Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Chronic Obstructive Pulmonary Disease Influence on Lung Cancer Risk Through Blood Metabolite Mediation: A Two-Sample Mendelian Randomisation and Mediation Analysis
Authors Cao YN, Wang L, Fan HS, Zhang ZJ ![]()
Received 10 March 2026
Accepted for publication 15 June 2026
Published 20 June 2026 Volume 2026:21 608125
DOI https://doi.org/10.2147/COPD.S608125
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Zijing Zhou
Yan-Nan Cao,1,2 Long Wang,1,2 Hu-Sen Fan,1,2 Zhen-Jiang Zhang2
1School of Clinical Medicine, Shandong Second Medical University, Weifang, Shandong, People’s Republic of China; 2Department of Thoracic Surgery, Weifang People’s Hospital, Weifang, Shandong, People’s Republic of China
Correspondence: Zhen-Jiang Zhang, Department of Thoracic Surgery, Weifang People’s Hospital, Weifang, Shandong, People’s Republic of China, Tel +86 176 5801 2401, Email [email protected]
Purpose: Lung cancer (LC) risk is increased by chronic obstructive pulmonary disease (COPD) through metabolic and inflammatory processes. In addition to identifying possible inflammatory or blood metabolite mediators, this study explores the causal link between LC and COPD.
Patients and Methods: To determine whether COPD and LC are causally related, as well as whether inflammatory variables and blood metabolites mediate this relationship, we used two-sample and two-step Mendelian randomization (MR) analyses based on summary data from published genome-wide association studies (GWAS). Directionality and robustness were investigated using reverse MR and sensitivity analyses, primarily using the inverse variance weighted (IVW) method.
Results: Two-sample MR analyses revealed that COPD was a risk factor for the occurrence of LC (P = 0.002, OR = 1.114 [1.042– 1.191]). Moreover, 113 blood metabolites and six inflammatory factors showed nominally significant causal associations with LC risk (P < 0.05). Conversely, LC had no casual effect on COPD, while LC had significant causal associations with six blood metabolites. Mediation analysis demonstrated that three blood metabolites enhanced the promoted effect of COPD on the occurrence of LC, namely 1−stearoyl− 2−arachidonoyl−gpc (18:0/20:4) levels, Epiandrosterone sulfate levels, and 1−palmitoyl− 2−arachidonoyl−gpc (16:0/20:4n6) levels. However, there was no casual effect of COPD on inflammatory factors. Sensitivity analyses confirmed that all results were reliable.
Conclusion: These findings shed light on the underlying mechanisms that link COPD and LC and highlight the fact that COPD is a risk factor for the development of LC, with blood metabolites partially mediating this effect.
Keywords: inflammatory factors, lung cancer, blood metabolites, Chronic obstructive pulmonary disease, Mendelian randomisation analysis
Introduction
Lung cancer (LC) continues to be a major contributor to cancer mortality globally.1 Although screening and systemic therapies have improved, population-level outcomes remain unsatisfactory.2 Smoking is the strongest risk factor, yet a substantial proportion of cases occur in never-smokers and cannot be explained by established environmental or occupational exposures.3 Therefore, even with today’s treatments, we still lack a clean explanation for who develops LC and why—especially outside the classic exposure story.
Observational studies and meta-analyses demonstrate that chronic obstructive pulmonary disease (COPD) is consistently associated with higher LC risk.4–6 The burden of respiratory comorbidities extends beyond cancer outcomes. For instance, there is a close association among tuberculosis (TB), COPD, and COVID-19, with SARS-CoV-2 infection potentially increasing the morbidity and mortality of both respiratory conditions, and TB infection further contributing to the elevated risk of death in COPD patients following COVID-19 infection.7,8 Notwithstanding these interconnections, the independent contribution of COPD to LC risk remains uncertain, as both conditions share common risk factors, most importantly smoking intensity, making it difficult to isolate the independent effect of COPD. Reverse causation is also plausible: early LC can change symptoms and lung function, which may shift when COPD is detected or recorded.6 That leaves a simple but important question: if we reduce these biases as much as we can, does COPD still point toward higher LC risk?
Mechanistically, COPD is driven by chronic airway inflammation and imperfect repair, and inflammation is closely linked to cancer development.9,10 At the same time, blood inflammatory markers are an imperfect proxy for what happens in the lung microenvironment. In contrast, metabolism provides a additional, often more stable biological readout. Notably, cancer is characterized by metabolic reprogramming,11 with lipid/phospholipid metabolic pathways playing a particularly pivotal role. These pathways link substrate pools to inflammatory lipid mediators and tumor-microenvironment interactions.12–15 From a practical angle, metabolites and circulating inflammatory factors are measurable “middle layers.” Therefore, when attempting to decipher the complete pathological pathway from COPD to LC, these markers provide a reasonable starting point.
Although substantial evidence indicates a strong association between COPD and LC, along with multiple shared biological alterations, LC prevention strategies specifically targeting COPD patients remain limited. Conventional approaches have not adequately isolated the independent effect of COPD on LC incidence, particularly in populations with substantial background exposure to other risk factors. Mendelian randomization (MR) can strengthen causal inference by using genetic instruments to reduce confounding and reverse causation under standard assumptions.16 Two-sample MR enables large-scale analyses with genome-wide association study (GWAS) summary statistics and offers sensitivity checks to probe pleiotropy.17,18 On this basis, integrating mediation analysis into the MR framework can further quantify the mediating effects of specific blood metabolites and inflammatory factors in the COPD-LC pathway so as to distinguish direct and indirect effects and identify potential intervention targets. Adhering to STROBE-MR guidelines for methodological rigor,19 we utilized large-scale GWAS datasets to evaluate the causal link between COPD and LC, pinpoint blood metabolites and inflammatory factors causally linked to LC, and examine their mediating effects in the COPD-LC pathway.18
Material and Methods
Study Design
Figure 1 shows the particular study design and methodology. First, the causal link between LC and COPD was assessed with a two-sample MR analysis. Three fundamental presumptions underpin the MR approach in order to guarantee objective causal inference: (1) instrumental variables (IVs) must have a significant correlation with the exposure; (2) IVs only influence the outcome through the exposure; and (3) IVs are unaffected by confounding variables. Subsequently, mediation analysis was conducted to explore whether inflammatory factors or blood metabolites serve as intermediaries in the COPD-LC association. Additionally, reverse MR analysis was carried out to assess whether LC might exert causal influences on COPD, inflammatory markers, and blood metabolites. This study strictly adhered to the STROBE-MR guidelines, and a completed STROBE-MR checklist was provided in Supplementary Table 1.20
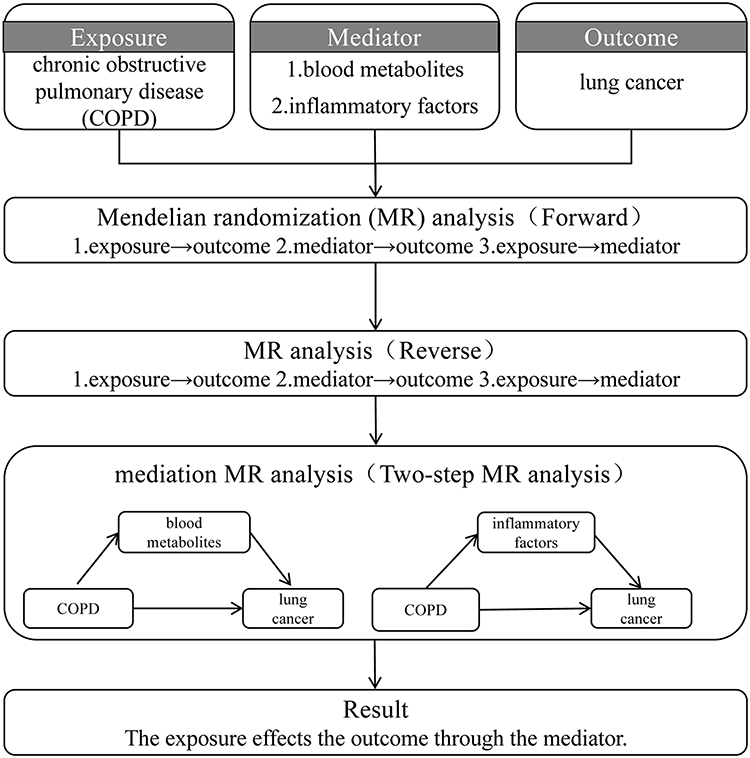 |
Figure 1 The flowchart of the study design. |
Data Sources
Publicly available GWAS summary results were utilized in this study. The data of COPD (finngen_R12_J10_COPD) comprises 433,208 samples (24,138 COPD and 409,070 control samples) and 20,462,603 single nucleotide polymorphisms (SNPs).21 The GWAS data of LC, ieu-a-987, contains 85,449 samples (29,863 LC and 55,586 control samples) and 10,439,018 SNPs. The data on blood metabolites was extracted from the Canadian Longitudinal Study on Aging (CLSA), including 1,400 blood metabolites from 8,299 randomly selected unrelated individuals.22 A total of 91 plasma inflammatory proteins were sourced from 14,824 individuals.23
Selection of IVs
Due to the limited number of genetic variants associated with COPD/LC/blood metabolites/inflammatory factors in GWAS, applying more stringent thresholds would result in an insufficient number of IVs for analysis. Therefore, we adopted the following criteria for IV selection. Firstly, the threshold of IV selection for COPD and LC was P < 5×10−6 and for inflammatory factors and blood metabolites was P < 1×10−5. The SNPs with LD were then eliminated by the ld_clump() function of the “ieugwasr” R package (r2 = 0.001, kb = 10,000). Thirdly, F-statistic values were applied to determine the strength of the IVs, and those with an F-value below 10 were eliminated. Fourth, the SNPs relevant to the outcome were removed with P < 1 ×.10–5 Fifth, to ensure unidirectionality of causation, IVs were screened by the Steiger method. Finally, we adjusted the strands of non-transcribed IVs and removed IVs containing transcribed sequences to ensure that the IVs corresponded to the same alleles in the same order in terms of exposure and outcome.
The Two-Sample MR Analysis
Five distinct methods were employed to evaluate causal associations: MR Egger, Inverse Variance Weighted (IVW), Simple Mode, Weighted Mode, and Weighted Median. IVW served as the primary method, while the others offered supplementary evidence. According to the results of the heterogeneity test, the IVW-fixed effects or IVW-random effects were chosen. When there is no heterogeneity (P > 0.05), the IVW-fixed effects is selected. When heterogeneity exists (P < 0.05), the IVW-random effects is utilized. Causal effects were expressed as odds ratios (OR) with 95% confidence intervals (CI).
Mediation Analysis Link “COPD-Blood Metabolites-LC” and “COPD-Inflammatory Factors-LC”
The two-step MR analysis was applied to examine the mediating influence of blood metabolites or inflammatory factors between COPD and LC. The causal influence of COPD on LC (c), the causal impact of blood metabolites or inflammatory factors on LC (b), and the causal connections of COPD with blood metabolites or inflammatory factors (a) were measured. The formula of mediated effect for blood metabolites or inflammatory factors was: mediated effect = a*b. The formula for the direct effect of COPD on LC was: direct effect = c - a*b. The formula for mediation proportion of blood metabolites or inflammatory factors was: mediation proportion = a*b/c. The delta method was used to estimate the standard errors of direct effects, mediated effects, and mediation proportions.
Sensitivity Analysis
Sensitivity analyses were performed to assess the reliability and stability of the Mendelian randomization (MR) findings. These included evaluations for heterogeneity, horizontal pleiotropy, and leave-one-out (LOO) analysis. Heterogeneity across instrumental variables was quantified using the `mr_heterogeneity` function. Horizontal pleiotropy was investigated via MR-Egger regression; a statistically significant deviation of the intercept from zero (P < 0.05) indicated the presence of directional pleiotropy. Additionally, the LOO approach was implemented to systematically evaluate the influence of each single-nucleotide polymorphism (SNP) on the overall causal estimate.
Reverse MR Analysis
To explore whether LC had a causal effect on COPD, blood metabolites, and inflammatory factors, the reverse MR analysis was carried out. The procedure for reverse MR analysis was the same as for forward MR analysis.
Statistical Analysis
The R package were applied to perform all statistical analyses. A P less than 0.05 was considered statistical significance.
Results
The Causal Association Between COPD and LC
A total of 87 SNPs were selected to estimate the causal association between COPD and LC. The F-statistics for all IVs exceeded 10, ranging from 22.572 to 45.531 (mean = 25.226), indicating no evidence of weak instrument bias. Since there was heterogeneity (P < 0.05), the IVW-RE was chosen. Results revealed a significant causal correlation between COPD and LC (P = 0.002), with COPD increasing the risk of LC (OR = 1.114 [1.042–1.191]) (Table 1). The scatter plot, forest plot, and funnel plot collectively confirmed the consistency and reliability of the results (Figure 2A–C). Moreover, the sensitivity analysis demonstrated the absence of horizontal pleiotropy, and the exclusion of the specific SNP did not impact the result (Table 2 and Figure 2D). However, reverse MR analysis indicated no causal association between LC and COPD (P = 0.923) (Supplementary Figure 1).
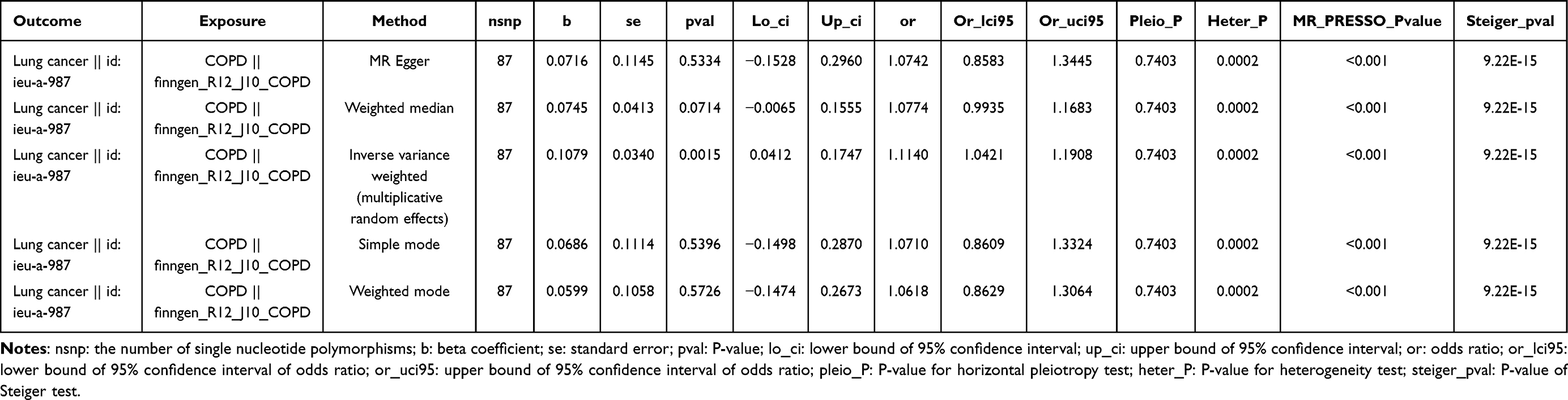 |
Table 1 The Causal Effect of Chronic Obstructive Pulmonary Disease (COPD) on Lung Cancer (LC) |
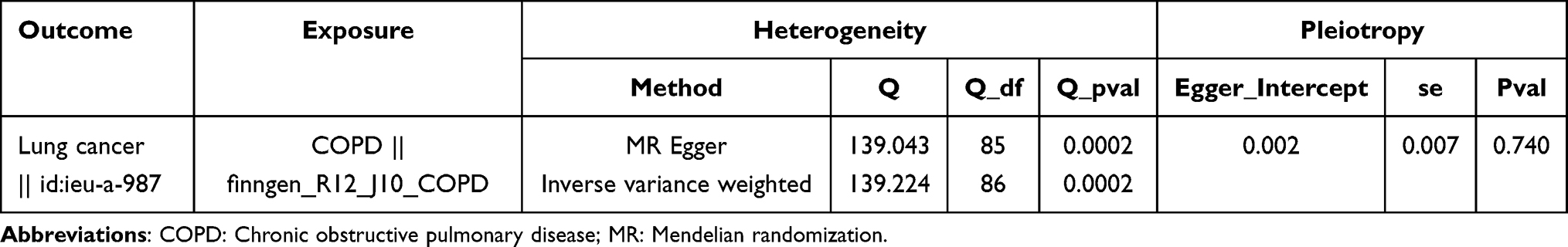 |
Table 2 Heterogeneity and Pleiotropy Analyses |
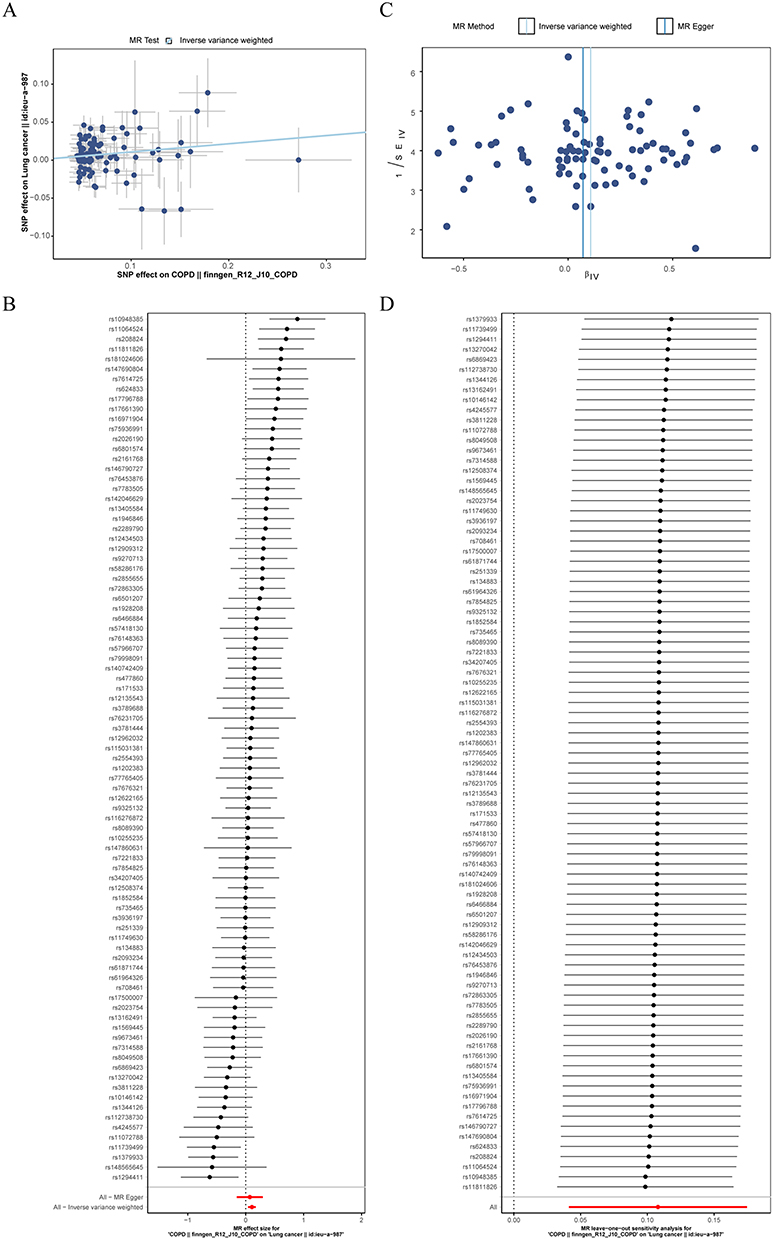 |
Figure 2 The causal effect of chronic obstructive pulmonary disease (COPD) on lung cancer (LC). (A) The scatter plot. (B) The forest plot. (C) The funnel plot. (D) The Leave-one-out (LOO) sensitivity analysis. |
The Causal Associations Between Blood Metabolites/Inflammatory Factors and LC
To screen the blood metabolites or inflammatory factors that were causally relevant to the occurrence of LC, two-sample MR analyses were conducted. All IVs had F-statistics above 10 (mean range: 21.583–170.295), indicating no evidence of weak instrument bias. Results illustrated that a total of 113 blood metabolites had nominally significant causal associations with LC, of which 56 suggested protective effects (P < 0.05, OR < 1) and 57 suggested risk effects (P < 0.05, OR > 1) (Figure 3A and Supplementary Table 2). Conversely, reverse MR analysis indicated that LC suggested a protective effect on 5−oxoproline to citrate ratio (P =0.021, OR = 0.938 [0.888–0.991]), and had risk effects on Glycine to serine ratio (P =0.003, OR = 1.088 [1.030–1.149]), Methionine sulfone levels (P =0.006, OR = 1.075 [1.021–1.133]), Methyl vanillate sulfate levels (P =0.035, OR = 1.087 [1.006–1.174]), Orotidine levels (P =0.015, OR = 1.067 [1.013–1.124]), and X−25420 levels (P =0.016, OR = 1.077 [1.014–1.145]) (Figure 3B and C). Furthermore, sensitivity analyses further validated the stability of the findings (Supplementary Tables 2 and 3).
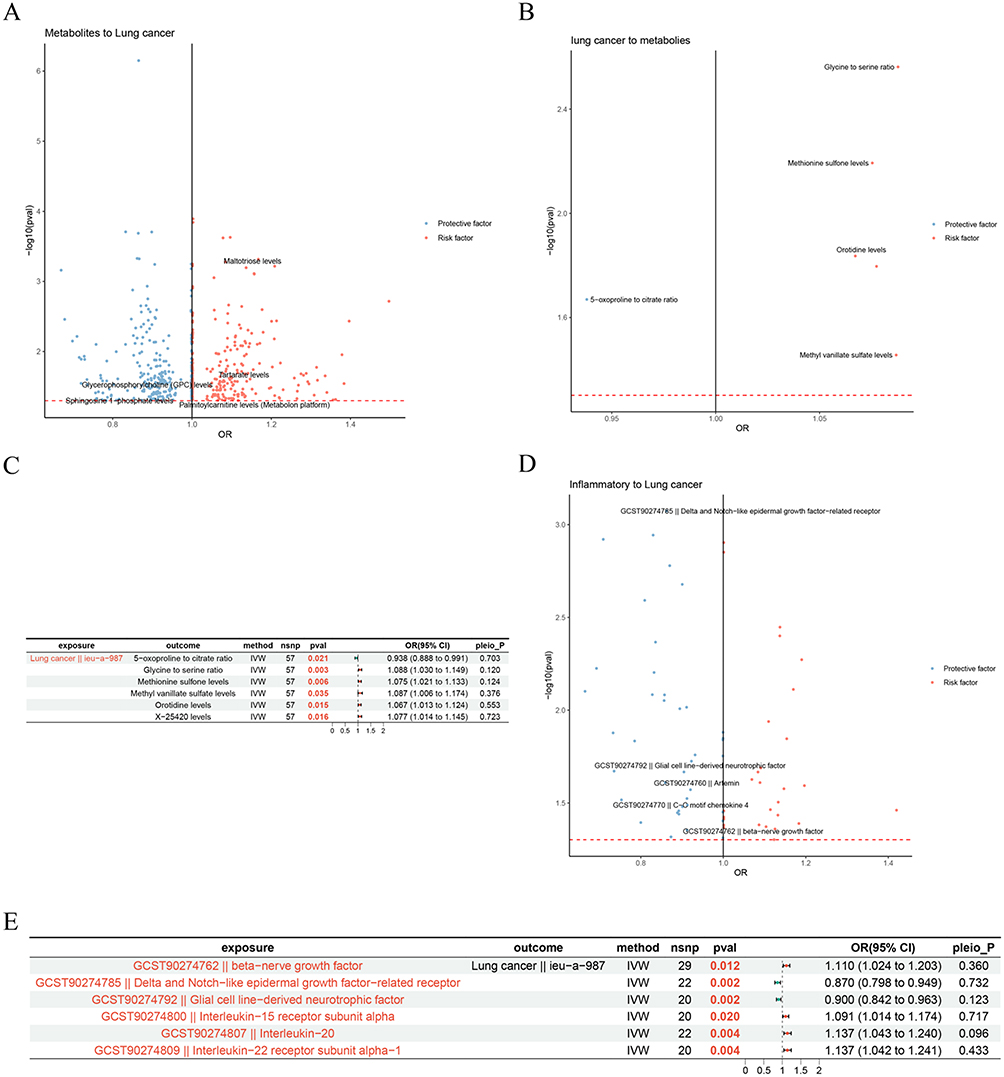 |
Figure 3 The causal effect of mediators on LC. (A) The causal effect of blood metabolites on LC. Each dot represents a blood metabolite. The x-axis showing the estimated causal effect (odds ratio, OR) of blood metabolite on LC risk, and the y-axis showing the significance level (-log10 P-value). Red points indicate blood metabolites with a significant positive association with LC risk, while blue points indicate metabolites with a significant negative association. (B and C) The causal effect of LC on blood metabolites. (B) The volcano diagram. Each dot represents a blood metabolite. The x-axis showing the estimated causal effect of LC on blood metabolite levels, expressed as OR, and the y-axis indicating the significance level of the effect (-log10 P-value). Red dots represent metabolites that are significantly increased by LC (positive association), while blue dots represent metabolites that are significantly decreased by LC (negative association). (C) The forest diagram. (D and E) The causal effect of inflammatory factors on LC. (D) The volcano diagram. Each dot represents a inflammatory factor. The x-axis showing the estimated causal effect (OR) of inflammatory factors on LC risk, and the y-axis showing the significance level (-log10 P-value). Red points indicate inflammatory factors with a significant positive association with LC risk, while blue points indicate inflammatory factors with a significant negative association. (E) The forest diagram. Abbreviations: nsnp: the number of single nucleotide polymorphisms; OR (95% CI): odds ratio (95% confidence interval); pleio_P: P-value for horizontal pleiotropy test. |
After that, the causal associations between inflammatory factors and LC were investigated. Similarly, all IVs for inflammatory factors had F-statistics above 10 (mean range: 21.004–99.921). Two inflammatory factors were nominally associated with lower LC risk: Delta and Notch−like epidermal growth factor−related receptor (P =0.002, OR = 0.870 [0.798–0.949]) and Glial cell line−derived neurotrophic factor (P =0.002, OR = 0.900 [0.842–0.963]) (Figure 3D and E). Four inflammatory factors were nominally associated with higher LC risk: beta−nerve growth factor (P =0.012, OR = 1.110 [1.024–1.203]), Interleukin−15 receptor subunit alpha (P =0.020, OR = 1.091 [1.014–1.174]), Interleukin−20 (P =0.004, OR = 1.137 [1.043–1.240]), and Interleukin−22 receptor subunit alpha−1 (P =0.004, OR = 1.137 [1.042–1.241]) (Figure 3D and E). Sensitivity analysis illustrated that the results were dependable and robust (Supplementary Table 4).
Mediation Analysis of Potential Blood Metabolites
Before screening potential blood metabolites as mediators between COPD and LC, the causal associations between COPD and 113 blood metabolites causally associated with LC were investigated. All instrumental variables had F-statistics above 10 (mean range: 26.385–26.536), indicating no weak instrument bias. Results indicated that COPD showed suggestive protective associations with nine blood metabolites (P < 0.05, OR < 1) and suggestive risk associations with 13 blood metabolites (P < 0.05, OR > 1) (Figure 4A and B) (Supplementary Table 5). Conversely, 57 blood metabolites showed suggestive protective associations with COPD risk (P < 0.05, OR < 1), while 70 blood metabolites showed suggestive risk associations with COPD risk (P < 0.05, OR > 1) (Figure 4C and Supplementary Table 6). These results were both robust and reliable (Supplementary Tables 5 and 6).
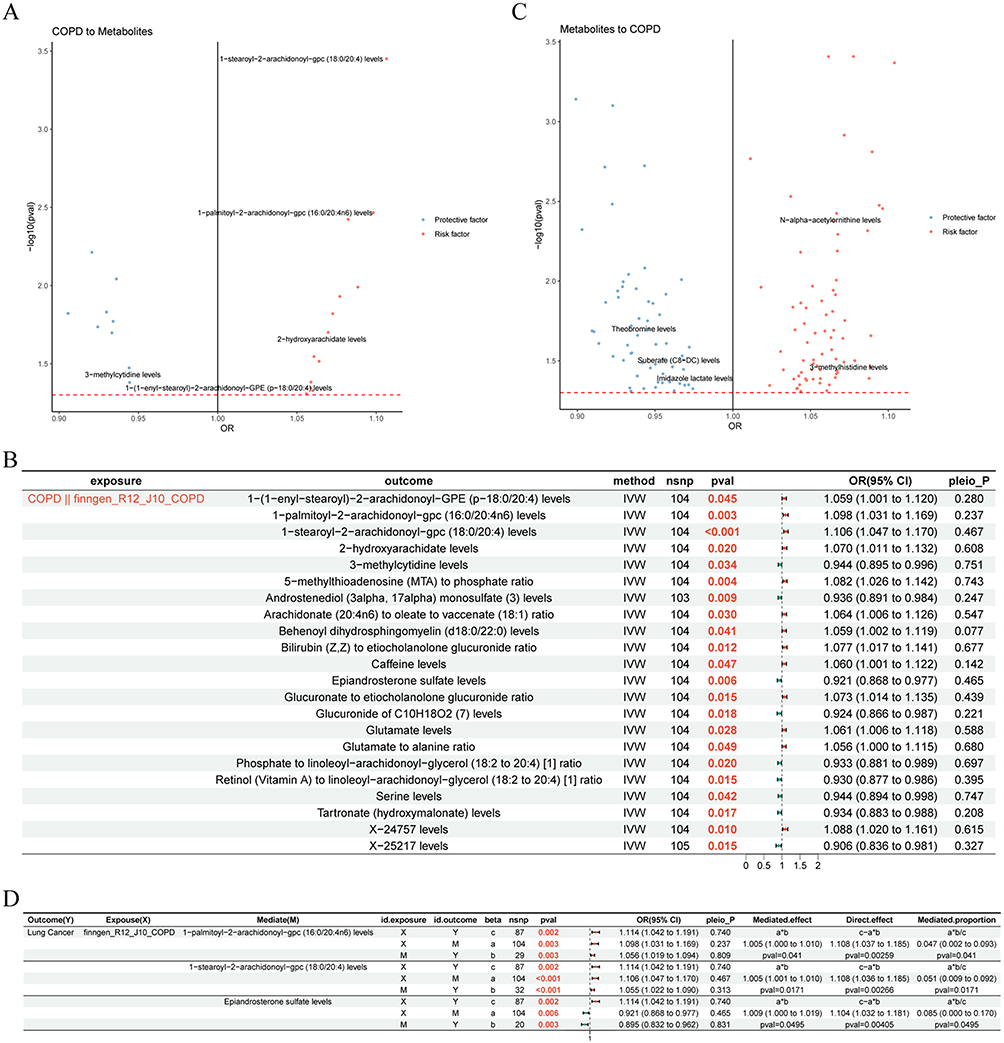 |
Figure 4 Causal effect of COPD on LC via blood metabolites. (A and B) The causal effect of COPD on blood metabolites. (A) The volcano diagram. Each dot represents a blood metabolite. The x-axis showing the estimated causal effect of COPD on blood metabolite levels, expressed as OR, and the y-axis indicating the significance level of the effect (-log10 P-value). Red dots represent metabolites that are significantly increased by COPD (positive association), while blue dots represent metabolites that are significantly decreased by COPD (negative association). (B) The forest diagram. (C) The causal effect of blood metabolites on COPD. Each dot represents a blood metabolite. The x-axis showing the estimated causal effect (OR) of blood metabolite on COPD risk, and the y-axis showing the significance level (-log10 P-value). Red points indicate blood metabolites with a significant positive association with COPD risk, while blue points indicate metabolites with a significant negative association. (D) The forest diagram illustrating effect of COPD on LC via blood metabolites. “*” denotes multiplication. Abbreviations: nsnp: the number of single nucleotide polymorphisms; OR (95% CI): odds ratio (95% confidence interval); pleio_P: P-value for horizontal pleiotropy test. |
Subsequently, mediation analysis demonstrated that three blood metabolites were potential mediators that impacted the causal effect of COPD on LC. Specifically, 1−palmitoyl−2−arachidonoyl−gpc (16:0/20:4n6) levels, Epiandrosterone sulfate levels, and 1−stearoyl−2−arachidonoyl−gpc (18:0/20:4) levels mediated a small but statistically significant proportion of the total COPD effect on LC risk, and the mediation proportion was 4.7%, 5.3%, and 9.3%, respectively (Figure 4D). Additionally, since there was no significant causal connection between LC (exposure) and COPD (outcome), the mediation analysis was not conducted with the blood metabolites as mediators.
Mediation Analysis of Potential Inflammatory Factors
To reveal the mediated role of inflammatory factors, we initially analyzed the causal effect of COPD on six inflammatory factors causally relevant to LC. Results illustrated that COPD had no significant casual correlation with these six inflammatory factors (P > 0.05) (Supplementary Figure 2). Therefore, the mediation analysis could not be conducted with the inflammatory factors as mediators.
Discussion
Our two-sample MR analyses provided evidence supporting a causal effect of genetically proxied COPD liability on LC risk. Mediation analysis then highlighted three blood metabolites—1-palmitoyl-2-arachidonoyl-gpc (16:0/20:4n6), epiandrosterone sulfate, and 1-stearoyl-2-arachidonoyl-gpc (18:0/20:4)—which showed measurable indirect effects. In contrast, mediation through circulating inflammatory factors was not supported because COPD showed no significant causal association with the six inflammatory factors that were causally linked to LC.
Epidemiologic studies have long reported higher LC risk in COPD, but concerns about residual confounding and reverse causality persist.4–6 MR cannot remove every limitation, but it offers a more robust route to causal inference than conventional observational approaches.16 Under this framework, our results suggest that genetic liability to COPD may contribute to LC independently of shared environmental exposures. Importantly, this does not imply that COPD directly “causes” LC in a single step; rather, the genetic evidence positions COPD as a contributing factor within a broader risk pathway. This direction also fits what we know biologically: COPD involves chronic inflammation, oxidative stress, and altered repair, which may create a more permissive setting for malignant change.4 Furthermore, our results were robust in sensitivity analyses: we detected no significant horizontal pleiotropy, LOO analyses confirmed that no single SNP drove the association, and reverse MR found no evidence for a causal effect of LC liability on COPD (P = 0.923), supporting the proposed direction of causality.
Three blood metabolisms, 1-palmitoyl-2-arachidonoyl-gpc (16:0/20:4n6), epiandrosterone sulfate, and 1-stearoyl-2-arachidonoyl-gpc (18:0/20:4), significantly mediated the association between COPD and LC. Although the mediation proportions were modest (each <10%), this is consistent with the multifactorial nature of the COPD–LC pathway, where individual mediators often contribute small yet biologically meaningful proportions. The 1-palmitoyl-2-arachidonoyl-gpc (16:0/20:4n6) and 1-stearoyl-2-arachidonoyl-gpc (18:0/20:4) are arachidonic-acid–containing glycerophosphocholines, which points to membrane lipid remodeling and arachidonic acid (AA) metabolism. AA-derived eicosanoids influence inflammation and immune signaling and can shape angiogenesis and tumor microenvironments.12–15 The cPLA2–AA–COX-2 axis has been discussed in LC contexts, including potential links to prognosis and detection.24 Thus, these findings are biologically tractable, linking COPD to a known lipid mediator axis rather than to an isolated metabolite. A plausible working model is that COPD-related alterations in lipid substrates or membrane composition may, over time, bias these downstream mediator networks, thereby nudging the lung microenvironment toward carcinogenic susceptibility. Additionally, the epiandrosterone sulfate points to the steroid-related metabolism. Androgen receptor signaling has been discussed across multiple cancers and may be relevant in LC biology as well.25 Importantly, we do not claim that these pathways fully explain the COPD–LC association; rather, we treat them as testable leads and hypothesis-generating findings that warrant further experimental validation. If we had to pick a next step, it would be to check whether these metabolite patterns replicate and whether they align with lung- or airway-level lipid/steroid pathways in independent datasets. In summary, from a genetic causal inference perspective, this study reveals that lipid remodeling and steroid metabolism may represent potential contributory pathways linking COPD to LC risk, providing novel and testable hypotheses for understanding the mechanisms underlying this comorbid relationship.
However, not seeing stable mediation by circulating inflammatory factors does not mean inflammation is irrelevant. Instead, this likely reflects a measurement discordance: systemic blood markers may not adequately capture the lung-specific inflammatory processes that are most directly involved in malignant transformation.9,10 In addition, inflammatory traits are complexly intertwined, which can make “one clean mediator” hard to identify. In this context, the negative mediation result is still informative: it tells us where blood-based signals are (and are not) showing up in the causal chain. In contrast, metabolites may provide a more stable and integrated readout of long-term physiological states.
A key strength is that we combined two-sample MR with mediation analysis, moving from causal evidence to specific pathway candidates, in line with established guidance.16–19 Several limitations merit consideration. First, our findings are primarily based on GWAS data from European ancestry populations, and their generalizability to other ancestries requires further testing. Second, while we applied rigorous sensitivity analyses, MR mediation relies on key assumptions, and residual pleiotropy cannot be entirely ruled out.16–19 Third, our analysis was confined to circulating biomarkers, which may not fully reflect pathological changes within the lung tissue. Moreover, we cannot exclude the possibility that the null mediation result for inflammatory factors is due to insufficient statistical power or the limitations of using blood-based rather than lung-specific inflammatory markers. Fourth, the GWAS summary statistics used in this study lack individual-level clinical information, such as COPD severity (GOLD stage), acute exacerbation status, or medication use. Consequently, we were unable to perform stratified analyses to assess potential effect heterogeneity across different disease states. The pooling of samples with varying severity or disease status may have introduced dilution bias towards the null, potentially reducing statistical power to detect causal effects. Finally, the identified blood metabolites only partially mediated the effect of COPD on lung cancer, leaving a substantial proportion of the total effect unexplained. This reflects the multifactorial nature of the COPD–lung cancer pathway, and other unmeasured mediators—such as additional metabolites, proteomic signatures, or epigenetic modifications—may also play important roles. Therefore, while our findings are hypothesis-generating, experimental studies—such as in vitro models or targeted interventions in animal models—are warranted to validate the biological mechanisms underlying these mediation effects.
Conclusion
In conclusion, this study provides genetic evidence supporting a causal role of COPD in lung cancer risk, with lipid remodeling and steroid metabolism identified as potential mediating pathways. While the mediation effects were modest, these findings shift the focus from epidemiological association toward testable metabolic mechanisms. Further experimental studies are warranted to validate whether modulating these pathways could mitigate lung cancer risk in patients with COPD.
Data Sharing Statement
The datasets used during our study are available in the IEU OpenGWAS project (https://gwas.mrcieu.ac.uk/) and the Canadian Longitudinal Study on Aging (CLSA) (https://www.clsa-elcv.ca/).
Ethics Approval
The data involved all originate from publicly published GWAS summary databases, which complies with the conditions for exemption from review as stated in the “Ethical Review Measures for Life Sciences and Medical Research Involving Humans”.
Acknowledgments
We would like to express our gratitude to all investigators and consortia for their generous contribution of valuable summary data.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received no specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Laversanne M, Sung H. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca a Cancer J Clin. 2024;74(3):229–12. doi:10.3322/caac.21834
2. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. Ca a Cancer J Clin. 2024;74(1):12–49. doi:10.3322/caac.21820
3. Thun MJ, Hannan LM, Adams-Campbell LL, et al. Lung cancer occurrence in never-smokers: an analysis of 13 cohorts and 22 cancer registry studies. PLoS Med. 2008;5(9):e185. doi:10.1371/journal.pmed.0050185
4. Young RP, Hopkins RJ, Christmas T, Black PN, Metcalf P, Gamble GD. COPD prevalence is increased in lung cancer, independent of age, sex and smoking history. Eur Resp J. 2009;34(2):380–386. doi:10.1183/09031936.00144208
5. Zhang X, Jiang N, Wang L, Liu H, He R. Chronic obstructive pulmonary disease and risk of lung cancer: a meta-analysis of prospective cohort studies. Oncotarget. 2017;8(44):78044–78056. doi:10.18632/oncotarget.20351
6. Powell HA, Iyen-Omofoman B, Baldwin DR, Hubbard RB, Tata LJ. Chronic obstructive pulmonary disease and risk of lung cancer: the importance of smoking and timing of diagnosis. J Thorac Oncol. 2013;8(1):6–11. doi:10.1097/JTO.0b013e318274a7dc
7. Muneeb Hassan M, Ameeq M, Jamal F, Tahir MH, Mendy JT. Prevalence of covid-19 among patients with chronic obstructive pulmonary disease and tuberculosis. Ann Med. 2023;55(1):285–291. doi:10.1080/07853890.2022.2160491
8. Hassan MM, Tahir MH, Ameeq M, Jamal F, Mendy JT, Chesneau C. Risk factors identification of COVID-19 patients with chronic obstructive pulmonary disease: a retrospective study in Punjab-Pakistan. Immun Inflamm Dis. 2023;11(8):e981. doi:10.1002/iid3.981
9. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi:10.1016/j.cell.2010.01.025
10. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi:10.1038/nature07205
11. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
12. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10(3):181–193. doi:10.1038/nrc2809
13. Horn L, Backlund M, Johnson DH. Targeting the eicosanoid pathway in non-small-cell lung cancer. Expert Opinion on Therapeutic Targets. 2009;13(6):675–688. doi:10.1517/14728220902915567
14. Johnson AM, Kleczko EK, Nemenoff RA. Eicosanoids in Cancer: new Roles in Immunoregulation. Front Pharmacol. 2020;11:595498. doi:10.3389/fphar.2020.595498
15. Sonkar K, Ayyappan V, Tressler CM, et al. Focus on the glycerophosphocholine pathway in choline phospholipid metabolism of cancer. NMR Biomed. 2019;32(10):e4112. doi:10.1002/nbm.4112
16. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Human Molecular Genetics. 2014;23(R1):R89–R98. doi:10.1093/hmg/ddu328
17. Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181(4):251–260. doi:10.1093/aje/kwu283
18. Carter AR, Sanderson E, Hammerton G, et al. Mendelian randomisation for mediation analysis: current methods and challenges for implementation. Eur j epidemiol. 2021;36(5):465–478. doi:10.1007/s10654-021-00757-1
19. Skrivankova VW, Richmond RC, Woolf BAR, et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization (STROBE-MR): a Korean translation of explanation and elaboration. Ewha Med J. 2025;48(4):e68. doi:10.12771/emj.2025.00934
20. Skrivankova VW, Richmond RC, Woolf BAR, et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: the STROBE-MR Statement. JAMA. 2021;326(16):1614–1621. doi:10.1001/jama.2021.18236
21. Kurki MI, Karjalainen J, Palta P, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613(7944):508–518. doi:10.1038/s41586-022-05473-8
22. Zhao G, Cai Y, Wang Y, Fang Y, Wang S, Li N. Genetically predicted blood metabolites mediate the association between circulating immune cells and pancreatic cancer: a Mendelian randomization study. J Gene Med. 2024;26(5):e3691. doi:10.1002/jgm.3691
23. Baranova A, Luo J, Fu L, Yao G, Zhang F. Evaluating the effects of circulating inflammatory proteins as drivers and therapeutic targets for severe COVID-19. Front Immunol. 2024;15:1352583. doi:10.3389/fimmu.2024.1352583
24. Xin C, Chu L, Zhang L, et al. Expression of Cytosolic Phospholipase A2 (cPLA2)-Arachidonic Acid (AA)-Cyclooxygenase-2 (COX-2) Pathway Factors in Lung Cancer Patients and Its Implication in Lung Cancer Early Detection and Prognosis. Med Sci Monitor. 2019;25:5543–5551. doi:10.12659/msm.915314
25. Chang C, Lee SO, Yeh S, Chang TM. Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene. 2014;33(25):3225–3234. doi:10.1038/onc.2013.274
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Exercise Rehabilitation and Chronic Respiratory Diseases: Effects, Mechanisms, and Therapeutic Benefits
Xiong T, Bai X, Wei X, Wang L, Li F, Shi H, Shi Y
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:1251-1266
Published Date: 19 June 2023
Physicians’ Knowledge of Pulmonary Rehabilitation in China: A Cross-Sectional Study
Pan F, Lu AT, Mao X, Hu F, Zhang H, Han B
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:121-131
Published Date: 16 January 2024
COPD and Immune Checkpoint Inhibitors for Cancer: A Literature Review
Lycan Jr TW, Norton DL, Ohar JA
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:2689-2703
Published Date: 9 December 2024
Incidence of Lung Cancer in COPD Patients in Western Yokohama Managed by Primary Care Physicians with Hospital Collaboration
Nishi Y, Tsuburai T, Komase Y, Yagihashi K, Nishida M, Kaneko S, Tanaka S, Muraoka H, Ueno J, Matsushima A, Shinozaki Y, Nishiyama K, Numata Y, Tsuruoka H, Oyama B, Hida N, Mineshita M, Inoue T
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:3461-3471
Published Date: 24 October 2025