Back to Journals » OncoTargets and Therapy » Volume 14
Carbonic Anhydrase XII is a Clinically Significant, Molecular Tumor-Subtype Specific Therapeutic Target in Glioma with the Potential to Combat Invasion of Brain Tumor Cells
Authors Li G, Chen TW, Nickel AC, Muhammad S, Steiger HJ ![]() , Tzaridis T, Hänggi D, Zeidler R
, Tzaridis T, Hänggi D, Zeidler R ![]() , Zhang W
, Zhang W ![]() , Kahlert UD
, Kahlert UD ![]()
Received 8 January 2021
Accepted for publication 5 February 2021
Published 4 March 2021 Volume 2021:14 Pages 1707—1718
DOI https://doi.org/10.2147/OTT.S300623
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Alberto Bongiovanni
Guanzhang Li,1,* Ting-Wei Chen,2,* Ann-Christin Nickel,2 Sajjad Muhammad,2 Hans-Jakob Steiger,2 Theophilos Tzaridis,3,4 Daniel Hänggi,2 Reinhard Zeidler,5 Wei Zhang,6– 8 Ulf Dietrich Kahlert1,2
1Department of Molecular Neuropathology, Beijing Neurosurgical Institute, Capital Medical University, Beijing, People’s Republic of China; 2Clinic for Neurosurgery, Medical Faculty, Heinrich-Heine University, Düsseldorf, Germany; 3Division of Clinical Neurooncology, Department of Neurology and Institute of Clinical Chemistry and Clinical Pharmacology, University of Bonn, Bonn, 53127, Germany; 4Tumor Initiation & Maintenance Program, NCI-Designated Cancer Center, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA, 92037, USA; 5Department for Otorhinolaryngology, Klinikum der Universität München (LMU), Munich, Germany; 6Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing, People’s Republic of China; 7China National Clinical Research Center for Neurological Diseases, Beijing, People’s Republic of China; 8Chinese Glioma Genome Atlas Network (CGGA) and Asian Glioma Genome Atlas Network (AGGA), Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ulf Dietrich Kahlert Moorenstrasse 5, Düsseldorf, 40225, Germany
Tel +49-211-810 8013
Fax +49-211-810 8012
Email [email protected]
Background: The metabolic enzyme carbonic anhydrase 12 (CA12/CAXII) emerges as a promising cancer therapeutic target with drug development projects underway. Previous reports proposed the relevance of CA12 in the context of glioma but are limited in patient data quantity, ignore ethnic diversity of patients or rely on semi-quantitative, thereby out of date, methodology. Moreover, little is known on the association of CA12 to brain tumor stemness or on the effect of anti-CAXII-directed monotherapies on glioma stem cells (GSCs), in particular their response regarding mesenchymal differentiation status.
Methods: We performed in silico analysis on three independent, large-scale patient datasets interrogating state of the art molecular diagnostics alongside clinical outcomes. We analyzed CAXII abundance on a collection of GSCs and functionally tested their response to exposure to CAXII blocking antibody 6A10.
Results: CA12 is highly expressed in glial tumors compared with normal tissue and predicts for poor clinical course of tumor patients. CA12 expression in glioblastoma significantly correlates with clinically established, molecular markers of IDH1WT DNA, WHO grade IV or absence of 1p/19q chromosome arm co-deletion. Furthermore, tumors with elevated CA12 cluster into the mesenchymal transcription subclass of the disease. CAXII abundance in different GSCs ranges from almost absent to high levels and does not correlate to stem cell marker CD133/AC133 cell surface expression. Moreover, aiming to pharmacologically block CAXII in our cells with antibody 6A10 caused significant functional response only in one of the tested GSCs models, featuring suppression of cell invasion accompanied by reduction of ZEB1 protein and other stem cell markers.
Conclusion: CA12 represents a clinically relevant and molecular brain tumor-subtype specific therapeutic target. Our correlative data from experimental and clinical samples does not support CA12/CAXII to be GSC specific. 6A10 possesses promising potential to impede the invasive capacity of glioma cells and supports the emerging concept that CAXII interacts with cancer EMT programs. However, further mechanistic studies are required to comprehensively assess the therapeutic potential of 6A10 and to identify different resistance mechanisms of GSCs.
Keywords: carbonic anhydrase 12, 6A10, glioma, therapeutic antibody, ZEB1, disease modeling
Introduction
Gliomas, amongst which glioblastoma (GBM) is the most aggressive form, represent a clinically challenging class of cancers. Their high malignancy is characterized by elevated levels of therapy resistance and invasive growth. It is believed that malignant features of GBM are promoted by cells with stem cell properties.1 Recent evidence proposes that GBM stem cells (GSCs) are tightly associated with the molecular reconversion into mesenchymal state, and epithelial-to-mesenchymal-like processes (EMT) are intertwined with GBM stemness in order to augment various malignant properties of the disease.2 Over recent years, the prominent transcriptional EMT activator ZEB1 (Zinc finger E-box-binding homeobox 1) has emerged as a potent regulator of GSCs.3–7 A pharmacological strategy to block the GSCs ZEB1 program is of high clinical need.
Carbonic anhydrases (CA), a family of isoforms of hypoxia-responsive cell metabolic enzymes, are discussed as potent anti-cancer targets in a wide variety of cancers,8 including GBM.9–13 Previous work indicates CA isoform 12 (CA12) may be of particular relevance. However, until now our knowledge has been restricted to a low number of analyzed patient samples, ignoring ethnic diversity of patients and exclusively based on semi-quantitative, thereby out of date, methodology to score CA12 activation.11,12 The presented results on over 1700 patient samples using various state of the art molecular diagnostics14–18 closes all these gaps of current knowledge in order to benchmark the clinical relevance of CA12 in the current scientific neuro-oncological environment. We also compare the clinical prognostic value of CA12 mRNA expression against counterparts of other CA isoforms. Overall, our data strongly position CA12 as a molecular target with significant clinical negative prognostic value of glioma patients, and with significant association with various WHO diagnostic guideline-accepted, molecular properties of GBM. Aiming to validate our computational-build hypothesis with functional data, we performed applied stem cell models of GBM, in order to recapitulate the most aggressive subtype of cells of the disease. In total, correlative data on clinical and experimental biosamples do not support CA12 to be highly stem cell specific nor predict for response to anti-CAXII-directed therapeutic antibody 6A10. Interestingly in a fraction of tested GSCs, 6A10 treatment caused significant suppression of ZEB1 associated with impaired cellular invasion. Since clinically feasible strategies to directly inhibit transcription factor activities are challenging, our data promote subsequent studies with 6A10 as a potential drug candidate. Resistance mechanisms and biomarkers predicting response need to be identified to facilitate 6A10’s clinical application.
Materials and Methods
Patient Data Resources and in silico Analysis
Molecular and clinical data of glioma patients were obtained from the publicly assessable sources, namely The Chinese Glioma Genome Atlas (CGGA, (https://www.cgga.org.cn, two independently acquired datasets with n=325 individuals, and a 2019 dataset with n=693 individuals) and The Cancer Genome Atlas (TCGA, with n=702 individuals, for verification). For CGGA, molecular testing of each patient was performed at the Molecular Pathology Testing Center of Beijing Neurosurgical Institute and the data acquisition was approved by the Beijing Tiantan Hospital institutional review board and tumor specimen quality control.
The statistical computations and figure drawing were performed with R package ‘ggplot2ʹ. Unsupervised cluster analysis was performed by pheatmap package in software environment R (version 4.0.0). The analysis processes were carried out under the default parameters. Student’s t test was used in the differential analysis of the two groups in this study. The drawing was performed by ggplot2 package in software environment R (version 4.0.0). The differential analysis of CA families between normal brain tissue and brain tumor tissue were performed in the GEPIA portal (http://gepia.cancer-pku.cn). The most significantly related genes of CA12 were obtained by Pearson correlation analysis. Genes with a correlation coefficient greater than 4.5 were used for enrichment analysis. Biological function and pathway enrichment analysis were performed by analysis tools in The Database for Annotation, Visualization and Integrated Discovery (DAVID, v6.8) (https://david.ncifcrf.gov). The Pearson correlation analysis method was used in the correlation analysis of this study. Glioma stem cell markers were obtained from the Cell Marker portal (http://biocc.hrbmu.edu.cn/CellMarker/). Kaplan–Meier survival analysis was used in the survival analysis of this study. Drawing was performed using ggplot2 and survival packages in software environment R (version 4.0.0). The analysis processes were carried out under the default parameters.
Cell Culture and Pharmacological Model
The in vitro cell models 407 and BTSC233 were kindly provided by M.S. Carro, Freiburg University, Germany; GBM1 by A. Vescovi, San Raffaele Hospital, Milano, Italy; NCH421k and NCh644 by G. Herold-Mende, Heidelberg, Germany; JHH520 by G. Riggins and SF188 by E.H. Raabe (both Johns Hopkins Hospital, Baltimore, USA). GSC models were grown under neurosphere conditions as described previously.20 6A10 was provided as previously described.21 Determination of the molecular concentration for testing the effect of the substance (25 μg/mL) was based on the wide experience with 6A10 from co-author R. Zeidler, Munich. Moreover, our experimental design was guided by published data of 6A10 impacting cancer cells in vitro using the same concentration.22 Duration for acquiring cell invasion potential was limited to time span of overnight to a maximum of 24 hours. For all assays, volume-adjusted medium was used as mock treatment. Ethical approval to conduct the in vitro studies with the used cell lines was granted by the ethics commission of the Medical Faculty of the Heinrich-Heine University (study ID 5841R).
Functional in vitro Assays
Cell growth quantification was executed using MTT assay similarly as described before.7 The following specific parameters were applied: assay format: 96 well, 4000 cells each well/100 μL culture media. Quantification of cell proliferation, apoptosis and cell cycle progression was performed using Muse® Cell Analyzer (Merck KGaA, Germany) instrumentation using relevance kits and following manufacturer protocols (Luminex, United States: Muse® Ki67 Proliferation Kit, Muse® Annexin V & Dead Cell Kit, Muse® Cell Cycle Kit). Invasion assay was performed using modified Boyden chamber assay similarly as described before.3 In brief, Matrigel was thawed at 4 °C overnight. Inserts were incubated with 1 mL Matrigel® at 37 °C for an hour. Harvested cells were adjusted to concentration of 3 x 105 in - FCS solution. The upper chamber was filled with 500 µL harvested cells, while 800 µL + FCS solution was added to the lower chamber. Non-invading cells that remained in the upper chamber were removed by cotton swab. Invasive cells in the lower chamber were fixed with 500 µL precooled methanol. After one wash, cell nuclei were stained with hematoxylin and washed with warm water. N=5 randomly selected areas in each insert were photographed under inverted light microscope (40× objective) and further quantified in Image J by manual counting of cells. For the in vitro assays, acquired data were presented as average and SEM. Welch’s t-test was used to check for significant differences (p≤0.05), with p-values showing to be significant (p<0.05, *), very significant (p<0.01, **) and highly significant (p<0.001, ***).
qPCR
Targeted gene expression quantification was performed according to established procedures in the lab using SYBR green-based nucleic acid intercalation assay, similar to those described previously.23 Suppl. Figure S1 lists all primer specifications relevant for this study. Acquired data were presented as average and SEM. Welch’s t-test was used to check significant difference (p≤0.05), with p-values showing to be significant (p<0.05), very significant (p<0.01) and highly significant (p<0.001).
Protein Quantification Assays
Western Blot
Standard procedures similar to those described previously were applied.24 Non-specific sites were blocked by 5% milk powder solution followed by primary antibody incubation at 4 °C overnight. Fluorescent secondary antibody was diluted in TBS-T and incubated at room temperature for an hour. Detected signals was acquired with Odyssey® CLx (LI-COR Biosciences, Germany). Further data analysis was processed in Image Studio Ver2.1. Suppl. Figure S2 lists all antibody specifications relevant for this study.
FACS Analysis
In this flow cytometry experiment, cells were prepared according to the direct flow cytometry protocol from Abcam. In brief, 5–10 × 105 cells were collected in a Falcon round bottom polystyrene test tube. After trypsinization into single cells, media was replaced by ice cold 10% FBS in PBS. Afterwards, cells were incubated with fluorescent-labelled antibodies on ice for 30 min (Suppl. Figure S2). Since there is overlap between two fluorescent emission spectra, single-stained samples were applied for correction as compensation control. Cells were washed with ice cold PBS 3x and re-suspended in 500 µL 10% FBS in PBS. Samples were analyzed within one hour on BD FACSCanto®. Bivariate histograms and statistical plots were generated by the BD FACSDiva® Software at the core facility setup.
Results
CA12 Transcription Level is Increased in Gliomas Compared with Non-Tumor Cells, Predict Poor Clinical Prognosis of Glioma Patients
We present the landscape of transcription of CA families and their relationship between diverse diagnostically relevant, clinicopathological factors in the CGGA Database and CGGA 2019 Database (Figure 1A and B). Amongst all CA isoforms, expression levels of CA12 and CA13 are the only family members that predict for significant poor prognosis in terms of overall patient survival time in both CGGA datasets (Figure 1C). Moreover, expression levels of those negative predictors are significantly increased in tumors compared with non-tumor tissue, both in low grade (LGG) and high grade gliomas (HGG) respectively (Figure 1D). Poor prognostic value of high CA12 mRNA levels was confirmed in additional datasets of Gravendeel et al.25 and Kawaguchi et al.26 (Suppl. Figure S3). To the best of our knowledge, this is the ethnically most diverse and largest number of cancer patient samples probed for clinical prognostic values of CA12. Given these strong indications, next we interrogated the data of state of the art molecular characterization of brain tumors to dissect any relevance of CA12 as a potential molecular-subtype specific therapeutic target.
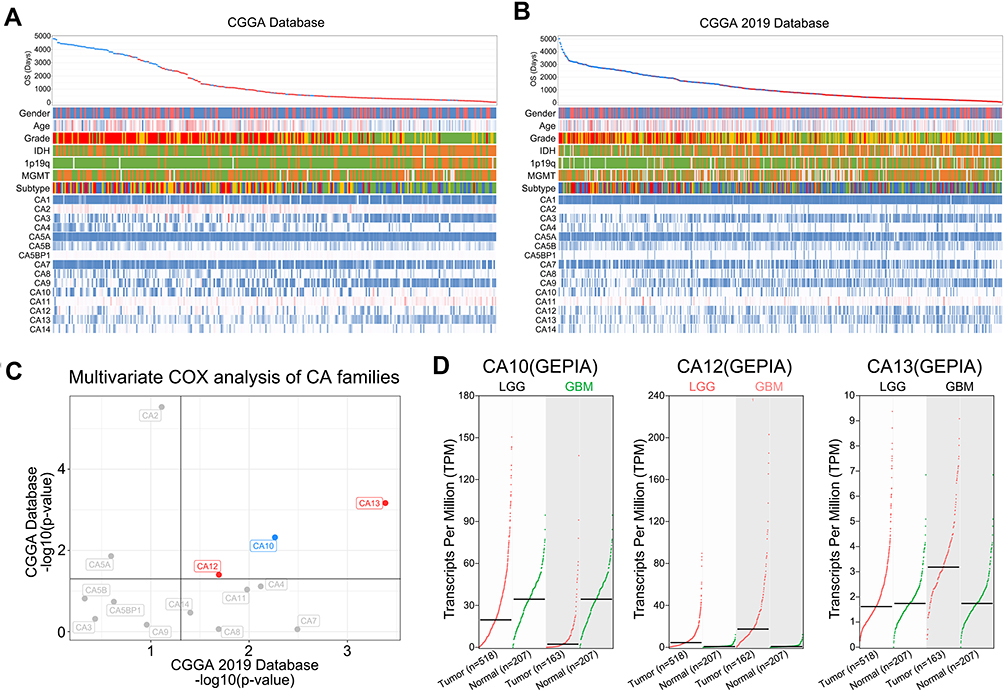 |
Figure 1 CA12, amongst all CA family members, represents the most promising therapeutic target in glioma. The landscape of CA families and their relationship between diverse diagnostic relevant clinicopathological factors in (A) CGGA database and (B) CGGA 2019 database. (C) CA10, CA12 and CA13 are independent prognostic factors in both CGGA and CGGA 2019 databases. Red: poor prognostic factors. Blue: good prognostic factors. (D) Comparison of the expression differences of CA10, CA12 and CA13 in normal brain tissue and brain tumor tissue. Green: significantly high expression in normal tissues. Red: significantly high expression in tumor tissues. Black: no significant difference. |
Elevated CA12 Transcriptional Levels in Glioma Correlate with Clinically Established, Molecular Properties
High mRNA levels of CA12 in glioma significantly correlate with clinically relevant, negative prognostic pathological and molecular markers of brain tumors. Those are tumors defined as WHO grade IV types (Figure 2A and F), tumors with IDH1 wildtype DNA according to WHO classification of 201627 (Figure 2B and G), and tumors without 1p19q chromosome co-deletion28 (Figure 2C and H). Of note, in one of CGGA datasets, high CA12 expression was found in tumors with methylated DNA promoter of O(6)-methylguanine-DNA methyltransferase (MGMT)29 (Figure 2D and I). In addition, the mesenchymal GBMs, known to be particularly aggressive,15 present significantly higher expression levels of CA12 than other transcriptional subtypes in both Asian datasets (Figure 2E and J). We validated our findings in the TCGA dataset and found high CA12 expression significantly associated to gliomas with WHO grade IV staging, to tumors with IDH1WT DNA, to tumors with 1p19q chromosome arm co-deletion as well as to cases classified as mesenchymal subtype of GBM (Figure 3A–F). In the TCGA dataset CA12 transcription is enriched in cases with unmethylated MGMT promoter DNA (Figure 3E).
 |
Figure 2 The relationship between CA12 and clinicopathological factors in CGGA and CGGA 2019 databases. High CA12 mRNA levels significantly associated with WHO IV grade (A and F), IDH1 wildtype DNA status (B and G) and tumors without 1p/19q chromosome arm co-deletion (C and H). CA12 mRNA levels are upregulated in tumors carrying methylated MGMT promoter region (significantly for CGGA) (D and I). CA12 mRNA is significantly upregulated in most aggressive mesenchymal subtype of GBM (E and J). |
 |
Figure 3 Validation of the identified association of CA12 transcription level with clinicopathological factors using TCGA database. The landscape of CA families and their relationship between diverse diagnostically relevant clinicopathological factors in TCGA Database (A). High CA12 mRNA levels significantly associate with WHO IV grade (B), IDH1 wildtype DNA status (C), tumors with 1p/19q chromosome arm co-deletion (D) and CA12 tumors carrying de-methylated MGMT promoter region (E). CA12 mRNA is significantly upregulated in the most aggressive mesenchymal subtype of GBM (F). |
Correlation of CA12/CAXII with Stem Cell Markers
To look for an association with malignancy promoting tumor stem cell signals, we performed targeted correlative assays in clinical and experimental biosamples. This rationale was further supported by the work of others in the context of lung cancer, where CAXII was described to serve as a marker for tumor stem cells.21 Our analysis does not unequivocally support this hypothesis to apply in the context of brain tumors. First, in silico analysis testing for correlative expression of CA12 with mRNA levels of previously reported stem cell molecules shows a mixed picture. Although prominent markers such as heme oxygenase 1,30 membrane transporter SLC16A131 or Nestin32 show positive correlation in all datasets, negative correlation between CA12 and likewise prominent stem cell markers SOX233 or ZEB14 exists (Figure 4A–C). Moreover, high CA12 expression revealed association to expression patterns of biological processes and molecular signaling pathways (using Kyoto Encyclopedia of genes/KEGG consensus items as determinates) both associated to cancer stem cells and to those previously not dominantly associated to tumor stemness (Suppl. Figure S4).
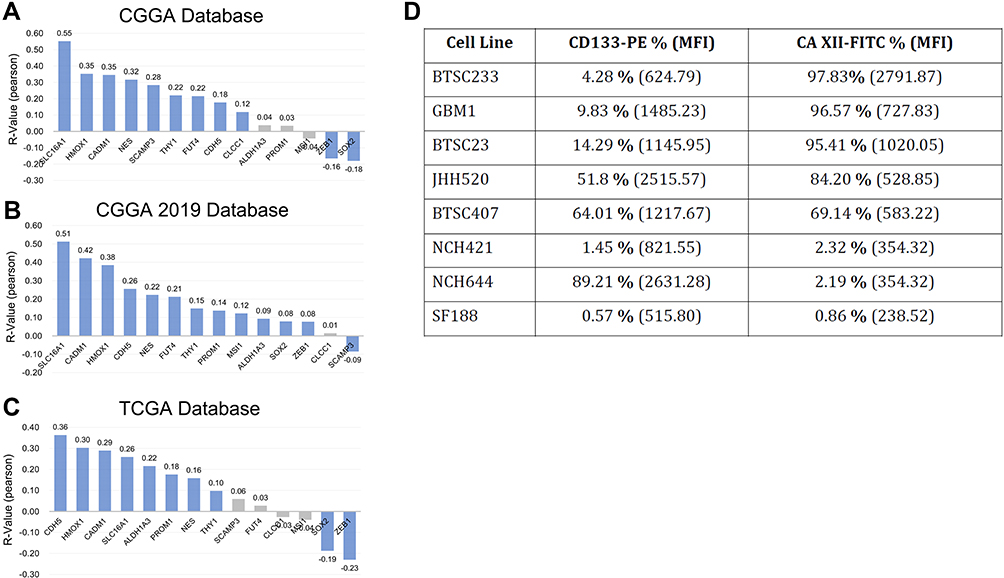 |
Figure 4 The relationship between CA12 and glioma stem cell markers. CA12 is significantly positively correlated with the expression of most glioma stem cell markers in CGGA (A), CGGA 2019 (B) and TCGA (C) databases. Blue: significantly correlated. Black: non-significantly correlate. Cell surface abundancy of CA12 does not correlate with relative cell surface abundance of bona fide brain tumor stem cell marker CD133 in our in vitro disease models as expressed by percentage of positive cells and mean fold intensity/MFI, one time experiment (D). |
In addition, our protein analysis on GSC in vitro models does not support CA12 to be GSC specific. FACS data show that the amount of cell surface CAXII does not correlate with the prevalence of cells positive for bona fide GSC marker CD133/AC13334 (Figure 4D). In addition, baseline abundance of total CAXII in our stem cell models varies from very low to high signals, as detected in Western blot (Figure 5). Together, our clinical and experimental data do not strongly support that high CA12/CAXII levels correlate with brain tumor stem cell signatures.
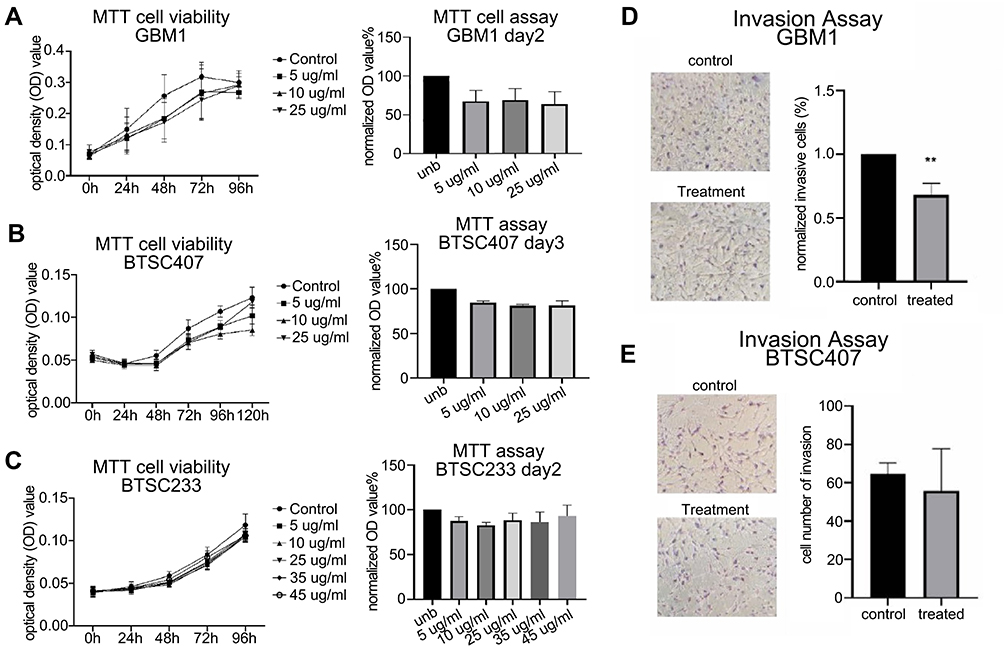 |
Figure 5 Pharmacological targeting of CAXII in glioma in vitro functional responses. 6A10 treatment did cause mild inhibitory effect on cell growth in subset of tested GSC models/GBM1 and BTSC407, and no effect on BTSC233, unb = control (A–C). All cells are IDH1wt and MGMT promoter methylated, GBM1 and 407 are classical subtype of GBM, BTSC233 is mesenchymal subtype of GBM (data not shown). 6A10 treatment caused suppression of cellular invasion in responder cell line GBM1 but not in non-responder model 407 (D and E), BTSC233 not tested. |
6A10 Treatment in vitro Can Cause Moderate Inhibition of Growth and Significant Reduction of Invasion Capacity
In our antibody treatment experiments, we observed a mild reduction of cell growth for GBM1 and 407 (Figure 5A and B), and no clear effect for BTSC233 (Figure 5C). This was accompanied by a trend towards induction of apoptosis in GBM1, abut no changes in apoptosis rate were observed in the other two models used (Suppl. Figure S5). None of the cell models showed a difference in cell proliferation or cell cycle analysis in response to the treatment (Suppl. Figure S5). Most notably, responder model GBM1 reacted with significantly diminished capacity of cellular invasion (Figure 5D), as 407 did not reduce its invasive properties (Figure 5E).
6A10 Treatment Can Suppress the Activation of EMT Marker ZEB1 and Leads to Induction of CA12 Expression and Protein
Next, we tested the change of stem cell markers in response to 6A10 exposure on mRNA level. Interestingly, GBM1 showed significantly suppressed expression of various stem cell markers, whereas no clear trend was seen for the other two tested cell models. All tested models showed signs of upregulation of CA12 expression upon antibody treatment, with GBM1 showing significant induction (Figure 6A).
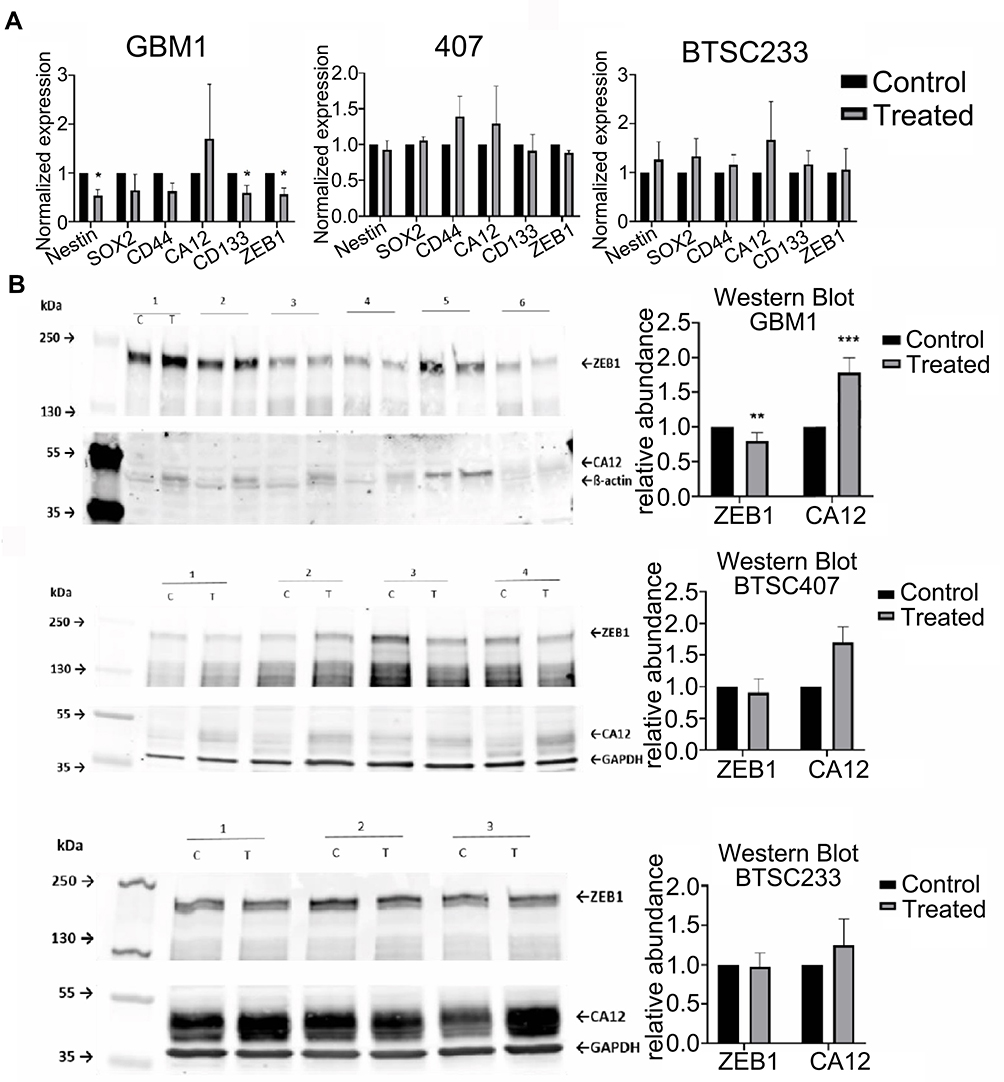 |
Figure 6 Pharmacological targeting of CAXII in glioma in vitro effects on stem cell markers, CAXII and ZEB1 protein 6A10 treatment on GBM1 caused significant reduction of stem cell molecules Nestin, CD133/PROM1, ZEB1 mRNA, and trend of reduction of CD44 and SOX2. Moderate effects are observed on models 407 and BTSC233 (A). Induction of 6A10 target CA12 on mRNA and protein was found in all models with significant upregulation on protein level in GBM1 (A and B). Reduction of stem cell marker ZEB1 in GBM1 upon 6A10 treatment was confirmed on protein level with no change detected in 407 and BTSC233 (B) (c – control/mock, T – treatment, 25µg/mL 6A10, for more details please see relevant section in Materials and Methods section). |
When testing the abundance of relevant factors on total protein level, to our surprise we identified CAXII to be virtually absent in naïve GBM1 and 407, and comparatively strongly active in BTSC233 (Figure 6B and C). Moreover, it seems that the cell surface protein level of CAXII differs from total CAXII, as 407, but foremost GBM1, are high on cells positive for cell surface CAXII (Figure 4D). Interestingly, for all models we identified stronger CAXII bands when exposed to 6A10, with significant induction in GBM1 (Figure 6B). Naïve ZEB1 levels are roughly similar between the tested cell models. Interestingly, we see reduction of protein in response to treatment in some of the biological repetition experiments in any of the investigated cell lines, although significance over all repetitions was reached in GBM1 only (Figure 6B and C).
Discussion
Our data show CA12 to serve as a clinically relevant anti-cancer therapeutic target and proposes its potential suitability for the development of neuro-oncology precision medicine. By using large-scale, molecular and clinical data retrieved from three different datasets, thereby ensuring ethnic and gender diversity, to the best of our knowledge this is the largest descriptive analysis of CA12 in tumor samples. In the context of brain tumor, a previous study on only 22 GBM samples identified CA12 mRNA levels to be associated with tumors carrying EGFR/RNF139 DNA abnormalities. The authors acknowledged the limitation of their results as they were retrieved on a small sample size only. Moreover, no data on clinical diagnostic molecular markers according to WHO guidelines are presented.11 Haapasalo et al. assessed CAXII protein abundance in a larger series of glioma cases (n=370) and found signal enrichment with increasing WHO staging of the tumors. Their survival studies (on n=269 GBM samples) revealed that high CAXII protein predicts for shorter survival of the patients.12 Although this study is indicative and our data can at least be seen as confirmative, we believe the respective part of our data is more convincing as Haapasalo et al. applied a semi-quantitative method, prone to subjective errors. In addition, we extended the analysis of CA12 activation to clinical molecular markers, namely IDH1 DNA mutation status, chromosome aberration status and MGMT-methylation status of the tumors. Moreover, we correlated CA12 expression to total transcriptome signatures of the tumors. In total, given our multi-method the acquired data are more comprehensive and more precise than previous reports, and our CA12 focused results are a superior contribution to the field of precision neuro-oncology.
Given GSCs are thought to be a main cause of tumors’ malignant properties, we extended our analysis to probe if CA12 can be associated with glioma stem cell maintenance. By and large, both our expression analysis in clinical samples, as well as protein analysis in naïve experimental models did not present a clear picture regarding this question. Expression of stem cell markers in patient samples is both positively and negatively associated to high CA12 mRNA abundance. On the other hand, inter-case comparison of our stem cell models showed that CAXII is activated to different extents, from high activity to virtual absence of signals in Western blot or FACS, depending on the cell line. We also did not notice any correlation between the level of cells positive for bona fide GSC marker of cell surface CD133/AC133 and the level of cells positive for surface CAXII. Interestingly, our cell line data revealed differences in relative abundance of total CAXII and cell surface CAXII, indicating differences in protein stability or protein processing amongst different GBMs. As our cell models only resemble IDH1WT cases, it would be interesting to see if there is a noticeable difference in correlation of CA12/CAXII with stem cell molecules when comparing GSC models with or without IDH1R132. However the field lacks overall availability of accepted models carrying IDH mutations27 and we cannot conduct this study at this point. Of note, a recent conference abstract reports that CA12 plays a role in CSG maintenance but no original data is assessable, such as how the authors define GSCs or what the IDH DNA mutation status of the used cells is.35
Next, aiming towards a clinical translational perspective for our anticipated results, we decided to use a pharmacological strategy to target CAXII in vitro. By choosing 6A10 as a previously validated, highly specific CA12 blocking antibody,21,22,36,37 we found that only GBM1 models show significant phenotypic responses to the treatment. Although all IDH1WT, our models represent different molecular subtypes of GBM (GBM1 and 407 are classical subtype, BTSC233 is mesenchymal subtype,24 data not shown). Thus, together with the known fact of the enormous inter-tumoral heterogeneity of the disease,15,28 our data suggest that the therapeutic potential of 6A10 may be limited to a subset of tumors only. In the context of Notch1 receptor, we recently showed that drug target protein abundance in GSCs is predictive for the cells’ responsiveness to the blocking antibody intervention.20 Our correlative data in this project do not support this rationale to be true in the context of CAXII and 6A10, however we acknowledge our study has severe technical limitations. First, we did not include an IgG control arm to test any off-target effects of the antibody setup. We acknowledge previous work of others that indicates that IgG treatment does not cause severe phenotypic effects of responder model GBM1,38 but differences in age of the cells and different IgG specifications may have varying influences. Moreover we designed our study on the assumption of ultra-high target specificity of 6A10 to apply in our tests, further justifying our chosen mock treatment as an acceptable experimental design for this project, especially given the main focus of this paper lies on the molecular diagnostics of patient samples. Second, a genetic model of CAXII suppression could be applied to discern if the phenotypic response of 6A10 is target specific
Our translational data position well in current drug development campaigns as the generation of CA-inhibitors is under heavy investigation worldwide.19,39–43 Given this active field of research, a comprehensive discussion of the therapeutic angle of our 6A10 data, adequately appreciating the entire activities in the field, is challenging. That is why we focus on direct competitor drug candidates in the context of neuro-oncology. In this regard, 6A10 has to withstand benchmarking against very promising recent results reported with natural-derived compound Psammaplin C and its derivatives. Those substances impair enzymatic activity of CA12 in glioma cells, reducing their resistance to the standard chemotherapeutic agent temozolomide (TMZ) that eventually leads to prolonged survival of mice bearing GBM models.44,45 Emergence of therapy resistance against CA12 inhibitors is discussed, with reports showing CA9 compensating altered enzymatic activity of CA12 to promote cancer malignancy;9,10 we sought to correlate the expression patterns of those isoforms in our clinical samples. Although we find positive correlation of CA12 with CA9 in our clinical datasets (Suppl. Figure S6), further studies testing enzymatic activity of CA12 and CA9 upon 6A10 treatment will be relevant to assess the potential of this antibody to achieve durable therapeutic effects. Moreover, testing the effect of 6A10 treatment on the resistance of cells to standard chemotherapy may be an additional set of experiments to benchmark the antibody against competitor compounds. Although not observed in all studied models, 6A10 can impair GSC growth to some extent. This observation is in line with reported effects of 6A10 being able to block the growth of cells from lung, gastric, ductal breast and colon cancers.37 However, to the best of our knowledge, for the first time we provide evidence that anti-CA12 directed treatment in glioma can effectively block cell invasion marker ZEB1 accompanied by significant suppression of cellular invasion. Given the relevance of ZEB1 in mediating the highly invasive growth of GBM,3,4 and given ZEB1 activation has been suggested to mediate the resistance of GBM cells to TMZ,4,24,46 we believe this result is of particular high translational interest requiring further exploration. Interestingly, CA12 activation was found to be a consequence of breast cancer cell EMT.47 Moreover, in a pan-cancer study, CA12 was prominently positioned as a top candidate of metabolic genes that is positively correlated to EMT transcription signature48 and TGF-b signaling in GBM, a known glioma EMT activator,2 augments CAXII signals49. Taken together, our functional data extend evidence that CAXII participates in - or at least is a reporter of - cancer EMT programs. We enforce the concept that generation of pharmacological anti-EMT directed strategies is feasible and that these represent a promising approach to develop future tumor-agnostic treatment regimes.
Conclusion
CA12 activation predicts for negative clinical course in Asian and TCGA brain tumor patients and is associated with clinical molecular marker of IDH1WTDNA and to 1p19q chromosome arm co-deletion. GBMs of the mesenchymal subtype are enriched for CA12. In total, our data fill the hitherto existing gap of knowledge of probing the emerging cancer target CA12 in the context of state of the art molecular neuropathology. Correlative analysis in clinical samples and corresponding protein studies on experimental models does not promote CA12/CAXII to present a specific glioma stem cell marker. However, anti-CAXII directed pharmacotherapy using 6A10 blocking antibody can cause suppression of growth and cellular invasion in a fraction of GSCs and supports the growing concept that CAXII interacts with cancer EMT programs. Further studies are required to delineate the risk of off-target events of 6A10 in GSCs and to identify the molecular background responsible for the different response rates of glioma cells to the antibody. We enforce the concept that generation of pharmacological anti-EMT directed strategies is feasible and that these represent a promising approach to develop future tumor-agnostic treatment regimes.
Acknowledgments
UDK wants to thank MVM Domínguez for her technical assistance in the completion of this project.
Disclosure
Prof. Reinhard Zeidler reports a patent, EP10004646.5, issued. The authors report no other potential conflicts of interest in this work.
References
1. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CLL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217. doi:10.1101/gad.261982.115
2. Kahlert UD, Joseph JV, Kruyt FAE. EMT- and MET-related processes in nonepithelial tumors: importance for disease progression, prognosis, and therapeutic opportunities. Mol Oncol. 2017;11:860–877. doi:10.1002/1878-0261.12085
3. Kahlert UD, Suwala AK, Raabe EH, et al. ZEB1 promotes invasion in human fetal neural stem cells and hypoxic glioma neurospheres. Brain Pathol. 2015;25:724–732. doi:10.1111/bpa.12240
4. Siebzehnrubl FA, Silver DJ, Tugertimur B, et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med. 2013;5:1196–1212. doi:10.1002/emmm.201302827
5. Rosmaninho P, Mükusch S, Piscopo V, et al. Zeb1 potentiates genome-wide gene transcription with lef1 to promote glioblastoma cell invasion. EMBO J. 2018;37:e97115. doi:10.15252/embj.201797115
6. Suzuki K, Kawataki T, Endo K, Miyazawa K, Kinouchi H, Saitoh M. Expression of ZEBs in gliomas is associated with invasive properties and histopathological grade. Oncol Lett. 2018;16:1758–1764. doi:10.3892/ol.2018.8852
7. Kahlert UD, Maciaczyk D, Doostkam S, et al. Activation of Canonical WNT/β-Catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and Other Activators of Epithelial-to-Mesenchymal Transition. Cancer Lett. 2012;325:42–53. doi:10.1016/j.canlet.2012.05.024
8. Mboge MY, Mahon BP, McKenna R, Frost SC. Carbonic Anhydrases: role in PH Control and Cancer. Metabolites. 2018;8. doi:10.3390/metabo8010019
9. Chiche J, Ilc K, Laferrière J, et al. Hypoxia-Inducible Carbonic Anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular PH. Cancer Res. 2009;69:358–368. doi:10.1158/0008-5472.CAN-08-2470
10. Doyen J, Parks SK, Marcié S, Pouysségur J, Chiche J. Knock-down of hypoxia-induced carbonic anhydrases ix and xii radiosensitizes tumor cells by increasing intracellular acidosis. Front Oncol. 2012;2:199. doi:10.3389/fonc.2012.00199
11. Beckner ME, Pollack IF, Nordberg ML, Hamilton RL. Glioblastomas with Copy Number Gains in EGFR and RNF139 show increased expressions of carbonic anhydrase genes transformed by ENO1. BBA Clin. 2015;5:1–15. doi:10.1016/j.bbacli.2015.11.001
12. Haapasalo J, Hilvo M, Nordfors K, et al. Identification of an alternatively spliced isoform of carbonic anhydrase XII in Diffusely Infiltrating Astrocytic Gliomas. Neuro Oncol. 2008;10:131–138. doi:10.1215/15228517-2007-065
13. Proescholdt MA, Merrill MJ, Stoerr E-M, Lohmeier A, Pohl F, Brawanski A. Function of Carbonic Anhydrase IX in Glioblastoma Multiforme. Neuro Oncol. 2012;14:1357–1366. doi:10.1093/neuonc/nos216
14. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555:469–474. doi:10.1038/nature26000
15. Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi:10.1016/j.ccr.2009.12.020
16. Louis DN, Perry A, Reifenberger G, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–820. doi:10.1007/s00401-016-1545-1
17. Louis DN, Wesseling P, Aldape K, et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020;30:844–856. doi:10.1111/bpa.12832
18. Reifenberger J, Reifenberger G, Liu L, James CD, Wechsler W, Collins VP. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol. 1994;145:1175–1190.
19. Singh S, Lomelino CL, Mboge MY, Frost SC, McKenna R. Cancer drug development of carbonic anhydrase inhibitors beyond the active site. Molecules. 2018;23:1045. doi:10.3390/molecules23051045
20. Herrera-Rios D, Li G, Khan D, et al. A computational guided, functional validation of a novel therapeutic antibody proposes notch signaling as a clinical relevant and druggable target in glioma. Sci Rep. 2020;10:16218. doi:10.1038/s41598-020-72480-y
21. Battke C, Kremmer E, Mysliwietz J, et al. Generation and characterization of the first inhibitory antibody targeting tumour-associated carbonic anhydrase XII. Cancer Immunol Immunother. 2011;60:649–658. doi:10.1007/s00262-011-0980-z
22. von Neubeck B, Gondi G, Riganti C, et al. An inhibitory antibody targeting carbonic anhydrase XII abrogates chemoresistance and significantly reduces lung metastases in an orthotopic breast cancer model in vivo. Int J Cancer. 2018;143:2065–2075. doi:10.1002/ijc.31607
23. Koch K, Hartmann R, Tsiampali J, et al. A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov. 2020;6:20. doi:10.1038/s41420-020-0258-3
24. Vargas-Toscano A, Nickel A-C, Li G, et al. Rapalink-1 targets glioblastoma stem cells and acts synergistically with tumor treating fields to reduce resistance against temozolomide. Cancers. 2020;12:3859. doi:10.3390/cancers12123859
25. Gravendeel LAM, Kouwenhoven MCM, Gevaert O, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69:9065–9072. doi:10.1158/0008-5472.CAN-09-2307
26. Kawaguchi A, Yajima N, Tsuchiya N, et al. Gene expression signature-based prognostic risk score in patients with Glioblastoma. Cancer Sci. 2013;104:1205–1210. doi:10.1111/cas.12214
27. Mehrjardi NZ, Hänggi D, Kahlert UD. Current biomarker-associated procedures of cancer modeling-a reference in the context of IDH1 Mutant Glioma. Cell Death Dis. 2020;11:998. doi:10.1038/s41419-020-03196-0
28. Wiestler B, Capper D, Sill M, et al. Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathol. 2014;128:561–571. doi:10.1007/s00401-014-1315-x
29. Riemenschneider MJ, Hegi ME, Reifenberger G. MGMT Promoter Methylation in Malignant Gliomas. Target Oncol. 2010;5:161–165. doi:10.1007/s11523-010-0153-6
30. Ghosh D, Ulasov IV, Chen L, et al. TGFβ-Responsive HMOX1 expression is associated with stemness and invasion in glioblastoma multiforme. Stem Cells. 2016;34:2276–2289. doi:10.1002/stem.2411
31. Kim E-J, Jin X, Kim OR, Ham SW, Park S-H, Kim H. Glioma stem cells and their non-stem differentiated glioma cells exhibit differences in mitochondrial structure and function. Oncol Rep. 2018;39:411–416. doi:10.3892/or.2017.6075
32. Neradil J, Veselska R. Nestin as a Marker of Cancer Stem Cells. Cancer Sci. 2015;106:803–811. doi:10.1111/cas.12691
33. Berezovsky AD, Poisson LM, Cherba D, et al. Sox2 promotes malignancy in glioblastoma by regulating plasticity and astrocytic differentiation. Neoplasia. 2014;16:193–206.e25. doi:10.1016/j.neo.2014.03.006
34. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi:10.1038/nature03128
35. Calvo KA, Scrideli CA, Moraes T, Da S, Teixeira SA. Silencing of CA12 by Interfering RNA sensitizes GBM cells to chemotherapy and reduces cell proliferation. Rev Med. 2020;99:14–15. doi:10.11606/issn.1679-9836.v99iSupplp14-15
36. Fiedler L, Kellner M, Gosewisch A, et al. Evaluation of 177Lu[Lu]-CHX-A″-DTPA-6A10 fab as a radioimmunotherapy agent targeting carbonic anhydrase XII. Nucl Med Biol. 2018;60:55–62. doi:10.1016/j.nucmedbio.2018.02.004
37. Gondi G, Mysliwietz J, Hulikova A, et al. Antitumor efficacy of a monoclonal antibody that inhibits the activity of cancer-associated carbonic anhydrase XII. Cancer Res. 2013;73:6494–6503. doi:10.1158/0008-5472.CAN-13-1110
38. Schreck KC, Taylor P, Marchionni L, et al. The Notch Target Hes1 Directly Modulates Gli1 Expression and Hedgehog Signaling: a Potential Mechanism of Therapeutic Resistance. Clin Cancer Res. 2010;16:6060–6070. doi:10.1158/1078-0432.CCR-10-1624
39. Stravinskiene D, Sliziene A, Baranauskiene L, Petrikaite V, Zvirbliene A. Inhibitory monoclonal antibodies and their recombinant derivatives targeting surface-exposed carbonic anhydrase xii on cancer cells. Int J Mol Sci. 2020;21. doi:10.3390/ijms21249411
40. Uda NR, Stenner F, Seibert V, et al. Humanized monoclonal antibody blocking carbonic anhydrase 12 enzymatic activity leads to reduced tumor growth in vitro. Anticancer Res. 2019;39:4117–4128. doi:10.21873/anticanres.13570
41. Aslam S, Gupta V. Carbonic Anhydrase Inhibitors. In StatPearls; StatPearls Publishing: treasure Island (FL). 2020.
42. Supuran CT. Carbonic Anhydrases: novel Therapeutic Applications for Inhibitors and Activators. Nat Rev Drug Discov. 2008;7:168–181. doi:10.1038/nrd2467
43. Williams KJ, Gieling RG. Preclinical evaluation of ureidosulfamate carbonic anhydrase ix/xii inhibitors in the treatment of cancers. Int J Mol Sci. 2019;20. doi:10.3390/ijms20236080
44. Salaroglio IC, Mujumdar P, Annovazzi L, et al. Carbonic anhydrase xii inhibitors overcome p-glycoprotein-mediated resistance to temozolomide in glioblastoma. Mol Cancer Ther. 2018;17:2598–2609. doi:10.1158/1535-7163.MCT-18-0533
45. Mujumdar P, Kopecka J, Bua S, Supuran CT, Riganti C, Poulsen S-A. Carbonic anhydrase xii inhibitors overcome temozolomide resistance in glioblastoma. J Med Chem. 2019;62:4174–4192. doi:10.1021/acs.jmedchem.9b00282
46. Koch K, Hartmann R, Schröter F, et al. Reciprocal regulation of the cholinic phenotype and epithelial-mesenchymal transition in glioblastoma cells. Oncotarget. 2016;7:73414–73431. doi:10.18632/oncotarget.12337
47. Vergara D, Ravaioli S, Fonzi E, et al. Carbonic anhydrase xii expression is modulated during epithelial mesenchymal transition and regulated through protein kinase c signaling. Int J Mol Sci. 2020:21. doi:10.3390/ijms21030715
48. Shaul YD, Freinkman E, Comb WC, et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell. 2014;158:1094–1109. doi:10.1016/j.cell.2014.07.032
49. Ghosh D, Funk CC, Caballero J, et al. A cell-surface membrane protein signature for glioblastoma. Cell Syst. 2017;4:516–529.e7. doi:10.1016/j.cels.2017.03.004
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.