or cognitive dysfunction, with the risk estimated to be over 50% over a decade of having RBD.23
Neurodegeneration and sleep disturbance
At a structural level, there is growing evidence for an underlying neuroanatomical basis for these observed sleep–wake disruptions. In a community-based study of AD patients, actigraphy was obtained prior to death, and postmortem analysis was performed looking at the number of galanin-positive neurons in the intermediate nucleus, the human equivalent to the ventrolateral preoptic area, the primary wake promoting nucleus in rodents. This investigation demonstrated a correlation between the loss of neurons in the intermediate nucleus and increased sleep fragmentation on actigraphy.24 More recently, it has also been demonstrated that there is a correlation between the circadian amplitude of rest–activity, and the number of vasoactive intestinal peptide expressing neurons in the suprachiasmatic nucleus, the primary circadian pacemaker in mammals.25 In PD, sleep disturbance frequently precede motor symptoms, and this is presumed to reflect the degeneration of areas such as the raphe nucleus and locus coeruleus that constitute preclinical stages 1 and 2 of the pathological staging proposed by Braak et al.26,27 These data suggest that the observed sleep–wake and circadian disturbances in neurodegenerative disease are secondary to pathological degeneration of primary brain regions important for regulating the sleep–wake and circadian cycles.
Melatonin changes with neurodegenerative disease
Melatonin is a hormone released by the pineal gland in a circadian pattern, with levels rising several hours before sleep onset, and peaking in the middle of the night. The pattern and amplitude of melatonin release is a reflection of the overall circadian state of the individual. As the sleep–wake cycle is closely tied to the circadian cycle, circadian disruptions are in turn closely linked to disrupted sleep. Many neurodegenerative disorders have been associated with sleep and circadian disruption as reflected in changes in the pattern of melatonin secretion.
In patients with PD, initial studies comparing untreated patients to treated patients with or without motor complications demonstrated an overall advance in the timing of melatonin secretion in treated patients compared to controls, as well as a decrease in the amplitude of melatonin secretion in treated patients with motor complications.28 Later studies have demonstrated that PD patients have a decrease in the amplitude of melatonin secretion compared to controls,29,30 and this effect is more pronounced in patients with excessive daytime sleepiness.29 In addition, the pattern of expression of clock genes, particularly Bmal1, appears to be altered in these individuals.30 Patients with AD have also exhibited a decrease in the amplitude of the melatonin rhythm.31,32
Interestingly, in addition to reflecting underlying sleep and circadian dysfunction, alterations in melatonin secretion patterns may contribute to the underlying pathophysiology of these disorders. Melatonin, known to have antioxidant properties, has been demonstrated in in vivo models to protect against oxidative stress,33 and can protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurotoxicity in mouse models of PD.34 In addition, in vitro models have demonstrated that melatonin can inhibit α-synuclein assembly and toxicity, which is believed to be an important step in the pathophysiology of PD.35 This again supports the idea that changes in melatonin release may not only reflect the observed disruption of sleep and circadian rhythms but also may contribute to further progression of the disease.
Orexin levels/neuronal loss in neurodegenerative disease
Orexin (hypocretin) is an important neuropeptide involved in the maintenance of wakefulness and also stabilizes sleep–wake transitions.36 While orexin represents the primary peptide that is deficient in patients with narcolepsy,37 changes in orexin levels may also play a role in the sleep fragmentation and excessive sleepiness frequently observed in neurodegenerative disorders. Orexin-A levels can be measured in the cerebral spinal fluid (CSF) of patients. In addition, at autopsy, the number of orexin-positive neurons can be assessed.
In HD, mouse models have demonstrated a significant decrease in the number of orexin-positive neurons in the hypothalamus.38,39 Similar results have also been found in autopsy studies from humans with HD.40 Interestingly, several studies looking at CSF orexin levels in patients with HD did not find any significant difference when compared with controls,41–44 suggesting that it takes substantial neuronal loss before significant changes are reflected in the CSF profile.
Similarly, several studies in patients with PD have demonstrated normal CSF orexin levels on average,45–47 though some differences have been noted if measured at the level of the ventricle in late-stage PD, with lower levels correlating with increased disease severity.48 Similar to patients with HD, even in PD patients with normal CSF orexin levels, there does appear to be a 25%–50% loss of orexin-containing cells on postmortem autopsy.49 In vitro, there is evidence that orexin can play a neuroprotective role in a cellular model of PD, suggesting that loss of these cells may play a role in the underlying sleep–wake disturbances, but in turn, cell loss may also contribute to the progression of disease.50
The story becomes more complicated in patients with AD, where results have been more variable. In at least one study, CSF levels of orexin were found to be no different than those in controls,51 while another group looking at postmortem brains in patients with advanced AD found both decrease in the number of orexin neurons and a decrease in the CSF orexin levels. Furthermore, lower CSF levels correlated with those who complained of greater daytime sleepiness.52 Of note, another recent study found increased CSF orexin levels in patients with moderate-to-severe AD when compared to controls. This patient population also had a decrease in sleep efficiency, with increased wake after sleep onset on polysomnography (PSG) that correlated with the increased orexin levels.53 In a mouse model of AD, knocking out orexin results in a decrease in amyloid-β deposition. This decrease seems to be related to effects on sleep, rather than direct effects of the orexin peptide, as selective rescue with local injections of orexin does not affect amyloid-β deposition, while sleep deprivation results in increased amyloid-β deposition in orexin knockout mice.54 Overall, this suggests that in neurodegenerative disorders the orexin-containing neurons in the lateral hypothalamus are often affected as part of the underlying neurodegenerative process. In turn, the loss of these neurons can accelerate the underlying pathology, either through loss of direct neuroprotective effects, as in PD, or by further disrupting and fragmenting sleep, resulting in increased amyloid-β deposition, as will be detailed in the following section.
Sleep disruption as a potential cause of neurodegenerative disease
While there is growing evidence that sleep disruption can be an early symptom in the development of neurodegenerative disease (as already discussed), sleep disturbances often predate the onset of cardinal diagnostic clinical symptoms, suggesting that the sleep disruption may actually also play a role in the development and progression of neurodegenerative diseases. This is an exciting concept because it positions the sleep–wake cycle as a novel, readily modifiable factor in its efforts to develop and test neuroprotective/disease-modifying strategies.
The glymphatic system and neurodegenerative disease
One of the primary factors thought to contribute to the development of AD is the accumulation of amyloid-β. This accumulation increases during wakefulness, with further increases observed following sleep deprivation.55 More recently, the mechanism behind these changes in levels has been identified. Throughout the body, systems have been developed to clear the waste products of normal metabolism and function. A process for promoting this clearance from the central nervous system was recently discovered, and named the “glymphatic system”. As implied by the name, this system works similarly to the previously well-described lymphatic system. The CSF circulates and removes by-products from the interstitial fluid, including proteins like amyloid-β that have been linked to neurodegeneration.56 Interestingly, it has recently been demonstrated that this process is up to two times more active during either natural or anesthetic-induced sleep when compared to wakefulness.57 In vivo studies have also demonstrated an increased buildup of insoluble tau protein in response to sleep deprivation.58 As most neurodegenerative disorders are associated with a toxic buildup of waste products in the nervous system, these findings support both the importance of sleep in the prevention of neurodegenerative disorders and a potential mechanism by which sleep disruption could be contributing to the development of these disorders.
Chronic sleep fragmentation, hypoxemia, and the development of neurodegenerative disease
Other factors that may contribute to the development of neurodegenerative disease include the presence of other comorbid sleep disorders. Obstructive sleep apnea (OSA) is a very common disorder affecting up to 19% of men and 15% of women.59 The obstructive events seen in this disorder result in intermittent hypoxemia, as well as sleep fragmentation secondary to the arousals related to respiratory events, both of which may contribute to the development of neurodegenerative disease. Sleep fragmentation has been associated with impaired performance on tasks of vigilance, to a similar degree as is seen with total sleep deprivation.60 In mouse models, chronic mild sleep restriction has been associated with impairments in contextual and cued memory, and an increase in amyloid-β and insoluble tau.61 Neuroimaging studies have demonstrated that the sleep fragmentation associated with OSA has been associated with decreased gray matter volume in the hippocampus, a brain region associated with memory formation.62
In mice, intermittent hypoxia has been associated with increased accumulation of both amyloid-β and insoluble tau protein, two factors that contribute to the development of neurodegenerative disease. These increases corresponded with impaired special learning and memory.63 In humans, in the Apnea Positive Pressure Long-term Efficacy Study (APPLES) it was demonstrated that neurocognitive performance deficits were weakly associated with the apnea–hypopnea index and severity of oxygen desaturation during events.64 Treatment of OSA with continuous positive airway pressure (CPAP) did result in mild improvement in executive and frontal-lobe function for those with severe disease at baseline, suggesting that at least some of the cognitive changes associated with OSA may be reversible.65
Sleep deprivation and motor performance in PD
Sleep deprivation may not be entirely bad when it comes to neurodegenerative disorders. Of interest is the observation from the Nurses’ Health study that individuals who worked night shifts for >15 years had a significantly decreased risk of developing PD. The risk of developing PD was also much lower in individuals sleeping <6 hours/night when compared to those sleeping >9 hours/night.66 Cause and effect are unclear however, as it is not clear at this point whether working night shifts and short sleep duration are protective for developing PD, or if those who are predisposed to develop PD have a lower tolerance for working the night shift. A few small studies have demonstrated that sleep deprivation transiently improves the motor deficits seen in PD, possibly mediated by potentiating dopaminergic activity, though these findings have been somewhat inconsistent, and may reflect different subtypes of the disease.67,68 These observations are certainly in contrast with “sleep benefit” in PD, a clinical phenomenon whereby improved motor function and responsiveness to dopaminergic treatments are noted among PD patients after a restful night of sleep.69 Regardless, these findings do suggest that the link between sleep and neurodegenerative disease is complex and more sleep may not always be better.
Conclusion
Disruptions of sleep–wake cycles are common, yet underdiagnosed and underreported manifestations of neurodegenerative disorders. Further research is needed to clarify whether sleep disruption is primarily a cause of, or a symptom of, the underlying neurodegenerative process. Regardless, it appears that efforts to normalize sleep–wake schedules in patients with neurodegenerative diseases should be beneficial for this population and further may have an impact in slowing the overall disease course.
Disclosure
Dr Videnovic received funding from NIH/NINDS (K23 NS072283). The authors report no other conflicts of interest in this work.
References
Espana RA, Scammell TE. Sleep neurobiology from a clinical perspective. Sleep. 2011;34(7):845–858. | |
Vitiello MV, Borson S. Sleep disturbances in patients with Alzheimer’s disease: epidemiology, pathophysiology and treatment. CNS Drugs. 2001;15(10):777–796. | |
Abbott SM, Zee PC. Irregular sleep-wake rhythm disorder. Sleep Med Clin. 2015;10(4):517–522. | |
Tractenberg RE, Singer CM, Kaye JA. Symptoms of sleep disturbance in persons with Alzheimer’s disease and normal elderly. J Sleep Res. 2005;14(2):177–185. | |
Wisor JP, Edgar DM, Yesavage J, et al. Sleep and circadian abnormalities in a transgenic mouse model of Alzheimer’s disease: a role for cholinergic transmission. Neuroscience. 2005;131(2):375–385. | |
Okawa M, Mishima K, Hishikawa Y, Hozumi S, Hori H, Takahashi K. Circadian rhythm disorders in sleep-waking and body temperature in elderly patients with dementia and their treatment. Sleep. 1991;14(6):478–485. | |
Witting W, Kwa IH, Eikelenboom P, Mirmiran M, Swaab DF. Alterations in the circadian rest-activity rhythm in aging and Alzheimer’s disease. Biol Psychiatry. 1990;27(6):563–572. | |
Lim AS, Yu L, Costa MD, et al. Increased fragmentation of rest-activity patterns is associated with a characteristic pattern of cognitive impairment in older individuals. Sleep. 2012;35(5):633B–640B. | |
Tranah GJ, Blackwell T, Stone KL, et al. Circadian activity rhythms and risk of incident dementia and mild cognitive impairment in older women. Ann Neurol. 2011;70(5):722–732. | |
Chiu HY, Lin EY, Wei L, et al. Hypnotics use but not insomnia increased the risk of dementia in traumatic brain injury patients. Eur Neuropsychopharmacol. Epub 2015 Oct 8. | |
Bohlken J, Schulz M, Rapp MA, Batzing-Feigenbaum J. Pharmacotherapy of dementia in Germany: results from a nationwide claims database. Eur Neuropsychopharmacol. Epub 2015 Oct 3. | |
Videnovic A, Golombek D. Circadian and sleep disorders in Parkinson’s disease. Exp Neurol. 2013;243:45–56. | |
Videnovic A, Lazar AS, Barker RA, Overeem S. ‘The clocks that time us’– circadian rhythms in neurodegenerative disorders. Nat Rev Neurol. 2014;10(12):683–693. | |
Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med. 2000;342(20):1484–1491. | |
PSG. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. JAMA. 2000;284:1931–1938. | |
Paus S, Brecht HM, Koster J, Seeger G, Klockgether T, Wullner U. Sleep attacks, daytime sleepiness, and dopamine agonists in Parkinson’s disease. Mov Disord. 2003;18(6):659–667. | |
Rye DB. Sleepiness and unintended sleep in Parkinson’s disease. Curr Treat Options Neurol. 2003;5(3):231–239. | |
Goodman AO, Morton AJ, Barker RA. Identifying sleep disturbances in Huntington’s disease using a simple disease-focused questionnaire. PLoS Curr. 2010;2:RRN1189. | |
Morton AJ. Circadian and sleep disorder in Huntington’s disease. Exp Neurol. 2013;243:34–44. | |
Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J Neurosci. 2005;25(1):157–163. | |
Pallier PN, Maywood ES, Zheng Z, et al. Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington’s disease. J Neurosci. 2007;27(29):7869–7878. | |
Zanigni S, Calandra-Buonaura G, Grimaldi D, Cortelli P. REM behaviour disorder and neurodegenerative diseases. Sleep Med. 2011;12(Suppl 2):S54–S58. | |
Postuma RB, Gagnon JF, Vendette M, Fantini ML, Massicotte-Marquez J, Montplaisir J. Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology. 2009;72(15):1296–1300. | |
Lim AS, Ellison BA, Wang JL, et al. Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer’s disease. Brain. 2014;137(Pt 10):2847–2861. | |
Wang JL, Lim AS, Chiang WY, et al. Suprachiasmatic neuron numbers and rest-activity circadian rhythms in older humans. Ann Neurol. 2015; 78(2):317–322. | |
Braak H, Del Tredici K. Invited article: nervous system pathology in sporadic Parkinson disease. Neurology. 2008;70(20):1916–1925. | |
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. | |
Bordet R, Devos D, Brique S, et al. Study of circadian melatonin secretion pattern at different stages of Parkinson’s disease. Clin Neuropharmacol. 2003;26(2):65–72. | |
Videnovic A, Noble C, Reid KJ, et al. Circadian melatonin rhythm and excessive daytime sleepiness in Parkinson disease. JAMA Neurol. 2014;71(4):463–469. | |
Breen DP, Vuono R, Nawarathna U, et al. Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol. 2014;71(5):589–595. | |
Ohashi Y, Okamoto N, Uchida K, Iyo M, Mori N, Morita Y. Daily rhythm of serum melatonin levels and effect of light exposure in patients with dementia of the Alzheimer’s type. Biol Psychiatry. 1999;45(12):1646–1652. | |
Skene DJ, Swaab DF. Melatonin rhythmicity: effect of age and Alzheimer’s disease. Exp Gerontol. 2003;38(1–2):199–206. | |
Saravanan KS, Sindhu KM, Mohanakumar KP. Melatonin protects against rotenone-induced oxidative stress in a hemiparkinsonian rat model. J Pineal Res. 2007;42(3):247–253. | |
Acuna-Castroviejo D, Coto-Montes A, Gaia Monti M, Ortiz GG, Reiter RJ. Melatonin is protective against MPTP-induced striatal and hippocampal lesions. Life Sci. 1997;60(2):PL23–PL29. | |
Ono K, Mochizuki H, Ikeda T, et al. Effect of melatonin on alpha-synuclein self-assembly and cytotoxicity. Neurobiol Aging. 2012;33(9):2172–2185. | |
Hagan JJ, Leslie RA, Patel S, et al. Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc Natl Acad Sci U S A. 1999;96(19):10911–10916. | |
Peyron C, Faraco J, Rogers W, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6(9):991–997. | |
Williams RH, Morton AJ, Burdakov D. Paradoxical function of orexin/hypocretin circuits in a mouse model of Huntington’s disease. Neurobiol Dis. 2011;42(3):438–445. | |
Petersen A, Gil J, Maat-Schieman ML, et al. Orexin loss in Huntington’s disease. Hum Mol Genet. 2005;14(1):39–47. | |
van Wamelen DJ, Aziz NA, Roos RA, Swaab DF. Hypothalamic alterations in Huntington’s disease patients: comparison with genetic rodent models. J Neuroendocrinol. 2014;26(11):761–775. | |
Bjorkqvist M, Petersen A, Nielsen J, et al. Cerebrospinal fluid levels of orexin-A are not a clinically useful biomarker for Huntington disease. Clin Genet. 2006;70(1):78–79. | |
Baumann CR, Hersberger M, Bassetti CL. Hypocretin-1 (orexin A) levels are normal in Huntington’s disease. J Neurol. 2006;253(9):1232–1233. | |
Gaus SE, Lin L, Mignot E. CSF hypocretin levels are normal in Huntington’s disease patients. Sleep. 2005;28(12):1607–1608. | |
Meier A, Mollenhauer B, Cohrs S, et al. Normal hypocretin-1 (orexin-A) levels in the cerebrospinal fluid of patients with Huntington’s disease. Brain Res. 2005;1063(2):201–203. | |
Overeem S, van Hilten JJ, Ripley B, Mignot E, Nishino S, Lammers GJ. Normal hypocretin-1 levels in Parkinson’s disease patients with excessive daytime sleepiness. Neurology. 2002;58(3):498–499. | |
Baumann C, Ferini-Strambi L, Waldvogel D, Werth E, Bassetti CL. Parkinsonism with excessive daytime sleepiness – a narcolepsy-like disorder? J Neurol. 2005;252(2):139–145. | |
Yasui K, Inoue Y, Kanbayashi T, Nomura T, Kusumi M, Nakashima K. CSF orexin levels of Parkinson’s disease, dementia with Lewy bodies, progressive supranuclear palsy and corticobasal degeneration. J Neurol Sci. 2006;250(1–2):120–123. | |
Drouot X, Moutereau S, Nguyen JP, et al. Low levels of ventricular CSF orexin/hypocretin in advanced PD. Neurology. 2003;61(4):540–543. | |
Fronczek R, Overeem S, Lee SY, et al. Hypocretin (orexin) loss in Parkinson’s disease. Brain. 2007;130(Pt 6):1577–1585. | |
Feng Y, Liu T, Li XQ, et al. Neuroprotection by Orexin-A via HIF-1alpha induction in a cellular model of Parkinson’s disease. Neurosci Lett. 2014;579:35–40. | |
Ripley B, Overeem S, Fujiki N, et al. CSF hypocretin/orexin levels in narcolepsy and other neurological conditions. Neurology. 2001;57(12):2253–2258. | |
Fronczek R, van Geest S, Frolich M, et al. Hypocretin (orexin) loss in Alzheimer’s disease. Neurobiol Aging. 2012;33(8):1642–1650. | |
Liguori C, Romigi A, Nuccetelli M, et al. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 2014;71(12):1498–1505. | |
Roh JH, Jiang H, Finn MB, et al. Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. J Exp Med. 2014;211(13):2487–2496. | |
Kang JE, Lim MM, Bateman RJ, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326(5955):1005–1007. | |
Nedergaard M. Neuroscience. Garbage truck of the brain. Science. 2013;340(6140):1529–1530. | |
Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373–377. | |
Di Meco A, Joshi YB, Pratico D. Sleep deprivation impairs memory, tau metabolism, and synaptic integrity of a mouse model of Alzheimer’s disease with plaques and tangles. Neurobiol Aging. 2014;35(8):1813–1820. | |
Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. New Engl J Med. 1993;328(17):1230–1235. | |
Bonnet MH, Arand DL. Clinical effects of sleep fragmentation versus sleep deprivation. Sleep Med Rev. 2003;7(4):297–310. | |
Rothman SM, Herdener N, Frankola KA, Mughal MR, Mattson MP. Chronic mild sleep restriction accentuates contextual memory impairments, and accumulations of cortical Abeta and pTau in a mouse model of Alzheimer’s disease. Brain Research. 2013;1529:200–208. | |
Macey PM, Henderson LA, Macey KE, et al. Brain morphology associated with obstructive sleep apnea. Am J Respir Crit Care Med. 2002;166(10):1382–1387. | |
Zhang X, Li L, Zhang X, et al. Prenatal hypoxia may aggravate the cognitive impairment and Alzheimer’s disease neuropathology in APPSwe/PS1A246E transgenic mice. Neurobiol Aging. 2013;34(3):663–678. | |
Quan SF, Chan CS, Dement WC, et al. The association between obstructive sleep apnea and neurocognitive performance – the Apnea Positive Pressure Long-term Efficacy Study (APPLES). Sleep. 2011;34(3):303B–314B. | |
Kushida CA, Nichols DA, Holmes TH, et al. Effects of continuous positive airway pressure on neurocognitive function in obstructive sleep apnea patients: the Apnea Positive Pressure Long-term Efficacy Study (APPLES). Sleep. 2012;35(12):1593–1602. | |
Chen H, Schernhammer E, Schwarzschild MA, Ascherio A. A prospective study of night shift work, sleep duration, and risk of Parkinson’s disease. Am J Epidemiol. 2006;163(8):726–730. | |
Hogl B, Peralta C, Wetter TC, Gershanik O, Trenkwalder C. Effect of sleep deprivation on motor performance in patients with Parkinson’s disease. Mov Disord. 2001;16(4):616–621. | |
Reist C, Sokolski KN, Chen CC, Coskinas E, Demet EM. The effect of sleep deprivation on motor impairment and retinal adaptation in Parkinson’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 1995;19(3):445–454. | |
van Gilst MM, van Mierlo P, Bloem BR, Overeem S. Quantitative motor performance and sleep benefit in Parkinson disease. Sleep. 2015;38(10):1567–1573. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.