Back to Archived Journals » Pathology and Laboratory Medicine International » Volume 8
CD30: receptor, marker, target
Authors Gottesman SR
Received 2 February 2016
Accepted for publication 9 March 2016
Published 10 June 2016 Volume 2016:8 Pages 27—36
DOI https://doi.org/10.2147/PLMI.S89466
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Paul Zhang
Susan RS Gottesman,1,2
1Department of Pathology, 2Department of Cell Biology, SUNY Downstate Medical Center, Brooklyn, NY, USA
Abstract: CD30 is a member of the tumor necrosis factor receptor family and is expressed on activated lymphocytes and a few other normal cells. Its signaling activates NF-κB transcription factor, resulting in pleiotropic regulation of gene function. CD30 is the characteristic marker of classical Hodgkin lymphoma, anaplastic large-cell lymphoma, and embryonal cell carcinoma, and it is expressed on a subset of aggressive T- and B-cell neoplasms. Its restricted expression on normal cells makes it an attractive candidate for targeted therapy. The underlying molecular alterations in these malignancies serve to explain the need for a toxin-bound therapeutic reagent.
Keywords: NF-κB, Hodgkin lymphoma, TNF-R, ALCL
Introduction
An objective when diagnosing a malignancy is to characterize the tumor by the origin of the cell that has gone awry due to the acquisition of mutations. This cell of origin is sometimes determined by characteristic markers or cell products, such as cytokeratin, when morphological identification proves difficult. In some instances, these markers provide a target for therapy, as in the case of estrogen receptors, and/or a hint to the pathophysiology or behavior of the tumor as in the case of Her2neu, for breast cancers.
The use of markers for malignant cell characterization is particularly important for lymphoid neoplasms whose classification is based not only on the normal cell counterpart it resembles but also on its stage of differentiation. Each stage of lymphocyte differentiation, state of activation, and subset classification have been well characterized for decades. Information includes not only the presence of a marker but also the biological function of that molecule in the normal immune system.
CD30 is therefore somewhat unique in that it was first identified on the malignant cells of Hodgkin lymphoma.1 The representation of CD30 and CD30L (CD153) on normal immune cells and their functions are only recently being elucidated; this is in spite of the fact that the genes had been cloned years earlier.2 The importance of identifying a malignancy as CD30+ and an educated approach to using the marker as a target for therapy can only be fully realized when we have knowledge of the normal function of CD30. Therefore, this review begins with a summary of the state of knowledge of normal CD30–CD30L interaction in the immune response and the downstream effects of CD30 signaling in the cell, before discussing the CD30+ malignancies.
Molecular characteristics and function of CD30
CD30 is a member of the tumor necrosis factor receptor (TNFR) superfamily, specifically TNFR8, and CD30L is correspondingly a member of the TNF superfamily.3,4 Both are cell surface glycoproteins; the receptor family is characterized by cysteine-rich extracellular domains. The TNFR superfamily exists as type 1 transmembrane monomers that associate as trimers upon interaction with their ligand.
It is generally recognized that both CD30 and CD30L are activation markers demonstrable on certain normal T- and B-cell subsets and that they are not found on resting naïve lymphocytes.5–7 In addition to being expressed on activated lymphocytes and some dendritic cell subsets, CD30L may also be expressed on eosinophils and mast cells.8,9 CD30 has a more restricted distribution, and the few CD30+ cells found in the circulation have other markers indicative of memory cells. In lymph node tissue sections, immunohistochemical staining shows rare CD30+ immunoblasts just outside and circling the germinal centers.7,10 The expression of CD30 on T-cells occurs fairly late after activation in vitro, leading some to suggest that it has a role in promoting cell survival or the generation of memory in the T-cell population, while others suggest that the CD30–CD30L interaction plays a role in terminating the immune response of T-cells. CD30 expression is also found on some virally infected lymphocytes, including Epstein–Barr virus (EBV)-infected B-cells and human T cell leukemia virus-1-infected T-cells (HTLV). Low levels of soluble CD30 can be detected in the serum, and it is suggested that this may be used as a marker of tumor burden for a CD30+ tumor or a marker of disease activity for some autoimmune diseases.11–13
The expression of CD30 is restricted to the lymphoid lineage among normal cells and during embryogenesis. Large lymphoid blasts in the lymph nodes and thymic medulla at 14 weeks of gestation and beyond are CD30+, whereas human fetal lung, kidney, liver, gastrointestinal tract, adrenal gland, spleen, gonads, and brain are negative when assayed at 6 weeks of gestation and beyond.14
Studies of mice in which the CD30 gene was knocked out (CD30−/− mice) at first showed few, if any, immune deficiencies.15–17 Although there were some reports of an increase in autoimmune diseases in the absence of CD30 (an observation at variance with the suggestion of high soluble CD30 in autoimmune diseases), these reports have not been substantiated.18 Subsequent studies from several groups demonstrated defects in secondary humoral immune responses in the CD30 knockout mice.9,19 This would fit with the concept that CD30 expression on activated B-cells is a requirement for maximum response. The CD30L+ cells supplying those signals could be the activated T-cells or accessory cells such as dendritic cells associated with the germinal center. However, the results could also be explained by a deficiency in T-cell memory in the absence of CD30. As both activated T- and B-cells normally express CD30, in the absence of CD30, neither would be capable of receiving CD30 signaling. Therefore, it is difficult to separate the importance of CD30 signaling in T-cells versus B-cells in these studies. Seemingly, disparate results were found with human lymphocytes tested in vitro.8 In this study, upregulation of CD30L on immunoglobulin (Ig)M+ IgD+ B-cells occurred during the course of stimulation through the B-cell receptor and CD40. The CD30L on the B-cells in turn stimulated the upregulation of CD30 on T-cells, and the subsequent interaction between the cells actually decreased isotype class switching. The effects of CD30–CD30L interactions on cell-mediated immunity are even less well delineated; however, the expression of the ligand on those cells of the innate immune system that initiates the cell-mediated adaptive immune response points to a role.
CD30 signaling
To understand the significance of CD30 signaling in a cell, the mechanism of action of the transcription factor NF-κB, its major downstream mediator, needs to be considered. NF-κB, a pleiotropic regulator of gene function, is actually multiple transcription factors, each composed of two polypeptides (out of a total of seven) as homo- or heterodimers.20 The proteins p50 and p65/RelA are ubiquitously expressed, and as a dimer respond to inflammatory mediators. cRel protein (also called Rel) is expressed in hematopoietic cells and more highly expressed in lymphocytes. The NF-κB dimers are normally kept in inactive form by inhibitor proteins (IκB) which are bound to them. The canonical and noncanonical pathways of NF-κB activation are distinguished by the kinases (IKK) employed to degrade the IκB inhibitory proteins, and thereby release the NF-κB from inhibition. In the canonical pathway, IKK1 (also called IKKα), IKK2 (also called IKKß), and the scaffold protein NF-κB essential modulator (NEMO, also called IKKγ) rapidly degrade Iκ proteins, releasing NF-κB and leading to proliferation and survival as well as apoptosis as part of the inflammatory response of lymphocytes. This response is both rapid and rapidly reversible. Any signaling through the noncanonical pathway leads to the activation of IKKα dimer, degradation of Iκ proteins, and a slow but long-lasting activation of NF-κB.21
How does this relate to CD30 signaling? CD30 exists as a monomer and forms a homotrimer once bound to its ligand. CD30 does not itself have a kinase domain. Rather, the C-terminal end, which is part of the intracytoplasmic tail, recruits TNFR-associated factors (TRAF), particularly TRAF2 and TRAF5. These associate with other proteins into E2 and E3 ligase complexes leading to polyubiquitination of receptor-activating protein kinase (RIP1) and thus recruitment and activation of IKK.22 Eventually, this signaling results in downstream NF-κB signaling, which can have various effects on the cell. In actuality, there is still conflicting data on the route taken between CD30 and NF-κB, whether it works through the canonical path, noncanonical path, or both, with most data supporting at least canonical pathway signaling. Overall, CD30 signaling has some role in proliferation, differentiation, and survival (anti-apoptotic).23 This is a common feature of signal transduction through TNFR superfamily cascades. They activate inhibitors of apoptosis, which, in addition to inhibiting caspases, also modulate cell proliferation and activate the NF-κB transcription factor family.21 A second pathway of CD30 signaling may occur through TRAF2 activation of the mitogen-activated protein kinase (MAPK) cascade.24 The MAPK pathway can then activate c-Fos which combines with c-Jun to form the heterodimeric transcription factor activator protein-1 (AP-1). This is required for progression through the G1 phase of the cell cycle and can cooperate with NF-κB to prevent apoptosis. Aryl hydrocarbon nuclear translocator functions as part of a negative feedback loop for CD30, turning off signaling.25
Interestingly, a variant form of CD30 (CD30v) has been identified. This variant form is the cytoplasmic tail of CD30 only. It does not require ligand for signaling but does require TRAFs for NF-κB activation and is found to co-localize to the nucleus with TRAFs.26 Its presence in Hodgkin lymphoma cells and anaplastic large-cell lymphoma (ALCL) cells may partially explain the results showing a lack of dependency of the malignant cells on intact CD30 signaling (refer to the following sections).
Identification of CD30
The identification of CD30 is intimately tied to the progress made in the diagnosis and definition of classical Hodgkin lymphoma, the “poster child” of CD30+ neoplasms. In Hodgkin lymphoma, a neoplasm of lymphoid tissue, the origin of its malignant cell, the Reed–Sternberg cell, and Hodgkin variants was unknown for more than a century after the description of the disease in 1832. This void of information had several causes: the morphological dissimilarity of the malignant cell to any normal cell, the rarity of the malignant cells in relation to the background reactive cells, the absence of lineage-specific markers, and the inability to culture the cells in vitro. In vitro cell lines of Hodgkin lymphoma were produced and reported on in 1980.27 The cell line, L428, was used to generate Reed–Sternberg-specific mouse antibodies,1 followed by the production of the monoclonal antibody (moAb), Ki-1, against the malignant cells. This antigen, specifically found on the malignant cells and initially not found on any cells in normal tonsil, was named Ki-1 antigen after the antibody.28 The designation CD30 was assigned to Ki-1 at the Third International Workshop and Conference on Human Leukocyte Differentiation Antigens in 1986. The identification of the Ki-1 antigen on cells was hampered by the requirement for frozen tissue for analysis. In addition, the Ki-1 antibody was subsequently found to react with a second molecule called Ki-1/57, a nucleic kinase. In 1989, a new hybridoma line, BerH2, was reported.29 This cell line produced an antibody, BerH2 moAb, that was able to stain formalin-fixed paraffin-embedded tissue as a result of its specificity for a different epitope on CD30. In addition to its broader applicability to tissue, BerH2 moAb was of higher affinity and therefore able to detect the CD30 antigen expressed at lower density on different cell and tumor types. In situ hybridization for CD30 transcripts confirmed the staining pattern of BerH2. For Reed–Sternberg cells and variants, the characteristic staining pattern is surface membrane (actually an intracellular epitope) and Golgi dot. Normal immunoblasts generally show lighter staining, often with a diffuse pattern and cytoplasmic blush.
Although the detection and visualization of CD30 on Reed–Sternberg cells and variants did not lead to identification of the lineage of the malignant cells, it did provide both a diagnostic tool and a tool to isolate the cells from tissue. Using microdissection of individual CD30+ cells from frozen tissue of Hodgkin lymphoma, followed by molecular analysis of T-cell receptor (TCR) and Ig genes, a group headed by Kuppers and Rajewsky identified Reed–Sternberg cells and variants as members of the B-cell lineage.30–32 This analysis also further suggested that they may be germinal center B-cells, which would normally have undergone apoptosis (refer to the following sections).
CD30+ malignancies: overview
CD30 is found on the malignant cells of classical Hodgkin lymphoma, a B-cell neoplasm; CD30+ ALCL, usually T-cell in origin; occasional high-grade lymphomas of T- or B-cell origin; testicular embryonal carcinoma, a nonhematopoietic tumor; and possibly undifferentiated pleomorphic sarcoma (UPS), previously called malignant fibrous histiocytoma, and inflammatory myofibroblastic tumor. Although reported as being weakly expressed on a minor percentage of virtually all solid tumors, none of these reports have been confirmed. CD30 expression on nonlymphoid malignancies may be regarded as true aberrant expression, since the normal counterparts of these tumors do not express the antigen.
Testicular embryonal carcinoma
Embryonal carcinoma is a germ cell tumor, nonseminomatous type, which may present in pure form or more often may be mixed with other malignant germ cell elements. The embryonal component is composed of undifferentiated cells, most similar to embryonic stem cells. CD30 expression is documented, not only on the vast majority of embryonal carcinomas but also on the vast majority of cells within the tumor. This strong and consistent staining is in contrast to other germ-cell-derived tumors and in contrast to staining on normal fetal gonadal tissue. Thus, this staining allows the distinction between embryonal carcinoma and atypical seminoma and the identification of embryonal elements in mixed germ cell tumors.14,33 This was confirmed by in situ hybridization for CD30 mRNA and is of significant therapeutic and prognostic importance.34 Whereas following initial surgery, pure seminomas were successfully treated with radiation, depending on the stage, mixed germ cell tumors have a worse prognosis and were and still are treated with chemotherapy. Recently, seminoma may also be treated with single-agent chemotherapy to avoid the side effects of radiation.35 CD30 expression on embryonal carcinoma should also be kept in mind when a young male presents with a mediastinal mass and no known primary testicular tumor.
Expression of CD30 on other nonlymphoid tumors
CD30 may be represented on a subset of UPS and/or inflammatory myofibroblastic tumors.36,37
UPS is a dilemma unto itself with no clear understanding of the precursor of this high-grade mesenchymal tumor. It may represent the end point of several different sarcomas and therefore may have multiple possible cell types of origin. These tumors are usually in the deep soft tissue of extremities.
Inflammatory myofibroblastic tumor is another lesion of uncertain origin, cellular composition and, in this case, even clinical consequences. Alternatively considered an inflammatory and reactive lesion versus a benign tumor or a tumor with a small potential for metastasis, its nature is still not well defined. As indicated from its name, it consists of numerous myofibroblasts along with inflammatory cells. CD30 expression, if present, may be related to the inflammatory background. In contrast to UPS, inflammatory myofibroblastic tumors may arise in multiple organs. It is generally regarded as a distinct entity whose separation from an inflammatory pseudotumor at one extreme and an inflammatory fibrosarcoma on the other extreme can be difficult. The rearrangement and expression of anaplastic lymphoma kinase (ALK) in a subset of cases, particularly pediatric cases, would lead to confusion with some ALK+ lymphomas; lesions which are also CD30+, were the morphologies not so distinct.
Although the consistency and significance of CD30 expression on a subset of these two lesions are unknown, the possibility of using targeted therapy still exists, particularly in the case of UPS positive for the antigen.
Expression of CD30 on lymphoid neoplasms
Without question, the expression of CD30 is most relevant to neoplasia of the lymphoid system. Beyond the fact that any large-cell T- or B-cell lymphoid neoplasm may express CD30 on few or all of its malignant cells, some neoplasms are characterized and classified by its expression. The following sections review those lesions in which its expression is characteristic of the tumor and a brief description of those known to sometimes express the antigen. Keep in mind that not all CD30 overexpression is created equal. The mechanisms underlying the overexpression and the downstream mutations and results of signaling or blocking signaling through CD30 differ. This had consequences when developing CD30-targeted therapy.
Hodgkin lymphoma
Although the use of Ki-1 and BerH2 antibody, in combination with other markers (or lack thereof), to aid in the recognition of Reed–Sternberg cells and Hodgkin variants practically revolutionized the diagnosis of Hodgkin lymphoma for pathologists, it did not provide insight as to the normal counterpart of the Reed–Sternberg cells. That finding had to await the elegant studies of a group headed by Kuppers and Rajewsky30–32 who microdissected individual Reed–Sternberg cells from frozen tissue and employed polymerase chain reaction technology to detect Ig and TCR gene rearrangements. They not only showed that Ig genes were rearranged in Reed–Sternberg cells but also demonstrated similar rearrangements among individual Reed–Sternberg cells from the same node, from multiple nodes in the same patient, and from the same patient upon relapse. The rearranged Ig genes had variable regions which appeared to have undergone somatic mutations which were nonproductive. These mutations should have led to apoptosis of the B-cells in the germinal center. Although on rare occasions, T-cell gene rearrangements have been found, the vast majority of Hodgkin lymphomas are considered to be B-cell in origin, a post-germinal center B-cell that normally would have died in the germinal center. The malignant cell is characterized as CD30+, CD15+/−, and CD20−/+. All other lineage-specific markers are absent as is CD45 (at least in formalin-fixed tissue) and epithelial membrane antigen (EMA). Many of the transcription factors characteristic of B-cells and hence responsible for B-cell gene expression programs are downregulated and are not detected in Reed–Sternberg cells. Examples include the absence of expression of OCT2, BOB1, and PU.1 with partial decrease in PAX5.38,39 The absence of the B-cell-specific transcription factors accounts for the lack of expression of B-cell-specific markers. This, combined with the crippling changes in the Ig genes, accounts for the lack of B cell receptor (BCR) expression. The question then becomes, what allows for the continued survival of the cell particularly in the absence of BCR signaling. Perhaps the inflammatory background is providing the survival signals.
Classical Hodgkin lymphoma, in addition to having diagnostic Reed–Sternberg cells and Hodgkin variants, has an inflammatory background which dictates the subtype of Hodgkin lymphoma: lymphocyte rich, nodular sclerosis, mixed cellularity, and lymphocyte depleted (Figure 1A and B). Nodular lymphocyte-predominant Hodgkin lymphoma is a separate entity whose malignant cells are most often but not always CD30−, are CD15−, and express CD20, the classic B-cell lineage marker.
Although now recognized for most malignancies, it has been known for a long time that in classical Hodgkin lymphoma the malignant cells and background cells are engaged in cross talk. CD30 is in the middle of that fray. For some subsets of Hodgkin lymphoma, mixed cellularity, in particular, EBV infection of the malignant cells plays a role. We will not go into that in this review except to point out that EBV infection of normal B-cells leads to upregulation of CD30 expression. Even in EBV– Hodgkin cases, CD30 is overexpressed. This may be due to an increase in AP-1 or the constitutive activation of other mediators such as STAT5A and STAT5B that can induce the upregulation of CD30 in normal B-cells.40 There is conflicting evidence as to whether CD30 on Hodgkin cells needs ligand binding for activation, whether it is constitutively active, or whether its activation is necessary at all. If ligand binding is needed, there is no shortage of CD30L+ cells in the environment (Figure 1A). Recall that CD30L is expressed on eosinophils, mast cells, and some dendritic cells. Mixed cellularity Hodgkin lymphoma and most nodular sclerosis Hodgkin lymphoma in particular are characterized by eosinophils in their environment. The interaction may work both ways with CD30 providing a survival signal for those CD30L-expressing inflammatory cells.
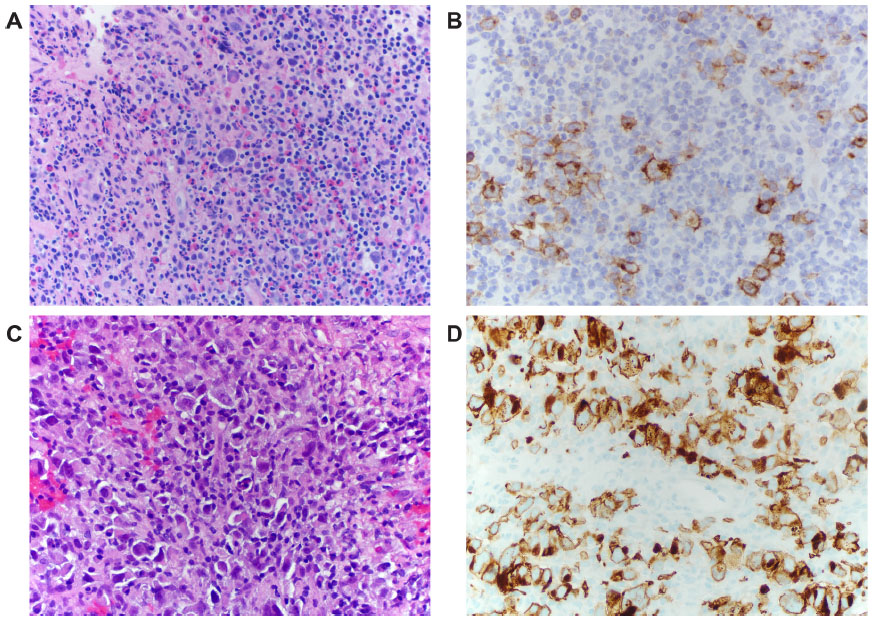 | Figure 1 CD30 staining of Hodgkin Lymphoma vs ALCL. |
CD30 on the Hodgkin cells, whether it requires a ligand or not, activates NF-κB, certainly through the canonical pathway at least, as well as being one route for phosphoinositide 3-kinase (PI3K) activation. The latter will act as an anti-apoptotic signal. In general, the activation of CD30, as well as other stimulatory pathways (CD40–CD40L, CD80–CD28), results in increased survival, cytokine production, and increased proliferation of Reed–Sternberg cells when investigated experimentally. But are they necessary? The malignant cells may also harbor mutations downstream of these receptors which allow the continued survival independent of receptor signaling. Is survival partly a positive feedback loop that includes CD30? Does this change in programming which leads to increased CD30 expression also result in increased NF-κB and thus a survival and proliferative signal? Perhaps the answer is partially.
It is doubtful that the cell is dependent on stimulation starting at CD30 (with or without ligand). In fact, multiple investigators have found no effect of either stimulating through CD30 or downregulating CD30 on Hodgkin cell lines.41–43 This is in contrast to similar studies on ALCL lines by the same investigators (refer to the following sections).43 Rather, Hodgkin cells have constitutive NF-κB activity quite independent of CD30 signaling.41,44 This constitutive activity is likely the result of mutations downstream of CD30 signaling or mutations in NF-κB components themselves. Some examples that have been found include mutations in Janus kinase (JAK)-STAT, which are most frequently activating mutations.45 As previously noted, CD30v, the cytoplasmic tail of CD30, has been identified in Hodgkin lymphoma and ALCL cells and it functions independently of ligand in the cytoplasm and nucleus of the cell.26 More than 50% of Hodgkin lymphomas have been reported to have an amplification or genomic gain of REL, coding for a component of NF-κB heterodimer which results in its increased transcription.46 In addition, the loss-of-function mutations have been discovered in multiple inhibitors of NF-κB such as mutations in IκBα impairing its ability to inhibit NF-κB.47,48 Thus, there are many routes to the final effect of increased NF-κB, many of which are downstream of CD30. Constitutively active NF-κB in Hodgkin cells then activates STAT5a; its overexpression adds to the progression through the cell cycle and inhibition of apoptosis.49 This of course implies that blocking CD30 or other signaling from the environment may have little effect on the malignant cells, even if there is much cross talk occurring. On the other hand, the overexpression of CD30 on the cell surface may contribute to the attraction of the inflammatory cells in the background. These cells may then have additional means via other stimulatory molecules, of supporting the survival of the malignant cells. Certainly, they contribute to the clinical symptoms we see in Hodgkin lymphoma: fever, suppressed cell-mediated immunity, and eosinophilia.
Anaplastic large-cell lymphoma
ALCL is actually a group of CD30+ lymphoid malignancies whose cells of origin were elusive until the advent of modern molecular techniques and which have both overlapping and important distinctive characteristics. There are three subtypes: ALCL, ALK+; ALCL, ALK− (provisional entity); and primary cutaneous ALCL (C-ALCL). All are CD30+ T-cell neoplasms (Figure 1C and D).50
The two systemic lymphomas, ALK+ and ALK− both strongly express CD30, have a paucity or absence of T-lineage markers, and may express EMA, particularly in the ALK+ group. This aberrant expression of an epithelial antigen, along with its propensity, in some patients, to infiltrate the lymph nodes in a sinusoidal fashion originally led to some misdiagnoses of metastatic carcinoma.
The ALK+ ALCL, by definition, overexpresses ALK as the result of a translocation to one of several partner genes. The most common partner is nucleophosmin (NPM) that allows the transport of ALK to the nucleus, where it acts as an oncoprotein. CD30 is expressed strongly on the large cells but may be absent from the smaller malignant cells. The expression of ALK distinguishes this lymphoma from peripheral T-cell lymphomas (PTCL) which may express CD30 on some of their malignant cells. On the other hand, the expression of CD30 helps distinguish this from the rare ALK+ diffuse large B-cell lymphoma (DLBCL) which also loses lineage markers.51
Ninety percent of ALK+ ALCL have TCR gene rearrangements, the remaining 10% have neither TCR nor Ig gene rearrangements. All express proteins characteristic of cytotoxic T-cells, TIA1, granzyme B, or perforin, and are considered by the World Health Organization classification system as having an activated cytotoxic T-cell as the normal cell counterpart; yet most are CD4+, rather than CD8.50
ALK− ALCL is a morphologically and immunophenotypically similar CD30+ T-cell lymphoma in that CD30 overexpression is strong and positive in virtually all tumor cells. This uniform CD30 positivity distinguishes this entity from PTCL, NOS, in which only some cells express CD30.
ALK+ and ALK− ALCLs are two different entities as evidenced by the fact that the ALK+ ALCL is more frequent in children and young adults, whereas ALK− ALCL is most frequent in an older adult age group although there is overlap. The distinction is important as the response to therapy and prognosis is much more favorable in the ALK+ group.
Primary C-ALCL, as the name implies, is a CD30+ ALCL neoplasm that is limited to the skin. The distinction from the abovementioned two entities is complicated by the fact that both ALK+ and ALK− ALCL, noncutaneous types, can involve the skin as their primary site. C-ALCL does not harbor ALK translocations and often expresses cutaneous lymphocyte antigen, a skin-homing receptor.
In contrast to Hodgkin lymphoma, studies on ALCL cell lines demonstrated an effect of signaling through CD30. Although investigators agree that there is an effect if one stimulates or blocks CD30 activation, there is confusion in the results obtained. This may rest in the pleiotropic and seemingly disparate effects of CD30 signaling even in nonmalignant cells. There are both prosurvival and proliferative signals promoted through NF-κB stimulation. Increased apoptosis may be the result of prolonged signaling from CD30.
Atsaves et al52 reported that CD30 overexpression is the result of the upregulation and/or amplification of JUNB, which in turn binds to the CD30 promoter. The absence of JUNB, decrease in CD30 expression, or antibody blocking of CD30 signaling all resulted in cell cycle arrest. In contrast, Mir et al41 and Wright et al43 showed that stimulation of CD30 on ALCL cells (but not Hodgkin lymphoma cells) resulted in rapid apoptosis of at least a portion of the malignant cells. However, the surviving cells showed activation of NF-κB which, with prolonged CD30 stimulation, resulted in cell cycle arrest.43 Hirsch et al42 suggest that CD30 stimulation results in two opposing effects: NF-κB activation leading to increased proliferation and survival and caspase activation leading to apoptosis. They also agree that CD30 modulation on Hodgkin lymphoma cells is without effect. For ALCL and CD30, at least, it seems that timing is everything.
C-ALCL and lymphomatoid papulosis
When the CD30+ lesions are limited to the skin, the question becomes: Are we dealing with C-ALCL or lymphomatoid papulosis (LyP)? In actuality, the lesions may be considered a spectrum with the clinicians treating LyP as a reactive or benign rather than a malignant entity. Startlingly, in both, the infiltrate of inflammatory cells and ominous appearing large cells may spontaneously regress or at least may be self-limiting. For both the normal counterpart is an activated skin-homing T-cell. As noted earlier, the abnormal cells do not have ALK translocations and they do not express EMA. They express cytotoxic proteins but only a minority expresses CD8. C-ALCL and two of the three subtypes of LyP (types A and C) express CD30 on the malignant/abnormal cells. Not surprisingly, there is a gray zone between the lesions where distinction is not possible. Demonstration of monoclonality based on TCR gene rearrangements is not considered proof of malignancy nor used to favor one diagnosis over the other.50
Enteropathy-associated T-cell lymphoma
This very aggressive lymphoma of activated intraepithelial T-cells is usually composed of large malignant cells and is associated with celiac disease evident prior to or at the time of diagnosis of the lymphoma. A subset of enteropathy-associated T-cell lymphoma is composed of small T-cells and has no relationship with celiac disease. This may in fact be a totally separate entity. In the large cell type, almost all cases are CD30+, but the antigen is expressed on only some of the malignant cells. The cells contain cytotoxic granule-associated proteins and are either “double negative” (CD4−, CD8−) or partial CD8+. The αß TCR is expressed.53
Mature T- and B-cell lymphomas: a CD30 survey
Given that normal activated T and B immunoblasts may express CD30, it is not surprising that the antigen can be found on a large number of individual cases of aggressive peripheral lymphomas. That the expression of CD30 seems to be limited to the more aggressive lymphomas is in keeping with their normal cell counterpart being activated lymphocytes. Thus, expression of CD30 on malignant cells may be found on numerous types of PTCL including PTCL, NOS54 and some DLBCL. Even within an individual lesion, CD30 is often on the more anaplastic looking of the malignant cells. In general, it is only seen on a portion of the cells and often not as intensely as is characteristic of classical Hodgkin lymphoma or ALCL. Examples are DLBCL associated with chronic inflammation and DLBCL of the elderly, both of which may be EBV associated; DLBCL, anaplastic variant; and plasmablastic lymphoma. Most consistent is the expression of CD30 on primary mediastinal large B-cell lymphoma. In contrast to the staining in classical Hodgkin lymphoma, the staining for CD30 is weak and the staining for CD20 is strong and consistent. There are “gray-zone” lymphomas showing features, including immunophenotype, characteristic of both classical Hodgkin lymphoma, and primary mediastinal large B-cell lymphoma.55
In addition to the expression of CD30 on some PTCL, NOS and those T-cell lymphomas characteristically positive for the antigen, CD30 may be expressed on adult T-cell leukemia/lymphoma cells, the human T-cell leukemia virus-associated malignancy. Here, CD30 is distinctly present on the large cells and may first appear when the patient relapses with a more aggressive looking lesion. These T-cells are most often CD4+ with features of T-regulatory cells.56
Making use of CD30 expression for more than just phenotyping
The first attempts to use CD30 as a target for therapy employed unconjugated anti-CD30 on Hodgkin cell lines, but this had little or no effect.57 Neither agonistic nor antagonistic actions on CD30 would be expected to produce apoptosis when one considers that the malignant cells often have mutations downstream of CD30 signaling which result in constitutive activation of the pathways. Given that anti-CD20 moAb therapy is so successful for the treatment of B-cell lymphomas via its stimulation of antibody-dependent cell-mediated cytotoxicity (ADCC), the Fc portion of the CD30 moAb was altered to allow for more effective ADCC. Unfortunately, the use of this altered moAb was also unsuccessful in Phase I trials although the preclinical studies had looked promising.58
The clear difference in experimental results between Hodgkin lymphoma and ALCL cell lines with regard to CD30 modulation strongly suggests that CD30’s involvement in the oncogenic process of these CD30+ neoplasms is different. The insensitivity of Hodgkin lymphoma cells to CD30 signaling or downregulation suggests that this molecule is a bystander, a convenient marker with only limited biological significance to the malignant cell. Perhaps we need to change our “poster child” for CD30 to ALCL, where modulating CD30 has significant, though difficult to interpret, results. Attempted therapy with anti-CD30 antibody alone was unsuccessful in Hodgkin lymphoma patients, not stimulating enough ADCC to get the job done. It is probably wise that these antibodies were not tested in patients with ALCL, given that the tumor might actually have been stimulated by agents binding CD30. Thus, use of CD30 as a target for both lymphomas was attempted with a toxin incorporated into the reagent, and this was found to be successful.
Brentuximab vedotin has been approved for use in primary refractory and relapsed Hodgkin lymphoma.59 This anti-CD30 moAb is conjugated to a toxin, monomethyl auristan E which has anti-microtubule effect. The antibody–drug conjugate binds specifically to the CD30+ cell and is endocytosed; the toxin is released by the actions of lysosomal enzymes on the linker.60 The toxin then causes cell cycle arrest and apoptosis. A recently reported Phase III trial employed brentuximab as consolidation therapy following autologous stem cell transplant in primary refractory and relapsed Hodgkin lymphoma.61 Results of Phase I and II clinical trials using brentuximab prior to standard chemotherapy as first-line treatment are being evaluated.
Brentuximab vedotin has been, or is being, evaluated in the treatment of virtually all CD30+ T-cell lymphomas. A Phase II trial showed promising results in relapsed and refractory ALCL leading to immediate US Food and Drug Administration approval.62 Trials are also underway for CD30+ cutaneous T-cell lymphoma, primary C-ALCL, LyP, CD30+ mycosis fungoides, and Sézary syndrome. Its specificity for CD30, the scarcity of CD30 on most normal cells, and the release of the toxin only following lysosomal action make use of this drug attractive in combination with standard therapy for these difficult diseases.
What started as a mere marker to assist in the pathological diagnosis of Hodgkin lymphoma is now the basis of successful targeted therapy in several CD30+ diseases. One caveat will be the question of its effectiveness in neoplasms demonstrating the CD30 antigen on only a portion of the malignant cells. Certainly, many of these are aggressive malignancies in urgent need of additional therapeutic modalities. Using CD30 to deliver toxin to the malignant cell while being able to avoid attacking normal cells should increase options for these patients.
Disclosure
The author reports no conflicts of interest in this work.
References
Stein H, Gerdes J, Kirchner H, Schaadt M, Diehl V. Hodgkin and Sternberg-Reed cell antigen(s) detected by an antiserum to a cell line (L428) derived from Hodgkin disease. Int J Cancer. 1981;28(4):425–429. | |
Dürkop H, Latza U, Hummel M, Eitelbach F, Seed B, Stein H. Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin’s disease. Cell. 1992; 68(3):421–427. | |
Smith CA, Gruss HJ, Davis T, et al. CD30 antigen, a marker for Hodgkin’s lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell. 1993;73(7):1349–1360. | |
Gedrich RW, Gilfillan MC, Duckett CS, Van Dongen JL, Thompson CB. CD30 contains two binding sites with different specificities for members of the tumor necrosis factor receptor-associated factor family of signal transducing proteins. J Biol Chem. 1996;271(22):12852–12858. | |
Ellis TM, Simms PE, Slivnick DJ, Jäck HM, Fisher RI. CD30 is a signal-transducing molecule that defines a subset of human activated CD45RO+ T cells. J Immunol. 1993;151(5):2380–2389. | |
Gilfillan MC, Noel PJ, Podack ER, Reiner SL, Thompson CB. Expression of the costimulatory receptor CD30 is regulated by both CD28 and cytokines. J Immunol. 1998;160(5):2180–2187. | |
Stein H, Gerdes J, Schwab U, et al. Identification of Hodgkin and Sternberg-Reed cells as a unique cell type derived from a newly-detected small-cell population. Int J Cancer. 1982;30(4):445–459. | |
Cerutti A, Schaffer A, Goodwin R, et al. Engagement of CD153 (CD30L) by CD30+ T cells inhibits class switch DNA recombination and antibody production in Human IgD+ IgM+ B cells. J Immunol. 2000;165(2):786–794. | |
Kennedy MK, Willis CR, Armitage RJ. Deciphering CD30 ligand biology and its role in humoral immunity. Immunology. 2006;118(2):143–152. | |
Falini B, Pileri S, Pizzolo G, et al. CD30 (Ki-1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood. 1995;85(1):1–14. | |
Pizzolo G, Vinante F, Chilosi M, et al. Serum levels of soluble CD30 molecule (Ki-1 antigen) in Hodgkin’s disease: relationship with disease activity and clinical stage. Br J Haematol. 1990;75(2):282–284. | |
Nadali G, Vinante F, Stein H, et al. Serum levels of the soluble form of CD30 molecule as a tumor marker in CD30 | |
Caligaris-Cappio F, Bertero MT, Converso M, et al. Circulating levels of soluble CD30, a marker of cells producing Th2-type cytokines, are increased in patients with systemic lupus erythematosus and correlate with disease activity. Clin Exp Rheumatol. 1995;13(3):339–343. | |
Latza U, Foss HD, Dürkop H, et al. CD30 antigen in embryonal carcinoma and embryogenesis and release of the soluble molecule. AmJ Pathol. 1995;146(2):463–471. | |
Amakawa R, Hakem A, Kundig TM, et al. Impaired negative selection of T cells in Hodgkin’s disease antigen CD30-deficient mice. Cell. 1996;84(4):551–562. | |
Kishimoto H, Sprent J. Several different cell surface molecules control negative selection of medullary thymocytes. J Exp Med. 1999;190(1):65–73. | |
Flórido M, Borges M, Yagita H, Appelberg R. Contribution of CD30/CD153 but not of CD27/CD70, CD134/OX40L, or CD137/4-1BBL to the optimal induction of protective immunity to Mycobacterium avium. J Leukoc Biol. 2004;76(5):1039–1046. | |
Kurts C, Carbone FR, Krummel MF, Koch KM, Miller JF, Heath WR. Correction: signalling through CD30 protects against autoimmune diabetes mediated by CD8 T cells. Nature. 2000;407(6802):413. | |
Gaspal FM, Kim MY, McConnell FM, Raykundalia C, Bekiaris V, Lane PJ. Mice deficient in OX40 and CD30 signals lack memory antibody responses because of deficient CD4 T cell memory. J Immunol. 2005;174(7):3891–3896. | |
Hoffmann A, Baltimore D. Circuitry of nuclear factor κB signaling. Immunol Rev. 2006;210:171–186. | |
Kocab AJ, Duckett CS. Inhibitor of apoptosis proteins as intracellular signaling intermediates. FEBS J. 2016;283(2):221–231. | |
Aizawa S, Nakano H, Ishida T, et al. Tumor necrosis factor receptor associated factor (TRAF)5 and TRAF2 are involved in CD30-mediated NFKappaB activation. J Biol Chem. 1997;272(4):2042–2045. | |
Buchan SL, Al-Shamkhani A. Distinct motifs in the intracellular domain of human CD30 differentially activate canonical and alternative transcription factor NF-κB signaling. PLOS One. 2012;7(9):e45244. | |
Wendtner CM, Schmitt B, Gruss HJ, et al. CD30 ligand signal transduction involves activation of a tyrosine kinase and of mitogen-activated protein kinase in a Hodgkin’s lymphoma cell line. Cancer Res. 1995;55(18):4157–4161. | |
Wright CW, Duckett CS. The aryl hydrocarbon nuclear translocator alters CD30-mediated NF-κB-dependent transcription. Science. 2009; 323(5911):251–255. | |
Thakar NY, Ovchinnikov DA, Hastie ML, Kobe B, Gorman JJ, Wolvetang EJ. TRAF2 recruitment via T61 in CD30 drives NF-κB activation and enhances hESC survival and proliferation. Mol Biol Cell. 2015;26(5):993–1006. | |
Schaadt M, Diehl V, Stein H, Fonatsch C, Kirchner H. Two neoplastic cell lines with unique features derived from Hodgkin’s disease. Int J Cancer. 1980;26(6):723–731. | |
Schwab U, Stein H, Gerdes J, et al. Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature. 1982;299(5878):65–67. | |
Schwarting R, Gerdes J, Dürkop H, Falini B, Pileri S, Stein H. Ber-H2: a new anti-Ki-1 (CD30) monoclonal antibody directed at a formol-resistant epitope. Blood. 1989;74(5):1678–1689. | |
Küppers R, Rajewsky K, Zhao M, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A. 1994;91(23):10962–10966. | |
Kanzler H, Küppers R, Hansmann ML, Rajewsky K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med. 1996;814(4):1495–1505. | |
Vockerodt M, Soares M, Kanzler H, et al. Detection of clonal Hodgkin and Reed-Sterberg cells with identical somatically mutated and rearranged VH. Blood. 1998;92(8):2899–2907. | |
Leroy X, Augusto D, Leteurtre E, Gosselin B. CD30 and CD117 (c-kit) used in combination are useful for distinguishing embryonal carcinoma from seminoma. J Histochem Cytochem. 2002;50(2):283–285. | |
Dürkop H, Foss HD, Eitelbach F, et al. Expression of CD30 antigen on non-lymphoid tissues and cells. J Pathol. 2000;190(5):613–618. | |
NCCN [homepage on the Internet]. NCCN Guidelines Testicular Cancer version 1.2016. Available from: www.nccn.org. Accessed March 24, 2016. | |
Mechtersheimer G, Möller P. Expression of Ki-1 antigen (CD30) in mesenchymal tumors. Cancer. 1990;66(8):1732–1737. | |
Mariño-Enríquez A, Wang WL, Roy A, et al. Epithelioid inflammatory myofibroblastic sarcoma: an aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol. 2011;35(1):135–144. | |
Stein H, Marafioti T, Foss HD, et al. Downregulation of BOB.1 and Oct2 in classical Hodgkin disease but not in lymphocyte predominant Hodgkin disease correlates with immunoglobulin transcription. Blood. 2001;97(2):496–501. | |
Küpper R. The biology of Hodgkin’s lymphoma. Nat Rev Cancer. 2009;9(1):15–27. | |
Scheeren FA, Diehl SA, Smit LA, et al. IL-21 is expressed in Hodgkin lymphoma and activates STAT5; evidence that activated STAT5 is required for Hodgkin lymphomagenesis. Blood. 2008; 111(9):4706–4715. | |
Mir SS, Richter BWM, Duckett CS. Differential effects of CD30 activation in anaplastic large cell lymphoma and Hodgkin disease cells. Blood. 2000;96(13):4307–4312. | |
Hirsch B, Hummel M, Bentink S, et al. CD30-induced signaling is absent in Hodgkin’s cells but present in anaplastic large cell lymphoma cells. Am J Pathol. 2008;172(2):510–520. | |
Wright CW, Rumble JM, Duckett CS. CD30 activates both the canonical and alternative NF-kB pathways in anaplastic large cell lymphoma cells. J Biol Chem. 2007;282(14):110252–110262. | |
Bargou RC, Leng C, Krappmann D, et al. High-level nuclear NF-kB and Oct-2 is a common feature of cultured Hodgkin/Reed-Sternberg cells. Blood. 1996;87(10):4340–4347. | |
Joos S, Küpper M, Ohl S, et al. Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30 | |
Barth TFE, Martin-Subero JI, Joos S, et al. Gains of 2p involving the REL locus correlate with nuclear c-Rel protein accumulation in neoplastic cells of classical Hodgkin lymphoma. Blood. 2003; 101(9):3681–3686. | |
Krappmann D, Emmerich F, Kordes U, Scharschmidt E, Dörken B, Scheidereit C. Molecular mechanisms of constitutive NF-κB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene. 1999;18(4):943–953. | |
Jungnickel B. Clonal deleterious mutations in the IκBα gene in the malignant cells in Hodgkin’s disease. J Exp Med. 2000;191(2):395–401. | |
Hinz M, Lemke P, Anagnostopoulos I, et al. Nuclear factor κB-dependent gene expression profiling of Hodgkin’s disease tumor cells, pathogenetic significance, and link to constitutive signal transducer and activator of transcription 5a activity. J Exp Med. 2002;196(5):605–617. | |
Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Geneva: WHO Press; 2008. | |
Stein H, Foss HD, Dürkop H, et al. CD30+ anaplastic large cell lymphoma: a review of its histopathologic, genetic, and clinical features. Blood. 2000;96(12):3681–3695. | |
Atsaves V, Lekakis L, Drakos E, et al. The oncogenic JUNB/CD30 axis contributes to cell cycle deregulation in ALK+ anaplastic large cell lymphoma. Br J Haematol. 2014;167(4):514–523. | |
Isaacson PG. Enteropathy-associated T-cell lymphoma and other primary intestinal T-cell lymphomas. In: Jaffe ES, Harris NL, Vardiman JW, Campo E, Arber DA, editors. Hematopathology. Missouri: Elsevier Saunders; 2011:580–587. | |
Sabattini E, Pizzi M, Tabanelli V, et al. CD30 expression in peripheral T-cell lymphomas. Haematologica. 2013;98(8):e81–e82. | |
Harris NL. Shades of gray between large B-cell lymphomas and Hodgkin lymphomas: differential diagnosis and biological implications. Mod Pathol. 2013;26(Suppl 1):S57–S70. | |
Yao J, Gottesman SRS, Ayalew G, Braverman AS, Axiotis CA. Loss of Foxp3 is associated with CD30 expression in the anaplastic large cell subtype of ATLL in US Caribbean patients. Am J Surg Pathol. 2013;37(9):1407–1412. | |
Ansell SM, Horwitz SM, Engert A, et al. Phase I/II study of an anti-CD30 monoclonal antibody (MDX-060) in Hodgkin’s lymphoma and anaplastic large cell lymphoma. J Clin Oncol. 2007;25(19):2764–2769. | |
Bartlett NL, Younes A, Carabasi MH, et al. A phase I multidose study of SGN-30 immunotherapy in patients with refractory or recurrent CD30+ hematological malignancies. Blood. 2008;111(4):1848–1854. | |
Younes A, Gopal AK, Smith SC, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol. 2012;30(18):2183–2189. | |
Francisco JA, Cerveny CG, Meyer DL, et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102(4):1458–1465. | |
Moskowitz CH, Nademanee A, Masszi T, et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomized, double-blind, placebo-controlled, phase 3 trial. Lancet. 2015;385(9980):1853–1862. | |
Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol. 2012;30(18):2190–2196. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.