Back to Journals » Journal of Inflammation Research » Volume 15
CASPorter: A Novel Inducible Human CASP1/NALP3/ASC Inflammasome Biosensor
Authors Zou C ![]() , Beard JA, Yang G
, Beard JA, Yang G ![]() , Evans WE, Bonten EJ
, Evans WE, Bonten EJ
Received 25 August 2021
Accepted for publication 28 December 2021
Published 19 February 2022 Volume 2022:15 Pages 1183—1194
DOI https://doi.org/10.2147/JIR.S333725
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Chan Zou,1,2 Jordan A Beard,1 Guoping Yang,2– 4 William E Evans,1 Erik J Bonten1,5
1Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, Memphis, TN, USA; 2Center for Clinical Pharmacology, The Third Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China; 3Department of Pharmacy, The Third Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China; 4Research Center for Drug Clinical Evaluation of Central South University, Changsha, Hunan, People’s Republic of China; 5Department of Chemical Biology and Therapeutics, St. Jude Children’s Research Hospital, Memphis, TN, USA
Correspondence: Erik J Bonten, Department of Chemical Biology and Therapeutics, St. Jude Children’s Research Hospital, Memphis, TN, USA, Tel +1 901 595-3980, Fax +1 901 5955715, Email [email protected] Guoping Yang, Center for Clinical Pharmacology, the Third Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China, Tel/Fax +86 731 88618933, Email [email protected]
Background: Following our 2015 elucidation of the CASP1/NALP3 inflammasome mechanism of glucocorticoid (GC)-resistance in pediatric acute lymphoblastic leukemia (ALL) patients, we engineered a cell-based CASP1/NALP3 reporter system suitable for high-throughput screening (HTS) of small molecule libraries, with the purpose of identifying compounds capable of inhibiting the CASP1/NALP3 inflammasome and synergizing with GC drugs for the treatment of GC-resistant ALL patients and various autoinflammatory diseases.
Methods: A Dox-controlled system was utilized to induce the expression of the ASC transgene in HEK293 cells while simultaneously overexpressing NLRP3 and CASP1. ASC/CASP1/NALP3 inflammasome complex formation was confirmed by co-immunoprecipitation (co-IP) experiments. Next, a LV fluorescence-based biosensor (CASPorter) was transduced in the HEK293-iASC-NLRP3/CASP1 cell line to monitor the real-time activation of CASP1/NALP3 inflammasome in live cells. The applicability and effectiveness of the CASPorter cell line were tested by co-treatment with Dox and four known CASP1/NLRP3 inhibitors (MCC950, Glyburide, VX-765 and VRT-043198). Inflammasome activation and inhibitions were assessed by Western blotting, fluorescence microscopy and flow cytometry (FC) methods.
Results: Dox treatment significantly induced ASC expression and increased levels of cleaved and catalytically active CASP1, co-IPs further demonstrated that CASP1 was pulled-down with NLRP3 in HEK293-iASC-NLRP3/CASP1 cells after induction of ASC by Dox treatment. In HEK293-iASC-NLRP3/CASP1-CASPorter cell system, cleavage of the CASP1 consensus site (YVAD) in the CASPorter protein after Dox treatment causing excitation/emission of green fluorescence and the 71% GFP+ cell population increase quantified by FC (78.1% vs 6.90%). Dox-induced activation of the NLRP3 inflammasome was dose-dependently inhibited by Dox co-treatment with four known CASP1/NLRP3 inhibitors.
Conclusion: We have established a cell-based CASP1/NLRP3 inflammasome model, utilizing a fluorescence biosensor as readout for qualitatively observing and quantitatively determining the activation of caspase 1 and NLRP3 inflammasomes in living cells and easily define the inhibitory effect of inhibitors with high efficacy.
Keywords: cell-based biosensor, NALP3 inflammasome, CASP1, ASC
Introduction
Glucocorticoids (GCs) are anti-inflammatory drugs used for the treatment of multiple diseases including pediatric acute lymphoblastic leukemia (ALL).1–3 GC resistance is a major challenge in the implementation of multi-drug therapy, and ALL patients that present with de novo or acquired GC resistance have a less favorable prognosis.4,5 Based on multiple causative mechanisms related to steroid resistance in ALL patients, we reason that synergistic co-treatment of GCs with small molecule inhibitors targeting these pathways could potentially reduce or prevent GC resistance. This may lead to possible combination therapeutic approaches in GC-resistant ALL patients.6–8 In addition, such small molecule inflammasome inhibitors could also be developed for the treatment of various autoinflammatory diseases.
In 2015, we reported that the activation of the CASP1/NALP3 (NACHT, LRR and PYD domains-containing protein 3) inflammasome in primary ALL cells caused CASP1-mediated cleavage and inactivation of the GR and subsequent GC resistance.9 The CASP1/NALP3 inflammasome is a large multiprotein complex comprised NLRP3, the adaptor protein ASC, and the cysteine protease pro-caspase 1.10 The CASP1/NALP3 inflammasome can be activated both by exogenous stimuli and host-derived molecules, including extracellular ATP, glucose or monosodium urate crystals, or chemicals such as nigericin.11 Generally, the activation of the CASP1/NALP3 inflammasome complex requires 2 steps.12,13 The first step, priming, involves the binding of bacterial lipopolysaccharide (LPS) to Toll-like receptor 4 (TLR4), which, in turn, leads to NF-kB activation, critical for transcriptional upregulation of both pro-IL-1β and NLRP3. This priming step is essential because pro-IL-1β is not constitutively expressed and basal levels of NLRP3 are inadequate for efficient inflammasome formation.12 The second or activation step is induced by a wide range of structurally dissimilar NLRP3-activating stimuli (including ATP, nigericin, etc.), resulting in the assembly of the 3-component inflammasome, consisting of the adaptor protein ASC, NLRP3, and pro-Caspase 1. Finally, inflammasome formation results in the autoproteolytic activation of caspase 1. Once enzymatically active, caspase 1 can process the cytokines pro-IL-1β and pro-IL-18 into their mature secreted forms.12 These related cytokines cause a wide variety of biological effects associated with infection, inflammation and autoimmune processes.14
Following our 2015 elucidation of the NALP3 inflammasome mechanism of GC-resistance in pediatric ALL patients, we decided to engineer a cell-based model suitable for high-throughput screening (HTS) of small molecule libraries, with the aim to identify compounds capable of inhibiting the CASP1/NALP3 inflammasome and synergizing with GC drugs for the treatment of GC-resistant ALL patients. We adopted a Tetracycline (Tet)-controlled system to be able to induce the expression of the ASC transgene in human embryonic kidney (HEK293) cells, while simultaneously overexpressing NLRP3 and pro-CASP1. Without induction of ASC, constitutively overexpressed CASP1 and NLRP3 are incapable of inflammasome formation and activation. Thus, instead of the priming/activating stimuli mentioned above, inducible ASC functions as the off/on switch, operated by Tetracycline or the orthologous drug Doxycycline (Dox). Finally, to be able to monitor and measure inflammasome activation or inhibition in the ASC-inducible HEK293 cell model, we engineered a fluorescent inflammasome Biosensor (CASPorter, GFP-based caspase 1 activity sensor). The CASPorter is designed with a Caspase 1 consensus tetrapeptide cleavage site (YVAD) integrated within GFP, which prevents fluorescence. Cleavage of the caspase 1 cleavage site by activated Caspase 1 causes conformational changes in GFP, resulting in green fluorescence which can be measured/monitored in live cells. Ultimately, the effectiveness of (potential) NLRP3/CASP1 inhibitors on the inflammasome activity can be determined by measuring the reduction in GFP fluorescence.
Materials and Methods
Gene Design
The synthetic NLRP3-CASP1 dual expression plasmid consisted of 7039 bp, of which 4329 bp encodes NLRP3-CASP1. Restriction sites for Nhel at the 5′ end and BamHI at the 3′ end to facilitate cloning (Supplementary Figure 1). The synthetic Myc-ASC gene consisted of 672 bp. The salient features of the codon‐optimized Myc-ASC gene include a Kozak consensus (GCCACCATGG) 5′ to the ATG start codon (Kozak 1991), and restriction sites for EcoRV at the 5′ end and DraI at the 3′ end to facilitate cloning. The sequence encoding the Myc-ASC gene was optimized for mammalian cell codon usage (Supplementary Figure 2).
Plasmids
Plasmid of pLenti CMV TetR Blast (716-1) (#17492, Addgene, USA) was purchased from Addgene. pUC57 plasmid containing NLRP3-CASP1 gene (PUC57-CASP1-T2A-NLRP3-R260W), pUC57 plasmid contains ASC gene (pUC57-MYC-ASC) and CASPorter plasmid was synthesized and purchased from GenScript. CD514B-1/NLRP3-CASP1 construct was generated by inserting CASP1-T2A-NLRP3-R260W cDNA from PUC57-CASP1-T2A-NLRP3-R260W plasmid as a Nhel-BamHI fragment into the lentiviral backbone pCDH-CMV-MCS-EF1α-Neo/CD514B-1 (empty vector; System Biosciences, USA). pLenti-puro/myc-ASC construct was generated by inserting MYC-ASC cDNA from pUC57-MYC-ASC as an EcoRV-DraI fragment into the tetracycline-inducible pLenti-Puro lentiviral vector (#39481, Addgene, USA). The correctness of insertion was confirmed by digestion with EcoRV/DraI and DNA sequencing (Hartwell center, St. Jude Children’s Research Hospital, USA).
Cell Culture
Hek293 and Hek293T cell lines were purchased from the American Type Culture Collection (ATCC, USA). HEK293, HEK293T cells were cultured in Dulbecco’s modified Eagle’s media (DMEM) supplemented with 10% fetal bovine serum (FBS). Tetracycline-Inducible HEK293 cells were cultured in DMEM supplemented with 10% Tet system approved FBS (Takara, Japan) in a humidified 5% CO2 incubator at 37°C. All cells were passaged every 2 or 3 days, and viability was assessed by staining with trypan blue each time at passage.
Lentivirus Package and Transduction of HEK293 Cells
Viral packaging was done in HEK293T cells follow standard protocol, lentiviral constructs pLenti CMV TetR Blast, CD514B-1/NLRP3-CASP1, pLenti-puro/myc-ASC, CASPorter were co-transfected with packing plasmid CAG-KGP1-1R, HDM-G and CAG4-RTR2 into HEK293T cells using lipofectamine 2000 (Invitrogen, Carlsbad, California, USA). After 48 h, virus particles were harvested and concentrated with Lenti-X™ GoStix™ (clontech, PT5185-2). For transduction, HEK293 cells were plated in 60 mm2 dishes at 8×105 cells per dish one day before, then transduce cells with low, medium, or high concentration of virus with and 8ug/mL polybrene and then the cells were selected with blasticidin (TetR), puromycin (ASC), neomycin (NLRP3/CASP1) and FACS (CASPorter). HEK293T, HEK293 cells were routinely authenticated and screened for mycoplasma.
Cell Treatment
Unless otherwise specified, HEK293 cells were seeded in six-well plates one day before treatment at 3.0×105 cells per well and incubated overnight at 37°C in a humidified 5% CO2 incubator. After removing culture media, 25 ng/mL Dox in 5 mL fresh media was added to for 24 h to induce ASC expression. For inhibition test, cells were seeded and co-treatment with MCC950 (0–50 μM), Glyburide (0–200 μM), VX-765 (0–50 μM), VRT-043198 (0–50 μM) and Dox 25 ng/mL for 24 h. Doxycycline hyclate, MCC950, and VX-765, blasticidin, neomycin, puromycin were purchased from Sigma-Aldrich (USA).
Western Blotting
After desired treatment, cells were pelleted by centrifugation washed once with PBS, and lysed with RIPA buffer (Sigma, R0278-50ML) added cOMPLETE Protease inhibitors (Roche, 11836170001), 1×PhosStop (Roche, 04906845001) and Benzonase (EMD Millipore, EM71205-3, 5 μL/mL RIPA buffer). Proteins were loaded onto 4–12% Novex Bis-Tris gels (Life Technologies, USA) and then transblotted to PVDF membranes (Life Technologies, USA). Antibody to TetR monoclonal antibody (Clontech, 631132), Caspase 1 (Adipogen, AG-20B-0048), NLRP3 (Adipogen, AG-20B-0014-C100), ASC (Santa Cruz Biotechnology, sc-514414), LacZ (Santa Cruz Biotechnology, sc-377257), GAPDH (Santa Cruz Biotechnology, sc-47724) was used as primary antibody. All primary antibodies were used with a dilution of 1:1000, followed by incubation with appropriate horseradish peroxidase (HRP)-conjugated secondary IgG (Dako). The secondary antibodies were used with a dilution of 1:10,000. Signal intensities for all proteins were quantified using Image Studio Lite (Li-Cor, Version 5.2).
Co-Immunoprecipitation Assay
Co-immunoprecipitation (Co-IP) assays were performed based on Samir Parimal’s protocol.15 Basically, after desired treatment, cells were lysed in NP-40 lysis buffer (1% NP-40, 150 mM NaCl, 50 mM HEPES), and lysates were cleared by centrifugation at maximum speed for 15 min. Whole-cell lysates were incubated with 4 μg of the indicated primary antibodies on a rocking platform for 4–5 h at 4°C. Protein A/G PLUS-Agarose (Santa Cruz, USA) beads were washed, incubated for 30 min with 3% BSA and then added to the samples. Incubation was continued for an additional 2 h on the rocking platform. The agarose beads were collected by centrifugation and washed three times with the NP-40 lysis buffer. After washes, immunoprecipitates were eluted in sample buffer and used for immunoblotting analysis.
Imaging and Flow Cytometric Analysis
After desired treatment, the fluorescence was captured with Fluorescence Imaging System (Olympus, USA). Then, cells were harvested and resuspended in PBS and immediately analyzed using BD FACS Melody (BD Biosciences, U.S.A.). Results were analyzed with FlowJo software (TreeStar; Ashland, OR). HEK293 were gated first on live RFP+ cells followed by GFP+ population.
FACS Sorting
After desired treatment, cells were collected and washed with PBS for once. Cell then resuspended in fresh medium and filtered through a 40‐μm mesh. GFP-RFP+ expressing cells were isolated using BD FACS Melody (BD Biosciences, USA) into 96-well plate. Approximately 200 cells were sorted per population for each experiment. All analyzed populations were collected simultaneously.
IncuCyte ZOOM
HEK293-NLRP3/CASP1-iASC or iLacZ cells were seeded in 96-well plate (2 × 104/well) one day before treatment and incubated overnight at 37°C in a humidified 5% CO2 incubator. Following desired treatment, microtiter plates were imaged with the IncuCyte ZOOM instrument (Essen BioScience). The default software parameters for a 96 well plate (Corning) with a 10× objective was used for imaging. The IncuCyte software was used to calculate mean confluence from four non-overlapping bright phase images of each well. The mean number of GFP+ and RFP+ cells per image was calculated from four non-overlapping fluorescent images; this value was then to calculate the GFP/RFP ratio per well.
Statistical Analyses
Data are presented as average values from multiple individual experiments each carried out in triplicate or as average values ± standard deviation (sd) from triplicate measurements in a representative experiment. Nonlinear regression analysis of inhibitor versus normalized response (variable slope) was performed using Prism Software (GraphPad). Statistical analysis was carried out using a nonparametric Mann–Whitney t-test, an unpaired two-tailed t-test, or a Log rank test using Prism software (GraphPad). Data were considered significant when P ≤ 0.05 (*), P ≤ 0.01 (**), P ≤ 0.001 (***) or P ≤ 0.0001 (****).
Results
Construction of a Dox-Inducible NLRP3/CASP1-iASC-HEK293 Cell Line
To establish a cell-based model for high-throughput (HTP) screening of small molecule inhibitors of the NLRP3 inflammasome with high efficiency and stability, we performed Lentivirus (LV) transductions to generate a stepwise series of human embryonic kidney (HEK293) cell lines. First, we constitutively overexpressed the Tet repressor (TetR), followed by the constitutive co-expression of CASP1 and NLRP3, and, finally, we added ASC, whose expression remains silent unless induced by Tet or the ortholog drug Dox (CASP1/NLRP3-iASC-HEK293; Figure 1A).
 |
Figure 1 Protein expression profile in manipulated HEK293 cell line and schematic depiction of Caspase 1 activation. (A) Full expression profile of proteins (TetR, ASC, LacZ, Pro-caspase-1, Cleaved-caspase-1, NLRP3 and house keeping gene GAPDH) in a range of manipulated HEK 293 cell lines in the presence or absence of 25 ng/mL Dox treatment for 24h; (B) schematic illustration of the CASP1/NLRP3-iASC-HEK293 model works in the presence or absence of Dox in the system. The TetR protein was expressed using a TetR-containing plasmid capable of interacting with the Tet responsive element (TRE) of the Tet operator (TetO) in the tetracycline-dependent promoter upstream of the target gene ASC, which interfered the expression of ASC protein although NLRP3/CASP1 simultaneously expressed stably in the cells, the inflammasome was not assembly (inactive). Once Dox is added in this system, it will bind to TetR and cause conformational changes in the repressor, making it unable to bind to the TetO. The TetR-Tet complex then dissociates from the TetO and allows induction of transcription from ASC. Increased ASC protein together with the high levels of NLRP3/CASP1 lead to the assembly of the Nlrp3 inflammasome. The activation of NLRP3 inflammasome subsequently cleaved pro-caspase 1 and initiated downstream reactions (active). Data is representative of three independent experiments. 1b was created with BioRender.com. Abbreviations: Dox, doxycycline; TetR, tetracycline repressor; TetO, tetracycline operator. |
For the first engineering step, we transduced HEK293 cells with a TetR LV expression plasmid, followed by selection of blasticidin-resistant cell population. Constitutive TetR expression levels were confirmed on Western blots (Figure 1A). Subsequently, these cells were used for the second genetic engineering step, the LV transduction of co-expressed CASP1/NLRP3 to obtain HEK293-TetR-NLRP3/CASP1 cells (Figure 1A). In the final LV transduction step, dox-inducible-ASC (iASC) or a dox-inducible-LacZ (iLacZ) control plasmid, were cloned downstream of the Tet Operator (TetO) in the LV expression plasmid pLenti-Puro, and subsequently transduced in HEK293-TetR-NLRP3/CASP1 cells. In the control cell line, ASC was replaced by LacZ, and although these cells also constitutively co-express NLRP3 and CASP1, lacking ASC, these cells are inflammasome deficient. In these cell lines, expression of ASC or LacZ is repressed unless TetR is released from the TetO by treatment by Dox. To induce expression, the resulting cell lines, HEK293-iASC-NLRP3/CASP1 and HEK293-iLacZ-NLRP3/CASP1, were treated with Dox for 24 hours, and analyzed on Western blots for the expression of ASC and LacZ (Figure 1A). A schematic illustration of the mechanism of the in vitro model of Dox-inducible iASC/NLRP3/CASP1 inflammasome is shown in Figure 1B. The constitutively expressed TetR protein interacts with the Tet responsive element (TRE) of the TetO in the Tet-dependent promoter upstream of the target gene ASC, or lacZ in the control cells, and prevents transcriptional activation of the transgenes.16 Once added to the cells, Dox will bind to TetR, causing it to conformationally change, thereby rendering it incapable to bind to the TetO, allowing transcriptional activation of ASC or LacZ. Dox-induced expression of ASC leads to the assembly of functional ASC/NLRP3/CASP1 inflammasome complex, whereby the pro-caspase 1 zymogen is autoactivated into a p10/p20 catalytically active heterodimer. In HEK293-iASC-NLRP3/CASP1 cells inflammasome activation was confirmed by significantly increased level of cleaved CASP1 (Figure 1A and Supplementary Figure 6), indicating that the NLRP3 inflammasome complex was formed by Dox treatment, independent of the addition of LPS and exogenous stimuli such as ATP or nigericin, typically needed for inflammasome activation (Figure 1B).
NLRP3 Interaction with ASC and CASP1 During Dox-Induced NALP3 Inflammasome Activation
To test whether Dox-induced ASC expression resulted in CASP1/NALP3 inflammasome complex formation, we performed co-immunoprecipitation (co-IP) experiments in HEK293-iASC-NLRP3/CASP1 and HEK293-iLacZ-NLRP3/CASP1 control cells after treatment with Dox for 24 h. As shown in Figure 1A (second panel from the top), ASC was induced after Dox treatment in HEK293-iASC-NLRP3/CASP1 cells but not in iLacZ control cells. Co-IPs in both cell lines using an anti-NLRP3 antibody demonstrated that after 24 hours of Dox treatment, CASP1 was pulled-down with NLRP3, but only in HEK293-iASC-NLRP3/CASP1 cells and not in the corresponding iLAcZ control cells (Figure 2, top panel). This indicates that the expression of ASC is a prerequisite for the ASC-induced inflammasome formation between CASP1 and NLRP3.
 |
Figure 2 Co-immunoprecipitation (co-IP) of CASP 1 with NLRP3 in HEK293-iASC-NLRP3/CASP1 cells after Dox-induced ASC expression. HEK293-iASC-NLRP3/CASP1 or HEK293-iLacZ-NLRP3/CASP1 cells were seeded in six-well plates overnight and treated with Dox 25 ng/mL for 24h. Cell lysates were extracted and co-immunoprecipitation was performed using anti-NLRP3 antibody. Lysates (inputs) and immunoprecipitated proteins were analyzed by immunoblotting using CASP1, NLRP3 and GAPDH antibodies. GAPDH was used as a loading control for the inputs while iLacZ control cells (right 2 lanes) were used as the negative control for the co-immunoprecipitation experiments. |
Expression of a Caspase 1 Biosensor – CASPorter – in HEK293-iASC-NLRP3/CASP1 Cells
To monitor the activation of CASP1/NALP3 inflammasome in live cells, we engineered a LV fluorescence-based biosensor (CASPorter) expressed in HEK293-NLRP3/CASP1-iASC or iLacZ control cells. Two fluorescent proteins (RFP and GFP) were included in the CASPorter construct. Constitutively expressed RFP was utilized to monitor cell populations stably expressing the biosensor, whereas a GFP-based caspase 1 Activity Biosensor was obtained by modifying a previously reported Caspase 3 biosensor.17 We replaced the Caspase 3 DEVD tetrapeptide cleavage site, which was engineered within the GFP protein to prevent green fluorescence unless cleaved by Caspase 3, with the Caspase 1-specific YVAD tetrapeptide cleavage site (Figure 3A and B). Caspase 1-cleavage at the YVAD site within GFP protein results in a conformational change allowing the excitation/emission of green fluorescence (Figure 3C). This was tested by adding Dox to HEK293-NLRP3/CASP1-iASC/CASPorter cells, resulting in CASP1/NLRP3 inflammasome activation, and subsequent activation of the CASPorter as indicated by acquisition of GFP signal and increased GFP/RFP ratio over time. Next, we used known small molecule Caspase 1 and NLRP3 inhibitors to prevent Caspase 1 cleavage of the YVAD site in GFP and measured a reduction in GFP signal and a decrease in GFP/RFP ratio (Figure 3C). The validity and functionality of our in vitro HEK293 CASP1/NLRP3 inflammasome Biosensor was confirmed by real-time detection of inflammasome activity, measuring increased levels of GFP/RFP ratio after Dox treatment (0 to 24 hours), and by measuring inhibition of GFP fluorescence intensity after co-treatment with Dox and the NLRP3 inhibitor MCC950 in HEK293-NLRP3/CASP1-iASC cells but not in HEK293-NLRP3/CASP1-iLacZ cells (Figure 3D).
 |
Figure 3 Schematic illustration of CASPorter generation in HEK293-iASC-NLRP3/CASP1 cells. (A) Scheme for the CASPorter and the GFP-based caspase-1 activity indicator (GC1A1); (B) cleavage site of GC1AI versus the reported GFP-based caspase-3 activity indicator (GC3AI); (C) schematic representation of the switch-on or switch-off of the GFP intensity (reflected by GFP:RFP ratio), indicating the inhibitory effect of small molecule inhibitors on the activation of the NALP3 inflammasome. (D) GFP intensity increased overtime with Dox 25 ng/mL treated for 24h, and co-treatment with the reported NLRP3 inhibitor MCC950 showed dose-dependent inhibition by in HEK293-NLRP3/CASP1-iASC cells but not in HEK293-NLRP3/CASP1-iLacZ cells. ##p < 0.001 compared to control, *p < 0.01 compared to Dox treatment at the same time point, **p < 0.001 compare to Dox treatment at the same time point. |
Single Cell Cloning of HEK293-iASC-NLRP3/CASP1-CASPorter
To improve the GFP/RFP in HEK293-NLRP3/CASP1-iASC/CASPorter cells after treatment with Dox, we obtained single cell clones by fluorescence activated cell sorting (FACS) for RFP intensity into 96-well culture plates. After sorting, single cell clones were cultured for 2–3 weeks to allow for cell growth and expansion and subsequently analyzed the clonal cell lines on Western blots to select clones with the highest expression levels of GFP after Dox-induction. After clone selection, when assessed by fluorescent microscopy, the GFP+ population and GFP/RFP ratio were significantly increased after Dox treatment, but only in HEK293-iASC-NLRP3/CASP1-CASPorter cells, and not in either of two control cell lines, HEK293-iLacZ-NLRP3/CASP1-CASPorter and HEK293-iASC-NLRP3/CASP1-control, that had also been single cell-sorted (Figure 4A). The GFP+ population quantified by flow cytometric analysis showed a 71% GFP+ cell population increase in HEK293-iASC-NLRP3/CASP1-CASPorter cells after Dox treated for 24 h (78.1% vs 6.90%), but no difference in GFP was detected in either control cell lines HEK293-iLacZ-NLRP3/CASP1-CASPorter or HEK293-iASC-NLRP3/CASP1-control (Figure 4B and C). Correspondingly, there was a substantially increased level of cleaved Caspase 1, consistent with decreased pro-Caspase 1 in HEK293-iASC-NLRP3/CASP1-CASPorter (Figure 4D, lanes 2 and 6).
 |
Figure 4 Identification of HEK293-NLRP3/CASP1-iASC-CASPorter clones by fluorescent microscope and flow cytometry. (A) Changes of GFP+ population after Dox 25 ng/mL treatment for 24h in HEK293-NLRP3/CASP1-iASC-CASPorter, HEK293-NLRP3/CASP1-iLacZ-CASPorter and HEK293-NLRP3/CASP1-iASC-Control under fluorescent microscope; (B) quantification of GFP+ population in the presence or absence of Dox for iASC-CASPorter, iLacZ-CASPorter and iASC-Control with flow cytometry. (C) statistical results of GFP+ population in iASC-CASPorter, iLacZ-CASPorter and iASC-Control with flow cytometry. (D) Protein expression (ASC, LacZ, Pro-caspase-1, Cleaved-caspase-1, NLRP3 and house keeping gene GAPDH) of iASC-CASPorter, iLacZ-CASPorter and iASC-Control with or without Dox 25 ng/mL treatment for 24h. Images are representative of three independent experiments. Values represent mean ± s.d. N3, NLRP3 C1, CASP1. |
Effect of Known NALP3 Inflammasome Inhibitors on Dox-Induced Fluorescent Intensity in HEK293-iASC-NLRP3/CASP1-CASPorter Cells
We further assessed the inhibitory effect of four know NLRP3/CASP1 inhibitors (Glyburide, MCC950, VX-765, VRT-043198).18–20 in HEK293-NLRP3/CASP1-iASC-CASPorter cells. After 24 h Dox co-treatment with each of the individual inhibitors, we showed that Dox-induced activation of NLRP3 inflammasome was dose-dependently inhibited (Figure 5A and Supplementary Figure 3). The IC50 values were 62.75, 0.46,1.85, and 4.66 μM for Glyburide, MCC950, VX-765 and VRT-043198, respectively (Figure 5C). As expected, there was no inhibition detected in HEK293-NLRP3/CASP1-iLacZ -CASPorter control cells for any of the inhibitors, confirming that without ASC expression, constitutively expressed CASP1 and NLRP3 are unable to form an active inflammasome (Figure 5B and Supplementary Figure 4).
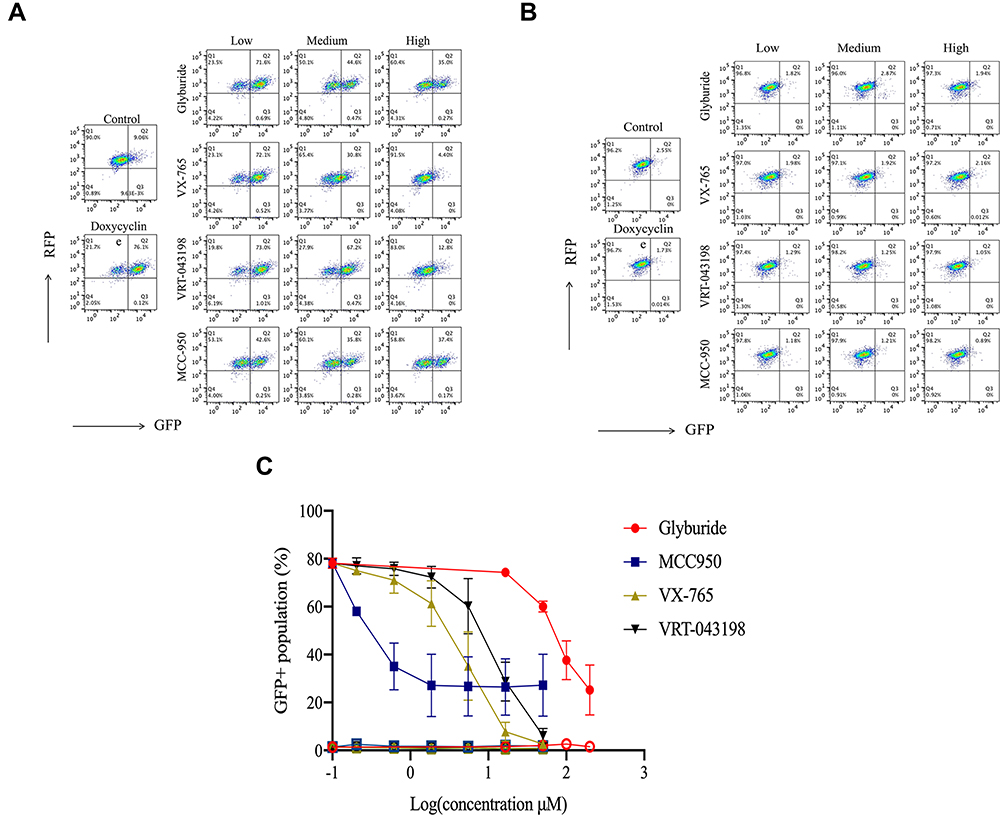 |
Figure 5 Effect of representative inhibitors of NLRP3 inflammasome on the fluorescent intensity in HEK293-NLRP3/CASP1-iASC-CASPorter and HEK293-NLRP3/CASP1-iLacZ-CASPorter. (A and B) The Dox-induced activation of NLRP3 inflammasome was dose-dependently inhibited by co-treatment with four reported inhibitors (MCC950 0–50 μM, Glyburide 0–200 μM, VX-765 0–50 μM, VRT-043198 0–50 μM) in HEK293-NLRP3/CASP1-iASC-CASPorter cells (A), but not in HEK293-NLRP3/CASP1-iLacZ-CASPorter cells (B). (C) Flow cytometric analysis of GFP+ population in these two cell models after co-treatment with Dox 25ng/mL and inhibitors (MCC950, Glyburide, VX-765, VRT-043198) for 24h. The solid and hollow symbols indicate HEK293-NLRP3/CASP1-iASC-CASPorter or -iLacZ cells, respectively. Values represent mean ± s.d. |
Discussion
It is well established that the NALP3 inflammasome plays a crucial role in the development and progression of multiple human diseases, eg, autoimmune disease, autoinflammatory syndromes, cancer, neurodegenerative disorder, metabolic disease, etc.21,22 In addition, we showed previously that a novel mechanism of GC resistance in primary ALL cells was related to the activation of the CASP1/NALP3 (NACHT, LRR and PYD domains-containing protein 3) inflammasome, expanding the understanding of NLRP3 inflammasome.9 In ALL blastocysts, treatment with GCs (dexamethasone, prednisolone) functions by activating the glucocorticoid receptor (GR) and inducing apoptosis in glucocorticoid-sensitive ALL patients.23 However, for about 20% of ALL patients that present with de novo or acquired GC resistance the prognosis is less favorable.24 Through genome wide association study (GWAS) of 444 newly diagnosed ALL leukemia patients, we showed that in newly diagnosed and relapsed patients the transcription and expression of two key components of the CASP1/NALP3 inflammasome pathway, Caspase 1 (CASP1) and NLR (NOD-like receptor) family pyrin domain containing 3 (NLRP3) where significantly increased in GC resistant ALL compared to sensitive ALL patients.9 We elucidated the mechanism of GC resistance by showing that constitutively activated NALP3 inflammasome caused CASP1-mediated cleavage and inactivation of GR.9 Moreover, in ALL cell lines and ALL xenograft mice, knockdown and inhibition of Caspase 1 restored GR levels, and sensitized leukemia cells to GC treatment.9,25 A later genome-wide CRISPR knockout screen further validated the critical role of NLRP3 in GC resistance in ALL patients.26 All these physiological and pharmacological processes involved in the NLRP3 inflammasome provides this target with huge potential for future treatment options of single or combination therapy.27
To date, several approaches have been used for the identification of small molecule CASP1/NLRP3 inhibitors.28–30 Conventional Western blot detection of cleaved caspase 1 and NLRP3 protein and by ELISA of released caspase 1 and the downstream production of pro-inflammatory factors (eg, IL-1β, IL-18) in cell lysates and supernatants (cell culture media, serum, etc.) are the most commonly used methods, but are not ideal for HTP screening of small molecule compound libraries. Other relatively simple and more sensitive methods involve the detection of caspase 1 catalytic activity by fluorometric/luminescence measurement, available in various commercial kits, such as Caspase-Glo® 1 Kit (Promega), and Caspase-1 Assay Kit (Abcam). These kits utilize synthetic peptide substrates containing the specific caspase 1 cleavage sites (eg, Z-WEHD, YVAD-AFC) which enable detection of catalytically active caspase 1 in cells or culture media by release of the light-emitting tags, which can be quantified by fluorometry or luminometry to evaluate the inflammasome activity. However, most of these commercially available kits are very expensive especially when used in large-scale HTP screens. Another caveat of these reagents for Caspase 1 activity detection is that it can only be used in cell lysates and supernatants. We were interested in developing an in vitro inflammasome model that would allow us to measure CASP1/NLRP3 inflammasome activity in real-time in live cells without the need of expensive reagents. Therefore, we developed an easy to use and highly efficient cell-based CASP1/NLRP3 model for HTP screening and validation of NALP3 inflammasome inhibitors.
Many studies have been published to explore the underlying mechanisms and therapeutic effects of novel NALP3 inhibitors in vitro and in vivo. In most studies, the activation of NALP3 inflammasomes requires two steps. First, a priming signal (eg, LPS) induces immune cells to upregulate the expression of NLRP3 and IL-1β through binding to the Toll-like receptor (TLR) in the plasma membrane, stimulating the downstream NF-κB signaling pathway, which contributes to a massive increase expression of both NLRP3 and pro-IL-1β. Second, an activating signal (eg, ATP) to the membrane P2X7R (a ligand-gated acid sensing ion channel receptor) leads to the assembly and activation of the inflammasome. Subsequently, the NLRP3 inflammasome cleaves pro-caspase 1 to the bioactive form (caspase-1 p20), which in turn is essential for the cleavage of pro-IL-1β into its mature form IL-1β, as well as the cleavage of full-length Gasdermin D (FL-GSDMD) into the active N-terminal fragment (GSDMD-N), inducing pore formation in the plasma membrane, finally leading to pyroptosis, an inflammatory form of cell death (details in Supplementary Figure 5). A limitation is that the required LPS and ATP and other exogenous stimuli such as nigericin and monosodium urate not only activate the NALP3 inflammasome, causing cell death by pyroptosis, but are very toxic to cells, confounding the measurement of Caspase 1 activity and the effect of inflammasome inhibitors on cell viability (Supplementary Figure 7). Therefore, we decided to use a tetracycline-controlled (Tet-On) gene expression system to regulate the activity of a key gene, ASC, essential for NALP3 inflammasome activation.31 Finally, we demonstrated that tetracycline-dependent induction of ASC, co-expressed with NLRP3 and CASP1 in HEK293 cells, resulted in active inflammasome formation without the need of toxic inflammasome inducers such as ATP or nigericin. Moreover, this in vitro cell system responded well to several typical NALP3 inhibitors.
Glyburide is an adenosine triphosphatase (ATP)-sensitive potassium (KATP) channel inhibitor for treatment of patients with type II diabetes, and it was approved by US FDA in 198432 and has become one of the most widely used sulfonylurea drug in the world.33 Recently, Glyburide was reported to be a NALP3 inhibitor, with good inhibitory effects at 200 µM in LPS/ATP (or Nigericin)-induced mouse bone marrow-derived macrophages (BMDMs and protection against LPS-induced lethality in C57BL/6 mice.20 However, the exact mechanism involved in inflammasome inhibition of glyburide was not fully elucidated, with the hypothesis that glyburide acts upstream of NLRP3.20 We tested glyburide in Dox-treated NLRP3/CASP1-iASC-CASPorter cells, and showed dose-dependent inhibition of NALP3 inflammasome at concentrations up to 200 µM, with an IC50 of 62.75 µM. Two other inflammasome inhibitors, the prodrug VX-765 and its active metabolite VRT-043198, which are potent and selective caspase 1 inhibitors, with an IC50 of ~0.7 µM and 1.9 µM in human peripheral blood mononuclear cells (PBMCs) from familial cold autoinflammatory syndrome (FCAS) patients, respectively,19,34 whereas MCC950, a specific NLRP3 inhibitor, had an IC50 of 7.5 nM in BMDMs and 8.1 nM in human monocyte-derived macrophages (HMDMs)18 (for details, see Supplementary Table 1). All three inhibitors showed potent inhibitory effects on the activation of the NALP3 inflammasome in HEK293-NLRP3/CASP1-iASC-CASPorter cells, suggesting this model is not only suitable for the identification of CASP1 and NLRP3 inhibitors, but also for screening small molecules that target upstream activators of the NALP3 inflammasome complex.
Conclusion
In summary, we have developed an in vitro cell-based Biosensor Inflammasome model in HEK293 cells, HEK293-NLRP3/CASP1-iASC-CASPorter, which utilizes tetracycline (Tet)-induced ASC while simultaneously overexpressing NLRP3 and pro-CASP1, and a fluorescence biosensor as readout for monitoring the activation and inhibition of inflammasome in live cells. Treatment of cells with four known CASP1/NLRP3 inhibitors (Glyburide, MCC950, VX765, VRT-043198) showed that this model is very accurate and efficient. The model can qualitatively observe and quantitatively determine the activation of caspase 1 and NLRP3 inflammasomes in living cells and easily define the inhibitory effect of inhibitors with high efficiency. We conclude that this cell-based model may be utilized for HTP screenings of specific NALP3 inflammasome inhibitors and may also be used to validate and perform kinetic analyses of such inflammasome inhibitors.
Acknowledgments
Research reported in this publication was supported in part by funds from the NIH Grants R01 CA36401 (W.E.E), P50 GM115279 (W.E.E.), U01 GM92666 (W.E.E.), St. Jude Comprehensive Cancer Center grant CA21765 from the US National Cancer Institute and by the American Lebanese Syrian Associated Charities. The authors appreciate sponsorship from the China Scholarship Council (CSC Grant No: 201706370069). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure
Dr Chan Zou reports grants from China Scholarship Council, outside the submitted work. Dr Guoping Yang reports personal fees from Third Xiangya Hospital, outside the submitted work. Dr William E Evans serves as Chair of the Scientific Advisory Board of Princess Máxima Center (Utrecht, Netherlands) and Board Member of BioSkryb (US). The author reports no other conflicts of interest in this work.
References
1. Gaynon PS, Lustig RH. The use of glucocorticoids in acute lymphoblastic leukemia of childhood. Molecular, cellular, and clinical considerations. J Pediatr Hematol Oncol. 1995;17(1):1–12. doi:10.1097/00043426-199502000-00001
2. Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350(15):1535–1548. doi:10.1056/NEJMra023001
3. Pui C-H, Evans WE. A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin Hematol. 2013;50(3):185–196. doi:10.1053/j.seminhematol.2013.06.007
4. Kaspers G, Pieters R, Klumper E, De Waal F, Veerman A. Glucocorticoid resistance in childhood leukemia. Leuk Lymphoma. 1994;13(3–4):187–201. doi:10.3109/10428199409056282
5. Haarman EG, Kaspers GJL, Veerman AJ. Glucocorticoid resistance in childhood leukaemia: mechanisms and modulation. Br J Haematol. 2003;120(6):919–929. doi:10.1046/j.1365-2141.2003.04189.x
6. Goossens S, Van Vlierberghe P. Overcoming steroid resistance in T cell acute lymphoblastic leukemia. PLoS Med. 2016;13(12):e1002208. doi:10.1371/journal.pmed.1002208
7. Delgado-Martin C, Meyer LK, Huang BJ, et al. JAK/STAT pathway inhibition overcomes IL7-induced glucocorticoid resistance in a subset of human T-cell acute lymphoblastic leukemias. Leukemia. 2017;31(12):2568–2576. doi:10.1038/leu.2017.136
8. Serafin V, Capuzzo G, Milani G, et al. Glucocorticoid resistance is reverted by LCK inhibition in pediatric T-cell acute lymphoblastic leukemia. Blood. 2017;130(25):2750–2761. doi:10.1182/blood-2017-05-784603
9. Paugh SW, Bonten EJ, Savic D, et al. NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nat Genet. 2015;47(6):607–614. doi:10.1038/ng.3283
10. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10(3):241. doi:10.1038/ni.1703
11. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP 3 inflammasome activation and assembly. Immunol Rev. 2015;265(1):35–52. doi:10.1111/imr.12286
12. Swanson KV, Deng M, Ting JP-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489. doi:10.1038/s41577-019-0165-0
13. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. doi:10.1038/nri3452
14. Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10(2):89–102. doi:10.1038/nri2691
15. Samir P, Kesavardhana S, Patmore DM, et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature. 2019;573(7775):590–594. doi:10.1038/s41586-019-1551-2
16. Gossen M, Freundlieb S, Bender G, et al. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268(5218):1766–1769. doi:10.1126/science.7792603
17. Zhang J, Wang X, Cui W, et al. Visualization of caspase-3-like activity in cells using a genetically encoded fluorescent biosensor activated by protein cleavage. Nat Commun. 2013;4(1):2157. doi:10.1038/ncomms3157
18. Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–255. doi:10.1038/nm.3806
19. Wannamaker W, Davies R, Namchuk M, et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoy l)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J Pharmacol Exp Ther. 2007;321(2):509–516. doi:10.1124/jpet.106.111344
20. Lamkanfi M, Mueller JL, Vitari AC, et al. Glyburide inhibits the cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187(1):61–70. doi:10.1083/jcb.200903124
21. Davis BK, Wen H, Ting JP-Y. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29(1):707–735. doi:10.1146/annurev-immunol-031210-101405
22. Menu P, Vince J. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol. 2011;166(1):1–15. doi:10.1111/j.1365-2249.2011.04440.x
23. Distelhorst C. Recent insights into the mechanism of glucocorticosteroid-induced apoptosis. Cell Death Differ. 2002;9(1):6–19. doi:10.1038/sj.cdd.4400969
24. Inaba H, Pui C-H. Glucocorticoid use in acute lymphoblastic leukaemia. Lancet Oncol. 2010;11(11):1096–1106. doi:10.1016/S1470-2045(10)70114-5
25. Paugh SW, Bonten EJ, Evans WE. Inflammasome-mediated glucocorticoid resistance: the receptor rheostat. Mol Cell Oncol. 2016;3(1):e1065947. doi:10.1080/23723556.2015.1065947
26. Autry RJ, Paugh SW, Carter R, et al. Integrative genomic analyses reveal mechanisms of glucocorticoid resistance in acute lymphoblastic leukemia. Nat Cancer. 2020;1(3):329–344. doi:10.1038/s43018-020-0037-3
27. Schwaid AG, Spencer KB. Strategies for targeting the NLRP3 inflammasome in the clinical and preclinical space. J Med Chem. 2021;64(1):101–122. doi:10.1021/acs.jmedchem.0c01307
28. Angosto-Bazarra D, Molina-López C, Peñín-Franch A, Hurtado-Navarro L, Pelegrín P. Techniques to study inflammasome activation and inhibition by small molecules. Molecules. 2021;26(6):1704. doi:10.3390/molecules26061704
29. Zito G, Buscetta M, Cimino M, et al. Cellular models and assays to study NLRP3 inflammasome biology. Int J Mol Sci. 2020;21(12):4294. doi:10.3390/ijms21124294
30. Nizami S, Millar V, Arunasalam K, et al. A phenotypic high-content, high-throughput screen identifies inhibitors of NLRP3 inflammasome activation. Sci Rep. 2021;11(1):15319. doi:10.1038/s41598-021-94850-w
31. Yamamoto M, Yaginuma K, Tsutsui H, et al. ASC is essential for LPS‐induced activation of procaspase‐1 independently of TLR‐associated signal adaptor molecules. Genes Cells. 2004;9(11):1055–1067. doi:10.1111/j.1365-2443.2004.00789.x
32. FDA approved drug products: Micronase (Discontinued). https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=017498.
33. Sola D, Rossi L, Schianca GPC, et al. Sulfonylureas and their use in clinical practice. Arch Med Sci. 2015;11(4):840–848. doi:10.5114/aoms.2015.53304
34. Stack JH, Beaumont K, Larsen PD, et al. IL-converting enzyme/caspase-1 inhibitor VX-765 blocks the hypersensitive response to an inflammatory stimulus in monocytes from familial cold autoinflammatory syndrome patients. J Immunol. 2005;175(4):2630–2634. doi:10.4049/jimmunol.175.4.2630
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.