Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 12
CAR T-Cell Therapy for Patients with Multiple Myeloma: Current Evidence and Challenges
Authors Rendo MJ, Joseph JJ, Phan LM, DeStefano CB
Received 5 July 2022
Accepted for publication 20 August 2022
Published 29 August 2022 Volume 2022:12 Pages 119—136
DOI https://doi.org/10.2147/BLCTT.S327016
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Wilson Gonsalves
Matthew J Rendo,1 Jacinth J Joseph,2 Liem Minh Phan,3 Christin B DeStefano4
1Department of Hematology/Oncology, Brooke Army Medical Center, San Antonio, TX, USA; 2Blood and Marrow Transplant Center, Methodist Le Bonheur Healthcare, Memphis, TN, USA; 3Clinical Investigation Facility, David Grant USAF Medical Center, Travis Air Force Base, CA, USA; 4Department of Hematology/Oncology, Walter Reed National Military Medical Center, Bethesda, MD, USA
Correspondence: Christin B DeStefano, Email [email protected]
Abstract: The therapeutic landscape of multiple myeloma (MM) has benefited from an emergence of novel therapies over the last decade. By inducing T-cell kill of target cancer cells, chimeric antigen receptor (CAR) T-cell therapies have improved outcomes of patients with hematologic malignancies. B-cell maturation antigen (BCMA) is the current target antigen of choice for most CAR T-cell products under investigation for MM. However, their shortcomings deal with logistical and clinical challenges, including limited availability, manufacturing times, and toxicities. This article provides an overview of recently developed and investigational CAR T-cell therapies for MM, highlighting current evidence and challenges.
Keywords: multiple myeloma, CAR T-cell, immunotherapy
Introduction
Multiple myeloma (MM) is the second most common hematologic malignancy diagnosed in the United States (U.S.).1 Although relapses are frequent, novel therapies have improved long-term outcomes as a median survival of over 10 years has been observed.2 Chimeric antigen receptor (CAR) T-cells have recently gained US Food and Drug Administration (FDA) approval for use in MM. CAR T-cells serve as a promising therapy in the treatment landscape of MM given their unprecedented responses in a refractory population, yet their logistical and clinical limitations must be acknowledged to further improve patient outcomes.
Background on CAR T-Cells
Genetically engineered T-cells were initially studied in 1989 to exploit cytotoxic T-cell effector functions.3 Cytotoxic T-cells can induce targeted cell killing through expression of FasL, release of granzyme, perforin, and pro-inflammatory cytokines, all of which activate target cell mitochondrial caspases and trigger apoptosis.4 T-cell activation requires binding of a T-cell receptor to its cognate antigen and class 1 major histocompatibility complex (MHC) as well as binding of a T-cell costimulatory receptor to its ligand.5 Once the T-cell receptor and its costimulatory receptor bind their targets, the T-cell receptor cytoplasmic domain (CD3ζ) triggers intracellular signaling pathways, inducing T-cell activation and robust cell-kill (Figure 1). CAR T-cells possess the lethal functions of cytotoxic T-cells but are engineered to be highly specific to their target antigen(s) and do not require MHC or costimulatory binding.6 Current FDA approved CAR T-cells for hematologic malignancies are second-generation CAR T-cells, composed of a single chain variable fragment (scFv) extracellular domain, a transmembrane domain (CD3ζ), and an intracytoplasmic T-cell costimulatory domain (Figure 2).6,7 After infusion, CAR T-cells bind their target antigen(s), proliferate, release tumor antigens, induce epitope spreading, and recruit other immune cells, ultimately orchestrating a multi-faceted immune response resulting in robust cell kill.6,8
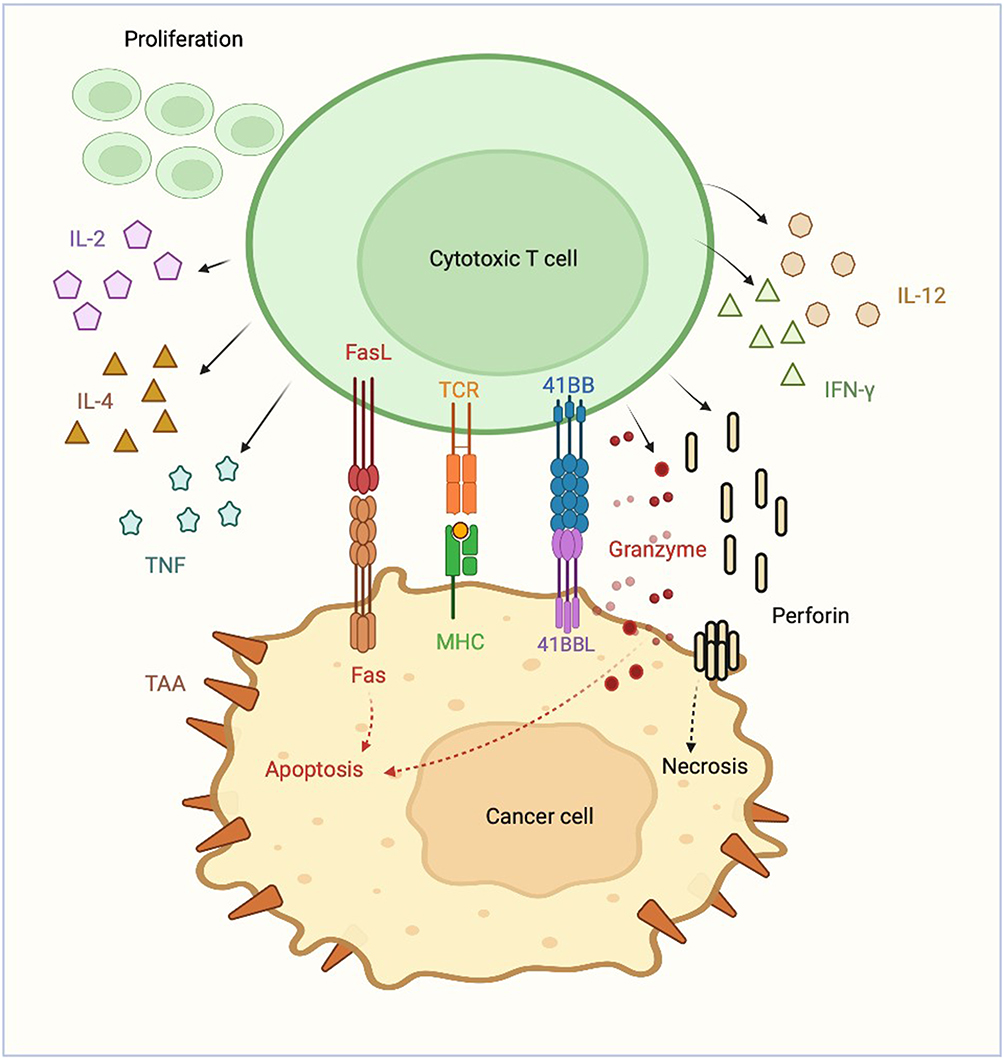 |
Figure 1 Cytotoxic T-cell. |
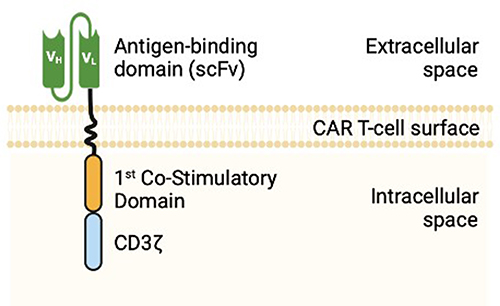 |
Figure 2 2nd Generation CAR T-cell Structure. |
Autologous CAR T-cell treatment requires leukapheresis of host T-cells followed by CAR T-cell manufacturing, bridging therapy if needed for disease control during manufacturing, administration of lymphodepleting drugs, and CAR T-cell infusion (Figure 3). Although hospitalization is standard after infusion, outpatient CAR T-cells are being delivered at select centers in highly selected patients. CAR T-cell manufacturing may take up to 6 weeks. During this time, ex-vivo T-cell stimulation and expansion occur. Isolated T-cells are stimulated to proliferate and expand via exposure to various pro-growth cytokines to include anti-CD3 antibodies and interleukin (IL)-2.9 Afterward, a lentiviral or gamma retroviral vector is used to transfer the gene encoding the CAR product into the T-cell genome.10 Of note, gene transfer can occur through other mechanisms as described in Investigational CAR T-Cell Therapies.
 |
Figure 3 Autologous CAR T-cell Process. |
FDA Approved CAR T-Cells in MM
The first B-cell maturation antigen (BCMA)-targeting CAR T-cells were studied in MM in 2013.11 The target antigen, BCMA, is a tumor necrosis factor receptor (TNFR) superfamily member 17 surface protein found predominantly on plasma cells.11 Overexpression of BCMA by its ligand, a proliferation-inducing ligand (APRIL), induces MM progression and survival through induction of protein kinase B (AKT), MAPK, and nuclear factor (NF)-κB signaling cascades (Figure 4).12 There are currently two FDA approved CAR T-cell products for treatment of relapsed/refractory MM (RRMM): idecaptagene vicleucel (ide-cel) and ciltacabtagene autoleucel (cilta-cel). Both drugs are approved for use in RRMM after four or more lines of therapy, including a proteasome inhibitor (PI), immunomodulatory agent and anti-CD38 directed therapy. Ide-cel and cilta-cel are both second generation autologous CAR T-cell products targeting BCMA, use a third-generation lentiviral vector for CAR gene transfer, share a similar method of CAR T-cell expansion, and use the 4–1BB intracytoplasmic costimulatory domain. Each is administered 5–7 days after lymphodepletion with cyclophosphamide 300 mg/m2 and fludarabine 30 mg/m2 daily for three days. Cilta-cel has dual-BCMA receptors which bind two distinct BCMA epitopes, whereas ide-cel has a single BCMA binding domain.
 |
Figure 4 BCMA Signaling Pathway. |
Idecaptagene Vicleucel
Ide-cel (formerly bb2121) first demonstrated impressive clinical activity in the Phase 1 CRB-401 trial.13 Thirty-three patients received single infusions at target doses of 50×106, 150×106, 450×106 or 800×106 CAR T-cells per kilogram in the dose-escalation phase. Very good partial responses (VGPR) and complete responses (CR) were attained only at doses of 150×106 or greater. Therefore, 150×106 to 450×106 CAR T-cells per kilogram were used in the expansion phase. At higher target doses, 100% of patients achieved a response, and the median progression-free survival (PFS) was 11.8 months. CAR T-cells persisted for up to 1 year after the infusion.
The Phase 2 KarMMa study (NCT03361748) enrolled 140 patients with RRMM who had received at least 3 prior lines of therapy including a PI, immunomodulatory agent, and anti-CD38 directed antibody.14 The primary endpoint was overall response rate (ORR), and the secondary endpoint was CR including stringent CR (sCR). Of the 140 patients enrolled, 128 were treated with ide-cel with a median follow-up of 13.3 months, median age of 61 (range 33–78) and 59% of whom were male. Thirty-five percent of treated patients had high-risk cytogenetics with a median of 6 prior lines of therapy (range 3–16), 94% had received a prior autologous stem cell transplant (ASCT), 84% were triple-refractory, and 26% were penta-refractory. Eighty-eight percent received bridging therapy during the manufacturing period, which consisted of various combinations of drugs including dexamethasone, cyclophosphamide, daratumumab, carfilzomib, bortezomib and pomalidomide. Responses to bridging therapy were observed in only 4% of patients.
The primary endpoint, ORR, was 73%. Rates of CRs, minimal residual disease (MRD) negativity (as measured by next-generation sequencing with a minimum cutoff of 10−5 nucleated cells), and median PFS among the entire cohort was 33%, 26%, and 8.8 months, respectively. A dose–response relationship was demonstrated, with ORRs, CR rates and PFS reaching 81%, 39%, and 12.1 months at the 450×106 target dose, respectively. Depth of response correlated with length of response; of the 42 patients with a CR or sCR, duration of response (DOR) and median PFS extended to 19 and 20.2 months, respectively. The median time to first response was 1 month (range, 0.5 to 8.8 months), and the median time to a CR or better was 2.8 months (range, 1.0 to 11.8 months). An updated analysis of the KarMMa trial revealed an overall survival (OS) of 24.8 months.15 Hematologic adverse events were common, including high rates of grade 3 or 4 neutropenia (89%), thrombocytopenia (52%), anemia (60%) and leukopenia (39%). Neurotoxicity was also common (18%), albeit grade 3 in only 3% of patients with no grade 4 toxicity reported. Although cytokine release syndrome (CRS) occurred in 84% of patients, only 5% were grade 3 or higher. Among the 128 treated patients, there were 44 deaths (34%), of which 35 were due to progressive disease or complications of progression. Three percent of deaths were treatment-related, including pulmonary aspergillosis, gastrointestinal hemorrhage, CRS, and cytomegalovirus pneumonia.
Ciltacabtagene Autoleucel
Cilta-cel (formerly JNJ-68284528) was FDA approved based on the CARTITUDE-1 study (NCT03548207), a phase 1b/2 open-label study in patients with RRMM who received at least 3 prior lines of therapy (PI, immunomodulatory agent, and anti-CD38 therapy) or were double-refractory to a PI and immunomodulatory drug.16 Of the 113 patients who enrolled in the study, 97 were treated with a single cilta-cel infusion at a target dose of 0.75×106 CAR T-cells per kilogram (range 0.5–1.0×106). The median follow-up was 12.4 months. The median age of treated patients was 61 (range 43–78), 59% were male, 24% had high-risk cytogenetics, and the median number of prior lines of treatment was 6 (range 3–18). Eighty-eight percent were triple-class refractory, 42% were penta-drug refractory, and 99% were refractory to their last line of therapy. Seventy-three of the 97 patients (75%) received bridging therapy which consisted mostly of various combinations of corticosteroids, bortezomib, cyclophosphamide, pomalidomide, carfilzomib, and daratumumab. Only 45% of patients responded to bridging therapy, whereas 49% experienced increased tumor burden while receiving bridging therapy.
The ORR was 98%, sCR rate was 80%, and VGPR rate was 95%. The median time to first response and best response were 1 month (range, 0.9 to 1.0 months) and 2.6 months (range, 1.0 to 6.1 months-) respectively. The 12-month PFS and OS were 77% (95% CI, 66–84.3) and 89% (95% CI, 80.2–93.5), respectively. Updated results from the CARTITUDE-1 trial with longer follow-up (median 28 months) revealed a 27-month PFS and OS of 55% (95% CI, 44.0–64.6) and 70% (95% CI, 60.1–78.6), respectively.17 The 27-month PFS and OS among patients who achieved sustained MRD (as measured by next-generation sequencing with a minimum cutoff of 10−5 nucleated cells) for ≥ 12 months was 79% (95% CI, 51.5–91.8) and 91% (95% CI, 67.7–97.6), respectively. Hematologic side effects were common with 95%, 68%, 61%, and 60% of patients experiencing grade 3 or 4 neutropenia, anemia, leukopenia, and thrombocytopenia, respectively. Neurotoxicity was the most common non-hematologic grade 3 or 4 toxicities, affecting 9% of patients. Neurotoxicity of any grade occurred in 21% of patients, including immune effector cell-associated neurotoxicity syndrome (ICANS) in 16%. Although CRS occurred in 95% of patients, it was mostly grade 1 or 2 with a median time to onset of 7 days, median duration of 4 days, with only 4% of patients experiencing grade 3 or 4 CRS, and one patient experiencing grade 5 toxicity. After 28-months of follow-up, there were 30 deaths (31%), of which 28 occurred after day +100.17 Fourteen deaths were due to progressive disease, and the remaining 10 deaths were due to adverse events unrelated to therapy.17 Six deaths were felt to be therapy related to sepsis/septic shock, CRS and hemophagocytic lymphohistiocytosis (HLH), lung abscess, respiratory failure, and neurotoxicity.16
Outcomes of Ide-Cel and Cilta-Cel in Real-World Patients
In the KarMMa and CARTITUDE-1 trials, 12 (9%) and 16 (14%) patients were enrolled but did not receive CAR T-cell therapy, respectively, and subsequently were excluded from efficacy and safety analyses. Some of these patients (2 in KarMMa and 9 in CARTITUDE-1) did not receive therapy due to death before CAR T-cell administration.14,16 Because these patients were not included in the efficacy analyses, efficacy results from these studies are likely inflated.18,20 Further, it is unknown how many patients could not enroll in the KarMMa and CARTITUDE-1 studies due to strict eligibility criteria. Therefore, it is unclear whether eligible real-world patients with multi-refractory disease will be able to survive during the waitlist and manufacturing period and experience outcomes as favorable as those reported in KarMMa and CARTITUDE-1. Nevertheless, multiple real-world studies suggest a benefit to the use of idel-cel or cilta-cel when compared to conventional therapies. A multicenter retrospective study assessed characteristics and outcomes of 108 patients with RRMM who had received standard of care ide-cel under the commercial FDA label.20 Despite that 67% of real-world patients would not have met eligibility criteria for KarMMa, comparable toxicities were noted as well as efficacy with an ORR of 83% including 34% CRs and median PFS of 8.9 months (95% CI, 8.5-NR). Further, although there are limitations to indirect comparisons, multiple other studies have compared data from KarMMa and CARTITUDE-1 with patient-level or aggregate data from RRMM registries, revealing a significant benefit favoring CAR T-cells over conventional regimens.21–26
Without direct comparisons, it is difficult to tell whether real-world patients benefit more from one commercial CAR T-cell product over the other. The KarMMa and CARTITUDE-1 studies enrolled similar patients with comparable baseline demographics. However, in the KarMMa trial, only 4% of patients who received bridging therapy responded, whereas 45% of patients in the CARTITUDE-1 trial responded to bridging therapy.14,16 Therefore, it is possible that the CARTITUDE-1 patients had more favorable tumor biology or lower tumor burden prior to CAR T-cell infusion. More time and data are needed to better understand subtle differences in efficacy between ide-cel and cilta-cel.
Logistical Challenges
The most formidable challenges to the administration of ide-cel or cilta-cel are logistical, dealing with lengthy manufacturing times, limited manufacturing slots, risk of manufacturing failure, access to care and costs, as outlined below.
Manufacturing Time
The manufacturing time of ide-cel and cilta-cel is around 28 days. For timely treatment, one should consider availability of manufacturing slots, timing the salvage treatment, pre-CAR T-cell work-up and testing and apheresis at the treating facility. In the KarMMa trial, all enrolled patients underwent apheresis, but 12 out of 140 (9%) enrolled patients did not receive CAR T-cell treatment.14 Similarly, 16 out of 113 (14%) enrolled patients in CARTITUDE-1 did not receive cilta-cel.16 The percentage of myeloma patients eventually not receiving their planned CAR T-cell treatment in the real-world scenario could be expected to be higher.
CAR T-Cell Availability
The approval of myeloma CAR T-cell therapies was met with high demand from patients whose disease had failed multiple lines of therapy. The bottle-neck due to manufacturing capabilities related to lentiviral vectors has led to limited slots provided at approved centers and delayed onboarding of other centers. At facilities where treatment is available, the treating physicians must choose which patients to offer a slot, which can pose an ethical challenge.18 They have to decide between those with the most aggressive and refractory disease who are in the greatest need but less likely to have a favorable outcome and those with less aggressive disease biology who are more likely to fare a better outcome.18 In the future, the availability of more products would help supply meet demand, as could use of novel manufacturing methods such as non-viral vectors or allogeneic CAR T-cell products.19 At the same time, approval of agents in earlier lines will be expected to further strain the system due to higher demand.
Manufacturing Failure
The wide range of variability in lymphocyte count, lymphocyte function, medication use, mononuclear cell collection by apheresis and CAR T-cell manufacturing leads to risk of failure to achieve an efficacious product. Myeloma is associated with complex immunoregulatory alterations including a decreased CD4+ T-cell subgroup, enhanced T-cell exhaustion, increased T-cell senescence, and abnormal sensitivity to mitogens.27–29 Prior use of lymphodepleting chemotherapy and current or recent use of corticosteroids further decrease T-cell yield and can lead to dysfunctional T-cells.30,31 In general, for CAR T-cell products that fall out of regulatory specifications, a special program such as MAP (managed access program) may allow use of the manufactured product, under the discretion of the treating provider. Furthermore, supply chain shortages have provided constraints on treatment modalities, notably tocilizumab and fludarabine. When approaching CAR T-cell therapies under such limitations, physicians may defer utilization of CAR T-cell therapy until necessary conditioning and supportive medications are available. Conversely, alternative lymphodepleting conditioning regimens may be utilized, albeit fludarabine and cyclophosphamide have been standardly used for most regimens.
Access to Care
In the US, an important factor affecting access to care is the availability of treatment centers close to home. About a quarter of patients who received CAR T-cell treatment on trials for myeloma lived over 2 hours away.32 Most centers require local stay for up to 30 days after CAR T-cell infusions and this is an important barrier for some patients. Many programs utilize grants, donations, or other funding to help overcome this cost, while local lodging resources such as Hope Lodge (provided by American Cancer Society) have made CAR T-cell treatment available to patients who cannot afford local lodging. The ability to have a caregiver can be a hurdle due to social and financial implications. A recent study utilizing the Vizient® Clinical Database showed multiple disparities in access to CAR T-cell treatment on clinical trials.32 Despite African Americans representing close to 20% of new myeloma cases, they made up only 1% of patients who received CAR T-cell treatment. Conversely, white individuals are over-represented in clinical trials and made up 65% of patients with MM treated with CAR T-cell therapy. Further, only 6% of CAR T-cell admissions were from neighborhoods with a mean income under $40,000, compared to 12% of non-CAR T-cell treatments, highlighting disparity in access to CAR T-cell treatment.
Cost
The approved myeloma CAR T-cell treatments carry a list price of $419,500 (ide-cel) and $465,000 (cilta-cel) for a one-time infusion. The cost of inpatient treatment must include hospital stay and specialized care, particularly in case of serious toxicity where the hospitalization can be both intensive and prolonged. Most commercial payers cover this treatment and often require a single-case agreement. In August 2019, CMS (Centers for Medicare and Medicaid Services) finalized the decision for Medicare to cover CAR T-cell therapies under FDA-approved indications. This important treatment remains elusive to the uninsured who represent 1% and Medicaid beneficiaries who represent 2.3% of patients treated with myeloma CAR T-cells in trials, in keeping with non-CAR T-cell treatments.32
Clinical Challenges
CAR T-Cell Toxicities
One of the biggest challenges of CAR T-cell therapy is the management of short- and long-term toxicities. Long-term safety data on ide-cel and cilta-cel are lacking; however, long-term follow-up and continued monitoring of patients who completed therapy is ongoing (NCT03435796) which will assess rates of late treatment complications such as second primary malignancies as well as other safety signals. Common BCMA CAR T-cell toxicities are discussed below.
CRS
CRS is a systemic toxicity marked by an increase in pro-inflammatory cytokines and may manifest along a spectrum from mild nausea and fever to life-threatening capillary leak syndrome with hypotension, cardiac arrest, and end-organ failure.33 A meta-analysis of the KarMMa and LEGEND-2 trials revealed up to 80% of patients experience CRS, with 14.1% experiencing grade 3 toxicities or higher with a median duration of 7 days for bb2121 and 9 days for LCAR-B38M.34 The rates and severity of CRS correlate with the dose of infused CAR T-cells.14 Other risk factors for severe CRS include large baseline tumor burden, CRS onset within 3 days of CAR T-cell infusion, multiple medical comorbidities, and an elevated IL-6 level.33,35
While most patients can tolerate low-grade CRS, interventions may be required. The American Society of Blood and Marrow Transplantation CRS Consensus Grading requires baseline fever for diagnosis with a degree of hypoxia and hypotension determining subsequent grades.36 Tocilizumab, an IL-6 receptor antagonist, was FDA approved in 2017 to treat CRS and can rapidly reverse symptoms.37 If rapid reversal does not occur, corticosteroids are also often administered. While most institutions provide anti-IL-6 therapy at the onset of grade 2 CRS, optimal timing and administration has not been established. Upon development of grade 3 toxicities, most patients will require intensive care unit (ICU) support. While early data have indicated that the use of tocilizumab or corticosteroids does not portend increased risk of relapse, the data are still maturing.6
Hematologic Toxicity
Hematologic toxicity after CAR T-cell infusion can be complex and multifactorial with lymphodepleting chemotherapy largely contributing, as well as inflammation, HLH, infections, disease relapse, and secondary neoplasms including myelodysplastic syndrome.38 Furthermore, patients are often heavily pretreated, leaving them with limited marrow reserve. Among trial and real-world patients treated with BCMA CAR T-cells, grade 3–4 hematologic adverse events are common and can be early (<30 days post-infusion), short-term (30–60 days post infusion), and prolonged (persistent beyond 90 days).38 Therefore, close monitoring of blood counts, prompt transfusion support, and appropriate supportive care with transfusion support, growth factors, and infection prophylaxis for severe neutropenia are required for prolonged periods of time post-CAR T-cell infusion.
Infections
As a product of lymphodepleting conditioning regimens, patients are at an elevated risk of acquiring serious infections. Moreover, this is not simply a function of the chemotherapy regimen, but the nature of BCMA CAR T-cells, which also neutralize antibody-producing healthy plasma cells and therefore induce hypogammaglobulinemia. Infections developed in 69% of patients enrolled in KarMMa of which 22% were grade 3 or 4.14 Fifty-eight percent of CARTITUDE-1 participants developed infections with 20% exhibiting grades 3 or 4, the most common of which were pneumonia (8%) and sepsis (4%).16 Furthermore, CAR T-cell recipients who develop Covid-19 infection are at a particularly high risk of mortality of up to 50%.39 Vigilance, prophylactic antimicrobials, vaccinations, intravenous immunoglobulin, and prompt treatment of infections are necessary in this heavily immunocompromised patient population, particularly in the era of Covid-19.
ICANS
ICANS is a neuropsychiatric syndrome that is believed to be a manifestation of CRS and typically occurs during CRS or just days following its resolution. ICANS carries a heterogeneous and variable clinical presentation ranging from altered consciousness or cognitive impairment to seizures and cerebral edema.40 Ide-cel and cilta-cel are believed to carry less risk and severity of ICANS relative to CD28 carrying CAR T-cell products as their 4–1BB domains are believed to result in slower T-cell expansion which may mitigate ICANS severity.41 In CARTITUDE-1, the median onset of ICANS was 8 days after T-cell infusion and occurred in 17% of patients.16 Comparatively, KarMMa observed neurologic toxicities in 45% of patients, 6% of whom received glucocorticoids.14 Factors that appear to be associated with increased ICANS risk include pre-existing neurologic or medical comorbidities, high disease burden, early onset of severe CRS, and younger age.33,42 Treatment of ICANS remains largely supportive and requires close vigilance for the progression of neurologic toxicities. Patients may require ICU level care with the development of progressive altered sensorium. Utilization of steroids as a treatment adjunct is common; however, optimal timing, duration and dosage are not established. Typical CRS and ICANS protocols call for dexamethasone 10 mg every six to twelve hours with tapering over two to five days in the absence of seizures or cerebral edema with the onset of grade 2 symptoms.37 In the setting of early onset ICANS and utilization of tocilizumab, 1–2 doses of dexamethasone are often given concurrently. While tocilizumab for moderate-to-severe ICANS is common, whether it decreases symptomatic severity remains controversial. While steroids are well tolerated by most, they may aggravate delirium, confounding the ICANS grade. A short course of steroids may effectively suppress ICANS without dampening CAR T-cell effectiveness.43 Seizure prophylaxis is also not standardized, albeit early levetiracetam prophylaxis is commonplace with the development of ICANS.37
Movement and Neurocognitive Treatment-Emergent Adverse Events (MNTs)
Like ICANS and other forms of neurotoxicity post-CAR T-cell infusion, MNTs are heterogeneous with variable clinical manifestations. MNTs can resemble Parkinsonism, as they are characterized by movement, cognitive, and personality changes, including tremors, micrographia, memory loss and flat affect.44 The pathophysiology of MNTs remains poorly understood but may have to do with neuronal BCMA expression within the basal ganglia.45,46 However, carbidopa/levodopa did not work in an affected patient from the CARTITUDE-1 trial, and post-mortem autopsy studies have revealed intact substantia nigricans in affected patients.44 Unlike ICANS, MNTs have a late onset, long duration, normal immune effector cell encephalopathy scores, and poor responses to steroids and other supportive measures. MNTs have been found to occur after a period of recovery from CRS and/or ICANS and may arise insidiously. In the CARTITUDE-1 study, 5 patients experienced MNTs (5%), including one grade 5 event.16 All five patients achieved a treatment response to cilta-cel. The median time to onset of MNTs was 27 days, and 17 days after recovery of CRS and ICANS. Patients who experienced MNTs were more likely to have a combination of at least two variables: high tumor burden, grade 2 or higher CRS, any grade ICANS, and high CAR T-cell expansion and persistence. Preventative, monitoring, and management strategies have resulted in a decreased incidence of MNTs across the cilta-cel program, and include enhanced bridging therapy to reduce tumor burden, early aggressive treatment of CRS and ICANS, handwriting assessments for early symptom detection, and extended monitoring for neurotoxicity up to a year post-infusion.44
Progression After CAR T-Cell Therapy
Outcomes after disease progression and optimal subsequent treatment approaches are poorly understood and require further study. In the KarMMa trial, the median PFS and OS were 8.8 months and 24.8 months, respectively.14 The 16-month gap between PFS and OS suggests favorable survival outcomes after progression of disease. Patients enrolled in the KarMMa trial who experienced progressive disease (n=104) were subsequently treated with various approaches, including ide-cel retreatment (n=28), conventional anti-myeloma therapy comprised mainly of dexamethasone and carfilzomib-based regimens (n=68), and BCMA-directed therapy (n=11). Subsequent treatment with conventional therapies or BCMA-directed therapies demonstrated modest activity, with an associated PFS2 of 13.6 months and 15.5 months, respectively.47 However, among the 28 patients retreated with ide-cel, activity seemed minimal with an ORR of only 21% (composed of 1 VGPR and 5 PRs) and median PFS of only 1-month (range, 1–2.1 months).17 Further, only 3 patients enrolled in the CARTITUDE-1 trial underwent cilta-cel retreatment.16 Responses were also minimal, with 2 patients having stable disease and one with progressive disease, and none of the 3 patients having CAR T-cell re-expansion. Real-world retrospective studies have demonstrated mixed outcomes after disease progression, with some data revealing poor responses to subsequent conventional and BCMA-directed therapies and a poor median OS of 5 months or less including zero responses to the BCMA antibody–drug conjugate, belantamab mafodotin.48,49 However, other data have revealed more positive responses to therapies such as multiagent chemotherapy, stem-cell boost, venetoclax- and selinexor-based treatments, and bispecific antibodies with an ORR of 46%, median time to progression of 105 days, and median OS of 15 months.50
CAR T-Cell Resistance
Multiple drug resistance mechanisms can lead to disease progression, including clonal and subclonal evolution.51 Regarding CAR T-cell therapy, cells with higher BCMA expression may be preferentially targeted, resulting in selection of cells with low or no BCMA expression.52,53 Downregulation of BCMA through homozygous deletion of the gene encoding TNFRSF17 has previously been described in patients who have received BCMA CAR T-cell therapies.53,54 However, only 1 of 71 relapses exhibited this resistance modality in the KarMMa trial.14 Another potential resistance mechanism is poor CAR T-cell persistence. In preclinical studies, bb21217, a derivative of bb2121 cultured with a phosphatidylinositol 3-kinase (PI3K) inhibitor, displayed elevated memory-like T-cell functionality with higher expression of CCR7 and CD27 and lower levels of the T-cell exhaustion marker CD57, compared to bb2121.55–57 Additionally, bb21217 exhibited myeloma clearance during a second treatment challenge in mouse models, whereas bb2121 did not.55 CAR T-cell persistence likely also depends on the costimulatory domain, as CD28-containing cells are associated with quick T-cell exhaustion and less durable responses compared to those maintaining the 4–1BB domain.57 A third potential resistance mechanism is trogocytosis, an active process in which the target antigen is transferred to T-cells (Figure 5). This phenomenon not only results in decreasing target density on tumor cells but abates T-cell activity by promoting fratricide.58
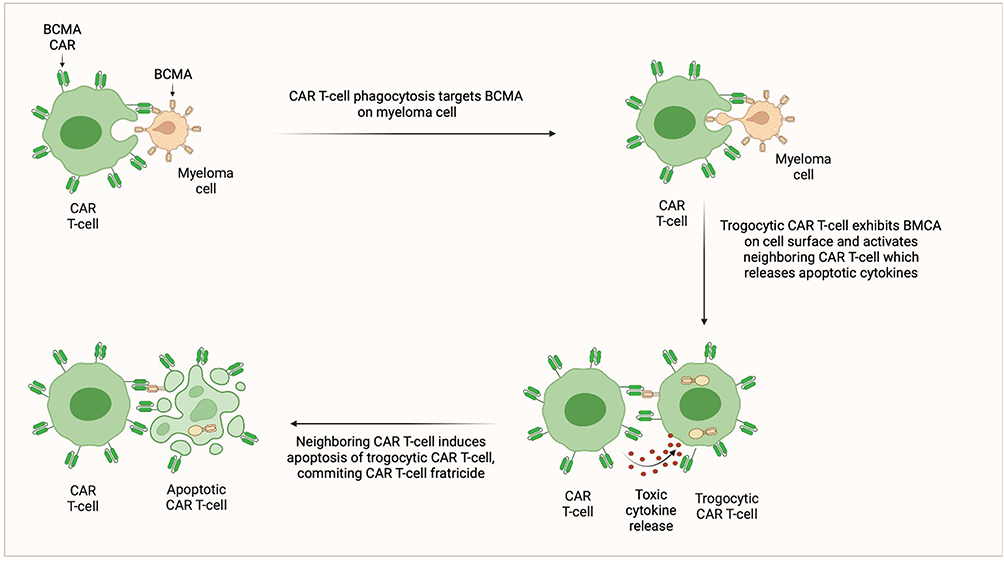 |
Figure 5 CAR T-cell Fratricide. |
Patient Selection for Commercial CAR T-Cell Therapy
Optimal patients for CAR T-cell therapy must meet FDA eligibility criteria, maintain the overall stable health required to survive wait list and manufacturing times, as well as CAR T-cell toxicities, and have adequate T-cell health to achieve manufacturing success. The chances of successful CAR T-cell therapy are likely impeded by prior use of lymphodepleting regimens (such as bendamustine containing regimens), poorly controlled disease despite bridging therapy, high tumor burden, and prior use of BCMA-directed therapies.21,59 However, those with high-risk disease features such as high-risk cytogenetics are likely to benefit.60
CAR T-Cells Vs Bispecific Antibodies
It is unclear whether currently FDA approved BCMA CAR T-cells outperform BCMA T-cell redirecting bispecific antibodies in terms of toxicity, efficacy, convenience, timing, costs, and quality of life. Currently available CAR T-cells are administered as a single infusion, whereas T-cell redirecting bispecifics are administered until unacceptable toxicity or disease progression. T-cell redirecting bispecifics do not require bridging therapy as they are “off-the-shelf”, do not require waitlists or manufacturing delays, and do not require lymphodepletion which carries risks such as prolonged hematologic toxicity and development of therapy-related myeloid neoplasms. Toxicities are similar, mostly comprising grade 1–2 CRS, hematologic toxicity, and infections, but neurotoxicities such as ICANS are less severe with bispecifics. In terms of efficacy, without direct comparisons, it is difficult to draw conclusions. However, compared to the BCMA T-cell redirecting bispecific antibody teclistamab, ide-cel and cilta-cel appear to induce deeper responses with similar rates of PFS.61
Investigational CAR T-Cell Therapies
A comprehensive list of key clinical trials of emerging CAR T-cell studies in MM is listed in Table 1 and described below.
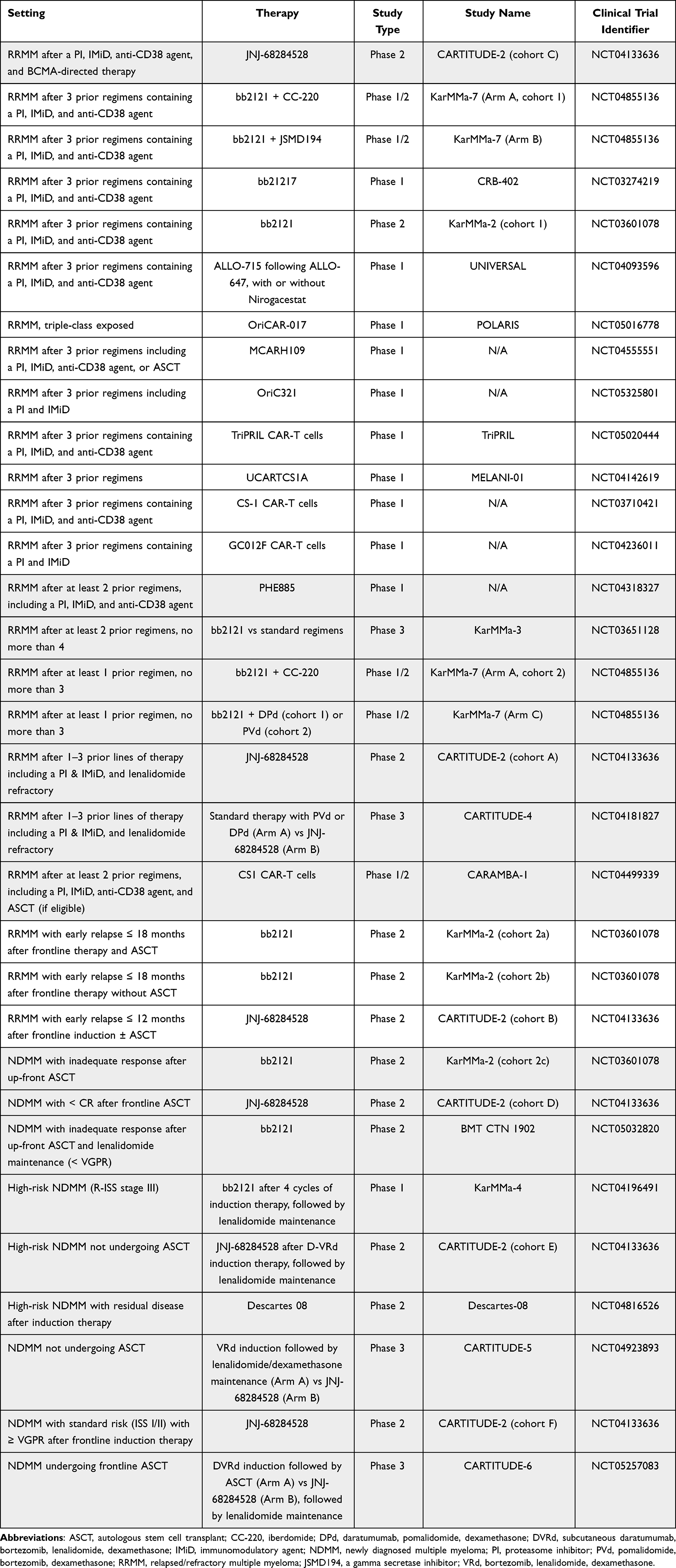 |
Table 1 Key Investigational CAR-T Cell Therapies for MM |
Autologous BCMA CAR T-Cells in Earlier Settings
Less than 10% of newly diagnosed adults with MM will survive to be eligible for 4th line CAR T-cell therapy under current FDA approval criteria.62 Additionally, CAR T-cells might be more efficacious given in earlier settings when it is easier to minimize tumor burden. As a result, multiple studies are assessing CAR T-cells at earlier times during the myeloma disease course, including after 1–3 lines of therapy, after early relapse to induction therapy, and in newly diagnosed myeloma (NDMM). Some key Phase 3 trials such as KarMMa-3 and CARTITUDE-4 are comparing ide-cel and cilta-cel with standard triplet regimens for RRMM. Furthermore, CARTITUDE-5 will randomize patients with NDMM not undergoing ASCT to either cilta-cel or lenalidomide and dexamethasone maintenance therapy after frontline induction with VRd (bortezomib, lenalidomide, dexamethasone). Finally, CARTITUDE-6 will randomize transplant-eligible patients with NDMM to either ASCT or cilta-cel after frontline induction with DVRd (subcutaneous daratumumab, bortezomib, lenalidomide, dexamethasone).
Improving CAR T-Cell Efficacy
Gamma secretase (GS) cleaves BCMA from the plasma cell membrane resulting in a reduction of BCMA-mediated NF-kB activation.63 As a result, increasing BCMA expression on plasma cells via GS inhibitors (GSI) has been shown to increase CAR T-cell targeted functioning in vitro and in preclinical models.64 Because soluble BCMA levels correlate with tumor burden and adverse outcomes, patients with lower baseline disease burden have better outcomes after CAR T-cell administration.14,65 A phase 1 first-in-human trial of escalating doses of BCMA CAR T-cells in combination with the oral GSI, JSMD194, in heavily pretreated RRMM patients was recently reported.66 Eighteen patients received JSMD194 as a run-in prior to lymphodepletion and BCMA CAR T-cell infusion, followed by 3 weeks of post-infusion JSMD194 treatment. Despite 4/18 (22%) patients previously receiving BCMA CAR T-cells, the ORR was 89% and median PFS was 11 months, which was greater for those with no prior exposure to BCMA-directed therapies. The ongoing KarMMa-7 study (NCT04855136) is a phase 1/2 study evaluating the safety and efficacy of ide-cel in combination with various agents including JSMD194 (arm B).
Preclinical studies have shown that culturing BCMA CAR T-cells with a PI3KCA inhibitor can enhance CAR T-cell expansion and persistence by increasing memory-like T-cell functions and decreasing differentiated or senescent T-cells.55 The CRB-402 phase 1 study (NCT03274219) utilizes bb21217. Updated results were reported on 72 patients after a median follow up of 9-months.67 Patients with higher 15-day post-infusion CD8+ CAR T-cells expressing CD27 and CD28 had a longer DOR, suggesting that more proliferative, less differentiated CAR T-cells at peak expansion correlate with a longer treatment response.
Non-BCMA Targets
A promising non-BCMA CAR T-cell target is the transmembrane receptor GPRC5D (G-protein coupled receptor family C group 5 member D), which is predominantly expressed on MM cells.68 MCARH109 is the first-in-class GPRC5D CAR T-cell product, and preliminary results have been reported on 18 enrolled patients, of whom 12 completed MCARH109 treatment and 6 received prior BCMA CAR T-cells.69 All patients received and were refractory to bridging therapy. Toxicities were as to be expected, with most patients experiencing grade 1–2 CRS and no neurologic toxicities. However, several patients experienced nail changes possibly related to treatment. Early treatment efficacy was modest, with 2 minimal responses, 3 PRs, 3 VGPRs, and 2 sCRs. All patients who received prior BCMA CAR T-cells achieved a treatment response. Another GPRC5D CAR T-cell product, OriCAR-017, is a CAR T-cell product with an additional proprietary Ori element that enhances expansion and durability. The phase 1 dose-escalation POLARIS study evaluates the safety, tolerability, cellular kinetics, and initial efficacy of OriCAR-017 in patients with triple-class exposed RRMM. Preliminary results have been reported on 8 patients, revealing an ORR of 100% including responses in 3 patients who had received prior BCMA CAR T-cells.70
Another potential non-BCMA target is CS1 (SLAMF7) which is highly expressed on MM cells.71 Preclinical studies of CS1 CAR T-cells have demonstrated in-vitro and in-vivo anti-myeloma cell kill in mouse models, although expression of CS1 on CD8+ T-cells led to fratricide.71 Several early phase clinical trials are assessing the maximum tolerated dose, safety, and efficacy of CS1 CAR T-cells in RRMM. The CARAMBA-1 trial (NCT04499339) is a first-in-human phase 1–2 study that manufactures SLAMF7 CAR T-cells using the virus-free Sleeping Beauty transposon system for gene-transfer. Similarly, the MELANI-01 study (NCT041426619) utilizes an off-the-shelf allogeneic CS1 CAR T-cell manufactured with TALEN gene editing technology to knock out the endogenous T-cell receptor and SLAMF7 expression to minimize the risk of fratricide.
Overexpression or activation of BCMA by its ligand, APRIL, promotes MM progression through various downstream mechanisms (Figure 4). CAR T-cells using a trimeric APRIL binding domain (TriPRIL) have enhanced in-vitro and in-vivo activity in both BCMA positive and negative MM cells in mouse models, and therefore might overcome antigen escape.72 Further, in addition to BCMA, TriPRIL CAR T-cells can also target transmembrane activator and CAML interactor (TACI), a TNFR that is highly expressed on MM cells, thus offering dual-specificity. TriPRIL CAR T-cells are currently being studied in a phase 1 trial (NCT05020444).
Some additional potential CAR T-cell targets include but are not limited to CD38, CD19, BAFF, and TACI. Targeting of CD38 would be challenging given unclear efficacy after treatment with commercial anti-CD38 products such as daratumumab and isatuximab. Further, CD38 is also expressed on hematopoietic stem cells which could pose problematic toxicities. Although not typically thought of as a myeloma marker, CD19 has been reported to be expressed in subsets of RRMM cells that have a B-cell phenotype.73 As a result, CD19 CAR T-cells have been utilized for treatment of RRMM after ASCT, in combination with BCMA CAR T-cells, and as part of a BCMA/CD19 dual-targeting CAR T-cell product utilizing the FasT CAR T-cell platform which allows for overnight manufacturing (NCT04236011), all of which have shown promising preliminary results.74,75
Allogeneic CAR T-Cells
Allogeneic CAR T-cells could potentially increase access to CAR T-cell therapy and overcome some logistical challenges associated with autologous CAR T-cell therapies. The phase 1 UNIVERSAL trial (NCT04093596) is assessing the safety, efficacy, and cellular kinetics/pharmacodynamics of ALLO-715 among patients with RRMM. ALLO-715 is a genetically modified BCMA AlloCAR TTM with a disrupted T-cell receptor alpha constant gene and a CD52 gene to minimize graft-versus-host disease (GvHD). Preliminary data have been promising, with a median time from enrollment to lymphodepletion of only 5 days, and therefore no patients requiring bridging therapy.76 Among patients receiving dose levels 3 or 4, the ORR was 61.5% including 38.5% VGPRs, and median DOR was 8.3 months. Toxicities were as to be expected, with no episodes of GvHD but grade 3 or higher infections occurring in 13% of patients.
Future Directions: Could CAR T-Cells Replace ASCT as Frontline Consolidation Therapy?
Historical data utilizing older induction regimens have shown deeper responses, improved PFS, and OS benefits with the use of ASCT.77–81 In the era of modern induction regimens, PFS benefits have also been demonstrated with the use of up-front ASCT, although to date OS benefits have not been demonstrated except in patients with high-risk cytogenetics.82–85 It is possible that it might take more time for an OS benefit to emerge after use of up-front ASCT.78 Notwithstanding, patients with NDMM treated with lenalidomide, bortezomib and dexamethasone, with or without upfront ASCT, followed by lenalidomide maintenance, showed significantly longer PFS (67.5 months versus 46.2 months), albeit 5-year OS was similar between the two arms.85 It is also possible that any benefits of up-front ASCT in the modern era hinge on the use of lenalidomide maintenance.86 Meanwhile, current FDA approved and investigational CAR T-cell products are likely better tolerated and with less mutational genotoxic damage to hematopoietic stem cells and malignant plasma cells.87 Ongoing clinical trials will address the benefits of BCMA CAR T-cell consolidation after induction therapy in the ongoing phase 3 CARTITUDE-5 and CARTITUDE-6 studies.
Conclusions
CAR T-cells are a powerful form of immunotherapy that will likely play an increasing role in the treatment of MM. The two currently FDA approved CAR T-cells in RRMM, ide-cel and cilta-cel, have demonstrated unprecedented responses in a highly refractory population, yet are associated with challenges such as prolonged manufacturing times, limited manufacturing slots, the potential for manufacturing failure, need for bridging therapy and lymphodepleting conditioning therapy, and high rates of hematologic, infectious, inflammatory, and neurologic toxicities. Further, a better understanding of resistance mechanisms, outcomes and optimal treatment approaches after progression, and ideal sequencing with other anti-BCMA directing therapies are needed. Ongoing studies are assessing the efficacy of CAR T-cells in earlier lines of therapy, as well as novel targets and methods to decrease manufacturing time.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There are no funding sources to report.
Disclosure
The opinions and assertions expressed herein are those of the authors and do not necessarily reflect the official policy or position of the US Air Force or the Department of Defense.
None of the authors have any financial or non-financial conflicts of interest to disclose.
References
1. National Cancer Institute. Cancer stat facts: myeloma 2021. Available from: https://seer.cancer.gov/statfacts/html/mulmy.html.
2. Joseph NS, Kaufman JL, Dhodapkar MV, et al. Long-Term Follow-Up Results of Lenalidomide, Bortezomib, and Dexamethasone Induction Therapy and Risk-Adapted Maintenance Approach in Newly Diagnosed Multiple Myeloma. J Clin Oncol. 2020;38(17):1928–1937.
3. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–10028.
4. Raskov H, Orhan A, Christensen JP, Gogenur I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br J Cancer. 2021;124(2):359–367.
5. Sharpe AH, Abbas AK. T-cell costimulation–biology, therapeutic potential, and challenges. N Engl J Med. 2006;355(10):973–975.
6. June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med. 2018;379(1):64–73.
7. Yu S, Li A, Liu Q, et al. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J Hematol Oncol. 2017;10(1):78.
8. Teoh PJ, Chng WJ. CAR T-cell therapy in multiple myeloma: more room for improvement. Blood Cancer J. 2021;11(4):84.
9. Makita S, Yoshimura K, Tobinai K. Clinical development of anti-CD19 chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma. Cancer Sci. 2017;108(6):1109–1118.
10. Jin C, Fotaki G, Ramachandran M, Nilsson B, Essand M, Yu D. Safe engineering of CAR T cells for adoptive cell therapy of cancer using long-term episomal gene transfer. EMBO Mol Med. 2016;8(7):702–711.
11. Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19(8):2048–2060.
12. Tai YT, Acharya C, An G, et al. April and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood. 2016;127(25):3225–3236.
13. Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med. 2019;380(18):1726–1737.
14. Munshi NC, Anderson LD, Shah N, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med. 2021;384(8):705–716.
15. Larry D, Anderson J, Munshi NC, et al. Idecabtagene vicleucel (ide-cel, bb2121), a BCMA-directed CAR T cell therapy, in relapsed and refractory multiple myeloma: updated KarMMa results. J Clin Oncol. 2021;39(15_suppl):8016.
16. Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398(10297):314–324.
17. Martin T, Usmani SZ, Berdeja JG, et al. Ciltacabtagene Autoleucel, an Anti-B-cell Maturation Antigen Chimeric Antigen Receptor T-Cell Therapy, for Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 2-Year Follow-Up. J Clin Oncol. 2022;JCO2200842.
18. Al Hadidi S, Zangari M, van Rhee F. Chimeric Antigen Receptor T-Cell Therapy in Multiple Myeloma-Challenges and Potential Solutions. JAMA Oncol. 2022;8(6):823–824.
19. Kourelis T, Bansal R, Patel KK, et al. Ethical challenges with CAR T slot allocation with idecabtagene vicleucel manufacturing access. J Clin Oncol. 2022;40(16_suppl):e20021.
20. Mohyuddin GR, Atieh T, Ahmed N, et al. Intention to treat versus modified intention-to-treat analysis in B-cell maturation antigen and CD19 chimeric antigen receptor trials: a systematic review and meta-analysis. Eur J Cancer. 2021;156:164–174.
21. Hansen DK, Sidana S, Peres L, et al. Idecabtagene vicleucel (Ide-cel) chimeric antigen receptor (CAR) T-cell therapy for relapsed/refractory multiple myeloma (RRMM): real-world experience. J Clin Oncol. 2022;40(16_suppl):8042.
22. Mateos M-V, Weisel K, Martin T, et al. Ciltacabtagene Autoleucel for Triple-Class Exposed Multiple Myeloma: adjusted Comparisons of CARTITUDE-1 Patient Outcomes Versus Therapies from Real-World Clinical Practice from the LocoMMotion Prospective Study. Blood. 2021;138(Supplement 1):550.
23. Shah N, Mojebi A, Ayers D, et al. Indirect treatment comparison of idecabtagene vicleucel versus conventional care in triple-class exposed multiple myeloma. J Comp Eff Res. 2022;11(10):737–749.
24. Jagannath S, Lin Y, Goldschmidt H, et al. KarMMa-RW: comparison of idecabtagene vicleucel with real-world outcomes in relapsed and refractory multiple myeloma. Blood Cancer J. 2021;11(6):116.
25. Costa LJ, Lin Y, Martin TG, et al. Cilta-cel versus conventional treatment in patients with relapse/refractory multiple myeloma. J Clin Oncol. 2021;39(15_suppl):8030.
26. Hari P, Berdeja JG, De Stefano V, et al. Meta-Analysis of Ciltacabtagene Autoleucel Versus Physician’s Choice in the Treatment of Patients with Relapsed or Refractory Multiple Myeloma. Blood. 2021;138(Supplement 1):1676.
27. Ozer H, Han T, Henderson ES, Nussbaum A, Sheedy D. Immunoregulatory T cell function in multiple myeloma. J Clin Invest. 1981;67(3):779–789.
28. Petersen J, Drivsholm A, Brandt M, Ambjornsen A, Dickmeiss E. B lymphocyte function in multiple myeloma: analysis of T cell- and monocyte-dependent antibody production. Eur J Haematol. 1989;42(2):193–201.
29. Zelle-Rieser C, Thangavadivel S, Biedermann R, et al. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol. 2016;9(1):116.
30. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–167.
31. Roddie C, O’Reilly M. Manufacturing chimeric antigen receptor T cells: issues and challenges. Cytotherapy. 2019;21(3):327–340.
32. Ahmed N, Shahzad M, Shippey E, et al. Socioeconomic and Racial Disparity in Chimeric Antigen Receptor T Cell Therapy Access. Transplant Cell Ther. 2022;1:6548.
33. Santomasso B, Bachier C, Westin J, Rezvani K, Shpall EJ. The Other Side of CAR T-Cell Therapy: cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am Soc Clin Oncol Educ Book. 2019;39:433–444.
34. Roex G, Timmers M, Wouters K, et al. Safety and clinical efficacy of BCMA CAR-T-cell therapy in multiple myeloma. J Hematol Oncol. 2020;13(1):164.
35. Teachey DT, Lacey SF, Shaw PA, et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016;6(6):664–679.
36. Lee DW, Santomasso BD, Locke FL, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. 2019;25(4):625–638.
37. Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62.
38. Taneja A, Jain T. CAR-T-OPENIA: chimeric antigen receptor T-cell therapy-associated cytopenias. eJHaem. 2022;3(Suppl. 1):32–38.
39. Busca A, Salmanton-Garcia J, Corradini P, et al. COVID-19 and CAR T cells: a report on current challenges and future directions from the EPICOVIDEHA survey by EHA-IDWP. Blood Adv. 2022;6(7):2427–2433.
40. Siegler EL, Kenderian SS. Neurotoxicity and Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy: insights Into Mechanisms and Novel Therapies. Front Immunol. 2020;11:1973.
41. Boyiadzis MM, Dhodapkar MV, Brentjens RJ, et al. Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: clinical perspective and significance. J Immunother Cancer. 2018;6(1):137.
42. Rubin DB, Danish HH, Ali AB, et al. Neurological toxicities associated with chimeric antigen receptor T-cell therapy. Brain. 2019;142(5):1334–1348.
43. Karschnia P, Jordan JT, Forst DA, et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood. 2019;133(20):2212–2221.
44. Cohen AD, Parekh S, Santomasso BD, et al. Incidence and management of CAR-T neurotoxicity in patients with multiple myeloma treated with ciltacabtagene autoleucel in CARTITUDE studies. Blood Cancer J. 2022;12(2):32.
45. Van Oekelen O, Aleman A, Upadhyaya B, et al. Neurocognitive and hypokinetic movement disorder with features of parkinsonism after BCMA-targeting CAR-T cell therapy. Nat Med. 2021;27(12):2099–2103.
46. Mohyuddin GR, Banerjee R, Alam Z, Berger KE, Chakraborty R. Rethinking mechanisms of neurotoxicity with BCMA directed therapy. Crit Rev Oncol Hematol. 2021;166:103453.
47. Rodriguez-Otero P, San-Miguel JF, Anderson LD, et al. Subsequent Anti-Myeloma Therapy after Idecabtagene Vicleucel (ide-cel, bb2121) Treatment in Patients with Relapsed/Refractory Multiple Myeloma from the KarMMa Study. Blood. 2021;138(Supplement 1):2743.
48. Parrondo RD, Sam K, Rasheed A, et al. Subsequent anti-myeloma therapy after idecabtagene vicleucel treatment in patients with relapsed/refractory multiple myeloma: a single center analysis. Blood Cancer J. 2022;12(4):66.
49. Arora S, Giri S, Godby KN, Ravi G, Costa LJ, Bal S. Beyond BCMA: outcomes of relapsed/refractory multiple myeloma after progression on anti-BCMA T cell redirecting therapy. J Clin Oncol. 2022;40(16_suppl):e20026.
50. Van Oekelen O, Mouhieddine TH, Pan D, et al. Clinical Outcomes and Treatment Strategies for Relapsed/Refractory Myeloma Patients after Relapse on BCMA-Targeted CAR T. Blood. 2021;138(Supplement 1):2704.
51. Keats JJ, Chesi M, Egan JB, et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120(5):1067–1076.
52. Friedman KM, Garrett TE, Evans JW, et al. Effective Targeting of Multiple B-Cell Maturation Antigen-Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum Gene Ther. 2018;29(5):585–601.
53. Da Via MC, Dietrich O, Truger M, et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat Med. 2021;27(4):616–619.
54. Samur MK, Fulciniti M, Aktas Samur A, et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat Commun. 2021;12(1):868.
55. Feinberg D, Paul B, Kang Y. The promise of chimeric antigen receptor (CAR) T cell therapy in multiple myeloma. Cell Immunol. 2019;345:103964.
56. Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–499.
57. Abate-Daga D, Davila ML. CAR models: next-generation CAR modifications for enhanced T-cell function. Mol Ther Oncolytics. 2016;3:16014.
58. Hamieh M, Dobrin A, Cabriolu A, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568(7750):112–116.
59. Shah N, Munshi NC, Berdeja JG, et al. Baseline Correlates of Complete Response to Idecabtagene Vicleucel (ide-cel, bb2121), a BCMA-Directed CAR T Cell Therapy in Patients with Relapsed and Refractory Multiple Myeloma: subanalysis of the KarMMa Trial. Blood. 2021;138(Supplement 1):1739.
60. Jakubowiak A, Usmani SZ, Berdeja JG, et al. Efficacy and Safety of Ciltacabtagene Autoleucel in Patients With Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 Subgroup Analysis. Blood. 2021;138(Supplement 1):3938.
61. Moreau P, Garfall AL, van de Donk N, et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N Engl J Med. 2022.
62. Giri S, Bal S, Godby KN, et al. Real-world applicability of commercial chimeric antigen receptor T-cell therapy among older adults with relapsed and/or refractory multiple myeloma. Am J Hematol. 2022;97(4):E153–E155.
63. Laurent SA, Hoffmann FS, Kuhn PH, et al. gamma-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun. 2015;6:7333.
64. Pont MJ, Hill T, Cole GO, et al. gamma-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood. 2019;134(19):1585–1597.
65. Sanchez E, Li M, Kitto A, et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br J Haematol. 2012;158(6):727–738.
66. Cowan AJ, Pont M, Sather BD, et al. Safety and Efficacy of Fully Human BCMA CAR T Cells in Combination with a Gamma Secretase Inhibitor to Increase BCMA Surface Expression in Patients with Relapsed or Refractory Multiple Myeloma. Blood. 2021;138(Supplement 1):551.
67. Raje NS, Shah N, Jagannath S, et al. Updated Clinical and Correlative Results from the Phase I CRB-402 Study of the BCMA-Targeted CAR T Cell Therapy bb21217 in Patients with Relapsed and Refractory Multiple Myeloma. Blood. 2021;138(Supplement 1):548.
68. Smith EL, Harrington K, Staehr M, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. 2019;11:485.
69. Mailankody S, Diamonte C, Fitzgerald L, et al. Phase I First-in-Class Trial of MCARH109, a G Protein Coupled Receptor Class C Group 5 Member D (GPRC5D) Targeted CAR T Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma. Blood. 2021;138(Supplement 1):827.
70. Huang H, Hu Y, Zhang M, et al. Phase I open-label single arm study of GPRC5D CAR T-cells (OriCAR-017) in patients with relapsed/refractory multiple myeloma (POLARIS). J Clin Oncol. 2022;40(16_suppl):8004.
71. O’Neal J, Ritchey JK, Cooper ML, et al. CS1 CAR-T targeting the distal domain of CS1 (SLAMF7) shows efficacy in high tumor burden myeloma model despite fratricide of CD8+CS1 expressing CAR-T cells. Leukemia. 2022;36(6):1625–1634.
72. Schmidts A, Ormhoj M, Choi BD, et al. Rational design of a trimeric April-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Adv. 2019;3(21):3248–3260.
73. Hajek R, Okubote SA, Svachova H. Myeloma stem cell concepts, heterogeneity and plasticity of multiple myeloma. Br J Haematol. 2013;163(5):551–564.
74. Garfall AL, Maus MV, Hwang WT, et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N Engl J Med. 2015;373(11):1040–1047.
75. Wang Y, Cao J, Gu W, et al. Long-Term Follow-Up of Combination of B-Cell Maturation Antigen and CD19 Chimeric Antigen Receptor T Cells in Multiple Myeloma. J Clin Oncol. 2015;1:
76. Mailankody S, Liedtke M, Sidana S, et al. Universal Updated Phase 1 Data Validates the Feasibility of Allogeneic Anti-BCMA ALLO-715 Therapy for Relapsed/Refractory Multiple Myeloma. Blood. 2021;138(Supplement 1):651.
77. Attal M, Harousseau JL, Stoppa AM, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. N Engl J Med. 1996;335(2):91–97.
78. Barlogie B, Attal M, Crowley J, et al. Long-term follow-up of autotransplantation trials for multiple myeloma: update of protocols conducted by the intergroupe francophone du myelome, southwest oncology group, and university of Arkansas for medical sciences. J Clin Oncol. 2010;28(7):1209–1214.
79. Palumbo A, Cavallo F, Gay F, et al. Autologous transplantation and maintenance therapy in multiple myeloma. N Engl J Med. 2014;371(10):895–905.
80. Gay F, Oliva S, Petrucci MT, et al. Chemotherapy plus lenalidomide versus autologous transplantation, followed by lenalidomide plus prednisone versus lenalidomide maintenance, in patients with multiple myeloma: a randomised, multicentre, phase 3 trial. Lancet Oncol. 2015;16(16):1617–1629.
81. Cavo M, Gay F, Beksac M, et al. Autologous haematopoietic stem-cell transplantation versus bortezomib-melphalan-prednisone, with or without bortezomib-lenalidomide-dexamethasone consolidation therapy, and lenalidomide maintenance for newly diagnosed multiple myeloma (EMN02/HO95): a multicentre, randomised, open-label, phase 3 study. Lancet Haematol. 2020;7(6):e456–e468.
82. Attal M, Lauwers-Cances V, Hulin C, et al. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N Engl J Med. 2017;376(14):1311–1320.
83. Dhakal B, Szabo A, Chhabra S, et al. Autologous Transplantation for Newly Diagnosed Multiple Myeloma in the Era of Novel Agent Induction: a Systematic Review and Meta-analysis. JAMA Oncol. 2018;4(3):343–350.
84. Chakraborty R, Siddiqi R, Willson G, et al. Impact of autologous transplantation on survival in patients with newly diagnosed multiple myeloma who have high-risk cytogenetics: a meta-analysis of randomized controlled trials. Cancer. 2022;128(12):2288–2297.
85. Richardson PG, Jacobus SJ, Weller EA, et al. Triplet Therapy, Transplantation, and Maintenance until Progression in Myeloma. N Engl J Med. 2022.
86. McCarthy PL, Holstein SA, Petrucci MT, et al. Lenalidomide Maintenance After Autologous Stem-Cell Transplantation in Newly Diagnosed Multiple Myeloma: a Meta-Analysis. J Clin Oncol. 2017;35(29):3279–3289.
87. Maura F, Weinhold N, Diamond B, et al. The mutagenic impact of melphalan in multiple myeloma. Leukemia. 2021;35(8):2145–2150.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Evaluating the Therapeutic Potential of Idecabtagene Vicleucel in the Treatment of Multiple Myeloma: Evidence to Date
Mann H, Comenzo RL
OncoTargets and Therapy 2022, 15:799-813
Published Date: 22 July 2022