Back to Journals » International Journal of Nanomedicine » Volume 16
Chitosan/Gold Hybrid Nanoparticles Enriched Electrospun PVA Nanofibrous Mats for the Topical Delivery of Punica granatum L. Extract: Synthesis, Characterization, Biocompatibility and Antibacterial Properties
Authors Mohamady Hussein MA ![]() , Ulag S, Abo Dena AS
, Ulag S, Abo Dena AS ![]() , Sahin A
, Sahin A ![]() , Grinholc M, Gunduz O, El-Sherbiny I
, Grinholc M, Gunduz O, El-Sherbiny I ![]() , Megahed M
, Megahed M
Received 1 March 2021
Accepted for publication 20 June 2021
Published 30 July 2021 Volume 2021:16 Pages 5133—5151
DOI https://doi.org/10.2147/IJN.S306526
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Mohamed Ahmed Mohamady Hussein,1,2 Songul Ulag,3 Ahmed S Abo Dena,4 Ali Sahin,5 Mariusz Grinholc,6 Oguzhan Gunduz,3,7 Ibrahim El-Sherbiny,4 Mosaad Megahed1
1Clinic of Dermatology, University Hospital of RWTH Aachen, Aachen, 52074, Germany; 2Department of Pharmacology, Medical Research Division, National Research Center, Dokki, Cairo, 12622, Egypt; 3Center for Nanotechnology & Biomaterials Application and Research (NBUAM), Marmara University, Istanbul, 34722, Turkey; 4Nanomedicine Laboratory, Center for Materials Science (CMS), Zewail City of Science and Technology, 6th of October, Giza, 12578, Egypt; 5Department of Biochemistry, School of Medicine, Genetic and Metabolic Diseases Research and Investigation Center, Marmara University, Istanbul, 34722, Turkey; 6Laboratory of Molecular Diagnostics, Department of Biotechnology, Intercollegiate Faculty of Biotechnology, University of Gdansk, Gdansk, Poland; 7Department of Metallurgical and Materials Engineering, Faculty of Technology, Marmara University, Istanbul, 34722, Turkey
Correspondence: Mohamed Ahmed Mohamady Hussein
University Hospital of Aachen, Aachen, 52074, Germany
Email [email protected]
Purpose: Intending to obtain Punica granatum L. extract (PE)-loaded drug delivery system of better impact and biomedical applicability, the current study reports the use of crosslinked PVA nanofibers (NFs) as platforms incorporating different amounts of biosynthesized PE-CS-gold nanoparticles (PE-CS-Au NPs).
Methods: PE-conjugated CS-Au nanoparticles (PE-CS-Au NPs) were synthesized via green chemistry approach. The formation of PE-CS-Au NPs was confirmed by UV spectroscopy, DLS, SEM and STEM. PE-CS-Au NPs were then dispersed into polyvinyl alcohol (PVA) solution at different ratios, where the optimized ratios were selected for electrospinning and further studies. Crosslinking of PE-CS-Au NPs loaded PVA nanofibers (NFs) was performed via glutaraldehyde vapor. The morphology, chemical compositions, thermal stability and mechanical properties of PE-CS-Au NPs loaded NFs were evaluated by SEM, FTIR and DSC. Swelling capacity, biodegradability, PE release profiles, release kinetics, antibacterial and cell biocompatibility were also demonstrated.
Results: By incorporating PE-CS-Au NPs at 0.6% and 0.9%, the diameters of the nanofibers decreased from 295.7± 83.1 nm in neat PVA to 165.6± 43.4 and 147.8± 42.7 nm, respectively. It is worth noting that crosslinking and incorporation of PE-CS-Au NPs improved thermal stability and mechanical properties of the obtained NFs. The release of PE from NFs was controlled by a Fickian diffusion mechanism (n value ˂0.5), whereas Higuchi was the mathematical model which could describe this release. The antibacterial activity was found to be directly proportional to the amount of the incorporated PE-CS-Au NPs. The human fibroblasts (HFF-1) showed the highest viability (123%) by seeding over the PVA NFs mats containing 0.9% PE-CS-Au NPs.
Conclusion: The obtained results suggest that the electrospun PVA NFs composites containing 0.9% PE-CS-Au NPs can be used as antibacterial agents against antibiotic-resistant bacteria, and as suitable scaffolds for cell adhesion, growth and proliferation of fibroblast populations.
Keywords: gold nanoparticles, chitosan, Punica granatum, nanofiber, antibacterial
Introduction
Polyvinyl alcohol (PVA) is a widely used polymer in biomedical applications either in the form of membranes or nanofibrous mats owing to its low toxicity, biodegradability and thermal stability. However, the major drawback of PVA nanofiber (NF) mats is their relatively rapid dissolution in biological media that results in poor durability, low tensile strength and fast degradation. Such rapid decomposition makes these polymeric nanofibrous mats undesirable for controlling drug release in drug delivery applications. These drawbacks made the biomedical applications of PVA alone very limited.1 Therefore, polyhydroxy PVA is used as an alternative for PVA to improve the physical characteristics. Crosslinking is considered one of the promising ways that make such polymers more appropriate for the intended application not only by rendering them water insoluble but also through improving their mechanical, physical and chemical characteristics.2 Many methods were employed to obtain crosslinked PVA NFs such as thermal crosslinking,2 solution method,3 crosslinking via heating with poly(acrylic acid) (PAA),4 sulfosuccinic acid (SSA),5 citric acid6 and glutaraldehyde (GA) vapor7 (Scheme 1).
 |
Scheme 1 Schematic illustration of the crosslinking reaction of PVA with GA vapor. |
Some plant extracts have proved their potential enhancing effect for fibroblast cell proliferation and differentiation. Berberine, a benzylisoquinoline plant alkaloid obtained from Coptidis rhizoma, was applied for periodontal regeneration, and it was found that berberine showed osteogenesis by promoting the differentiation of bone marrow mesenchymal stem cells (BMSCs). Furthermore, berberine could eradicate the growth of Porphyromonas gingivalis which is the main suspect in the process of periodontal tissue destruction.8
Punica granatum L. (pomegranate) is one of the ancient fruits, which is well known for its wide range of biomedical applications due to its antioxidants,9 anti-inflammatory,10 antibacterial,11 and anti-cancer activity,12 as well as its use in cardiovascular diseases,13 and wound healing.14 It was demonstrated that Punica granatum extract (PE) could enhance dermis and epidermis regeneration by promoting keratinocytes, fibroblasts and collagen synthesis. Moreover, PE showed the capability to inhibit the interstitial collagenase enzyme (ie, the major degrading enzyme in the skin).15 The extracts of plants such as Butea monosperma, Punica granatum and Cissus quadrangularis have been studied for skin regeneration and wound healing.16–19
Chitosan (CS) is the main derivative of chitin, a natural structural polysaccharide mainly extracted from the exoskeletons of arthropods (including crustaceans and insects), endoskeleton of cephalopods and fungal cell walls.20,21 CS is a linear polysaccharide of β-(1-4)-linked D-glucosamine and N-acetyl-D-glucosamine. It is well known for its low toxicity, biocompatibility, biodegradability, low immunogenicity and antibacterial activity.21
To the best of our knowledge, this is the first study that reports the use of such novel design composed of CS-AuNPs in PVA nanofiber mats for the delivery of PE. In 2011, Gomathi et al fabricated CS nanofibers/AuNPs composite to enhance the performance of a cholesterol voltammetric sensor.22 Ahmed et al suggested the application of electrospun CS/PVA/ZnO nanofibers as antibacterial and antioxidant mats for wound healing in diabetic rabbits.23 Moreover, a green method was used to synthesize carboxymethyl chitosan-PVA electrospun NFs enriched with gold nanoparticles to be used in biomedical applications.24 A very interesting review article summarizing the use of CS electrospun membranes and their composites with bioactive and therapeutic agents was reported by Augustine and coworkers.25 Nanosecond laser ablation is a promising strategy for fabricating nanofibrous films. Menazea and Ahmed synthesized CS-PVA films embedded with AuNPs with nanosecond laser ablation and investigated the in-vitro antibacterial activity of the prepared material.26 Yet, the study did not include any in-vitro cell proliferation experiments. Another study reported the use of silver nanoparticles embedded in bacterial cellulose gel membranes as an antibacterial wound dressing.27 Recently, Martinez et al synthesized gold nanoparticles via chemical reduction using CS as a reducing agent, and functionalized the produced nanoparticles with calreticulin.28 The functionalized nanoparticles were used to induce cell proliferation and wound healing in diabetic mice. In one of our recent studies, we reported the preparation of gold nanoparticles capped with a CS layer bearing PE in order to boost the antibacterial properties of CS-Au NPs against different antibiotic-resistant bacteria.29 Therein, the absence of a support medium that allows for topical delivery hiders topical application for purposes such as wound healing and/or skin care. In addition, the cell-proliferation improvement activity has not been investigated.
The current study aims to obtain a PE-loaded drug delivery system of better impact and applicability, through the use of GA-crosslinked PVA NFs as a platform incorporating different amounts of biosynthesized PE-CS-gold nanoparticles (PE-CS-Au NPs). Such structure is hypothesized to act as a sustained-release drug delivery system for the bioactive components of PE. To the best of our knowledge, the current study is the first to report the production of nanofibrous mats incorporating PE-loaded CS-Au NPs as an antibacterial and a cell proliferative agent. The proposed nanofibrous composite is expected to promote fibroblasts differentiation. Besides, the antibacterial effect of the embedded PE-CS-Au NPs in the produced crosslinked NFs will be a beneficial characteristic to eradicate infectious antibiotic-resistant pathogens.
Materials and Methods
Materials
Punica granatum (PE) was obtained from the local market. CS of molecular weight (Mw) 45 KDa and 80% of the degree of acetylation was purchased from Sigma-Aldrich (448877–250G). Glacial acetic acid (Panreac, Spain) and HAuCl4.3H2O (99.5%, Sigma Aldrich, Germany) were used throughout the experimental procedure. A 87–89% hydrolyzed PVA (Mw = 85–124 kDa) and glutaraldehyde (GA, 25%) were purchased from Sigma-Aldrich. Ultrapure Milli-Q water (18.2 Ω cm−1) was used for aqueous solution preparations. All chemicals were of analytical grade and were used without further purification.
Plant Materials
Punica granatum (pomegranate) fruits were collected from a local market in Cairo, Egypt. The fruits were taxonomically identified and authenticated by Dr. Aleksandra Krolicka, Associate Professor, from Laboratory of Biologically Active Compounds at University of Gdansk in Poland. The voucher specimen was deposited at Intercollegiate Faculty of Biotechnology Repository with a voucher number UG-IFB-2020/20.
Microorganism Strains
Clinical Pseudomonas aeruginosa (ATCC 27853), Staphylococcus aureus (ATCC 25923) and methicillin-resistant Staphylococcus aureus (MRSA, ATCC 43300) were cultivated overnight in trypticase soy broth (TSB) (bioMérieux) medium at 37°C in an orbital incubator (Innova 40, Brunswick, Germany) operating at 150 rpm.
Cell Culture
Human fibroblast cells (HFF-1) were purchased from the American Type Culture Collection (ATCC® SCRC-1041™). Cells were cultured in Dulbecco’s modified Eagle medium (DMEM, Sigma Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (Gibco) under standard culture conditions (humidified atmosphere, 5% CO2 and 95% air, at 37°C).
Preparation of P. granatum Extract and Chitosan Solution
The active components of P. granatum L. peel and seeds were extracted with water (1:10 ratio w/v) and centrifuged at 15,000 rpm for 30 min to get rid of the impurities. Eventually, seed extract was mixed with the peel extract in a ratio of 3:1 (seed/peel) and the mixture was left for 4 h at room temperature before storing at 4°C for further utilization.
CS solution was prepared by mixing a known amount of CS with 100 mL of acetic acid (1% v/v) and kept under stirring overnight to allow the complete dissolution of and to obtain a clear solution.
Synthesis of PE-CS-Au NPs
PE-CS-Au NPs were synthesized by mixing a definite amount of PE with CS solution to get a final volume of 50 mL. The mixture was stirred for 2 h to obtain PE-CS homogenous solution, which was then heated up to 80°C. After that, a suitable amount of HAuCl4.3H2O solution was added to get a final concentration of 1.25 mmol/l. The resulting mixture was left for 15 min whereafter it was cooled. The obtained CS-Au NPs were collected by ultracentrifugation at 28,000 rpm at 25°C for 30 min. The supernatants recovered from centrifugation were discarded and the pellets were re-suspended in milli-Q water for further characterization and utilization.
Physicochemical Characterization of the Nanoparticles
UV-Vis spectroscopy was used to record the electronic absorption spectra of the developed PE-CS-Au NPs, dynamic light scattering (DLS) was conducted to record zeta potential and size of NPs. Wet scanning transmission electron microscopy imaging technique (Wet-STEM) was used for morphological characterization to determine the shape and size of the obtained PE-CS-Au NPs. The Wet-STEM imaging has been performed with a FEI QUANTA FEG SEM 450. As a copper grid, Ted Pella, support films, carbon type A and 300 mesh were used. The STEM holder was cooled to 2°C and the pressure was set between 700 and 1300 Pa. A drop of the sample was added to the grid, and the samples were scanned in the dark field area with the Wet-STEM detector. Scanning electron microscope integrated with energy-dispersive X-ray spectroscopy (SEM-EDX) were employed to characterize and confirm the biosynthesis of PE-CS-Au NPs.
Preparation and Characterization of Electrospinning Solutions
A 12% (w/v) PVA solution was prepared by dissolving PVA in distilled water at 90°C with stirring for 4 h. Thereafter, the polymer solution was left for cooling at room temperature. Then, different amounts of PE-CS-Au NPs were incorporated into the PVA solution. All solutions were stirred for 2 h at room temperature to obtain a homogenous mixture of PVA/PE-CS-Au NPs. The conductivity of the prepared electrospinning solutions (Figure 1) was determined using an electrical conductivity meter (Cond 3110 SET 1 WTW, 82362 Weilheim, Germany), and their viscosity was detected with a digital viscometer (DV-E, Brookfield AMETEK, USA). The composition of the prepared nanofibrous mats is summarized in Table 1.
 |
Table 1 Composition of the Synthesized PE-CS-Au NPs-Loaded PVA NFs |
 |
Figure 1 Schematic representation of the fabrication and electrospinning preparation steps of PE-CS-Au NPs and the proposed nanofibrous PE-delivery system. |
Characterization of the Rheological Properties
The conductivity and viscosity of the prepared F0 solutions or PVA/NPs suspensions at the different ratios, were investigated at ambient temperature using an electroconductivity meter and a digital viscometer (DV-E, Brookfield AMETEK, USA), respectively. Each measurement was repeated three times, and the average value was calculated. The PVA/NPs suspensions with the optimum conductivity and viscosity values were then selected for the subsequent investigations.
Electrospinning
During electrospinning, a high-voltage was applied to the polymer solution contained in a syringe through an alligator clip attached to the syringe needle (internal diameter 0.9 mm). A laboratory-scale electrospinning unit (NS24, Inovenso Co., Turkey) was used for electrospinning. Different applied voltages were applied to monitor their effects on the electrospun NFs. The optimum applied voltage was adjusted after applying different voltages. A syringe pump (NE-300, New Era Pump Systems, Inc., USA) was used to control the flow rate (0.02 mL/h) of the polymer solution. The fibers were collected on electrically grounded aluminum foil placed 12 cm from the needle tip. Subsequently, the produced NFs were collected for further physicochemical characterizations.
Crosslinking
The PVA/NPs NFs were crosslinked in 25% GA vapor at room temperature for several hours. The cross-linking procedure was carried out in a sealed vessel with heating in a vacuum oven at 40°C for 12 h. After crosslinking, all samples, including the non-crosslinked samples, were exposed in an oven for 2 h at 50°C to remove residual GA and water. From now on, the codes of the non-crosslinked samples will be given capital NC letters (eg, NC-F3) and those of the crosslinked ones will be given a capital C letter (eg, C-F3) (Table 1).
Mechanical, Chemical and Morphological Properties
The samples were cut into dimensions of 1 cm × 5 cm, then the thickness of the electrospun NFs was measured by using a digital micrometer (Mitutoyo MTI Corp., USA). Mechanical tests were performed by recording the tensile strength and elongation at break using a tensile test machine (SHIMADZU, EZ-LX, China). The upper and lower portions of each sample were located horizontally in the relevant section of the device. Both ends of each specimen were compressed by the top and bottom grip and subjected to a tensile test under conditions of 5 mm/min test speed. The results were analyzed using Bluehill 2 software (Elancourt, France). The samples were measured in triplicates. Morphology of the fibrous matrices was observed by using a scanning electron microscope (SEM, MA-EVO10, ZEISS) at an accelerating voltage of 20 kV. Samples were cut and coated with gold (60 s) before imaging. Nanofibers’ diameters were calculated with imageJ using SEM images. The distribution of PE-CS-AuNPs over the fibers was observed using a combined field emission SEM (FE-SEM)/scanning TEM (STEM) ultra-high resolution electron microscope (Hitachi S-5500). Chemical characterization was performed for PE-CS-Au NPs, unloaded and PE-CS-Au NPs loaded nanofibers using Fourier Transform Infrared (FTIR) spectroscopy (JASCO 4700, USA) in the range of 400–4000 cm–1. The data was normalized to the background spectrum recorded from a clean empty cell. The thermal behaviors of the crosslinked and the non-crosslinked NFs of both plain PVA, and PVA/NPs, were studied using differential scanning calorimetry (DSC; Shimadzu DSC-60 Plus instrument). A 10–12 mg sample was sealed in aluminum pans and heated up to 250°C at a heating rate of 10°C/min under nitrogen atmosphere.
Biodegradability and Swelling
Swelling profiles of the developed crosslinked and non-crosslinked NF mats were investigated. NFs were weighed (Wd), placed in PBS (pH 7.4), and left in a shaking incubator at room temperature. At specific time intervals, the samples were removed from the buffer solution, plotted with a dry filter paper to remove any excess PBS, and weighed again (Ws) until no change in weight was observed. The swelling percentage is calculated according to Equation 1.30
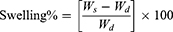
Biodegradability was studied by placing disc-shaped samples of the NFs in PBS (pH 7.4) in a shaking incubator for 7 days. The media were changed and replaced by fresh media every day. The sample discs were taken from the PBS solution, washed with distilled water and dried at 50°C in an oven until a constant weight was obtained. The remaining weight percentage was monitored daily using Equation 2, where Wi is the initial weight, and Wf is the measured final weight.30
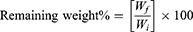
Encapsulation Efficiency and in-vitro Release
PE content in the NFs was quantified via spectrophotometric analysis through the complete dissolution of a certain amount of the NFs in a solvent. Loading dose (%) and encapsulation efficiency (EE%) were calculated using Equations 3 and 4, respectively:
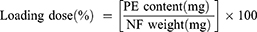
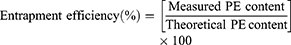
The release profile of PE from CF3 and CF4 was studied in PBS solution (pH = 7.4) over 7 days. The NFs (20 mg) were placed in 10 mL of PBS solution at 37°C with constant shaking. At determined time intervals, 1 mL samples were taken from the release medium and replaced with fresh PBS to maintain the original volume. The amount of PE released at selected time intervals was measured by UV spectroscopy (Shimadzu UV-3600) at 265 nm. The PE release percentage was calculated, and the release profile was plotted. All the measurements were performed in triplicates.
In-vitro Drug Release Kinetics
To study the mechanism of PE release from CF3 and CF4 NFs mats, five kinetics models were employed including: zero order, first order, Higuchi, Korsmeyer–Peppas, Hixson–Crowell using Equations (5–9), respectively. The model which displays the highest value of correlation coefficient (r2) is considered as the most fitting kinetic model to describe PE release behavior from either CF3 or CF4. GraphPad Prism Software Version 6 was used to analyze the obtained results.
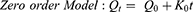
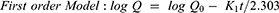
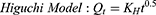
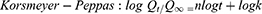
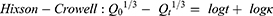
where Q is the fraction of drug released at time t, Q0 is initial fraction of drug in solution, K0 is zero order release constant, K1 is first-order release constant, and KH is Higuchi dissolution constant.
In the Korsmeyer-Peppas equation, Qt/Q∞ is the fraction of drug released at time t, Q∞ is the total drug released, k is a kinetic constant, and n is the exponent explaining the drug release mechanism. For instance, (n ≤ 0.5) corresponds a Fickian diffusion, (n = 1) represents case-II transport, (0.5 < n < 1) is anomalous transport, and (n > 1) is considered super case-II transport. In the Hixson–Crowell equation, κ (kappa) is a constant describing the surface–volume relation.
Cell Culture Assays
The potential cytotoxicity of the NFs was evaluated against HFF-1. To observe the cell-nanomaterial interactions and the influence of various amounts of the contained PE-CS-Au NPs, HFF-1s were used in passage number five. The medium for all cell culture tests was the same and consisting of DMEM, FBS (10%, v/v), penicillin/streptomycin solution (1%, v/v) and
The cell viability of the fibroblasts on the electrospun composite NFs were determined quantitatively by colorimetric MTT assay starting with the end of the first day of initial seeding and followed by the end of the third and seventh days. After the incubation period, all the media inside the wells were discarded and the composite NFs were rinsed with PBS solution thrice. An amount of 90 μL of the fresh medium and 10 μL of MTT solution (5 mg/mL in PBS solution) were added onto newly washed samples and kept in an incubator (37°C, 5% CO2) for 3 h. After the incubation period, purple media containing formazan crystals were obtained because of the reduction of MTT by living cells. The medium on the NFs was discarded, and the samples were taken to clean wells and 200 μL of DMSO were put onto the samples to dissolve the formed formazan crystals and kept in the incubator for an additional 1 h. Finally, the media from the wells were taken, and the absorbance values of the solutions were measured at 540 nm by Dynamica LEDETECT96 microplate reader.
Fluorescence microscopy analysis was performed to get information about cell attachment and proliferation on/into the composite NFs. The dedicated mats for the third and seventh days of the cell culture were taken, the growth medium was removed from the plates, and the nanofibrous mats were washed thrice with PBS. Subsequently, the cells on/inside the materials were fixed with 4% formaldehyde for 30 min at room temperature, and then washed with PBS. Moreover, 1 µg/mL of 4ʹ,6-Diamidino-2-Phenylindole Dihydrochloride (DAPI, invitrogen reagent) was added on each stroma construct to stain the nuclei of the cells, and incubated for 20 min at room temperature. Finally, DAPI solution was removed, and the constructs were imaged using an inverted fluorescence microscope (Leica).
SEM images were taken on the third and seventh days of the cell culture period. Briefly, pre-decided NFs were taken from the incubator, and all the media were discarded, and the materials were then washed with PBS solution. Thereafter, fixation was carried out by a 4% paraformaldehyde solution for 30 min. The nanofibrous mats lost their water contents via immersion into water/ethanol solutions with increasing amounts of ethanol up to 100% (v/v) for 2 min. Lastly, hexamethyldisilane (HMDS) was added onto the mats, and then they were left to dry in air. Dried mats were sputter-coated with gold and examined with SEM with 10 kV.
Antibacterial Assessment
Pseudomonas aeruginosa (ATCC 27853), Staphylococcus aureus (ATCC 25923) and methicillin-resistant Staphylococcus aureus (MRSA, ATCC 43300), were used as test organisms in the zone of inhibition test. All the nanofibrous mats were sterilized by UV light before the experiment. A bacterial solution (100 μL, with a concentration of ~106 CFU/mL) was dispensed onto an agar plate. Pieces of 1.5 cm × 1.5 cm of CF0, CF3 and CF4 were placed on the surface of the plate. The zone of inhibition was measured after 24-h incubation at 37°C.
Statistical Analyses
The results were analyzed using GraphPad Prism 5 software (version 5.01). The statistical comparison was carried out using Student’s t-test, with a p-value used to indicate significance. P values <0.05 were considered to be statistically significant.
Results and Discussion
Characterization of the Nanoparticles
PE-CS-Au NPs were characterized by UV spectroscopy, DLS, TEM and SEM-EDX. The reduction of Au+ to Au0 was confirmed by recording the electronic absorption spectra, where the PE-CS-Au NPs showed intense absorption bands at 532 nm. DLS measurements (Figure 2) revealed that the size of the prepared PE-CS-Au NPs is 65.3 ± 20.7 nm. In addition, the ζ-potential is +31.0 ± 6.0 mV. By investigation with TEM, it was observed that the PE-CS-Au NPs showed spherical shapes with an average diameter of 29 ± 5.9 nm. The surface morphology of the as prepared PE-CS-Au NPs was also recorded by SEM where NPs of spherical shape were observed. EDX analysis confirmed the elemental composition of the as synthesized hybrid nanocomposites as indicated from the signal corresponding to Au atoms observed at 2.3 keV.
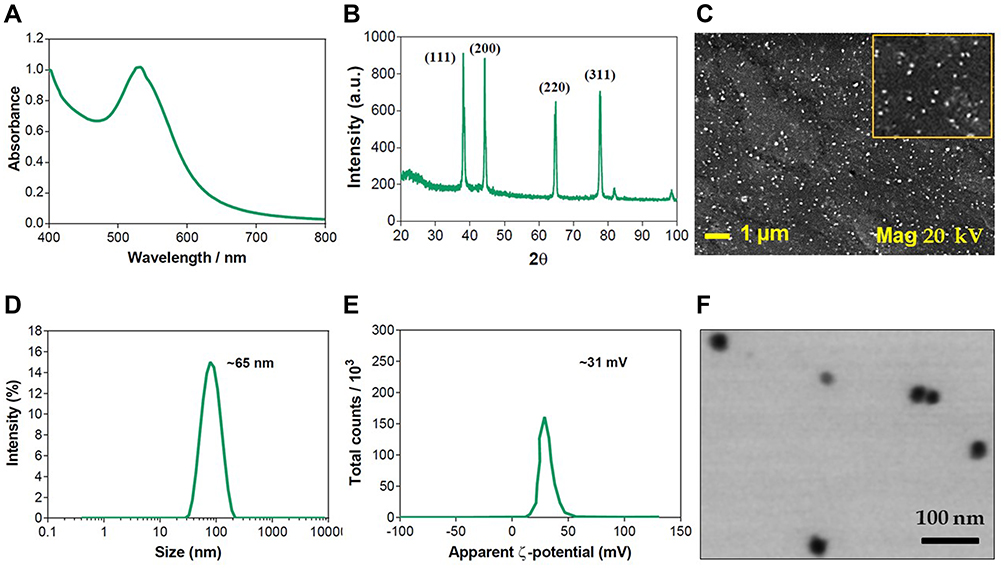 |
Figure 2 Characterization of PE-CS-Au NPs: (A) UV spectrum, (B) XRD, (C) DLS hydrodynamic diameter, (D) ζ-potential, (E) SEM image and (F) STEM image. |
Viscosity and Conductivity of the Electrospinning Solutions
The measured values of density and absolute viscosity were used to record the absolute viscosity over different compositions (F0, F1, F2, F3 and F4) of PVA/NPs blends. It has been demonstrated that compatible blends show linear plots, whereas incompatible ones show non-linear plots.31,32 Accordingly, it is noticed that the viscosity curve is almost linear (Figure 3), and thus it indicates that the corresponding blends are miscible. Furthermore, it is obvious that the viscosity of PVA/NPs blends was increased by increasing the proportion of PE-CS-Au NPs, which was likely attributed to the potential crosslinking through the interaction of NPs surface with hydroxyl groups of CS and PVA, forming hydrogen bonding and hence the viscosity was elevated.33
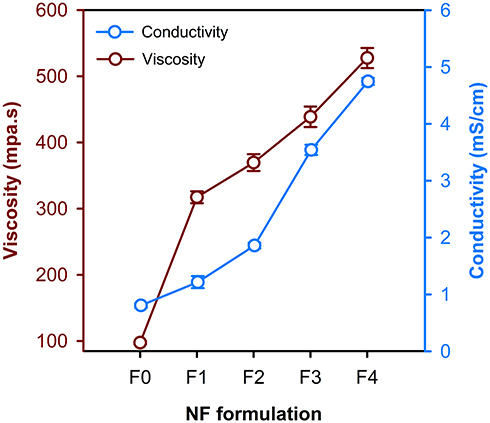 |
Figure 3 Variation of viscosity and electrical conductivity of the blends used for the synthesis of the different electrospun NFs. |
Conductivity is an important parameter during the electrospinning process. Figure 3 shows the effect of PVA mass ratio and hybrid NPs concentration on the conductivity of the electrospinning solutions. The conductivity significantly increased upon increasing the concentration of the hybrid NPs. It has been found that blending the polymer with the NPs could potentiate the conductivity of the mixture.34,35 These results are in consistence with previous reports which found that the properties of polymeric materials can be improved and qualified through dispersing metallic NPs into the polymer matrix to produce NPs-filled polymer.36–38 Based upon the obtained results of conductivity, F0, F3 and F4 formulations were chosen to conduct the rest of the study.
Effect of the Applied Voltage on the Nanofiber Diameter
The applied voltage is one of the most important parameters by which we can control and obtain uniform, smooth, ultrafine and well-distributed fibers in the nanoscale. Herein, the voltage was changed at constant flow rate and distance. Figure 4 shows SEM images of nanofibrous mats prepared by applying different voltages. The produced NFs were found to have diameters of 728.8 ± 235, 322.6 ± 105, 218.2 ± 71.6, 165.6 ± 43.4, 137.8 ± 44.9 and 230.8 ± 75.3 nm by applying voltages of 12, 16, 20, 23, 26 and 29 kV, respectively. These results depict that the increase in the applied voltage from 12 to 26 kV leads to producing NFs of smaller fiber diameters. However, at voltages above 26 kV the NFs diameter increased. Furthermore, above 23 kV (at 26 and 29 kV for instance) polymeric beads were observed in the produced NFs. Accordingly, the optimum voltage that results in the best NFs properties is 23 kV. Similar findings were demonstrated in previous literature studies.39 Nevertheless, it contradicts the results obtained by Liu et al.40 Our results can be explained by the fact that the reflex of their electrospinning bubbles to the applied voltage might be different from that of our solid NPs-immersed polymer. It was hypothesized that higher voltages may enhance stretching of the jets resulting in NFs with smaller diameters. Such effect continues until 26 kV, then the diameter starts to increase with some deformities such as the polymeric beads obtained at 26 and 28 kV (arrows in Figure 4).
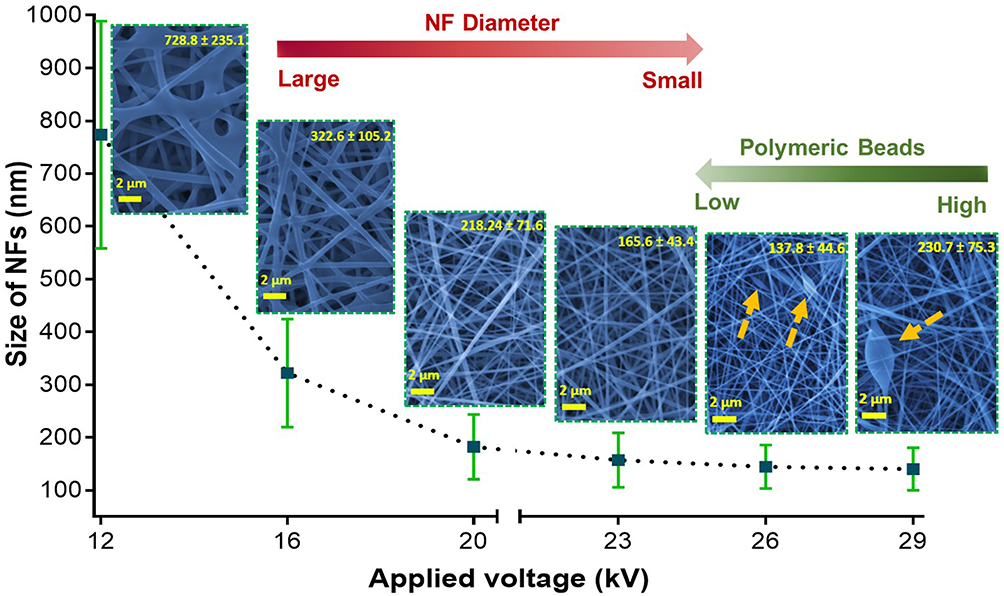 |
Figure 4 The dependence of the nanofiber diameter and uniformity on the applied electrospinning voltage. |
SEM of Neat PVA and PVA/NPs Nanofibers
The morphology of the prepared NFs was further studied by SEM imaging. SEM micrographs of hybrid F3 and F4 prepared by applying a voltage of 23 kV, an electrospinning distance of 12 cm and a flow rate of 0.02 mL/h are depicted in Figure 5A (original image is shown in Figure S1). The electrospinning of F0, F3 and F4 solutions produced nanofibrous mats with an average fiber diameter of ca. 296, 166 and 148 nm, respectively. The decrease in the diameter of the NFs can be attributed to the higher conductivity of the hybrid PVA/NPs solution (see Figure 3). Previous investigations demonstrated that the higher the conductivity of an electrospinnable solution, the smaller is the diameter of the NFs, and this phenomenon was explained by high charge capacities of the solution of the higher conductivity.41–43 Consequently, it creates elongation forces that exert more pressure upon the jet leading to a nanofiber with a reduced diameter.41 Moreover, it has been observed that increasing the content of NPs incorporated into the PVA solution had significantly increased the viscosity of the solution and enhances its spinnability as well. Abri et al reported that the addition of CS has decreased the NF diameter from 241 to 216 nm44 which is in an excellent agreement with our findings. In addition, Ganesh et al found that increasing CS/PVA ratio in the order 0/12, 2/8 and 3/7 w% has produced electrospun NFs with diameters of 200, 155 and 95 nm, respectively.45
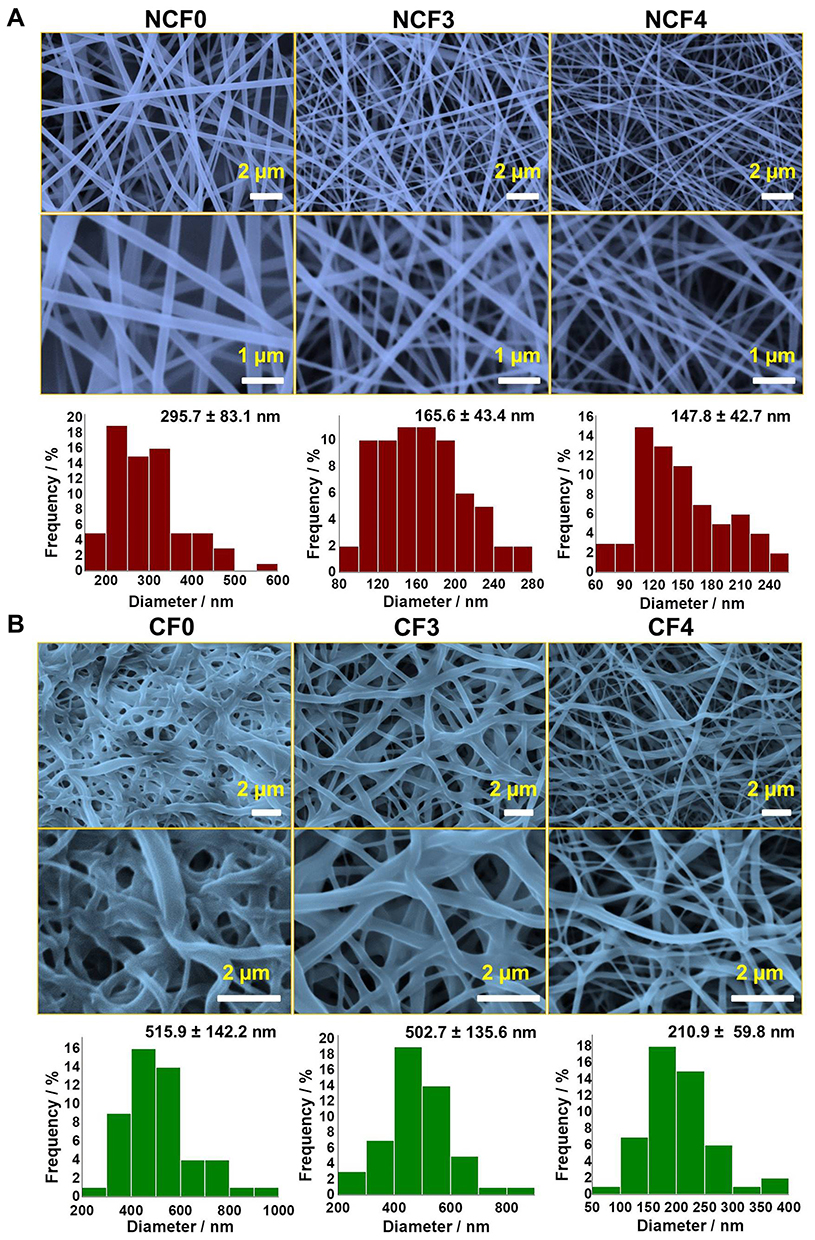 |
Figure 5 SEM micrographs of non-crosslinked (A) and crosslinked (B) PVA/NPs NFs. The corresponding size-distribution histograms are also depicted. The original SEM images with original scale bars are provided in the supporting information (Figure S1 and S2). |
SEM of Crosslinked Nanofibers
Owing to the immediate dissolution of the electrospun nanofibrous mats, the NFs swell and fuse leading to loss of porosity. Yet, porosity is considered the most important characteristic feature of PVA NFs because it presents a high surface-area-to-volume ratio. Therefore, GA vapor was used as an efficient crosslinking agent to avoid such consequence and maintain the porous structure and the morphology of the electrospun nanofibrous mats. It can be observed that crosslinking with GA resulted in swelled and flattened NFs as depicted in Figure 5B (original image is shown in Figure S2). The observed swelling and flattening are attributed to exposure of the NFs to the water content of the vapor phase of GA during the crosslinking process.7
The CF0 and CF3 NFs showed histograms displaced towards higher values compared to those of containing higher NPs content (CF4). CF0 and CF3 NFs showed average NF diameters of 515 and 503 nm, respectively, while the diameter CF4 NFs was about 211 nm. In addition, the porosity features were approximately maintained in both CF3 and CF4 NFs, while it was highly reduced in CF0 NFs. Such findings can be explained by the presence of CS as a polyelectrolyte which becomes highly protonated and positively charged in the acidic GA medium. Therefore, the repulsive forces among the protonated amino groups would prevent the unpleasant over chain entanglements during the crosslinking process.
Tensile Strength
The tensile strength tests were performed to characterize the mechanical properties of the electrospun NFs. Table 2 shows the tensile strength of the produced NFs through the elongation at break (also known as fracture strain). Demonstrating the tensile strength property of NFs is crucial to display the flexibility of the product.46 The ultimate tensile strength of the NCF0, NCF3 and NCF4 nanofiber mats was 3.8 ± 0.64, 5.6 ± 0.82 and 8.2 ± 1.4 MPa, respectively. After crosslinking, the tensile strength of the tested NFs was elevated to 5.2 ± 1.1, 6.1 ± 1.3 and 9.6 ± 0.94 MPa, respectively. The results revealed that CF4 has the highest tensile strength (ca. 9.6 MPa) compared to the other formulations. Therefore, the tensile strength was enhanced upon the inclusion of PE-CS-Au NPs in the PVA matrix.
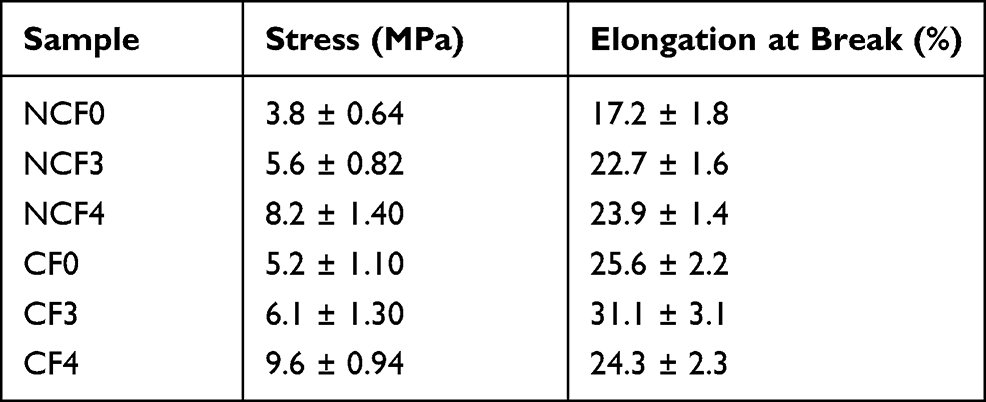 |
Table 2 Mechanical Properties of the Investigated NFs |
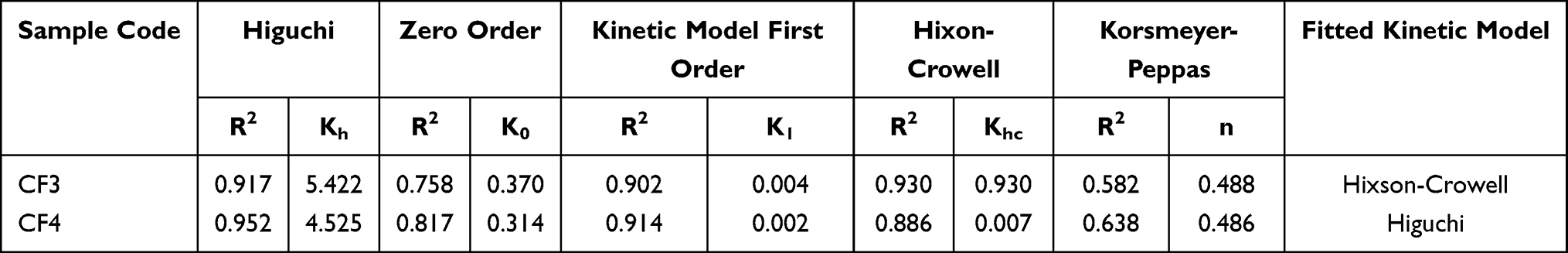 |
Table 3 Release Kinetics of PE from Crosslinked PVA NFs with Various Embedded Amounts of PE-CS-Au NPs |
Water Absorption and Biodegradability
The water absorption/swelling test was performed to estimate the hydrophilicity of the prepared NFs and to explore their behavior to load and release the incorporated extract. Figure 6 shows the results of the water absorption test of both plain and PE-loaded nanofibrous mats in the presence of PBS (pH 7.4) over 24 h at predetermined time intervals. The weight gain was calculated and plotted against time.
 |
Figure 6 STEM-FESEM image showing the distribution of PE-CS-Au NPs over the PVA NFs (A), water absorption (B) and biodegradability (C) properties of the as prepared NFs. |
During the first 3 h, all NFs showed a gradual increase in weight, where non-crosslinked NFs showed higher weight gain compared to their crosslinked counterparts. Non-crosslink NFs started to dissolve in PBS after 3, 6 and 9 h for NCF0, NCF3 and NCF4, respectively; hence their weight gain dropped abruptly. Meanwhile, CF0, CF3 and CF4 continued their gradual increase in swelling capacities until reaching the maximum capacity. Owing to crosslinking, plain and NPs loaded NFs showed swelling capacities lower than their non-crosslinked counterparts at the first few hours. Furthermore, it was demonstrated that the swelling capacities of NPs loaded NFs were higher than that of the plain ones. After crosslinking with GA, the mesoporous spaces have been diminished in CF0, and therefore, it relatively restricted the ability of the water molecules to diffuse throughout the network of the polymeric NFs. Accordingly, crosslinking could aid the nanofibrous mats to withstand dissolution and continue their gradual swelling. Moreover, it was observed that increasing the content of PE-CS-AuNPs maintained the uprising of swelling rate for a longer time before reaching the plateau (after 9, 12 and 24 h in case of CF0, CF3 and CF4, respectively).
By studying the biodegradability of the nanofibrous mats (Figure 6) it was observed that CF0, CF3 and CF4 displayed low degradation rates. Therefore, after 3-weeks incubation, the weight loss for CF0, CF3 and CF4 were found to be ca. 59%, 51% and 47%, respectively. On the other hand, NCF0 NFs exhibited a faster degradation rate relative to both NCF3 and NCF4 NFs. It can be demonstrated that the addition of PE-CS-AuNPs and crosslinking has influenced the degradation rate of PVA NFs via enhancing its hydrophobicity. These findings agree with a previous study where it was found that CS-stabilized PVA has shown lower weight loss compared to the non-stabilized one.47
Drug Release
The release profile of PE from CF3 and CF4 was conducted a long 168 h. The release profiles were investigated by immersing CF3 and CF4 in PBS solution (37°C, pH 7.4). Figure 7 depicts that an initial burst release of 37% and 26% occurred after 6 h in case of CF3 and CF4 NFs, respectively. This was followed by a second slow-release phase of PE. Furthermore, CF4 NFs shows a slower cumulative release profile (ca. 66%) in comparison to CF3 (ca. 82%), and this can be attributed to the higher degree of crosslinking between the PE-CS-Au NPs and the polymeric chains in case of CF4. The loading capacities were found to be 94% and 97% in CF3 and CF4 NFs, respectively. This might be caused by the loss of PE occurring during the electrospinning process.
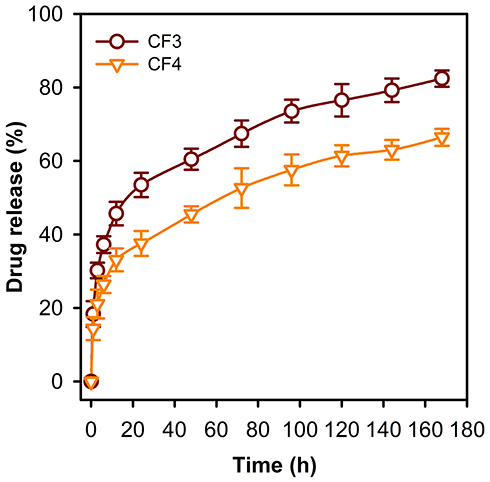 |
Figure 7 Release profile of PE from the CF3 and CF4 nanofibrous mats. It is obvious that the release rate is slower in case of CF4 compared to the CF3 formulation. |
Release Kinetic Study
To get a more close-up insight to the mechanism of PE release from the CF3 and CF4 NFs, the study allocated various release kinetic models including zero-order, first-order, Korsmeyer-Peppas and Higuchi models (Table 3). The model with the highest value of correlation coefficient (r2) was considered as the most fitting kinetic model.
Based on the “n” values in the Korsmeyer-Peppas model, the predominant release of PE from both CF3 and CF4 NFs was a Fickian diffusion pattern (n ˂ 0.5).30 The n values were 0.488 and 0.486 for CF3 and CF4 NFs, respectively. It was found that Hixson-Crowell model could describe the diffusion release of PE from CF3 with r2 > 0.93. While the PE diffusion release from CF4 NFs was governed by Higuchi model (r2 > 0.95). These findings confirmed that the release of PE from CF3 and CF4, was more consistent with a diffusion mechanism than with a matrix erosion mechanism.48
Eventually, it can be noted that the higher PE-CS-Au NPs content in the crosslinked PVA enhanced the hydrophobicity of the produced nanofibrous membranes, and thus it could delay the release of PE from the polymer matrix.
FTIR
The FTIR spectrum of PE (Figure 8) demonstrated characteristic bands at 3261 and 2934 cm−1 attributed to phenolic O-H and C-H stretching vibrations, respectively.49 The band appearing at 1697 cm−1 is attributed to C=O (carbonyls) stretching. The band observed at 1025 cm−1 is corresponding to C-O-C stretching vibration. The bands between 806 and 667 cm−1 are signed to alkyl halides (alkyl chloride and/or alkyl bromide) in PE.
 |
Figure 8 FTIR spectra (A) and DSC thermograms (B) of the different nanofibrous mats formulations under study. |
The characteristic band of CS located at 3300 cm−1 is attributed to stretching of O-H and N-H bonds (Figure 8). Moreover, the band observed at 2870 cm−1 is assigned to C-H stretching vibration. Carbonyl stretching of amid I, NH2 bending of amide II and C-N stretching of amide III resulted in the bands allocated at 1646, 1589 and 1375 cm−1, respectively. The bands corresponding to C-N stretching and β-(1-4) glycosidic linkage are located at 1150 and 1059 cm−1. The band due to stretching of C-O was determined at 1024 cm−1.49,50 It can be seen from the FTIR spectrum of PE-CS-Au NPs, that the addition of PE to CS during the synthesis of NPs led to a significant increase in band intensity in the area between 3200 and 3500 cm−1, which might be attributed to O-H stretching of both CS and the phenolic compounds of PE, indicating the incorporation of PE in the CS-Au NPs. The presence of IR bands between 817 and 688 cm−1, corresponding to alkyl halides of PE, also confirms the participation of PE in the reduction and capping of Au NPs.
The main characteristic bands of plain PVA NFs (NCF0) appeared at 3200–3680 cm−1 due to stretching of the hydroxyl group (Figure 8). In addition, other three peaks at 2923 cm−1 (C-H stretching), 1739 cm−1 (-C=O) and 1077 cm−1 (C-O stretching) were noticed.51,52 The increase in the intensity of the band observed at 3200–3680 cm−1 in NCF3 and NCF4, assigned to PE-CS-Au NPs interaction, was clearly demonstrated. The intensity of the band increased by increasing the content of the PE-CS-Au NPs.
In case of CF0, CF3 and CF4 NFs, a relative reduction of the intensity of the broad band (3200–3680 cm−1) was observed. This reduction may be due to the involvement of the hydroxyl groups of PVA with the aldehyde groups of GA to form an acetal bridge.3,7,53,54 Owing to the crosslinking reaction, a broadening of the band width at 1000−1176 cm−1, attributed to O−C−O vibration of the acetal group, can be clearly observed.7 Such broadening is clearer in the hybrid PVA/NPs NFs (CF3 and CF4), which might be attributed to the participation of the NPs in the crosslinking process.
Differential Scanning Calorimetry
PVA is a partially crystalline polymer with characteristic hydrogen bonds between its hydroxyl groups. It is expected that the incorporation of PE-CS-Au NPs will produce changes in the structure and the crystallinity of the polymer through interfering with the -OH groups associated with relative changes in the configuration of the polymer chains as well as their intermolecular and intramolecular interactions.55 Figure 8 shows DSC thermograms of plain PVA NFs, PVA/NPs NFs and their crosslinked counterparts. In case of pure PVA, the endothermic melting peak was recorded at 214°C. This peak has been shifted to 218°C after crosslinking with GA. NCF3 NFs showed an endothermic melting peak at 217°C; however, it was shifted to 221°C after crosslinking. For NCF4 NFs, the melting point was detected at 220°C which was elevated to reach 223°C after crosslinking. Other studies also reported the increase in the PVA melting point after crosslinking.6 Furthermore, NFs loaded with PE-CS-AuNPs exhibited higher melting peak, which could be attributed to the enhanced crosslinking caused by the hydrogen bond interactions of the NPs with PVA chains.45
Antibacterial Activity
The antibacterial activities of hybrid PVA/NPs NFs against Gram-positive, methicillin-resistant S. aureus (MRSA) and Gram-negative P. aeruginosa were evaluated by disc inhibition zone method. The abilities of the NFs to inhibit the growth of the tested microorganisms are shown in Figure 9A. The figure shows the incubated petri dishes containing the tested bacterial strains grown in the presence of the nanofibrous patches after 24 h. The appearance of clear zones around the nanofibrous patches loaded with the hybrid PE-CS-Au NPs is clearly noticed in the figure indicating the efficacy of the proposed NFs as antibacterial patches against the investigated strains. Meanwhile, plain nanofibrous patches showed no inhibition zones.
 |
Figure 9 Antibacterial activity of the proposed nanofibrous mats using the inhibition zone method (A), Cell viability test results. *p < 0.05 when compared to control (B), fluorescence images showing the proliferation of human fibroblasts (HFF-1) on the nanofibrous mats using DPAI staining after 3 and 7 days (C), and SEM micrographs of cells proliferation and adhesion on the nanofibrous mats (D). The red values in (A) represent the corresponding inhibition zone diameters in mm. The original SEM images with original scale bars are provided in the supporting information (Figure S3). |
The NFs mats CF3 and CF4 showed around 14.4 and 19.6 mm inhibition zones for S. aureus ATCC25923, respectively. Meanwhile, a slightly lower activity against P. aeruginosa ATCC27853 (11.1 and 13.2 mm) was demonstrated by CF3 and CF4, respectively. MRSA (S. aureus ATCC43300), showed moderate resistance where CF3 and CF4 exhibited 11.7 mm and 13.5 mm inhibition zones, respectively.
Such findings indicate that the hybrid NPs are responsible for the antibacterial activity of the composite fibers. The NPs are released steadily over 48-h incubation period, and exhibit zones of inhibition against the three selected bacterial strains. It can be noted that the Gram-negative P. aeruginosa showed higher resistance than S. aureus and MRSA. This might be attributed to the intrinsic difference in their cell wall structure. The Gram-negative bacterial cell wall is thick, hence acting as a strong biological barrier against biocides. It consists of lipids, proteins and lipopolysaccharides (LPS). However, the thinner and loose Gram-positive bacterial cell wall, due to missing of LPS, can be easily destroyed and penetrated by the NPs. Thus, the penetrated NPs might bind with DNA and cause denaturation.56 The incorporated hybrid NPs contain PE and CS. PE has proved its antibacterial efficacy through its bactericidal multi-components of polyphenols such as tannins (hydrophilic and hydrophobic part), and ellagic acid. Such components are likely to destroy the bacterial cell through more than one target including combination with sulfhydryl group (SH) of peptidoglycans, strong binding capacity with iron, and coupling complex with DNA gyrase. Furthermore, the induced antibacterial activity of the hybrid PVA/NPs NFs could be synergistically enhanced through CS, which destroys the bacterial cell wall and leads to leakage of cellular contents. CS has been reported to interact through electrostatically with negatively charged LPS of Gram-negative bacteria and teichoic acid of Gram-positive bacteria, leading to cell wall destruction.57
Cyto-Biocompatibility
The biocompatibility of the PVA/NPs nanofibrous mat with human fibroblasts (HFF-1) after 1, 3, and 7 days were observed using MTT assay (Figure 9B). It was noticed that the cell viability on CF0 increased and reached its peak on day 3. The cell viability on CF3 was slightly better than plain PVA (CF0) after 1 and 7 days, where it recorded a significant impact after 3 days. Cell viabilities were found to be around 76%, 113% and 86% after 1, 3 and 7 days, respectively. Meanwhile, cell viability on CF4 increased significantly over the whole period as after 3 and 7 days of cell culture the number of living cells was the highest amongst all of the tested groups. It reached 94% after 1 day and recorded a significant impact after 3 and 7 days with a cell viability of 114% and 123%, respectively. Such results indicate that PE-CS-Au NPs loaded NFs possess better biocompatibility and proliferative action than plain PVA. It can be concluded that the biocompatibility of the tested NFs follows the order CF4 > CF3 > CF0 based on the aforementioned cell viability results, since it is well known that scaffolds with the highest cell viability are the most biocompatible ones because they allow cellular proliferation on their surface. Furthermore, the fibroblasts were active on the surface of the crosslinked PVA nanofibrous mats making them suitable to be applied as effective/potential biocompatible nanofibrous scaffolds for cell proliferation. It was reported that mats with highly nanofibrous topographies and porosity are similar to ECM and are able to enhance the cellular responses.58
As shown in Figure 9C, DAPI staining was also used for visualizing of the increasing HFF-1 proliferation upon their seeding on CF0, CF3 and CF4. The obtained results demonstrated that the amount of cells on CF4 is significantly higher than the other NFs. Also, the number of cells in CF3 was slightly larger than the cells seeded on neat PVA NFs (CF0).
Figure 9D shows representative SEM micrographs of fibroblast cells on CF0, CF3 and CF4 mats (the original image is shown in Figure S3). The overall growth rate of fibroblasts on hybrid PVA/NPs NFs was significantly higher than those on CF0. Moreover, it was found that, at day 7 the cells attached strongly, proliferated and spread along the CF4 more than CF0 and CF3 (Figure S3). Thus, both the fabricated nanofibrous mats showed an acceptable cyto-biocompatibility, where cell proliferation was significantly higher upon incorporating 0.9% of PE-CS-AuNPs in the PVA matrix (ie, CF4). It can be claimed that the increase of the surface of the electrical polarity of PVA NFs upon embedding PE-CS-AuNPs could enhance the adhesion and proliferation of fibroblasts.59 In addition, PE was reported as a cosmeceutical component due to its ability to enhance dermal fibroblasts.15 The anti-inflammatory and antioxidant properties of PE could protect the HFF-1 from oxidative stress and maintain their normal growth.60 This reveals that the developed PE-CS-AuNPs loaded NFs fabricated in the current study could stimulate cells attachments and proliferation. Thus it can be utilized efficiently to regenerate damaged/injured skin cells.
Conclusions
In the present study, potential antibacterial topical nanofibrous mats based on newly synthesized PE-CS-Au NPs incorporated into crosslinked PVA nanofibers have been successfully prepared using the electrospinning technique. Conformational physicochemical and morphological characterizations have been ascertained by FTIR, DSC, SEM and EDX. The results indicated that the PE-CS-Au NPs containing nanofibrous mats conferred superior mechanical, thermal and antibacterial properties. The electrospun nanofibers demonstrated optimum porosity even after crosslinking, excellent cyto-biocompatibility, and provided a strong support for cell adhesion and proliferation. Incorporation of the PE-CS-Au NPs added the triple benefit of CS, Au NPs and PE in killing the investigated bacterial strains. In addition, the presence of PE-CS-Au NPs in the PVA nanofibers enhanced their mechanical properties, long-term stability, PE release profile and antimicrobial activity. This study confirms the applicability of the PVA nanofibers incorporated with PE-CS-Au NPs as potential long-acting drug delivery system for the delivery of PE in different biomedical applications, particularly in wound healing. However, further research is needed on the in-vivo assessment of the developed nanofibrous mats for wound healing.
Data Sharing Statement
The authors confirm that the data supporting the findings of this study are available within the article and the raw data will be available on request.
Acknowledgment
The authors are grateful to the Islamic Development Bank (IDB), Jeddah, Saudi Arabia, for financially supporting this work through the IDB Merit Scholarship Program (Ref. 36/11207330, File No. 23/EGT/P34). The work was supported also by National Science Centre in Poland with project no. 2018/30/Q/NZ7/00281 (M.G.)
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sayed MM, Mousa HM, El-Aassar MR, et al. Enhancing mechanical and biodegradation properties of polyvinyl alcohol/silk fibroin nanofibers composite patches for cardiac tissue engineering. Mater Lett. 2019;255:126510. doi:10.1016/j.matlet.2019.126510
2. Zhu M, Hua D, Pan H, et al. Green electrospun and crosslinked poly (vinyl alcohol)/ poly (acrylic acid) composite membranes for antibacterial effective air filtration. J Colloid Interface Sci. 2018;511:411–423. doi:10.1016/j.jcis.2017.09.101
3. Ullah S, Hashmi M, Hussain N, et al. Stabilized nanofibers of polyvinyl alcohol (PVA) crosslinked by unique method for efficient removal of heavy metal ions. J Water Process Eng. 2020;33:101111. doi:10.1016/j.jwpe.2019.101111
4. Kumeta K, Nagashima IS, Matsui S, Mizoguchi K. Crosslinking reaction of poly(vinyl alcohol) with poly(acrylic acid) (PAA) by heat treatment: effect of neutralization of PAA. J Appl Polym Sci. 2003;90(9):2420–2427. doi:10.1002/app.12910
5. Gil-Castell O, Galindo-Alfaro D, Sánchez-Ballester S, Teruel-Juanes R, Badia JD, Ribes-Greus A. Crosslinked sulfonated poly(vinyl alcohol)/graphene oxide electrospun nanofibers as polyelectrolytes. Nanomater. 2019;9(3):397. doi:10.3390/nano9030397
6. Nataraj D, Reddy R, Reddy N. Crosslinking electrospun poly (vinyl) alcohol fibers with citric acid to impart aqueous stability for medical applications. Eur Polym J. 2020;124:109484. doi:10.1016/j.eurpolymj.2020.109484
7. Destaye AG, Lin C-K, Lee C-K. Glutaraldehyde vapor cross-linked nanofibrous PVA mat with in situ formed silver nanoparticles. ACS Appl Mater Interfaces. 2013;5(11):4745–4752. doi:10.1021/am401730x
8. Zhang R, Yang J, Wu J, et al. Berberine promotes osteogenic differentiation of mesenchymal stem cells with therapeutic potential in periodontal regeneration. Eur J Pharmacol. 2019;851:144–150. doi:10.1016/J.EJPHAR.2019.02.026
9. Gil MI, Tomás-Barberán FA, Hess-Pierce B, Holcroft DM, Kader AA. Antioxidant activity of pomegranate juice and its relationship with phenolic composition and processing. J Agric Food Chem. 2000;48(10):4581–4589. doi:10.1021/jf000404a
10. Zarfeshany A, Asgary S, Javanmard SH. Potent health effects of pomegranate. Adv Biomed Res. 2014;3(1):100. doi:10.4103/2277-9175.129371
11. Satish S, Mohana DC, Raghavendra MP, Raveesha KA. Antifungal activity of some plant extracts against important seed borne pathogens of Aspergillus sp. J Agri Technol. 2007;3(1):109–119.
12. Syed DN, Chamcheu J-C, Adhami VM, Mukhtar H. Pomegranate extracts and cancer prevention: molecular and cellular activities. Anticancer Agents Med Chem. 2013;13(8):1149–1161. doi:10.2174/1871520611313080003
13. Aviram M, Rosenblat M. Pomegranate protection against cardiovascular diseases. Evid Based Complement Altern Med. 2012;2012:382763. doi:10.1155/2012/382763
14. Lukiswanto BS, Miranti A, Sudjarwo SA, Primarizky H, Yuniarti WM. Evaluation of wound healing potential of pomegranate (Punica granatum) whole fruit extract on skin burn wound in rats (Rattus norvegicus). J Adv Vet Anim Res. 2019;6(2):202–207. doi:10.5455/javar.2019.f333
15. Aslam MN, Lansky EP, Varani J. Pomegranate as a cosmeceutical source: pomegranate fractions promote proliferation and procollagen synthesis and inhibit matrix metalloproteinase-1 production in human skin cells. J Ethnopharmacol. 2006;103(3):311–318. doi:10.1016/j.jep.2005.07.027
16. Maurya R, Yadav D, Singh G, et al. Osteogenic activity of constituents from butea monosperma. Bioorg Med Chem Lett. 2008;19(3):610–613. doi:10.1016/j.bmcl.2008.12.064
17. Sreekumar S, Sithul H, Muraleedharan P, Azeez JM, Sreeharshan S. Pomegranate fruit as a rich source of biologically active compounds. Biomed Res Int. 2014;2014:686921. doi:10.1155/2014/686921
18. Spilmont M, Léotoing L, Davicco M-J, et al. Pomegranate peel extract prevents bone loss in a preclinical model of osteoporosis and stimulates osteoblastic differentiation in vitro. Nutrients. 2015;7(11):9265–9284. doi:10.3390/nu7115465
19. Singh N, Singh V, Singh RK, et al. Osteogenic potential of cissus quadrangularis assessed with osteopontin expression. Natl J Maxillofac Surg. 2013;4(1):52–56. doi:10.4103/0975-5950.117884
20. Raafat D, Sahl H-G. Chitosan and its antimicrobial potential – a critical literature survey. Microb Biotechnol. 2009;2(2):186–201. doi:10.1111/j.1751-7915.2008.00080.x
21. Mohamady Hussein MA, Baños FGD, Grinholc M, Abo Dena AS, El-Sherbiny IM, Megahed M. Exploring the physicochemical and antimicrobial properties of gold-chitosan hybrid nanoparticles composed of varying chitosan amounts. Int J Biol Macromol. 2020;162:1760–1769. doi:10.1016/j.ijbiomac.2020.08.046
22. Gomathi P, Ragupathy D, Choi JH, et al. Fabrication of novel chitosan nanofiber/gold nanoparticles composite towards improved performance for a cholesterol sensor. Sensors Actuators B Chem. 2011;153(1):44–49. doi:10.1016/j.snb.2010.10.005
23. Ahmed R, Tariq M, Ali I, et al. Novel electrospun chitosan/polyvinyl alcohol/zinc oxide nanofibrous mats with antibacterial and antioxidant properties for diabetic wound healing. Int J Biol Macromol. 2018;120:385–393. doi:10.1016/j.ijbiomac.2018.08.057
24. Ibrahim HM, Reda MM, Klingner A. Preparation and characterization of green carboxymethylchitosan (CMCS) – polyvinyl alcohol (PVA) electrospun nanofibers containing gold nanoparticles (AuNPs) and its potential use as biomaterials. Int J Biol Macromol. 2020;151:821–829. doi:10.1016/j.ijbiomac.2020.02.174
25. Augustine R, Rehman SRU, Ahmed R, et al. Electrospun chitosan membranes containing bioactive and therapeutic agents for enhanced wound healing. Int J Biol Macromol. 2020;156:153–170. doi:10.1016/j.ijbiomac.2020.03.207
26. Menazea AA, Ahmed MK. Wound healing activity of chitosan/polyvinyl alcohol embedded by gold nanoparticles prepared by nanosecond laser ablation. J Mol Struct. 2020;1217:128401. doi:10.1016/j.molstruc.2020.128401
27. Wu J, Zheng Y, Wen X, Lin Q, Chen X, Wu Z. Silver nanoparticle/bacterial cellulose gel membranes for antibacterial wound dressing: investigation in vitro and in vivo. Biomed Mater. 2014;9(3):35005. doi:10.1088/1748-6041/9/3/035005
28. Hernández Martínez SP, Rivera González TI, Franco Molina MA, et al. A novel gold calreticulin nanocomposite based on chitosan for wound healing in a diabetic mice model. Nanomater. 2019;9(1):75. doi:10.3390/nano9010075
29. Hussein MAM, Grinholc M, Dena ASA, El-Sherbiny IM, Megahed M. Boosting the antibacterial activity of chitosan–gold nanoparticles against antibiotic–resistant bacteria by Punica granatum L. extract. Carbohydr Polym. 2021;256:117498. doi:10.1016/j.carbpol.2020.117498
30. Ali IH, Khalil IA, El-Sherbiny IM. Single-dose electrospun nanoparticles-in-nanofibers wound dressings with enhanced epithelialization, collagen deposition, and granulation properties. Appl Mater Interfaces. 2016;8(23):14453–14469. doi:10.1021/acsami.6b04369
31. Singh YP, Singh RP. Compatibility studies on solid polyblends of poly(methyl methacrylate) with poly(vinyl acetate) and polystyrene by ultrasonic technique. Eur Polym J. 1983;19(6):529–533. doi:10.1016/0014-3057(83)90205-7
32. Haley J, Lodge T. Viscosity predictions for model miscible polymer blends: including self-concentration, double reptation, and tube dilation. J Rheol. 2005;49(3):1277–1302. doi:10.1122/1.2039847
33. Naveen Kumar HMP, Prabhakar MN, Venkata Prasad C, et al. Compatibility studies of chitosan/PVA blend in 2% aqueous acetic acid solution at 30°C. Carbohydr Polym. 2010;82(2):251–255. doi:10.1016/j.carbpol.2010.04.021
34. Bahuleyan BK, Induja C, Ramesan MT. Influence of titanium dioxide nanoparticles on the structural, thermal, electrical properties, and gas sensing behavior of polyaniline/phenothiazine blend nanocomposites. Polym Compos. 2019;40(11):4416–4426. doi:10.1002/pc.25303
35. Saravanan S, Sareen N, Abu-el-rub E, et al. Graphene oxide-gold nanosheets containing chitosan scaffold improves ventricular contractility and function after implantation into infarcted heart. Sci Rep. 2018(6):1–13. doi:10.1038/s41598-018-33144-0
36. Ahmad AA, Al-Juhani AA, Thomas S, De SK, Atieh MA. Effect of modified and nonmodified carbon nanotubes on the rheological behavior of high density polyethylene nanocomposite. J Nanomater. 2013;2013:731860. doi:10.1155/2013/731860
37. Jain S, Goossens JGP, Peters GWM, Duin V, Lemstra PJ. Strong decrease in viscosity of nanoparticle-filled polymer melts through selective adsorption. Soft Mat. 2008;4(9):1848–1854. DOI:10.1039/b802905a
38. Coetzee D, Venkataraman M, Militky J, Petru M. Influence of nanoparticles on thermal and electrical conductivity of composites. Polymers. 2020;12(4):742.
39. Beachley V, Wen X. Effect of electrospinning parameters on the nanofiber diameter and length. Mater Sci Eng C Mater Biol Appl. 2009;29(3):663–668. doi:10.1016/j.msec.2008.10.037
40. Liu Y, Dong L, Fan J, Wang R, Yu J. Effect of applied voltage on diameter and morphology of ultrafine fibers in bubble electrospinning. J Appl Poly Sci. 2010;120(1):592–608. doi:10.1002/app
41. Li C, Fu R, Yu C, et al. Silver nanoparticle/chitosan oligosaccharide/poly(vinyl alcohol) nanofibers as wound dressings: a preclinical study. Int J Nanomedicine. 2013;8:4131–4145. doi:10.2147/IJN.S51679
42. Barani H. Antibacterial continuous nanofibrous hybrid yarn through in situ synthesis of silver nanoparticles: preparation and characterization. Mater Sci Eng C. 2014;43:50–57. doi:10.1016/j.msec.2014.07.004
43. Maleki H, Mathur S. Antibacterial Ag containing core-shell polyvinyl alcohol- poly (lactic acid) nanofibers for biomedical applications. Poly Eng Sci. 2020;(3):1–10. doi:10.1002/pen.25375
44. Abri S, Mehrizi A, Ghaee A, Koosha M. Electrospinning of PVA/chitosan nanocomposite nanofibers containing gelatin nanoparticles as a dual drug delivery system. J Biomed Mater Res A. 2015;103(12):3852–3862. doi:10.1002/jbm.a.35529
45. Ganesh M, Aziz AS, Ubaidulla U, et al. Sulfanilamide and silver nanoparticles-loaded polyvinyl alcohol-chitosan composite electrospun nanofibers: synthesis and evaluation on synergism in wound healing. J Ind Eng Chem. 2016;39:127–135. doi:10.1016/j.jiec.2016.05.021
46. Cui S, Sun X, Li K, et al. Polylactide nanofibers delivering doxycycline for chronic wound treatment. Mater Sci Eng C. 2019;104:109745. doi:10.1016/j.msec.2019.109745
47. Çay A, Miraftab M, Perrin Akçakoca Kumbasar E. Characterization and swelling performance of physically stabilized electrospun poly(vinyl alcohol)/chitosan nanofibres. Eur Polym J. 2014;61:253–262. doi:10.1016/j.eurpolymj.2014.10.017
48. Derakhshandeh K, Erfan M, Dadashzadeh S. Encapsulation of 9-nitrocamptothecin, a novel anticancer drug, in biodegradable nanoparticles: factorial design, characterization and release kinetics. Eur J Pharm Biopharm. 2007;66(1):34–41. doi:10.1016/j.ejpb.2006.09.004
49. G. S, Jha PK, V. V; G. S, Jha PK, V. V, et al. Cannonball fruit (Couroupita guianensis, Aubl.) extract mediated synthesis of gold nanoparticles and evaluation of its antioxidant activity. J Mol Liq. 2016;215:229–236. doi:10.1016/j.molliq.2015.12.043.
50. Hasheminejad N, Khodaiyan F, Safari M. Improving the antifungal activity of clove essential oil encapsulated by chitosan nanoparticles. Food Chem. 2019;275(9):113–122. doi:10.1016/j.foodchem.2018.09.085
51. Dirican M, Yanılmaz M, Fu K, et al. Carbon-Confined PVA-derived silicon/silica/carbon nanofiber composites as anode for lithium-ion batteries. J Electrochem Soc. 2014;161(14):2197–2203. doi:10.1149/2.0811414jes]
52. Choo K, Ching YC, Chuah CH, Julai S, Liou N-S. Preparation and Characterization of polyvinyl alcohol-chitosan composite films reinforced with cellulose nanofiber. Mater. 2016;9(8). doi:10.3390/ma9080644
53. Kim K, Akada Y, Kai W, Kim B, Kim I. Cells attachment property of PVA hydrogel nanofibers incorporating hyaluronic acid for tissue engineering. J Biomater Nanobiotechnol. 2011;2011(10):353–360. doi:10.4236/jbnb.2011.24044
54. Wang Q, Fu Y, Yan X, Chang Y, Ren L, Zhou J. Preparation and characterization of underwater superoleophobic chitosan/poly(vinyl alcohol) coatings for self-cleaning and oil/water separation. Appl Surf Sci. 2017;412:10–18. doi:10.1016/j.apsusc.2017.03.216
55. Gaaz TS, Sulong AB, Akhtar MN, Kadhum AAH, Mohamad AB, Al-Amiery AA. Properties and applications of polyvinyl alcohol, halloysite nanotubes and their nanocomposites. Molecules. 2015;20(12):22833–22847. doi:10.3390/molecules201219884
56. Kong H, Jang J. Antibacterial properties of novel poly(methyl methacrylate) nanofiber containing silver nanoparticles. Langmuir. 2008;24(5):2051–2056. doi:10.1021/la703085e
57. Matica MA, Aachmann FL, Tøndervik A, Sletta H, Ostafe V. Chitosan as a wound dressing starting material: antimicrobial properties and mode of action. Int J Mol Sci. 2019;20(23):5889. doi:10.3390/ijms20235889
58. Yang X, Li Y, Liu X, Huang Q, Zhang R, Feng Q. Incorporation of silica nanoparticles to PLGA electrospun fibers for osteogenic differentiation of human osteoblast-like cells. Regen Biomater. 2018;5(4):229–238. doi:10.1093/rb/rby014
59. Azadian E, Arjmand B, Ardeshirylajimi A, Hosseinzadeh S, Omidi M, Khojasteh A. Polyvinyl alcohol modified polyvinylidene fluoride-graphene oxide scaffold promotes osteogenic differentiation potential of human induced pluripotent stem cells. J Cell Biochem. 2020;121(5–6):3185–3196. doi:10.1002/jcb.29585
60. Celiksoy V, Moses RL, Sloan AJ, Moseley R, Heard CM. Evaluation of the in vitro oral wound healing effects of pomegranate (Punica granatum) rind extract and punicalagin, in combination with zn (II) and. (Ii). 2020;10(9):1234.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.