Back to Journals » OncoTargets and Therapy » Volume 15
Evaluating the Therapeutic Potential of Idecabtagene Vicleucel in the Treatment of Multiple Myeloma: Evidence to Date
Authors Mann H ![]() , Comenzo RL
, Comenzo RL
Received 9 March 2022
Accepted for publication 8 July 2022
Published 22 July 2022 Volume 2022:15 Pages 799—813
DOI https://doi.org/10.2147/OTT.S305429
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Hashim Mann,1,2 Raymond L Comenzo1,2
1Division of Hematology/Oncology, Tufts Medical Center, Boston, MA, USA; 2The John Conant Davis Myeloma and Amyloid Program, Tufts Medical Center, Boston, MA, USA
Correspondence: Hashim Mann, Virginia Commonwealth University, 1001 East Leigh Street, Richmond, VA, 23219, USA, Tel +1 804-828-7999, Fax +1 804-828-5941, Email [email protected]
Abstract: Over the past two decades, significant progress has been made in the diagnosis, risk assessment and treatment of patients with multiple myeloma, translating into remarkable improvements in survival outcomes. Yet, cure remains elusive, and almost all patients eventually experience relapse, particularly those with high-risk and refractory disease. Immune-based approaches have emerged as highly effective therapeutic options that have heralded a new era in the treatment of multiple myeloma. Idecabtagene vicleucel (ide-cel) is one such therapy that employs the use of genetically modified autologous T-cells to redirect immune activation in a tumor-directed fashion. It has yielded impressive responses even in patients with poor-risk disease and is the first chimeric antigen receptor (CAR) T-cell therapy to be approved for treatment in relapsed or refractory multiple myeloma. In this review, we examine the design and pharmacokinetics of ide-cel, audit evidence that led to its incorporation into the current treatment paradigm and provide insight into its clinical utilization with a focus on real-life intricacies.
Keywords: CAR-T, immunotherapy, multiple myeloma, cytokine release syndrome, neurotoxicity, resistance
Introduction
Multiple myeloma (MM) is a malignancy of terminally differentiated plasma cells (PCs) that is always preceded by asymptomatic phases of clonal plasma cell expansion, known as monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM).1–3 It is the second most common hematologic malignancy and is estimated to account for 34,920 new cases and 12,410 deaths in the US in 2021.4 The current treatment paradigm for newly diagnosed patients involves the use of novel drugs and combinations, including the proteasome inhibitors (PIs) bortezomib and carfilzomib, immunomodulatory agents (IMiDs) such as lenalidomide and pomalidomide, the anti-CD38 monoclonal antibody (mAb) daratumumab, and if eligible, an autologous stem cell transplant (ASCT). These strategies have resulted in dramatic improvements in the outcomes of patients with newly diagnosed MM (NDMM), with a median overall survival (OS) that extends beyond 8–10 years in recent clinical studies.5–8 However, outcomes remain dismal in patients with high-risk disease and in those refractory to multiple agents, with a median OS that is typically less than 1–2 years despite the use of novel agents.9–12 This represents a major unmet need in the field, and employing newer strategies to improve outcomes in these groups of patients has been a focus of robust clinical and research efforts.
One avenue that has captivated the interest of researchers in this regard is manipulation of the immune system by means of chimeric antigen receptor (CAR) T-cells. CARs are genetically engineered receptors that allow autologous effector T-cells to be redirected towards a specific target, enabling CAR-modified T cells to recognize specific tumor antigens in an HLA-independent fashion, and leading to the generation of an amplified antitumor response.13 Although exploration of CAR-T cells for treatment of various oncologic disorders has been under investigation for several decades,14,15 the promise of this approach was exemplified by its success in the management of various B-cell malignancies, which led to approvals by the Food and Drug Administration (FDA) for four CD19-directed CAR-T cell therapies, including axicabtagene ciloleucel (axi-cel),16,17 tisagenlecleucel (tisa-cel),18,19 brexucabtagene autoleucel (brex-cel),20,21 and lisocabtagene maraleucel (liso-cel).22 In contrast, the only FDA-approved CAR-T cell treatments in MM are idecabtagene vicleucel (ide-cel), and more recently ciltacabtagene autoleucel (cilta-cel), both of which target B-cell maturation antigen (BCMA). In the ensuing discussion, we present a comprehensive overview of the rationale behind ide-cel use, examine relevant toxicities and mitigation strategies, discuss practical considerations, and provide a look into the future as it pertains to the clinical utilization of ide-cel in patients with MM.
BCMA as the Target
Since BCMA is the target of ide-cel, it is important to understand its relevance in MM and the reason behind its selection as a therapeutic target. Also referred to as tumor necrosis factor receptor superfamily member 17 (TNFRS17) or CD269, BCMA is a type III transmembrane protein that is induced in late memory B cells destined for plasmacytic differentiation.23–25 It is uniformly expressed on PCs but is virtually absent on naïve B-cells, hematopoietic stem cells and nonhematopoietic tissue, making it an attractive target for therapeutic intervention in MM while minimizing off-target effects.
Overactivation of BCMA by ligands of the tumor necrosis factor (TNF) family, a proliferation-inducing ligand (APRIL) and B cell-activating factor of the TNF family (BAFF), leads to downstream signaling that converges on activation of various intracellular pathways, including nuclear factor-kappa B (NF-κB), phosphatidylinositol 3-kinase (PI3K)/AKT, and mitogen-activated protein kinase (MAPK) pathways;26,27 upregulation of the antiapoptotic Bcl-2 family member proteins, Mcl-1 and Bcl-2;26,27 osteoclast activation;28 and in some instances, dysregulation of the immune microenvironment.29,30 This confers a survival advantage upon malignant PCs, leading to further proliferation of the abnormal clone(s). In MM, BCMA overexpression and activation is associated with progressive disease, large tumor burden, and worse survival.31,32 Moreover, membrane-bound BCMA can undergo γ-secretase-mediated cleavage, leading to shedding of soluble BCMA (sBCMA) into the circulation,33 which can then be detected in peripheral blood and allows for a rapid and more convenient diagnostic and prognostic assessment.34,35
Understanding of these principles enabled exploration of BCMA as a target for various anti-myeloma therapies. Anti-BCMA CAR-T cells showed encouraging preclinical efficacy,36,37 which was later confirmed in early clinical studies.38,39 However, support for targeting BCMA to clinical effect in MM was provided by the successful use of belantamab mafodotin (BLM) in treatment of patients with relapsed/refractory disease. BLM is a first-in-class, antibody–drug conjugate (ADC) that employs a humanized IgG1 anti-BCMA mAb, covalently linked to a microtubule-disrupting agent, monomethyl auristatin F (MMAF), via a noncleavable linker.40 In preclinical studies, it exhibited several tumoricidal mechanisms, including caspase 3-dependent cellular apoptosis, antibody-dependent cellular cytotoxicity (ADCC), and antibody-dependent cellular phagocytosis (ADCP).40 In clinical practice, this translated into impressive single-agent activity with overall response rates (ORR) ranging from 30% to 60% in heavily pretreated patients,41,42 resulting in FDA approval for its use in the fifth line of therapy.
CAR Design and Overview of Therapy
Deconstructing the Ide-Cel Construct
T-cell activation by T-cell receptors (TCRs) is a multistep process that is crucially dependent upon antigen presentation by major histocompatibility complex (MHC) proteins as well as subsequent signaling through costimulatory molecules.43–45 However, several factors limit the ability of naturally activated T-cells in mounting an enduring antitumor response, such as the need for repeated antigenic stimulation, immune exhaustion and antigen escape.46–48 CARs were designed to overcome some of these limitations while enhancing T-cell activation in a tumor-specific manner. First-generation CARs were composed of an extracellular antigen-recognition domain linked to an intracellular signaling domain via a transmembrane anchor. These molecules were able to activate T-cells independently of MHC proteins, but the lack of costimulatory domains resulted in limited in vivo efficacy.13,49 Second-generation CARs were then designed by addition a CD28 or 4-1BB costimulatory domain to the construct and showed more robust T-cell activation due to inflated cytokine production, modified intracellular signaling, enhanced T-cell proliferation, and delayed apoptosis.13,50
Ide-cel (also known BB2121) bears a second-generation CAR that was originally crafted by bluebird bio (Cambridge, MA) and is now co-developed and co-promoted by Bristol Myers Squibb. It consists of an extracellular BCMA-detecting murine single chain variable fragment (scFv) that is attached to transmembrane and intracellular domains via a human CD8 alpha hinge (Figure 1, insert).51 The intracellular domain comprises a costimulatory 4-1BB (CD137) molecule linked to a CD3 zeta (CD3ζ) T-cell activation domain.51 While the scFv imparts antigen-specificity, the hinge domain improves CAR–antigen interactions by conveying flexibility in epitope detection, enhancing cytokine production, and promoting CAR-T persistence, resulting in an amplified antitumor effect.52–54 Furthermore, compared to CD28, 4-1BB costimulation appears to be associated with enhanced antitumor activity, persistence of CAR-T cells and a favorable toxicity profile.55,56
 |
Figure 1 Overview of idecabtagene vicleucel therapy. Insert, ide-cel CAR design. Abbreviation: scFv, single chain variable fragment. Note: Created with BioRender.com. |
Overview of Therapy
The treatment scheme begins with leukapheresis for collection of peripheral blood mononuclear cells (PBMCs) (Figure 1). The collected cells are then transported to a central facility where T-cells are enriched, activated, and subjected to transduction by a lentiviral vector (LVV) that encodes the CAR protein.51,57 Quality of CAR expression in transduced cells (bb2121) is confirmed using immunophenotyping, and the cells of interest then undergo ex vivo expansion until optimal cell count is reached. The CAR-modified T-cells are then washed, concentrated, and cryopreserved prior to being shipped back to the treatment site for infusion. The ratio of CD4+ to CD8+ cells in the final product is variable, with a median of 85% (range, 42–98) CD4+, and 13% (2–47) CD8+ bb2121 cells in the Phase 1 experience.57 Overall, the manufacturing process can take up to 6 weeks to complete, and production failures are rare at less than 0.1%.57,58
Prior to CAR-T cell infusion, patients undergo lymphodepleting chemotherapy. With ide-cel, this was accomplished using a combination of fludarabine (Flu) 30 mg/m2 and cyclophosphamide (Cy) 300 mg/m2, administered on days –5, –4, and –3, followed by 2 days of rest, and CAR-T cell infusion on day 0.57,58 While lymphodepletion is not mandatory for in vivo CAR-T cell activity,59 its use has long been known to heighten the efficacy of adoptive T-cell therapy by supporting T-cell persistence.60,61 Since then, numerous additional mechanisms have also been elicited that may explain the therapeutic benefits of lymphodepleting chemotherapy prior to CAR-T cell therapy. These include depletion of immunosuppressive regulatory T-cells to allow for enhanced tumor antigen detection, increased CAR-T cell recruitment to tumor sites, accentuated CAR-T expansion, modulation of the immune microenvironment, and immune stimulation by the gut microbiome.62–66 Lymphodepletion also augments CAR T-cell activity by increasing the availability of cytokines such as IL-2, IL-7 and IL-15 that would otherwise be unavailable when sequestrated by homeostatic “sinks” established by naturally-occurring lymphocytes.67,68 Development of such a conducive cytokine milieu following Flu/Cy conditioning has been shown to correlate with favorable clinical outcomes,69,70 while subpar exposure to lymphodepleting therapy is associated with a higher risk of relapse.71 It is also worth noting that the optimal lymphodepleting regimen is not well-defined due to a lack of high-quality, randomized evidence. Instead, the selection of drugs for lymphodepletion in clinical use is primarily guided by expert consensus based on limited preclinical and clinical data. Therefore, standardizing the approach to pre-treatment conditioning remains a work in progress.
Another important consideration is that of the use of bridging therapy, which is often required to stabilize or debulk disease during the CAR-T manufacturing process, particularly in patients with aggressive disease. It is discussed at length in the subsequent text.
Preclinical Experience
The initial preclinical experience with ide-cel was reported by investigators from bluebird bio. In a study by Friedman et al,51 four different CARs – each with a unique anti-BCMA scFv – were evaluated in different MM and lymphoma models. Each scFv was sequentially linked to hinge, transmembrane, co-stimulatory, and T-cell signaling domains (Figure 1, insert). PBMCs obtained from healthy donors were transduced with LVVs encoding various CAR constructs, namely BB2120, BB2121, BB2122, and BB2123. An anti-CD19 CAR and a signaling-deficient anti-CD19Δ CAR (negative control) were also used for comparison. BCMA expression on different human myeloma cell lines (HMCLs) was analyzed using immunohistochemistry (IHC), while BCMA receptor density was assessed using flow cytometry and was noted to be high in all three HMCLs – NCI-H929, U266-B1, and RPMI-8226. Of the CAR-T cells of interest, bb2121 showed a high degree of transduction success, vector integration and cytokine production, leading to its selection for further evaluation. Cytotoxicity assessment demonstrated robust in vitro activity across all evaluated HMCLs when compared with anti-CD19 or negative control CAR-T cells.
In an NSG mouse model of human MM, a single bb2121 dose of 5×106 CAR-T cells resulted in complete tumor eradication by day 15, whereas merely tumor shrinkage was observed in mice treated with twice weekly bortezomib, and progressive disease noted in those treated with controls. Moreover, only the mice treated with bb2121 survived to the end of the 85-day study period. In terms of pharmacokinetics, bb2121 concentration in the peripheral blood peaked at day 11, while sBCMA declined sharply with a return to near-normal levels by day 8. Histological assessment of tumor tissue showed intense CAR-T cell infiltration by day 12 in parallel with a loss of BCMA expression by IHC. Interestingly, bb2121 also induced incomplete responses in NSG mouse models of human lymphoma. These observations paved way for additional studies to investigate the clinical efficacy of bb2121, as detailed below.
Clinical Experience
Phase I Study – CRB-401
CRB-401 was an open label, phase 1, dose-escalation and dose-expansion study that evaluated the use of bb2121 in patients with RRMM who had progressed after at least three lines of therapy.57 Different doses of the product were used, ranging from 50×106 to 800×106 CAR-T cells. The primary end point was safety, while secondary endpoints included response rates and duration. Adverse event (AE) grading was done using the 2010 National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4.03. Exploratory endpoints included minimal residual disease (MRD) evaluation by next-generation sequencing (NGS) to a sensitivity of 10−4, survival outcomes, cytokine profiling, and pharmacokinetic assessment.
At data cut-off in April 2018, 36 patients were enrolled in the study, of which 3 were unable to receive bb2121 due to progressive disease during the manufacturing period. The median age was 60 years (37–75), and most patients had an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1. Stage II or III disease was present in 67% of the patients, 45% had high-risk cytogenetics (defined by the presence of t(4;14), t(4;16) or del 17p), and about a quarter had extramedullary disease. Seven (3–23) median prior lines of therapy were used; and while all patients were exposed to PIs and IMiDs, 61% were refractory to bortezomib, 58% to carfilzomib, 73% to lenalidomide, and 79% to pomalidomide. Daratumumab had been previously used in 82% but only 55% were refractory, and almost all patients (97%) had received a prior ASCT. Bridging therapy was used in 42% during the manufacturing phase.
Safety
All patients experienced side effects, and 97% endured grade 3 or higher AEs. Hematologic toxicity was most common, and rates of grade ≥3 neutropenia were 85%, leukopenia 58%, anemia 45%, and thrombocytopenia 45%. The median time to absolute neutrophil count (ANC) improvement to more than 1000 cells/µL was 1.3 weeks, while that to platelet count of >50,000 cells/µL was 2 weeks, although delayed count recovery beyond 12 weeks was observed in some patients.
Cytokine release syndrome (CRS), graded according to the 2014 criteria proposed by Lee et al,72 was seen in 76% of the patients, and was predominantly grade 1 or 2 (70%). Grade 3 CRS was seen in 6% without any grade 4 events. Median time to CRS onset was 2 days (1–25), with a median duration of 5 days (1–32). Tocilizumab was used in seven patients and glucocorticoids in four. The incidence of CRS appeared to be higher with the use of CAR-T doses >150×106 cells, baseline ferritin elevation, as well as elevated peak levels of C-reactive protein (CRP), TNF-α, and IL-10. Peak CAR-T expansion was also higher in patients with CRS and did not appear to be dampened by tocilizumab or steroid use. Neurotoxicity was seen in 42% of the patients, with no grade 3 events, and 3% grade 4 toxicity. Other non-hematologic AEs were predominantly grade 1 or 2, with the most common being fatigue (42%), infections (42%), headache (30%), hypocalcemia (27%), constipation (27%), fever (24%), and hypokalemia (24%). One patient with a grade 3 AE later died of a cardiorespiratory event.
Efficacy
The overall response rate (ORR) was 85%, with stringent complete response (sCR) observed in 36%, and complete response (CR) in 9% of the patients. No very good partial responses or better (≥VGPR) were seen with the 50×106 CAR-T cell dose. Median time to first partial response or better (≥PR) was 1 month (0.5–3). Parallel reductions in serum free light chains (FLCs) and sBCMA were observed, albeit with a lag in the decrease in serum monoclonal protein (M-protein). Early improvements in bone marrow and extramedullary disease were also seen in some patients. The median duration of response (DoR) was 10.9 months. In the 18 patients evaluable for MRD status, 16 were MRD-negative at a threshold of 10−4, 15 were MRD-negative at 10−5, and 3 were MRD-negative at 10−6. In addition, 12 patients were MRD-negative at multiple time points, while 3 lost their MRD-negative status over time. With a median follow-up of 11.3 months (6.2–22.8), 52% experienced progressive disease, including six patients with a prior CR and six with prior MRD-negative disease. Median progression-free survival (PFS) was 12 months.
Pharmacokinetics
In vivo expansion of both CD4+ and CD8+ CAR-T cells was noted at doses greater than 150×106 cells, with significantly higher CAR+ T-cell numbers observed in responders as compared to non-responders. Persistent CAR-T cells were detected from the peripheral blood at 1, 3, 6, and 12 months in 96%, 86%, 57%, and 20% of the patients, respectively.
Phase II Study – KarMMa
The KarMMa study was conducted in follow-up to CRB-401.58 It was a Phase 2 trial that enrolled 140 patients with RRMM who had progressed after three prior lines of therapy, including a PI, an IMiD, and an anti-CD38 mAb. After receiving Flu/Cy lymphodepletion as described above, patients were treated with one of three target doses of ide-cel – 150×106, 300×106, and 450×106 CAR-T cells. The primary endpoint was ORR, while key secondary endpoints included rates of CR or better (≥CR), DoR, PFS, OS, MRD, and safety. Similar to CRB-401, AEs were assessed using NCI CTCAE, version 4.03 (2010), and CRS using the 2014 criteria proposed by Lee et al.72 MRD assessment was done using NGS to a sensitivity threshold of 10−5.
At data cut-off in November 2018, 128 of the 140 enrolled patients had received ide-cel. Twelve patients were unable to proceed with CAR-T treatment, primarily due to physician decision to discontinue enrollment (n=3), withdrawal of patient consent (n=3), death (n=2), progressive disease (n=1), and manufacturing failure (n=1). Of the patients who received study treatment, the 150×106 dose was administered in 4 (3%), 300×106 in 70 (55%), and 450×106 in 54 (42%) patients. Median age in the overall cohort was 61 (33–78), 59% were male, and the median time from initial diagnosis was 6 years (1–18). Most patients (98%) had an ECOG PS of 0 or 1. Revised International Staging System (R-ISS) stage II disease was seen in 70%, while stage III disease was observed in 16% of the patients (Table 1). High-risk cytogenetics by R-ISS criteria (t(4;14), t(14;16) or del 17p) were encountered in 35% of the patients, whereas patients with other high-risk aberrations were also included, such as amplification of 1q (35%) and deletion of 1p (6%). Extramedullary disease was present in 39% of the patients. Median number of prior therapies was 6 (3–16), and 94% of the patients had received a prior ASCT with 34% having undergone more than one transplant. Triple-refractory disease (refractory to a PI, an IMiD and an anti-CD38 mAb) was seen in 84%, while penta-refractory disease was encountered in 26% of the patients (Table 1). Bridging therapy was employed in 88%, with a median treatment duration of 15 days (1–33). Most common agents used for bridging were dexamethasone (70%), cyclophosphamide (37%), daratumumab (28%), carfilzomib (23%), bortezomib (20%), and pomalidomide (19%).
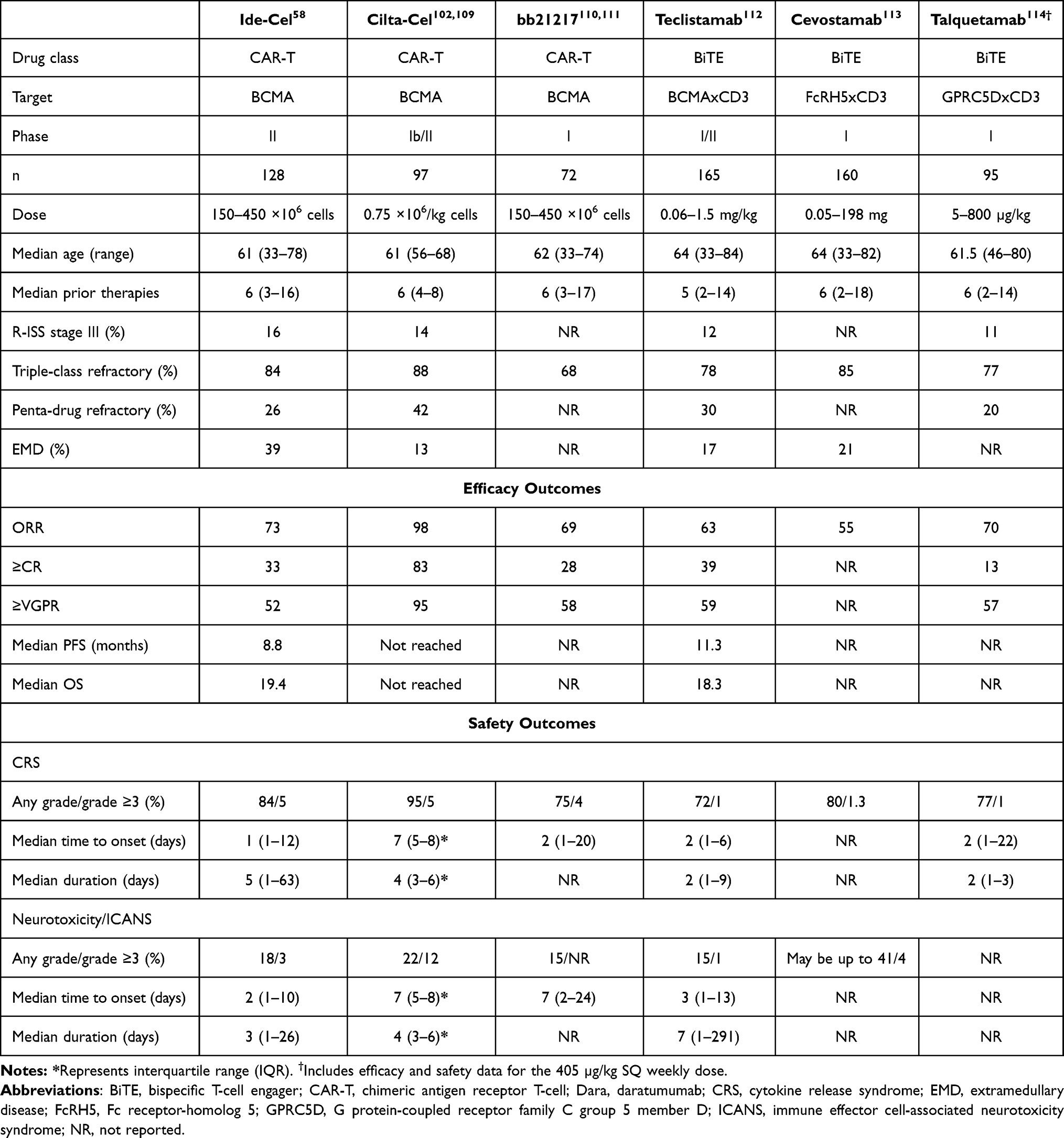 |
Table 1 Reported Outcomes in Different Trials Employing CAR-T Cell and BiTE Therapies in RRMM |
Efficacy
At a median of 13.3 months (0.2–21.2) of follow-up, the cumulative ORR was 73% in the study population, with ≥CR rate of 33% and ≥VGPR rate of 52%. At target doses of 150x106, 300x106, and 450x106, the respective ORRs were 50%, 69% and 81%; respective ≥CR rates were 25%, 29% and 39%; and respective ≥VGPR rates were 50%, 42% and 65% (Table 1). Of the patients who achieved ≥CR as response, 79% were MRD-negative to a threshold of 10−5 while MRD could not be evaluated in the remaining patients. In a subgroup analysis, higher ORRs were observed across most subgroups, including patients aged ≥65 years and those with extramedullary disease. The median time to first response was 1 month (0.5–8.8), and the median time to ≥CR was 2.8 months (1–11.8). The median DoR was 10.7 months (9–11.3), with longest responses seen in patients who had achieved ≥CR at 19 months. Median PFS for the overall cohort was 8.8 months, with 12.1 months (95% CI, 8.8–12.3) at the 450×106, 5.8 months (95% CI, 4.2–8.9) at the 300×106, and 2.8 months (95% CI, 1–not evaluable) at the 150×106 dose. The longest PFS of 20.2 months (95% CI, 12.3–not evaluable) was observed in patients who achieved ≥CR as a response. Median OS was 19.4 months (95% CI, 18.2–not evaluable), with 1-year survival of 78%. It is also important to note that of the patients who progressed after CAR-T, 28 underwent re-treatment with ide-cel; however, the best response in this group was VGPR (4%), while most patients continued to have disease progression (54%).
Safety
All patients experienced AEs, with 99% rate of grade 3 or higher toxicity. Hematologic AEs were most common, with grade ≥3 neutropenia observed in 89%, anemia in 60%, thrombocytopenia in 39%, and febrile neutropenia in 16%. Non-hematologic AEs were predominantly grade 1 or 2, most commonly being fatigue, GI toxicity, electrolyte imbalance, transaminitis, fever and hypogammaglobulinemia, although some grade 3 or 4 toxicity was also observed. Three hemorrhagic events were encountered involving the CNS, GI tract, and conjunctiva, while one was in the post-procedural setting. Infections occurred in 69% of the patients, with grade 3 or 4 in 22%, despite antibiotic and growth factor support in up to 95% of the patients. Most AEs were observed within the first 8 weeks following therapy with the notable exceptions of infections and hypogammaglobulinemia, which were frequently observed up to 6 months. In patients with grade ≥3 cytopenias, median time to ANC recovery was 1.9 months (1.2–5.6), while that for platelet recovery was 2.1 months (1.2–13.8), without significant impact from the CAR-T cell dose employed.
CRS occurred in 84% of the patients with a median time to onset of 1 day (1–12) and median duration of 5 days (1–63) (Table 1). Grade 3 or higher CRS was seen in only 5%, including one death. Rates of CRS correlated with the CAR-T cell dose used, although grade 3 or 4 CRS rates were similar at 6% between the 300×106 and 450×106 doses. Tocilizumab was used in 52% and glucocorticoids in 15% of the patients for management of CRS. All grade neurotoxicity was observed in 18% of the patients with a median time to onset of 2 days (1–10), and a median duration of 3 days (1–26). Grade 3 neurotoxic AEs were seen in 3%, with no grade 4 or 5 events, although rates of neurotoxicity were higher with increasing CAR-T cell doses. A total of 50 patients died on study, most commonly due to disease progression (42%) and treatment-emergent AEs (14%).
Pharmacokinetics
In the 127 patients in whom pharmacokinetics were evaluable, maximum CAR-T cell expansion was observed at a median of 11 days (7–30). Ide-cel peak exposure was higher in responders, and positively correlated with the depth of response and PFS. Patients who exhibited a response also demonstrated sharply declining sBCMA levels throughout the first 2 months following treatment, whereas no substantial change in sBCMA was appreciated in non-responders. Higher CAR-T cell expansion also inversely correlated with sBCMA levels. CAR-T cells were detectable in 59% of the patients at 6 months and in 36% at 12 months. While the risk of CRS or neurotoxicity did not correlate with baseline cytokine levels, patients who experienced CRS had higher levels of IL-6 and INF-γ compared to those who did not.
Health-Related Quality of Life
In data presented at the annual American Society of Hematology (ASH) meeting in 2021, assessment of health-related quality of life (HR-QoL) demonstrated clinically meaningful improvements in fatigue and pain in 40–70% of the patients at 24 months post-treatment. Physical and cognitive decline that occurred in many patients also seemed to improve at later timepoints. Furthermore, clinically meaningful improvements in symptoms and side effects were reported in 30–40% of the patients.73
Select Adverse Effects and Mitigation Strategies
With the increased utilization of CAR-T cell therapy across multiple oncologic disorders, the associated toxicity profile has also become indisputable While a comprehensive analysis of these AEs is beyond the scope of this review, it is worth briefly reviewing some of the representative toxicities in order to better understand the issues surrounding ide-cel use in clinical practice.
Cytokine Release Syndrome
CRS is the most common acute toxicity related to the use of CAR-T cell therapies. It was seen in 76–84% of the patients treated with ide-cel.57,58 While the relatively low rates of grade ≥3 CRS observed may be partly attributed to the grading criteria used in these studies, CRS carries the risk to evolve into fulminant hemophagocytic lymphohistiocytosis (HLH) and even death. It occurs due to an overproduction of cytokines, typically as a result of CAR-T cell expansion in vivo but also from T-cell mediated activation of bystander immune reactive cells, such as monocytes and macrophages.74,75 Various cytokines have been implicated, including INF-γ, IL-2, IL-2-receptor-α (IL2Rα), IL-6, IL6-receptor (IL6R), IL-8, IL-10, TNF-α, granulocyte-macrophage colony-stimulating factor (GM-CSF), CXCL9 (MIG), CXCL10 (IP-10), macrophage inflammatory protein (MIP)-1α, MIP-1β, and monocyte chemoattractant protein (MCP)-1,72,74–78 which give rise to a “cytokine storm” as is often seen with other infectious and inflammatory syndromes.79–81 Clinical manifestations are variable and can range from constitutional symptoms at milder end of the spectrum to multiorgan failure and death in extreme cases. Numerous tools have been used over time to assess the severity of CRS,72,77,82 but recent consensus guidelines recommend the use of 2019 criteria proposed by the American Society for Transplantation and Cellular Therapy (ASTCT) in order to standardize measurement and management across different clinical settings (Table 2).83–86 Prompt detection and timely administration of the IL6R antagonist, tocilizumab, as well as corticosteroids remain the cornerstone of management along with aggressive supportive care.83–85 The use of third-line agents such as anakinra and siltuximab may be considered if CRS persists despite two doses of tocilizumab.83
 |
Table 2 ASTCT Criteria for Grading CRS and ICANS. Adapted from Lee et al.86 |
Neurotoxicity
Neurotoxicity is frequently associated with CAR-T cell therapy and includes the syndrome of immune effector cell-associated neurotoxicity syndrome (ICANS) (Table 2). Neurotoxic symptoms can be as subtle as headache, lethargy, agitation, difficulty concentrating and tremors, or may be more dramatic, presenting with delirium, aphasia, encephalopathy, seizures, and posterior reversible encephalopathy syndrome (PRES).77,87,88 Late-onset movement disorders, cognitive impairment and personality changes have also been described, particularly in patients with high tumor burden, coexisting CRS (grade ≥2) or ICANS (any grade), and high CAR-T expansion.89 Rare instances of fatal cerebral edema have also been described.90,91 It is thought to result from an overproduction of various cytokines, including IL-2, IL-6, IL-10, IL-15, IFN-γ, TNF-α, GM-CSF, and MCP-1, as well as endothelial cell dysfunction and disruption of the blood–brain barrier (BBB).77,92,93 In severe cases, release of prothrombotic effectors such as von Willebrand factor (vWF) can ensue, leading to the development of a consumptive coagulopathy.77,92,93 More recently, BCMA expression on neurons and astrocytes in the basal ganglia has also been proposed as a potential mechanism for neurotoxicity94; however, current data are limited and further studies are required for validation.
We use the Immune Effector Cell-Associated Encephalopathy (ICE) score proposed by the ASTCT for early detection and grading of ICANS (Table 2),86 and perform the 10-point neurologic assessment at the bedside twice a day, in accordance with current guidelines.83,85,95 Management relies upon aggressive supportive care, including antiepileptic prophylaxis in patients at high risk for seizures, and the use of corticosteroids in patients with grade ≥2 ICANS. Tocilizumab has limited role in the treatment of isolated ICANS due to poor BBB penetration, but it should be administered in all patients who exhibit evidence of concurrent CRS.83–85
Cytopenias and Infectious Prophylaxis
Prolonged cytopenias and hypogammaglobulinemia are also common with ide-cel use and may be consequent to the underlying disease, prior anti-MM treatment, bridging therapy and/or lymphodepleting conditioning. While the severity and duration vary, most patients remain at an increased risk for infectious complications following CAR-T cell therapy, particularly in context of immune dysfunction and reconstitution. This necessitates adoption of a proactive approach to the use of appropriate supportive and prophylactic measures. In our practice, we routinely prescribe acyclovir or valacyclovir to prevent against varicella zoster virus (VZV) reactivation, and trimethoprim-sulfamethoxazole (TMP-SMX) for prophylaxis against Pneumocystis jirovecii pneumonia (PJP) for up to 1 year. In the immediate post-treatment phase; however, we prefer inhaled pentamidine over TMP-SMX for PJP prophylaxis in patients with significant coexisting cytopenias. Additionally, we employ fluoroquinolones for antibacterial, and fluconazole for antifungal prophylaxis, at least until the resolution of neutropenia, in accordance with the National Comprehensive Cancer Network (NCCN) guidelines. We also consider recombinant human granulocyte colony-stimulating factor (G-CSF) support in patients with profound neutropenia, although we prefer to delay administration for at least 14 days after CAR-T cell infusion in order to avoid potential interactions with CAR-T expansion and peak CRS risk.83
Practical Considerations
The use of ide-cel in clinical practice merits consideration of several additional factors. Patient selection is important, and outside of a clinical trial, ide-cel use should be reserved for those able to tolerate more intensive forms of therapy, at least until further data can establish its safety in the relatively “unfit” population. Ide-cel, like other CAR-T cell therapies, also has the benefit of being a one-time treatment; however, most patients are unlikely to enjoy enduring treatment-free intervals due to the substantial risk of relapse. Moreover, some patients with clinically aggressive disease are not able to wait for up to 6 weeks that can be required for manufacturing the CAR-T product. Others may require bridging therapy during this time for adequate disease control, which can also be challenging due to the theoretical risk of inducing ide-cel resistance, particularly if other BCMA-targeting agents are used.
Another important issue is that of cost. Ide-cel carries a price tag of $419,500 for a single CAR-T cell infusion, discounting the ancillary expenditures for lymphodepleting conditioning, supportive medications and hospital accommodation.96–100 Furthermore, the cost of managing associated toxicities can range anywhere from $18,497 to $121,535, depending on the severity and duration of AEs, which can significantly increase the price point.101 Therefore, it is important to take the financial burden into account when considering ide-cel use, particularly if alternate treatment options are available.
The use of ide-cel is currently also restricted by availability due to limited manufacturing capacity and factors pertaining to supply-chain management. Some of these impediments are expected to improve with time but will continue to present a challenge at least in the immediate short-term. And while rare, manufacturing failures can happen, rendering it imperative for treating physicians to have an alternate management strategy when planning ide-cel use in their patients.
And finally, ciltacabtagene autoleucel (cilta-cel) gained FDA approval on February 28, 2022, for treatment of RRMM. It carries the same indication as ide-cel and is approved for patients in the fifth line of therapy following progression on a PI, an IMiD, and an anti-CD38 mAb. Design of the cilta-cel CAR is unique as it contains a camelid-derived nanobody comprising two heavy chain variable regions that target two distinct BCMA epitopes. Presence of the dual epitope-binding anti-BCMA single-domain antibodies in the extracellular domain imparts antigen specificity and avidity. Like ide-cel, however, cilta-cel also contains 4-1BB costimulatory and CD3ζ T-cell signaling domains. Its efficacy was demonstrated in the open-label phase 1b/2 CARTITUDE-1 study,102 in which 97 patients with RRMM received cilta-cel infusion at a dose of 0.75×106 (range, 0.5–1.0 ×106) CAR-T cells per kg after a median of 6 (4–8) prior therapies. High-risk cytogenetic abnormalities (t(4;14), t(14;16) or del 17p) were observed in 24% of the patients, while 88% were triple-class and 42% were penta-drug refractory (Table 1). At a median follow-up of 12.4 months (10.6–15.2), ORR was 97% (95% CI, 91.2–99.4), with ≥VGPR rate of 93%, and a sCR rate of 67%. Median PFS was not reached, whereas PFS and OS rates at 12 months were 77% and 89%, respectively. Median time to first response was 1 month (0.9–1) while that to best response was 2.6 months (1–6.1). Of the 57 evaluable patients, 93% were able to achieve MRD-negativity at a threshold of 10−5 by NGS. Evaluation of safety demonstrated AEs in all patients, as assessed by the NCI CTCAE, version 5.0 (2017). All grade CRS occurred in 95% of the patients, although only 5% developed grade ≥3 CRS, including one death in a patient with prolonged CRS and concurrent HLH. Median time to CRS onset was 7 days (5–8) and median CRS duration was 4 days (3–6). Neurotoxicity was observed in 21% of the patients overall, ICANS in 17%, and both in 8%. While most cases were grade 1–2, the rate of grade ≥3 neurotoxic AEs was 10%, including one death. Median time to ICANS onset was 8 days (6–8), and the median ICANS duration was 4 days (3–6.5). All patients additionally experienced hematologic AEs, with high rates of grade 3–4 neutropenia (95%), anemia (68%), and thrombocytopenia (60%). Grade 3–4 non-hematologic toxicities were relatively infrequent.
It is noteworthy that direct comparisons between ide-cel and cilta-cel have not yet been performed. Therefore, any apparent incongruences in efficacy and safety measures of the two therapies may be purely coincidental or a product of numerous involved variables, such as differences in trial design or the study populations. As such, the choice of therapy in practice should be guided by availability and other practical considerations, at least until further data can guide clinical decision-making.
Future Directions
Incorporation of ide-cel as a therapeutic option in the treatment landscape of MM represents a major scientific and clinical advancement. Ide-cel has the potential to induce responses in patients with heavily pretreated and high-risk disease; however, it is also worth emphasizing that the rates of functional “cure” remain modest at best with current treatment strategies, as evidenced by almost half of the patients on the KarMMa study who progressed within the first year of treatment even at the target dose of 450×106 CAR-T cells.58 While efforts are ongoing, various resistance mechanisms have been identified. These include impaired CAR-T cell expansion and persistence, development of anti-CAR-T cell antibodies, dysregulation of the bone marrow microenvironment, and upregulation of pro-inflammatory chemokines, anti-apoptotic genes (MCL-1) and NF-κB signaling.58,59,95,103 Another important resistance mechanism is a deletion in chromosome 16p that leads to a loss of the BCMA-encoding gene, TNFRSF17.104,105 In clinical practice, a complete absence of PC membrane BCMA expression at disease progression following CAR-T treatment appreciated by flow cytometry would indicate the presence of a likely del 16p, and could be potentially utilized to predict response to subsequent therapies.
In order to overcome resistance and alleviate toxicity, numerous approaches are also currently under investigation. These include exploration of CAR-T cells directed against non-BCMA targets, such as G protein-coupled receptor, class C group 5 member D (GPRC5D),106 CD19, CD38, CD138 (SYND1), SLAMF7 (CS1), and NKG2D; development of dual-antigen targeting CARs, and concurrent use of drugs such as IMiDs for synergistic effect.95,107,108 Together with optimization of lymphodepleting regimens and streamlining production issues, these strategies will likely help enhance the efficacy and utilization of ide-cel in the near future. And lastly, with the emergence of newer therapies, such as bispecific T-cell engagers (BiTEs) and allogeneic CAR-T cell therapies, the optimal place for ide-cel in the treatment paradigm for MM will also become more discernable in the future. A summary of outcomes reported in different trials employing some of these novel therapeutic strategies is provided in Table 1.
Acknowledgments
This work was supported in part by the National Institutes of Health/National Institute of Aging grant R21-AG070502-01 (RLC). We also acknowledge the support by the Amyloidosis and Myeloma Research Fund at Tufts Medical Center, the Cam Neely and John Davis Myeloma Research Fund, the John C Davis Program for Myeloma and Amyloid at Tufts, the Sidewater Family Fund, the Lavonne Horowitz Trust, the Werner and Elaine Dannheisser Fund for Research on the Biology of Aging of the Lymphoma Foundation, David and Barbara Levine (in memoriam), and the Demarest Lloyd Jr Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mann H, Katiyar V, Varga C, Comenzo RL. Smoldering multiple myeloma - past, present, and future. Blood Rev. 2021;52:100869. doi:10.1016/j.blre.2021.100869
2. Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412–5417. doi:10.1182/blood-2008-12-194241
3. Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113(22):5418–5422. doi:10.1182/blood-2008-12-195008
4. American Society of Clinical Oncology. Multiple myeloma: statistics; 2021. Available from: https://www.cancer.net/cancer-types/multiple-myeloma/statistics.
5. Perrot A, Lauwers-Cances V, Cazaubiel T, et al. Early versus late autologous stem cell transplant in newly diagnosed multiple myeloma: long-term follow-up analysis of the IFM 2009 trial. Blood. 2020;136(Supplement 1):39. doi:10.1182/blood-2020-134538
6. Moreau P, Hulin C, Perrot A, et al. Maintenance with daratumumab or observation following treatment with bortezomib, thalidomide, and dexamethasone with or without daratumumab and autologous stem-cell transplant in patients with newly diagnosed multiple myeloma (CASSIOPEIA): an open-label, randomised, Phase 3 trial. Lancet Oncol. 2021;22(10):1378–1390. doi:10.1016/S1470-2045(21)00428-9
7. Facon T, Kumar SK, Plesner T, et al. Daratumumab, lenalidomide, and dexamethasone versus lenalidomide and dexamethasone alone in newly diagnosed multiple myeloma (MAIA): overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(11):1582–1596. doi:10.1016/S1470-2045(21)00466-6
8. Joseph NS, Kaufman JL, Dhodapkar MV, et al. Long-term follow-up results of lenalidomide, bortezomib, and dexamethasone induction therapy and risk-adapted maintenance approach in newly diagnosed multiple myeloma. J Clin Oncol. 2020;38(17):1928–1937. doi:10.1200/JCO.19.02515
9. Walker BA, Mavrommatis K, Wardell CP, et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33(1):159–170. doi:10.1038/s41375-018-0196-8
10. Chari A, Vogl DT, Gavriatopoulou M, et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N Engl J Med. 2019;381(8):727–738. doi:10.1056/NEJMoa1903455
11. Cornell R, Hari P, Tang S, et al. Overall survival of patients with triple-class refractory multiple myeloma treated with selinexor plus dexamethasone vs standard of care in MAMMOTH. Am J Hematol. 2021;96(1):E5–E8. doi:10.1002/ajh.26010
12. Tang D, Hari P, Ramasamy K, et al. Real-world treatment patterns and clinical, economic, and humanistic burden in triple-class refractory multiple myeloma: analysis of the connect® Multiple Myeloma (MM) disease registry. Blood. 2021;138:117. doi:10.1182/blood-2021-146830
13. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–398. doi:10.1158/2159-8290.CD-12-0548
14. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci. 1993;90(2):720–724. doi:10.1073/pnas.90.2.720
15. Kochenderfer JN, Wilson WH, Janik JE, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–4102. doi:10.1182/blood-2010-04-281931
16. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi:10.1056/NEJMoa1707447
17. Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31–42. doi:10.1016/S1470-2045(18)30864-7
18. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi:10.1056/NEJMoa1709866
19. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2018;380(1):45–56. doi:10.1056/NEJMoa1804980
20. Wang M, Munoz J, Goy A, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–1342. doi:10.1056/NEJMoa1914347
21. Shah BD, Ghobadi A, Oluwole OO, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. 2021;398(10299):491–502. doi:10.1016/S0140-6736(21)01222-8
22. Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839–852. doi:10.1016/S0140-6736(20)31366-0
23. Laabi Y, Gras M-P, Brouet J-C, Berger R, Larsen C-J, Tsapis A. The BCMA gene, preferentially expressed during B lymphoid maturation, is bidirectionally transcribed. Nucleic Acids Res. 1994;22(7):1147–1154. doi:10.1093/nar/22.7.1147
24. Madry C, Laabi Y, Callebaut I, et al. The characterization of murine BCMA gene defines it as a new member of the tumor necrosis factor receptor superfamily. Int Immunol. 1998;10(11):1693–1702. doi:10.1093/intimm/10.11.1693
25. O’Connor BP, Raman VS, Erickson LD, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. 2004;199(1):91–98. doi:10.1084/jem.20031330
26. Moreaux J, Legouffe E, Jourdan E, et al. BAFF and April protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood. 2004;103(8):3148–3157. doi:10.1182/blood-2003-06-1984
27. Shen X, Guo Y, Qi J, Shi W, Wu X, Ju S. Binding of B-cell maturation antigen to B-cell activating factor induces survival of multiple myeloma cells by activating Akt and JNK signaling pathways. Cell Biochem Funct. 2016;34(2):104–110. doi:10.1002/cbf.3169
28. Neri P, Kumar S, Fulciniti MT, et al. Neutralizing B-cell–activating factor antibody improves survival and inhibits osteoclastogenesis in a severe combined immunodeficient human multiple myeloma model. Clin Cancer Res. 2007;13(19):5903–5909. doi:10.1158/1078-0432.CCR-07-0753
29. Tai Y-T, Acharya C, An G, et al. April and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood. 2016;127(25):3225–3236. doi:10.1182/blood-2016-01-691162
30. Sanchez E, Gillespie A, Tang G, et al. Soluble B-cell maturation antigen mediates tumor-induced immune deficiency in multiple myeloma. Clin Cancer Res. 2016;22(13):3383–3397. doi:10.1158/1078-0432.CCR-15-2224
31. Shah N, Chari A, Scott E, Mezzi K, Usmani SZ. B-cell maturation antigen (BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia. 2020;34(4):985–1005. doi:10.1038/s41375-020-0734-z
32. Cho SF, Anderson KC, Tai YT. Targeting B cell maturation antigen (BCMA) in multiple myeloma: potential uses of BCMA-based immunotherapy. Front Immunol. 2018;9:1821. doi:10.3389/fimmu.2018.01821
33. Laurent SA, Hoffmann FS, Kuhn P-H, et al. γ-secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun. 2015;6(1):7333. doi:10.1038/ncomms8333
34. Sanchez E, Li M, Kitto A, et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br J Haematol. 2012;158(6):727–738. doi:10.1111/j.1365-2141.2012.09241.x
35. Godara A, Zhou P, Kugelmass A, et al. Presence of soluble and cell-surface B-cell maturation antigen in systemic light-chain amyloidosis and its modulation by gamma-secretase inhibition. Am J Hematol. 2020;95(5):E110–E113. doi:10.1002/ajh.25734
36. Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19(8):2048–2060. doi:10.1158/1078-0432.CCR-12-2422
37. Bu DX, Singh R, Choi EE, et al. Pre-clinical validation of B cell maturation antigen (BCMA) as a target for T cell immunotherapy of multiple myeloma. Oncotarget. 2018;9(40):25764–25780. doi:10.18632/oncotarget.25359
38. Ali SA, Shi V, Maric I, et al. T cells expressing an anti–B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128(13):1688–1700. doi:10.1182/blood-2016-04-711903
39. Brudno JN, Maric I, Hartman SD, et al. T cells genetically modified to express an Anti–B-Cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267–2280. doi:10.1200/JCO.2018.77.8084
40. Tai Y-T, Mayes PA, Acharya C, et al. Novel anti–B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123(20):3128–3138. doi:10.1182/blood-2013-10-535088
41. Trudel S, Lendvai N, Popat R, et al. Antibody–drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: an update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019;9(4):37. doi:10.1038/s41408-019-0196-6
42. Lonial S, Lee HC, Badros A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020;21(2):207–221. doi:10.1016/S1470-2045(19)30788-0
43. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27(1):591–619. doi:10.1146/annurev.immunol.021908.132706
44. Wieczorek M, Abualrous ET, Sticht J, et al. Major Histocompatibility Complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front Immunol. 2017;8. doi:10.3389/fimmu.2017.00292
45. Hwang J-R, Byeon Y, Kim D, Park S-G. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp Mol Med. 2020;52(5):750–761. doi:10.1038/s12276-020-0435-8
46. Kaminski H, Lemoine M, Pradeu T. Immunological exhaustion: how to make a disparate concept operational? PLoS Pathog. 2021;17(9):e1009892. doi:10.1371/journal.ppat.1009892
47. Allen CT, Clavijo PE, Van Waes C, Chen Z. Anti-tumor immunity in head and neck cancer: understanding the evidence, how tumors escape and immunotherapeutic approaches. Cancers. 2015;7(4):2397–2414. doi:10.3390/cancers7040900
48. Upadhyay R, Boiarsky JA, Pantsulaia G, et al. A critical role for fas-mediated off-target tumor killing in T-cell Immunotherapy. Cancer Discov. 2021;11(3):599–613. doi:10.1158/2159-8290.CD-20-0756
49. Kuwana Y, Asakura Y, Utsunomiya N, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149(3):960–968. doi:10.1016/0006-291X(87)90502-X
50. van der Stegen SJC, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499–509. doi:10.1038/nrd4597
51. Friedman KM, Garrett TE, Evans JW, et al. Effective targeting of multiple B-cell maturation antigen-expressing hematological malignancies by Anti-B-cell maturation antigen chimeric antigen receptor T cells. Hum Gene Ther. 2018;29(5):585–601. doi:10.1089/hum.2018.001
52. Qin L, Lai Y, Zhao R, et al. Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells. J Hematol Oncol. 2017;10(1):68. doi:10.1186/s13045-017-0437-8
53. Alabanza L, Pegues M, Geldres C, et al. Function of novel Anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. 2017;25(11):2452–2465. doi:10.1016/j.ymthe.2017.07.013
54. Jayaraman J, Mellody MP, Hou AJ, et al. CAR-T design: elements and their synergistic function. EBioMedicine. 2020;58:102931. doi:10.1016/j.ebiom.2020.102931
55. Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–1464. doi:10.1038/mt.2009.83
56. Ying Z, He T, Wang X, et al. Parallel comparison of 4-1BB or CD28 Co-stimulated CD19-targeted CAR-T cells for B cell non-Hodgkin’s lymphoma. Mol Ther Oncolytics. 2019;15:60–68. doi:10.1016/j.omto.2019.08.002
57. Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726–1737. doi:10.1056/NEJMoa1817226
58. Munshi NC, Anderson LD, Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–716. doi:10.1056/NEJMoa2024850
59. Cohen AD, Garfall AL, Stadtmauer EA, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. 2019;129(6):2210–2221. doi:10.1172/JCI126397
60. Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi:10.1126/science.1076514
61. Wallen H, Thompson JA, Reilly JZ, Rodmyre RM, Cao J, Yee C. Fludarabine modulates immune response and extends in vivo survival of adoptively transferred CD8 T cells in patients with metastatic melanoma. PLoS One. 2009;4(3):e4749. doi:10.1371/journal.pone.0004749
62. Antony PA, Piccirillo CA, Akpinarli A, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174(5):2591–2601. doi:10.4049/jimmunol.174.5.2591
63. Pittet MJ, Grimm J, Berger CR, et al. In vivo imaging of T cell delivery to tumors after adoptive transfer therapy. Proc Natl Acad Sci. 2007;104(30):12457–12461.
64. Neelapu SS. CAR-T efficacy: is conditioning the key? Blood. 2019;133(17):1799–1800. doi:10.1182/blood-2019-03-900928
65. Bechman N, Maher J. Lymphodepletion strategies to potentiate adoptive T-cell immunotherapy - what are we doing; where are we going? Expert Opin Biol Ther. 2021;21(5):627–637. doi:10.1080/14712598.2021.1857361
66. Murad JP, Tilakawardane D, Park AK, et al. Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Mol Ther. 2021;29(7):2335–2349. doi:10.1016/j.ymthe.2021.02.024
67. Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202(7):907–912. doi:10.1084/jem.20050732
68. Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26(2):111–117. doi:10.1016/j.it.2004.12.003
69. Hirayama AV, Gauthier J, Hay KA, et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood. 2019;133(17):1876–1887. doi:10.1182/blood-2018-11-887067
70. Kochenderfer JN, Somerville RPT, Lu T, et al. Lymphoma remissions caused by Anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J Clin Oncol. 2017;35(16):1803–1813. doi:10.1200/JCO.2016.71.3024
71. Fabrizio VA, Boelens JJ, Mauguen A, et al. Optimal fludarabine lymphodepletion is associated with improved outcomes following CAR T-cell Therapy. Blood Adv. 2021;5(2):496–503. doi:10.1182/bloodadvances.2020002735
72. Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. doi:10.1182/blood-2014-05-552729
73. Delforge M, Shah N, Rodríguez-Otero P, et al. Updated health-related quality of life results from the KarMMa clinical study in patients with relapsed and refractory multiple myeloma treated with the B-cell maturation antigen-directed chimeric antigen receptor T cell therapy idecabtagene vicleucel (ide-cel, bb2121). Blood. 2021;138:2835.
74. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127(26):3321–3330. doi:10.1182/blood-2016-04-703751
75. Wei J, Liu Y, Wang C, et al. The model of cytokine release syndrome in CAR T-cell treatment for B-cell non-Hodgkin lymphoma. Signal Transduct Target Ther. 2020;5(1):134. doi:10.1038/s41392-020-00256-x
76. Teachey DT, Lacey SF, Shaw PA, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6(6):664–679. doi:10.1158/2159-8290.CD-16-0040
77. Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy — assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62. doi:10.1038/nrclinonc.2017.148
78. Siegler EL, Kenderian SS. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: insights into mechanisms and novel therapies. Front Immunol. 2020;11. doi:10.3389/fimmu.2020.01973
79. Fajgenbaum DC, June CH, Longo DL. Cytokine Storm. N Engl J Med. 2020;383(23):2255–2273. doi:10.1056/NEJMra2026131
80. Schroeder C, Hachem H, Godara A, et al. 499. Rapid and sustained decline in CXCL-10 (IP-10) annotates clinical outcomes following TNF-α antagonist therapy in hospitalized patients with severe and critical COVID-19 respiratory failure. Open Forum Infect Dis. 2021;8(Supplement_1):S351–S352. doi:10.1093/ofid/ofab466.698
81. Hachem H, Godara A, Schroeder C, et al. Rapid and sustained decline in CXCL-10 (IP-10) annotates clinical outcomes following TNFα-antagonist therapy in hospitalized patients with severe and critical COVID-19 respiratory failure. J Clin Transl Sci. 2021;5(1):e146. doi:10.1017/cts.2021.805
82. Porter D, Frey N, Wood PA, Weng Y, Grupp SA. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J Hematol Oncol. 2018;11(1):35. doi:10.1186/s13045-018-0571-y
83. Maus MV, Alexander S, Bishop MR, et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune effector cell-related adverse events. J Immunother Cancer. 2020;8(2):e001511. doi:10.1136/jitc-2020-001511
84. Ibrahim Y-A, Christian C, Peter B, et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica. 2020;105(2):297–316. doi:10.3324/haematol.2019.229781
85. Ragoonanan D, Khazal SJ, Abdel-Azim H, et al. Diagnosis, grading and management of toxicities from immunotherapies in children, adolescents and young adults with cancer. Nat Rev Clin Oncol. 2021;18(7):435–453. doi:10.1038/s41571-021-00474-4
86. Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–638. doi:10.1016/j.bbmt.2018.12.758
87. Kansagra AJ, Frey NV, Bar M, et al. Clinical utilization of chimeric antigen receptor T cells in B cell acute lymphoblastic leukemia: an expert opinion from the European Society for Blood and Marrow Transplantation and the American Society for Transplantation and cellular therapy. Biol Blood Marrow Transplant. 2019;25(3):e76–e85. doi:10.1016/j.bbmt.2018.12.068
88. Garfall AL, Lancaster E, Stadtmauer EA, et al. Posterior Reversible Encephalopathy Syndrome (PRES) after infusion of Anti-BCMA CAR T cells (CART-BCMA) for multiple myeloma: successful treatment with cyclophosphamide. Blood. 2016;128(22):5702. doi:10.1182/blood.V128.22.5702.5702
89. Cohen AD, Parekh S, Santomasso BD, et al. Incidence and management of CAR-T neurotoxicity in patients with multiple myeloma treated with ciltacabtagene autoleucel in CARTITUDE studies. Blood Cancer J. 2022;12(2):32. doi:10.1038/s41408-022-00629-1
90. Turtle CJ, Hay KA, Hanafi L-A, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of ibrutinib. J Clin Oncol. 2017;35(26):3010–3020. doi:10.1200/JCO.2017.72.8519
91. Nastoupil LJ, Jain MD, Feng L, et al. Standard-of-care axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma: results from the US lymphoma CAR T consortium. J Clin Oncol. 2020;38(27):3119–3128. doi:10.1200/JCO.19.02104
92. Rice J, Nagle S, Randall J, Hinson HE. Chimeric antigen receptor T cell-related neurotoxicity: mechanisms, clinical presentation, and approach to treatment. Curr Treat Options Neurol. 2019;21(8):40. doi:10.1007/s11940-019-0580-3
93. Reagan PM, Neelapu SS. How I manage: pathophysiology and management of toxicity of chimeric antigen receptor T-cell therapies. J Clin Oncol. 2021;39(5):456–466. doi:10.1200/JCO.20.01616
94. Van Oekelen O, Aleman A, Upadhyaya B, et al. Neurocognitive and hypokinetic movement disorder with features of parkinsonism after BCMA-targeting CAR-T cell therapy. Nat Med. 2021;27(12):2099–2103. doi:10.1038/s41591-021-01564-7
95. Bruno B, Wasch R, Engelhardt M, et al. European Myeloma Network perspective on CAR T-Cell therapies for multiple myeloma. Haematologica. 2021;106(8):2054–2065. doi:10.3324/haematol.2020.276402
96. Bach PB. National coverage analysis of CAR-T therapies — policy, evidence, and payment. N Engl J Med. 2018;379(15):1396–1398. doi:10.1056/NEJMp1807382
97. Ran T, Eichmüller SB, Schmidt P, Schlander M. Cost of decentralized CAR T-cell production in an academic nonprofit setting. Int J Cancer. 2020;147(12):3438–3445. doi:10.1002/ijc.33156
98. Fiorenza S, Ritchie DS, Ramsey SD, Turtle CJ, Roth JA. Value and affordability of CAR T-cell therapy in the United States. Bone Marrow Transplant. 2020;55(9):1706–1715. doi:10.1038/s41409-020-0956-8
99. Powell K, Russler-Germain D, Prasad V. Idecabtagene vicleucel: questions regarding the appropriate role and cost. Br J Haematol. 2022;196(2):e15–e16. doi:10.1111/bjh.17784
100. Rebecca Borgert PB. Improving outcomes and mitigating costs associated with CAR T-cell therapy. Suppl Featured Publ. 2021;27(13):S253–S261.
101. Hari P, Nguyen A, Pelletier C, McGarvey N, Gitlin M, Parikh K. Healthcare resource utilization and economic burden of cytokine release syndrome (CRS) and neurologic events (NE) in patients (pts) with relapsed/refractory multiple myeloma (RRMM) receiving idecabtagene vicleucel (ide-cel, bb2121) in KarMMa. J Clin Oncol. 2020;38(29_suppl):61. doi:10.1200/JCO.2020.38.29_suppl.61
102. Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398(10297):314–324. doi:10.1016/S0140-6736(21)00933-8
103. Melnekoff DT, Ghodke-Puranik Y, Van Oekelen O, et al. Single-cell profiling reveals contribution of tumor extrinsic and intrinsic factors to BCMA-targeted CAR-T cell efficacy in multiple myeloma. Blood. 2021;138:326. doi:10.1182/blood-2021-150923
104. Da Vià MC, Dietrich O, Truger M, et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat Med. 2021;27(4):616–619. doi:10.1038/s41591-021-01245-5
105. Samur MK, Fulciniti M, Aktas Samur A, et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat Commun. 2021;12(1):868. doi:10.1038/s41467-021-21177-5
106. Smith EL, Harrington K, Staehr M, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. 2019;11(485):eaau7746. doi:10.1126/scitranslmed.aau7746
107. Sidana S, Shah N. CAR T-cell therapy: is it prime time in myeloma? Blood Adv. 2019;3(21):3473–3480. doi:10.1182/bloodadvances.2019000370
108. van de Donk N, Usmani SZ, Yong K. CAR T-cell therapy for multiple myeloma: state of the art and prospects. Lancet Haematol. 2021;8(6):e446–e461. doi:10.1016/S2352-3026(21)00057-0
109. Martin T, Usmani SZ, Berdeja JG, et al. Ciltacabtagene autoleucel, an Anti–B-cell maturation antigen chimeric antigen receptor T-cell therapy, for relapsed/refractory multiple myeloma: CARTITUDE-1 2-year follow-up. J Clin Oncol;2022.
110. Alsina M, Shah N, Raje NS, et al. Updated results from the Phase I CRB-402 Study of Anti-BCMA CAR-T cell therapy bb21217 in patients with relapsed and refractory multiple myeloma: correlation of expansion and duration of response with T cell phenotypes. Blood. 2020;136(Supplement 1):25–26. doi:10.1182/blood-2020-140410
111. Raje NS, Shah N, Jagannath S, et al. Updated clinical and correlative results from the Phase I CRB-402 Study of the BCMA-targeted CAR T cell therapy bb21217 in patients with relapsed and refractory multiple myeloma. Blood. 2021;138:548. doi:10.1182/blood-2021-146518
112. Moreau P, Garfall AL, van de Donk NWCJ, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022. doi:10.1056/NEJMoa2203478
113. Trudel S, Cohen AD, Krishnan AY, et al. Cevostamab monotherapy continues to show clinically meaningful activity and manageable safety in patients with heavily pre-treated Relapsed/Refractory Multiple Myeloma (RRMM): updated results from an ongoing Phase I study. Blood. 2021;138:157. doi:10.1182/blood-2021-147983
114. Krishnan AY, Minnema MC, Berdeja JG, et al. Updated Phase 1 results from MonumenTAL-1: first-in-human study of talquetamab, a G Protein-Coupled Receptor Family C Group 5 member D x CD3 bispecific antibody, in patients with relapsed/refractory multiple myeloma. Blood. 2021;138:158. doi:10.1182/blood-2021-146868
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
From Anti-HER-2 to Anti-HER-2-CAR-T Cells: An Evolutionary Immunotherapy Approach for Gastric Cancer
Sun J, Li X, Chen P, Gao Y
Journal of Inflammation Research 2022, 15:4061-4085
Published Date: 17 July 2022
CAR T-Cell Therapy for Patients with Multiple Myeloma: Current Evidence and Challenges
Rendo MJ, Joseph JJ, Phan LM, DeStefano CB
Blood and Lymphatic Cancer: Targets and Therapy 2022, 12:119-136
Published Date: 29 August 2022
Second-Line Treatment of Pancreatic Adenocarcinoma: Shedding Light on New Opportunities and Key Talking Points from Clinical Trials
Imperial R, Mosalem O, Majeed U, Tran NH, Borad MJ, Babiker H
Clinical and Experimental Gastroenterology 2024, 17:121-134
Published Date: 18 April 2024
Hydrogel-Based Immunomodulation of Tumor Immune Microenvironment in Hepatocellular Carcinoma: Current Strategies and Future Directions
Zhang S, Ding L, Hou T, Lin D, Qu Y
International Journal of Nanomedicine 2026, 21:576091
Published Date: 4 March 2026