Back to Journals » Breast Cancer: Targets and Therapy » Volume 9
Cholesterol biosynthesis inhibitor RO 48-8071 reduces progesterone receptor expression and inhibits progestin-dependent stem cell-like cell growth in hormone-dependent human breast cancer cells
Authors Liang Y, Goyette S, Hyder SM
Received 23 April 2017
Accepted for publication 1 June 2017
Published 7 July 2017 Volume 2017:9 Pages 487—494
DOI https://doi.org/10.2147/BCTT.S140265
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Yayun Liang,1,2,* Sandy Goyette,1,2,* Salman M Hyder1,2
1Department of Biomedical Sciences, 2Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO, USA
*These authors contributed equally to this work
Abstract: Clinical trials and studies have shown that postmenopausal women undergoing combination hormone replacement therapy containing estrogen and progestin have an increased risk of breast cancer compared with women taking estrogen or placebo alone. Using animal models, we have previously shown that synthetic progestins, including medroxyprogesterone acetate (MPA), which is widely used clinically, accelerate breast cancer tumor growth and promote metastasis. Furthermore, we have found that MPA elevates CD44 protein expression and aldehyde dehydrogenase (ALDH) activity, two markers of cancer stem cells (CSCs), and increases mammosphere formation, another hallmark of stem cells, in hormone-dependent T47-D human breast cancer cells. Herein, we show that RO 48-8071 (RO), an inhibitor of cholesterol synthesis, reduced MPA-induced CD44 protein expression in two hormone-dependent human breast cancer cell lines, T47-D and BT-474. Because we have previously shown that MPA induction of CD44 is progesterone receptor (PR) dependent, we examined RO’s effects on PR protein and mRNA expressions in T47-D cells. PR mRNA levels remained unchanged after RO treatment; however, RO significantly reduced the protein expression of both PR receptor isoforms, PR-A and PR-B. Using the proteasome inhibitor MG-132, we demonstrated that RO decreases PR protein expression in T47-D cells via the proteasomal degradation pathway. Importantly, treatment of T47-D cells with RO abolished MPA-induced mammosphere formation. Based on our observations, we contend that RO may represent a novel means of preventing MPA-induced CSC expansion. RO could be used clinically to both treat and prevent hormone-dependent breast cancers, which represent the majority of human breast cancers. RO may also have clinical utility in reducing resistance to antihormone therapy.
Keywords: breast cancer, cholesterol biosynthesis inhibitor, RO 48-8071, medroxyprogesterone acetate, cancer stem cells, CD44
Introduction
Millions of women undergo hormone replacement therapy (HRT) each year to alleviate the effects of menopause. HRT consists of either estrogen alone or a combination of estrogen and progestin, and increasing evidence suggests that women taking combination HRT have an increased risk of breast cancer compared with those taking either estrogen alone or a placebo.1–4 Combination HRT has also been linked to an increased risk of breast cancer recurrence, as well as metastasis.5 We have previously shown that synthetic progestins promote the growth and metastasis of human breast cancer tumors in vivo by increasing angiogenesis.6 Furthermore, we have reported that treatment of hormone-dependent breast cancer cells with synthetic progestins, such as medroxyprogesterone acetate (MPA), leads to an enrichment of the cancer stem cell (CSC)-like pool.7
CSCs have previously been linked to aggressive tumor growth, metastasis, and cancer recurrence.8,9 In cancers of the breast, the CSC subpopulation carries the phenotypic signature CD24low/-, CD44high, and aldehyde dehydrogenase (ALDH1+).10 CSCs have the ability to self-renew, initiate tumors in vivo, and generate heterogeneous and differentiated progeny.10 Enrichment of the CSC-like pool makes a cancer difficult to treat, as CSCs are resistant to chemotherapy and have been linked to the initiation of metastasis.9
In previous studies, we have shown that CD44 protein expression is elevated in two hormone-dependent human breast cancer cell lines, T47-D and BT-474, in response to both natural and synthetic progestins. We further showed that increases in the expression of CSC markers in T47-D cells may be related to the enrichment of CSCs.7 We also found that induction of CD44 protein expression in T47-D cells by both the naturally occurring hormone progesterone and the synthetic progestins, including MPA, the latter being widely used clinically, was fully attenuated by the antiprogestin RU-486. Furthermore, MPA did not induce CD44 protein expression in T47-Dco-Y cells, a progesterone receptor (PR)-negative T47-D subline, confirming that progestin-mediated CD44 induction is PR dependent.7
Statins are a class of drugs given widely to lower cholesterol. They generally reduce cholesterol production by inhibiting the enzyme HMG-CoA reductase, which catalyzes a key step in cholesterol biosynthesis.11 However, HMG-CoA reductase inhibition is often accompanied by adverse side effects that arise due to reduced levels of isoprenoids, resulting in defective post-translational modification of membrane proteins and impaired membrane structure and function.12 Unlike statins, RO 48-8071 (RO) inhibits cholesterol biosynthesis by inhibiting 2,3-oxidosqualene cyclase (OSC), an enzyme that acts downstream of HMG-CoA reductase.13 This specific targeting helps eliminate many of the negative side effects associated with statin use. Previous in vitro and in vivo data have revealed that RO possesses potent anticancer effects.14–16 RO reduces the levels of estrogen receptor (ER)α (which exerts a proliferative effect on hormone-dependent breast cancer cell lines), thereby reducing tumor cell proliferation.16 We hypothesized that RO treatment may similarly reduce levels of PR, therefore inhibiting progestin-induced CSC enrichment in breast cancer cell populations. Herein, we show that RO reduced MPA-induced CD44 protein expression by decreasing PR protein expression but not PR mRNA expression, via a ubiquitin-dependent degradation pathway. We further show that RO inhibited MPA-induced mammosphere formation, indicating that RO attenuates MPA’s ability to enrich CSCs or progenitor cells in hormone-dependent breast cancer cell populations.
Materials and methods
Cell lines and culture
Hormone-dependent T47-D and BT-474 human breast cancer cells were obtained from American Type Culture Collection (Manassas, VA, USA). Both cell lines are of the luminal subtype and express ER and PR, though they differ in their Her2/neu status. BT-474 cells overexpress Her2/neu. Cells were maintained and grown in a humidified atmosphere of 5% CO2 at 37°C in phenol red-free DMEM/F12 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St Louis, MO, USA). Cells were harvested using 0.05% trypsin-EDTA (Thermo Fisher Scientific). For all experiments involving MPA, cells were initially treated for 24 hours with DMEM/F12 supplemented with 5% dextran-coated charcoal (DCC; Sigma-Aldrich)-treated FBS. Subsequently, cells were washed with phosphate-buffered saline (PBS) and further incubated in fresh 5% DCC-treated FBS–DMEM/F12 for the different experimental treatments. MPA (Sigma-Aldrich), RO (Tocris, Bristol, UK), and MG-132 (EMD Millipore, Billerica, MA, USA) were diluted in ethanol, PBS, and dimethyl sulfoxide, respectively. Control cells were treated with an equal volume of vehicle.
Flow cytometry
Cells were cultured in 5% DCC-treated FBS–DMEM/F12 for 24 hours prior to cell treatment. Cells were subsequently treated with 10 nM MPA ±10 µM RO or 10 µM RO alone. After 24 hours, cells were washed once and harvested using Accutase (BD Biosciences, Franklin Lakes, NJ, USA). All subsequent reagents for flow cytometry were obtained from BD Biosciences. Cells were stained for 45 minutes on ice in 100 µL staining buffer containing phycoerythrin (PE)-mouse antihuman CD24 and allophycocyanin (APC)-mouse antihuman CD44 antibodies (BD Biosciences). Samples were washed twice and resuspended in 1 mL of staining buffer. Samples were analyzed by a Beckman Coulter CyAn ADP flow cytometer using Summit 5.2 software. Unstained and single-staining controls were used to define gates, and an equal number of cells were evaluated for each sample.
Western blotting
Whole-cell extracts were prepared with a nuclear extraction TransAM kit (Active Motif, Carlsbad, CA, USA). Nuclear extracts (25–40 μg per lane) were separated on a NuPAGE 10% Bis-Tris Gel (Thermo Fisher Scientific). Electrophoresis was performed at 100 V for 2 hours using NuPAGE® MOPS SDS Running Buffer (Thermo Fisher Scientific). Separated proteins were electrotransferred at 25 V for 25 minutes to a polyvinylidene difluoride membrane using the Trans-Blot semi-dry system (Bio-Rad, Hercules, CA, USA) in Tris/glycine buffer (Bio-Rad). Blots were blocked overnight at 4°C in 5% nonfat dry milk in Tris-buffered saline (TBS) containing 0.075% Tween-20 (TBS-T), after which they were incubated with mouse anti-PR (AB-52) (1:200 dilution; Santa Cruz Biotechnology Inc., Dallas, TX, USA) or mouse anti-β-actin (1:2000; EMD Millipore) for 2 hours at room temperature. Blots were then washed three times with TBS-T for 10 minutes each, incubated with antimouse IgG HRP-linked antibody (Cell Signaling Technology, Danvers, MA, USA) for 1 hour at room temperature, and washed seven more times with TBS-T for 10 minutes each. Immunoreactive bands were visualized using the ECL plus detection kit (Amersham Pharmacia Biotech, Arlington Heights, IL, USA) using the Bio-Rad ChemiDoc XRS system.
RNA preparation and reverse transcription-polymerase chain reaction (RT-PCR)
Total cellular RNA was isolated using the RNAzol reagent protocol (Molecular Research Center, Cincinnati, OH, USA). Briefly, samples were homogenized in RNAzol, RNAse-free water added to sediment DNA and proteins, and RNA precipitated in isopropanol. The pelleted RNA was washed with 75% ethanol, and RNA integrity was determined by 260/280 and 260/230 ratio using the NanoDrop 2000 system (Thermo Fisher Scientific). RT-PCR was performed on 1 µg of RNA, using the Thermo Fisher Scientific SuperScript III One-Step RT-PCR amplification kit. The following primers were used: PR-F: 5′-AGC CCT AAG CCA GAG ATT-3′ and PR-R: 5′-TAG GAT CTC CAT CCT AGA CC-3′ and GAPDH-F: 5′-ATG AGA AGT ATG ACA ACA GCC-3′ and GAPDH-R: 5′-TGA GTC CTT CCA CGA TAC C-3′. RT-PCR conditions were as follows: 60°C for 30 minutes; 94°C for 2 minutes; followed by 35 cycles of 94°C for 15 seconds, 55°C for 30 seconds, 68°C for 60 seconds, and a final elongation step at 68°C for 5 minutes. RT-PCR products were electrophoresed on 1.5% agarose gels containing ethidium bromide in 0.5 × TBE, pH 8.0, at 100 V, and then analyzed using the Bio-Rad ChemiDoc XRS system.
Mammosphere formation assay
T47-D cells were grown in 100 mm dishes to 60% confluence. Cells were washed twice in PBS and then incubated for 24 hours with 5% DCC-treated FBS–DMEM/F12 medium (8 mL). Cells were subsequently treated for 48 hours with either 5 nM MPA in 5% DCC-treated FBS–DMEM/F12 medium or the same medium alone. Following treatment, cells from each group were harvested separately and counted. Cells (5×103) in 0.1 mL of complete MammoCult medium (STEMCELL Technologies, Vancouver, BC, Canada) were seeded into low-attachment six-well plates. Each well contained 5 nM MPA, 1 µM RO, 5 µM RO, 5 nM MPA +1 µM RO, or 5 nM MPA +5 µM RO in 1.9 mL of complete MammoCult medium. For controls, an equal volume of ethanol was added to the medium. Cells were retreated every 48 hours by adding an additional 1 mL of fresh drug solution to appropriate wells. The number of mammospheres (>100 µm in diameter) in each group was counted on day 5 by viewing 25–45 images per well, and the mean determined using data from two to three wells. Representative pictures were taken on day 5.
Statistical analysis
Data were reported as mean ± standard error of the mean (SEM). Statistical significance was tested using one-way analysis of variance (ANOVA) or the Student t-test as appropriate, using SigmaPlot software. Nonparametric measure based on ranks was used, as needed. When ANOVA indicated a significant effect (F-ratio, P<0.05), the Student–Newman–Keuls multirange test was employed to compare the means of individual groups. When normality failed, significance was determined by Kruskal–Wallis test (one-way ANOVA by ranks) followed by the Student–Newman–Keuls test. For all comparisons, P≤0.05 was regarded as statistically significant.
Results
RO decreases MPA-induced CD44 protein expression in hormone-dependent human breast cancer cells
Flow cytometry analysis of CD44 density demonstrated that treatment of T47-D cells with a physiologically relevant concentration of MPA (10 nM) increased CD44 protein expression ~20-fold (Figure 1A). When 10 µM RO was added, MPA-induced CD44 expression was reduced by ~50%. MPA treatment also produced a significant, but less pronounced, increase in CD44 expression in BT-474 cells (Figure 1B). RO reduced MPA-induced CD44 expression in BT-474 cells by ~40%. Treatment with RO alone had no significant effect on CD44 expression in either cell line. Differences in CD44 induction between the two cell lines may be due to inherently higher levels of PR expression in T47-D cells.
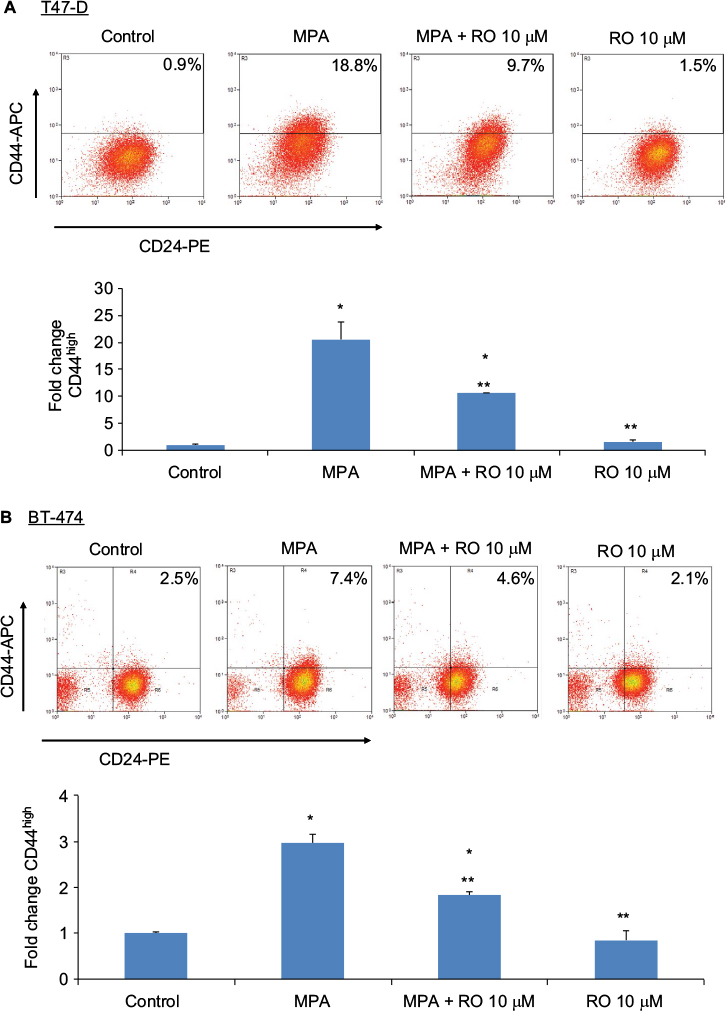 | Figure 1 RO decreases MPA-induced CD44 protein expression in hormone-dependent human breast cancer cells. Notes: T47-D (A) and BT-474 (B) cells were treated at 37°C for 24 hours with 10 nM MPA, 10 nM MPA +10 µM RO, or 10 µM RO alone in 5% DCC-treated FBS–DMEM/F12. Cells were labeled with CD44-APC and CD24-PE antibodies and analyzed using flow cytometry. Equal numbers of cells were analyzed in each treatment group. Bar graphs quantitate CD44 expression. Bars represent mean ± SEM (n=3); fold change is compared with control value (set at 1). *Significantly different compared with controls. **Significantly different compared with MPA-induced CD44 expression (ANOVA; P<0.05). Abbreviations: ANOVA, analysis of variance; APC, allophycocyanin; DCC, dextran-coated charcoal; FBS, fetal bovine serum; MPA, medroxyprogesterone acetate; PE, phycoerythrin; RO, RO 48-8071; SEM, standard error of the mean. |
RO decreases PR protein expression but not PR mRNA expression in T47-D cells
In a previous study, we established that MPA induction of CD44 protein expression in T47-D cells is PR dependent.7 Furthermore, we previously showed that RO degrades ERα, the primary receptor responsible for stimulating cell proliferation.16 Consequently, we hypothesized that RO-mediated decreases in MPA-induced CD44 expression (Figure 1) might arise due to PR degradation, which could occur in a manner similar to that previously reported for ERα. To examine if RO’s effects on CD44 expression that we observed in breast cancer cells were due to degradation of PR, we first investigated PR protein and mRNA expressions in RO-treated T47-D cells. Western blot analysis of T47-D cells demonstrated that protein expression of both human PR isoforms (transcribed from a single gene and differing in their N-terminal region; termed PR-A and PR-B17) was significantly reduced by RO treatment (Figure 2A and B). We next sought to determine whether regulation of PR by RO occurs at the transcriptional or post-transcriptional level. When we isolated RNA from RO-treated T47-D cells and then conducted RT-PCR analyses, we found that RO had no effect on PR mRNA expression, suggesting that RO effects on PR are exerted post-transcriptionally (Figure 2C and D).
 | Figure 2 RO decreases PR protein expression but not PR mRNA expression in T47-D cells. Notes: (A and B) T47-D cells were treated at 37°C for 24 hours with 10 µM RO in 5% DCC-treated FBS–DMEM/F12. (A) Whole-cell extracts were prepared and subjected to Western blotting. β-Actin was used as a loading control. A representative blot is shown. (B) Bar graphs quantitate Western blot data. Bars represent mean ± SEM (n=4); percent expression normalized with β-actin is compared with control value (set at 100%). *Significantly different compared with controls (Student t-test; P<0.05). (C and D) T47-D cells were treated at 37°C for 6 hours with 10 or 25 µM RO in 5% DCC-treated FBS–DMEM/F12. RNA was isolated from these cells and analyzed by RT-PCR for PR and GAPDH mRNA expressions. PCR products were then subjected to electrophoresis on ethidium bromide agarose gels. (D) Bar graphs quantitate PCR product data normalized with GAPDH expression. Bars represent mean ± SEM (n=7); fold change is compared with control value (set at 1). No significant difference was found between groups (ANOVA; P>0.05). Abbreviations: ANOVA, analysis of variance; DCC, dextran-coated charcoal; FBS, fetal bovine serum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PCR, polymerase chain reaction; PR, progesterone receptor; RT-PCR, reverse transcription-PCR; RO, RO 48-8071; SEM, standard error of the mean. |
RO decreases PR protein expression in T47-D cells via the proteasome degradation pathway
Ubiquitination is a process by which cells modify proteins for degradation via the proteasome complex.18 In order to explore the mechanisms by which RO may reduce PR protein expression in T47-D cells, we examined the effects of MG-132, a potent proteasome inhibitor, on PR expression. Western blot analysis of T47-D cells showed that reductions in PR protein expression observed in the presence of RO were “rescued” by MG-132, indicating that RO-mediated decreases in PR expression are indeed dependent on ubiquitination (Figure 3).
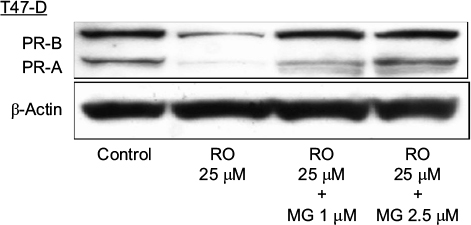 | Figure 3 RO reduces PR protein expression in T47-D cells in a ubiquitination-dependent manner. Notes: T47-D cells were treated at 37°C for 6 hours with 25 µM RO, 25 µM RO +1 µM MG-132, or 25 µM RO +2.5 µM MG-132 in 5% DCC-treated FBS–DMEM/F12. Whole-cell extracts were prepared and subjected to Western blotting. β-Actin was used as a loading control. A representative blot is shown, and the experiment was performed twice. Abbreviations: DCC, dextran-coated charcoal; FBS, fetal bovine serum; PR, progesterone receptor; RO, RO 48-8071. |
RO abolishes MPA-induced mammosphere formation in T47-D cells
We next conducted studies to functionally assess whether RO can reduce progestin-dependent CSC enrichment. To do so, we used mammosphere formation assays, which support anchorage-independent growth and are an excellent tool for examining CSCs and progenitor cells. Mammosphere formation was increased approximately threefold in MPA-treated T47-D cells compared with controls, whereas simultaneous treatment of T47-D cells with MPA and RO abolished the formation of mammospheres (Figure 4), suggesting that RO actively diminishes the CSC pool in T47-D cell populations.
 | Figure 4 RO abolishes MPA-induced mammosphere formation by T47-D cells. Notes: T47-D cells were pretreated at 37°C for 48 hours with 5 nM MPA. Cells were then subjected to the mammosphere assay with 5 nM MPA, 1 μM RO, 5 µM RO, 5 nM MPA +1 µM RO, or 5 nM MPA +5 µM RO in 5% DCC-treated FBS–DMEM/F12. Cells were retreated every 48 hours. Top panel, representative micrographs at day 5. Scale bar, 100 µm. Bottom panel, quantification of mammospheres formed. The number of mammospheres formed (100 mm) was counted after 5 days, and the number of mammospheres per 5000 cells was calculated. Bars represent mean ± SEM (n=3). *Significantly different compared with controls. **Significantly different compared with MPA and control groups (ANOVA, P<0.05). Abbreviations: ANOVA, analysis of variance; DCC, dextran-coated charcoal; FBS, fetal bovine serum; MPA, medroxyprogesterone acetate; RO, RO 48-8071; SEM, standard error of the mean. |
Discussion
Most breast cancers that are detected and treated clinically are of the hormone-dependent type.19,20 Multiple clinical trials have shown that undergoing combination HRT that includes a progestin component, such as MPA, elevates the risk of breast cancer.1–4 In previous studies using animal models, we showed that synthetic progestins accelerate breast cancer tumor growth and increase lymph-node metastasis.6 We also confirmed that progestin-dependent vascular endothelial growth factor (VEGF) production is a contributing factor to increased tumor formation in this model.21,22 Our most recent observations that progestin treatment can enrich the CSC-like pool in breast cancer cell populations may suggest another mechanism by which breast cancer tumor development is accelerated.7
In the present study, we showed that MPA significantly elevated the expression of CD44, an important CSC marker in breast cancer tumors, in T47-D cells, and that treatment with the cholesterol synthesis inhibitor RO reduced MPA-induced CD44 induction in these cells. CD44 is a cell-surface glycoprotein that is involved in a variety of important cellular functions, such as cell-to-cell communication, cell adhesion, and cell migration.23 It exists in many isoforms that arise from alternative splicing of the CD44 gene. Two splice variants, CD44v3 and CD44v6, play important roles in the activation of key CSC maintenance transcription factors and extracellular matrix degradation, respectively.24,25 We have previously established that RO suppresses breast and prostate cancer growth by downregulating ER and androgen receptor expressions, respectively.15,16 With this in mind, we sought to determine whether RO also downregulates PR and, if so, whether RO might then interfere with acquisition of stem cell-like properties by human breast cancer cells.
Cancers that resist treatment often display aberrant regulation of cholesterol homeostasis.26 Previous studies by Maione et al27 showed that targeting OSC activity with RO produced strongly antiangiogenic effects, resulting in reduced vascular density and increased pericyte coverage. In a previous study, we showed that hormone-dependent epithelial breast cancer cells respond to RO by decreasing their proliferative potential; that is, ERα was degraded, while the antiproliferative hormone receptor ERβ was upregulated.16 In this study, we demonstrated that exposure of hormone-dependent breast cancer cells to RO reduces PR protein expression.
Compared with ER, the potential of PR as a target through which to treat breast cancer has largely been ignored. However, as we now know, both naturally occurring progesterone and synthetic progestins promote the growth of breast tumors.6,21 Furthermore, PR has been shown to directly regulate VEGF, which is highly angiogenic and stimulates breast cancer cell proliferation and tumor growth.28 PR target genes, such as WNTs,29,30 play important roles in CSC maintenance. Blockage of the Wnt/β-catenin signaling pathway inhibits the CSC-like phenotype and suppresses breast cancer metastasis.31 In light of this, we propose that targeting PR-mediated effects such as upregulation of CD44 might be an effective way of preventing CSC enrichment in breast cancer tumors.
Our observation that RO inhibited MPA-dependent mammosphere formation in T47-D cells indicates that it interferes with progestin-dependent enrichment of CSCs and progenitor cells. We postulate that RO could therefore hamper progestin-dependent breast cancer tumor growth. Our findings are supported by those of Mejia-Pous et al,32 who report that OSC is not only required to maintain self-renewal in primary erythroid progenitors but is also highly upregulated in self-renewing cells. The results of our mammosphere formation assays may represent an additive effect of RO on OSC inhibition and PR degradation.
Based on our previous findings and the studies reported here, we contend that the OSC inhibitor RO may exert its potent anticancer effects in breast cancer in part through PR degradation, which, in turn, reduces progestin-induced expression of CD44, which is a PR-dependent process. RO’s ability to reduce both CD44 expression and the CSC pool in hormone-dependent breast cancer cell populations suggest that RO may provide an effective and novel means of preventing MPA-induced CSC expansion in hormone-dependent human breast cancer. Treatment with RO may also represent a novel strategy to reduce resistance to antihormone therapies, given that survival of stem cells following chemotherapy is the major cause of such treatment failures.33
Acknowledgments
The studies were supported by generous gifts from donors of Ellis Fischel Cancer Center, University of Missouri, Columbia, and by a faculty award from the College of Veterinary Medicine, University of Missouri, Columbia. SMH is the Zalk Missouri professor of tumor angiogenesis. We would also like to thank Dr Carolyn Henry for her invaluable support during the completion of this project.
Disclosure
The authors report no conflicts of interest in this work.
References
Rossouw JE, Anderson GL, Prentice RL, et al; Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321–333. | ||
Chlebowski RT, Hendrix SL, Langer RD, et al; WHI Investigators. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women’s Health Initiative Randomized trial. JAMA. 2003;289(24):3243–3253. | ||
Ross RK, Paganini-Hill A, Wan PC, Pike MC. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000;92(4):328–332. | ||
Beral V; Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the Million Women study. Lancet. 2003;362(9382):419–427. | ||
Holmberg L, Iversen O-EE, Rudenstam CM, et al. Increased risk of recurrence after hormone replacement therapy in breast cancer survivors. J Natl Cancer Inst. 2008;100(7):475–482. | ||
Liang Y, Benakanakere I, Besch-Williford C, Hyder RS, Ellersieck MR, Hyder SM. Synthetic progestins induce growth and metastasis of BT-474 human breast cancer xenografts in nude mice. Menopause. 2010;17(5):1040–1047. | ||
Goyette S, Liang Y, Mafuvadze B, Cook M, Munir M, Hyder S. Natural and synthetic progestins enrich cancer stem cell-like cells in hormone-responsive human breast cancer cell populations in vitro. Breast Cancer Targets Ther. 2017;9:347–357. | ||
Chen K, Huang Y-HH, Chen J-LL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34(6):732–740. | ||
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. | ||
Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555–567. | ||
Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292(5519):1160–1164. | ||
McTaggart SJ. Isoprenylated proteins. Cell Mol Life Sci. 2006;63(3):255–267. | ||
Thoma R, Schulz-Gasch T, D’Arcy B, et al. Insight into steroid scaffold formation from the structure of human oxidosqualene cyclase. Nature. 2004;432(7013):118–122. | ||
Mafuvadze B, Liang Y, Hyder SM. Cholesterol synthesis inhibitor RO 48-8071 suppresses transcriptional activity of human estrogen and androgen receptor. Oncol Rep. 2014;32(4):1727–1733. | ||
Liang Y, Mafuvadze B, Aebi JD, Hyder SM. Cholesterol biosynthesis inhibitor RO 48-8071 suppresses growth of hormone-dependent and castration-resistant prostate cancer cells. Onco Targets Ther. 2016;9:3223–3232. | ||
Liang Y, Besch-Williford C, Aebi JD, et al. Cholesterol biosynthesis inhibitors as potent novel anti-cancer agents: suppression of hormone-dependent breast cancer by the oxidosqualene cyclase inhibitor RO 48-8071. Breast Cancer Res Treat. 2014;146(1):51–62. | ||
Jacobsen BM, Horwitz KB. Progesterone receptors, their isoforms and progesterone regulated transcription. Mol Cell Endocrinol. 2012;357(1–2):18–29. | ||
Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458(7237):438–444. | ||
D’Abreo N, Hindenburg AA. Sex hormone receptors in breast cancer. Vitam Horm. 2013;93:99–133. | ||
Fisher B, Redmond C, Fisher ER, Caplan R. Relative worth of estrogen or progesterone receptor and pathologic characteristics of differentiation as indicators of prognosis in node negative breast cancer patients: findings from National Surgical Adjuvant Breast and Bowel Project Protocol B-06. J Clin Oncol. 1988;6(7):1076–1087. | ||
Liang Y, Besch-Williford C, Brekken RA, Hyder SM. Progestin-dependent progression of human breast tumor xenografts: a novel model for evaluating antitumor therapeutics. Cancer Res. 2007;67(20):9929–9936. | ||
Benakanakere I, Besch-Williford C, Schnell J, et al. Natural and synthetic progestins accelerate 7,12-dimethylbenz[a]anthracene-initiated mammary tumors and increase angiogenesis in Sprague-Dawley rats. Clin Cancer Res. 2006;12(13):4062–4071. | ||
Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4(1):33–45. | ||
Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11(4):254–267. | ||
Bourguignon LY, Wong G, Earle C, Chen L. Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J Biol Chem. 2012;287(39):32800–32824. | ||
Gorin A, Gabitova L, Astsaturov I. Regulation of cholesterol biosynthesis and cancer signaling. Curr Opin Pharmacol. 2012;12(6):710–716. | ||
Maione F, Oliaro-Bosso S, Meda C, et al. The cholesterol biosynthesis enzyme oxidosqualene cyclase is a new target to impair tumour angiogenesis and metastasis dissemination. Sci Rep. 2015;5:9054. | ||
Hyder SM, Chiappetta C, Stancel GM. Pharmacological and endogenous progestins induce vascular endothelial growth factor expression in human breast cancer cells. Int J Cancer. 2001;92(4):469–473. | ||
Faivre EJ, Lange CA. Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol. 2007;27(2):466–480. | ||
Brisken C, Heineman A, Chavarria T, et al. Essential function of Wnt-4 in mammary gland development downstream of progesterone signaling. Genes Dev. 2000;14(6):650–654. | ||
Jang GB, Kim JY, Cho SD, et al. Blockade of Wnt/beta-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci Rep. 2015;5:12465. | ||
Mejia-Pous C, Damiola F, Gandrillon O. Cholesterol synthesis-related enzyme oxidosqualene cyclase is required to maintain self-renewal in primary erythroid progenitors. Cell Prolif. 2011;44(5):441–452. | ||
Vidal SJ, Rodriguez-Bravo V, Galsky M, Cordon-Cardo C, Domingo-Domenech J. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene. 2014;33(36):4451–4463. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.