")
Back to Journals » International Journal of Nanomedicine » Volume 16
Cancer Therapy with Nanoparticle-Medicated Intracellular Expression of Peptide CRM1-Inhibitor
Authors Sui M, Xiong M, Li Y, Zhou Q, Shen X, Jia D, Gou M , Sun Q
Received 16 June 2020
Accepted for publication 29 September 2020
Published 14 April 2021 Volume 2021:16 Pages 2833—2847
DOI https://doi.org/10.2147/IJN.S266398
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Min Sui,1,* Meimei Xiong,2,* Yuling Li,1,* Qiao Zhou,1 Xiaofei Shen,3 Da Jia,3 Maling Gou,2 Qingxiang Sun1
1Department of Pathology and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, and Collaborative Innovation Centre for Biotherapy, Chengdu, 610041, People’s Republic of China; 2State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, and Collaborative Innovation Center for Biotherapy, Chengdu, 610041, People’s Republic of China; 3Key Laboratory of Birth Defects and Related Diseases of Women and Children, Department of Pediatrics, Division of Neurology, West China Second University Hospital, Sichuan University, Chengdu, 610041, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qingxiang Sun; Maling Gou Email [email protected]; [email protected]
Introduction: Peptides can be rationally designed as non-covalent inhibitors for molecularly targeted therapy. However, it remains challenging to efficiently deliver the peptides into the targeted cells, which often severely affects their therapeutic efficiency.
Methods: Herein, we created a novel non-covalent peptide inhibitor against nuclear export factor CRM1 by a structure-guided drug design method and targetedly delivered the peptide into cancer cells by a nanoparticle-mediated gene expression system for use as a cancer therapy.
Results: The nuclear export signal (NES)-optimized CRM1 peptide inhibitor colocalized with CRM1 to the nuclear envelope and inhibited nuclear export in cancer cell lines in vitro. The crystal structures of the inhibitors complexed with CRM1 were solved. In contrast to the covalent inhibitors, the peptides were similarly effective against cells harboring the CRM1 C528S mutation. Moreover, a plasmid encoding the peptides was delivered by a iRGD-modified nanoparticle to efficiently target and transfect the cancer cells in vivo after intravenous administration. The peptides could be selectively expressed in the tumor, resulting in the efficient inhibition of subcutaneous melanoma xenografts without obvious systemic toxicity.
Discussion: This work provides an effective strategy to design peptide-based molecularly targeted therapeutics, which could lead to the development of future targeted therapy.
Keywords: CRM1, non-covalent inhibitor, crystal structures, DNA nanocomplex, protein engineering
Introduction
Molecularly targeted therapy has been used extensively in cancer therapy. Structure-guided drug design is an important area of targeted therapy.1 Compared to small molecules, peptides often have the advantages of being more specific and potent in binding to a target, and able to bind to relatively flat surfaces of a target.2,3 The huge and ever-growing number of protein-protein interface structures provides enormous opportunities for peptide-based inhibitor design.4
CRM1 (chromosomal region maintenance 1, a.k.a. exportin-1, XPO1) is an essential and versatile nuclear export factor that binds and exports nuclear export signal (NES)-containing cargoes from the nucleus to the cytoplasm.5,6 CRM1-dependent nuclear export is implicated in various diseases, including cancer, viral infection, inflammation, and wound healing.7,8 Notably, CRM1 is overexpressed in a broad spectrum of cancers, and its overexpression is often an indicator of poor prognosis.8–10 CRM1 is additionally reported to mediate drug resistance in various contexts.11,12 We have proposed that CRM1 overexpression promotes nine out of the ten hallmarks of cancer, thereby explaining its oncogenic properties in a variety of cancers.13 Both knock down of CRM1 and use of CRM1 inhibitors have shown selective potency against a large panel of cancer cells.14–16 Leptomycin B, the most well-known CRM1 inhibitor, binds covalently to a cysteine residue (C528) in CRM1’s NES binding groove, unfortunately, it was very toxic in a clinical trial.17 The later-developed synthetic CRM1 inhibitors (including KPT-330, CBS9106, and S109) are reversible covalent inhibitors and much less toxic.18–20 KPT-330 was recently approved by the FDA to treat triple-class refractory multiple myeloma, despite an advisory panel’s concerns about its toxicity.21
We hypothesized that non-covalent inhibitors of CRM1 might also function as covalent inhibitors, but with further reduced toxicity, better efficacy, and resistance to C528 mutation of CRM1.13 It has been reported that the NS2 protein of the parvovirus Minute Virus of Mice (MVM) contains a supra-physiological NES that binds CRM1 with very high affinity, even in the absence of RanGTP.22 This might be exploited to design peptide inhibitors for non-covalent CRM1-targeted cancer therapy.
It remains challenging to efficiently deliver the peptides into the target cells, which often severely affects therapeutic efficiency. Targeted delivery of peptides into tumor cells could enhance anticancer activity in vivo.23 Nanoparticles provide a means for targeted delivery of biodrugs.24,25 Conventionally, the peptide would be directly loaded into a nanoparticulate delivery system before administration.26 However, this nanoparticle-mediated peptide delivery often elicits an immune response against the delivered peptide, limiting drug efficacy.27 Nanoparticle-mediated plasmid delivery to express the peptide in cells could effectively resolve the immune rejection problem and provide a versatile platform for intracellular delivery of peptides. Recently, many attempts have been performed to use nanoparticle-mediated gene therapy for multiple cancer treatments through intracellular expression of the encoded peptide or protein.28 Some gene therapy products have been approved for clinical study and clinical use.29 In our previous work, we developed an iDPP gene delivery system that could selectively deliver the DNAs into the tumor cells after intravenous administration.30 This platform could provide a tool for intracellular expression of a peptide discovered through structure-guided drug design.
In this work, we designed several high-affinity NESs as non-covalent CRM1 inhibitors and illustrated their mode of binding using crystal structures. The best peptides showed CRM1-binding affinities in the middle nM range in the presence of RanGTP/RanBP1. Cellular studies showed that the peptides were more potent than the wild-type (WT) peptide in binding to cellular CRM1, nuclear export inhibition, and the ability to kill cancer cells. We further generated an iDPP nanoparticle to selectively express the peptide inhibitor in tumors in vivo. Our results indicated that the DNA nano-complex significantly inhibited melanoma growth without observable toxicity.
Materials and Methods
Cloning, Protein Expression, and Purification
The NS2 NES (or its mutants) was cloned into a pGEX-4T1-based expression vector incorporating a TEV-cleavable N-terminal GST-tag fusion. The plasmid was transformed into E. coli BL-21 (DE3) and grown in LB Broth medium. Expression of protein was induced by the addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and the culture was grown overnight at 37°C. Cells were harvested and sonicated in lysis buffer (50 mM Tris pH 8.0, 200 mM NaCl, 10% glycerol, 2 mM DTT, 1 mM EDTA and 1 mM PMSF). GST-tagged NS2 NES was eluted with 20 mM Tris pH8.0, 200 mM NaCl, 1 mM EDTA, 2 mM DTT, 10 mM reduced glutathione and purified by Superdex 200 increase column. His-tagged human CRM1 (hCRM1) was expressed in E.coli grown in TB Broth medium. The protein was induced in the presence of 0.5 mM IPTG overnight at 25°C and purified by nickel beads. His-tagged hCRM1 were eluted with 300 mM imidazole 7.5, 300 mM NaCl, 10% glycerol and 2 mM BME. Saccharomyces cerevisiae CRM1 (yCRM1) was purified as previously described.31
Pull-Down Assay
All proteins used were purified by S200 prior to pull-down. To assess different interactions, we immobilized GST-tagged proteins (0.5 n mol) on GSH beads. Soluble proteins at indicated concentrations were incubated with the immobilized proteins in a total volume of 1 mL for 2 h at 4 °C. After two washing steps, bound proteins were separated by SDS/PAGE and visualized by Coomassie Blue staining. Each experiment was repeated at least twice and checked for consistency. Pull-down buffer contains 20 mM Tris pH 7.5, 200 mM NaCl, 10% glycerol, 2 mM MgCl2, 0.005% Triton-X100 and 2 mM DTT.
Isothermal Titration Calorimetry (ITC)
ITC experiments were conducted at 20°C using ITC200 (Microcal) in buffer containing 20 mM Tris pH 8.0, 200 mM NaCl, and 2 mM MgCl2. 125 µM GST-NES mutant was titrated into the sample cell containing 12 µM yRanBP1, 8 µM hCRM1, and 10 µM RanM189D. Each experiment was repeated at least twice. Data were processed by NITPIC and fitted by SEDPHAT.32,33
Crystallization of Peptides with yRanBP1-yCRM1ΔH9-RanY197A
Untagged RanY197A, GST-NESMVM (or mutants), GST-yRanBP1, and GST-yCRM1ΔH9 were expressed in E. coli separately and mixed, sonicated in lysis buffer (50 mM Tris pH 8.0, 200 mM NaCl,10% glycerol, 2 mM DTT, 5 mM MgCl2 and 1 mM PMSF). The complex was purified by GSH beads, cleaved off from beads by incubating the TEV enzyme overnight. The complex was further purified by gel filtration Superdex200 (GE Healthcare) column in buffer D (10 mM Tris 7.5, 100 mM NaCl, 5 mM Mg(OAc)2, 0.1 mM GTP, 2 mM BME). The complex was concentrated to 6 mg/mL using a Millipore spin concentrator (M.W. cutoff 10, 000). Crystals of different complexes were grown at room temperature by hanging drop vapor diffusion against 0.1 M Bis-Tris (pH 6.6), 0.2 M NH4NO3, and 18% PEG3350. Crystallization condition supplemented with glycerol (12% v/v) was used the as the cryoprotectant. X-ray diffraction data were collected at Shanghai Synchrotron Radiation Facility (SSRF) beamline BL17U1 and BL19U1.34 Coordinates of yCRM1-hRan-yRanBP1 (PDB code: 4HAT) were used as the search model and refined with rigid body refinement briefly then restrained refinement using the program Refmac5.35 Translation/Libration/Screw (TLS) refinement36 was used in the refinement process. The data collection and refinement statistics are provided in Table S1.
Cellular Nuclear Export Inhibition
Cells were maintained in Dulbecco’s modified Eagle’s medium (Hyclone) supplemented with 10% (v/v) fetal bovine serum (Biological Industries). Plasmids (2 µg each) encoding cytoplasm-localized mCherry-NES-MBP-NLS or either GFP-N1, GFP-WT, GFP-Nm15, GFP-Nm42 were co-transfected into cells (HeLa or A549 or 293T), followed by treatment with DMSO or KPT-330 (1 µM) for 10 hours. After 24 hours of transfection, cells were fixed and stained with Hoechst. Images were acquired by Olympus FV-1000 confocal microscope and analyzed using NIH ImageJ software.
Western Blot and Confocal Microscopy
HeLa cells were maintained and analyzed as previously described.37 Briefly, cells were maintained in Dulbecco’s modified Eagle’s medium (Hyclone) supplemented with 10% (v/v) fetal bovine serum (Biological Industries), and transfected with TurboFect transfection reagent (Thermo Scientific). GAPDH (ProteinTech) and mCherry (ProteinTech) antibodies were used at 1:5000 and 1:1000 dilution, respectively. Images were acquired by Olympus FV-1000 confocal microscope and analyzed using NIH ImageJ and Graphpad software.
Cell Culture and Animals
Mouse melanoma cell line B16-F10 was obtained from American Tissue Culture Collection and cultured in RPMI-1640 basal medium supplemented with 10% fetal bovine serum (FBS), 100 μg/mL streptomycin, 2 mM L-glutamine, and 100 U/mL penicillin at 37°C with 5% CO2 humidified air atmosphere. Female C57BL/6 mice (6–8 weeks) were obtained from the Laboratory Animal Center of Sichuan University (Chengdu, China). All the cell lines used has been approved by Sichuan University ethical review board. All animal procedures were performed according to the guidelines of Sichuan University and approved by the Animal Care Committee of Sichuan University.
Preparation and Characterization of iDPP/DNA Nanocomplex
iDPP was prepared by the previously described method.30 To prepare the iDPP/DNA nanocomplex, we mixed the iDPP nanoparticle with plasmid DNA at a mass ratio of 25:1 by gentle pipetting and then incubated at room temperature for 30 min. Particle size and zeta potential of the nanocomplex were determined by a dynamic light scattering detector (Malvern Nano-ZS90, UK) with a 2 min of equilibration time at 25 °C. The results were the mean of three test runs. The morphology of the nanocomplex was characterized by transmission electron microscopy (TEM, H-600, Hitachi, Japan).
In vitro Gene Transfection Efficiency
To determine the transfection efficiency of the iDPP nanoparticles in vitro, we seeded the B16-F10 cells into 6-well plates (2×105 cells per well) in 2 mL of 1640 medium containing 10% FBS. After 24 h of incubation, the medium in each well was replaced with fresh serum-free medium. The nanoparticles loaded with 2 μg of pGFP-N1 plasmid at the mass ratio of 25:1 were added to each well and incubated with cells for 6 h. Afterward, the serum-free medium was aspirated and replaced with the medium containing 10% FBS. Then, the cells were incubated at 37°C for another 24 h. The cell images were obtained using a fluorescence microscope (Olympus, Corp., Tokyo, Japan).
Cell Viability Assay
MTT assay was carried out to determine the cell viability of B16-F10 cells after treated with iDPP/DNA nanocomplexes. Cells were seeded in 96-well plates (5000 cells/well) and incubated for 24 h. Cells were then incubated with iDPP/DNA nanocomplex in a serum-free medium. After 6 h, the medium was replaced by complete medium and incubated at 37°C. After another 48 h, 20 µL of MTT was added into each well and reacted in the dark for 4 h. The absorbance value was determined by using a microplate reader at a wavelength of 570 nm, and the rate of viable cells was calculated.
Imaging of the Activity of the Luciferase in vivo
To investigate the in vivo gene transfection ability of iDPP, we used luciferase-encoding pGL6 (Beyotime Biotechnology, China) as a reporter plasmid. The B16-F10 melanoma tumor model was established in female C57BL/6 mice by subcutaneously injecting 5×105 B16-F10 cells. After the tumor diameter was about 1 cm, images of luciferase activity were taken by using the IVIS Lumia (Caliper Life Sciences, Mainz, Germany). The operation was as follows: 5 μg of pGL6 and iDPP at the mass ratio of 1:25 was incubated to form the iDPP/pGL6 nanocomplex and was subsequently injected intravenously into the mice. Seventy-two hours after the iDPP/pGL6 nanocomplex was administered, the mice were intravenously injected with 150 mg D-luciferin per kg body weight. Imaging data were obtained 20 min after D-luciferin injection from mice anesthetized with 2% isoflurane. Data were analyzed via Living Image Software.
Anticancer Efficacy of iDPP/DNA Nanocomplex in vivo
The mouse melanoma model was established by subcutaneous injection of B16-F10 cells at a dose of 5x105 per mouse (in 100 μL serum-free 1640 medium). Six days after inoculation, mice were randomly divided into three groups and then treated with NS (100 μL of 5% glucose solution) and iDPP/Nm42 nanocomplex (125 μg iDPP/5 μg pNm42) by intravenous injection once every two days for five times. The tumor volume and body weight of mice were recorded each other day. All mice were sacrificed by cervical dislocation on day 15. The tumor tissues were photographed and weighed. Vital organs were fixed in 4% paraformaldehyde.
Safety Evaluation
After received different treatments, the levels of alanine transaminase (ALT), aspartate transaminase (AST), and creatinine (CREA) of mice serum were acquired by serologic biochemical analyses. Histological analyses on the vital organs, including heart, liver, spleen, lung, and kidney, were performed using hematoxylin-eosin staining.
Results
Crystal Structure of NS2 NES in Complex with CRM1/Ran/RanBP1
Our initial work showed that WT MVM NS2 NES displayed little anti-cancer activity. In order to enhance the anti-cancer activity, a structure-based peptide design approach was taken. To view the mode of binding between NS2 NES and CRM1, we obtained the crystal structure of their complex in the presence of RanGTP/RanBP1. The H9 loop (residues 406–426) of CRM1 was deleted so that the NES groove was opened up for NS2 NES binding. The overall structure of the RanBP1-Ran-CRM1-MVM complex is highly similar to the previously reported RanBP1-Ran-CRM1 structure and the recently reported MVM complex structure (Figure 1A).38 NS2 NES follows the class 1a NES type and binds CRM1 similarly to the NES of PKI and hRio reversed (Figure 1B). The interaction between MVM and CRM1 is mainly hydrophobic, as expected. Of the five inter-molecular hydrogen bonds formed, two (between L89/G87MVM and K579CRM1) are strictly conserved by different NESs.39 The other three hydrogen bonds formed between H92/D93MVM and E586/G544CRM1 are not observed in other NESs (Figure 1C). The existence of new hydrogen bonds may explain why NS2 NES is the tightest-binding NES known.
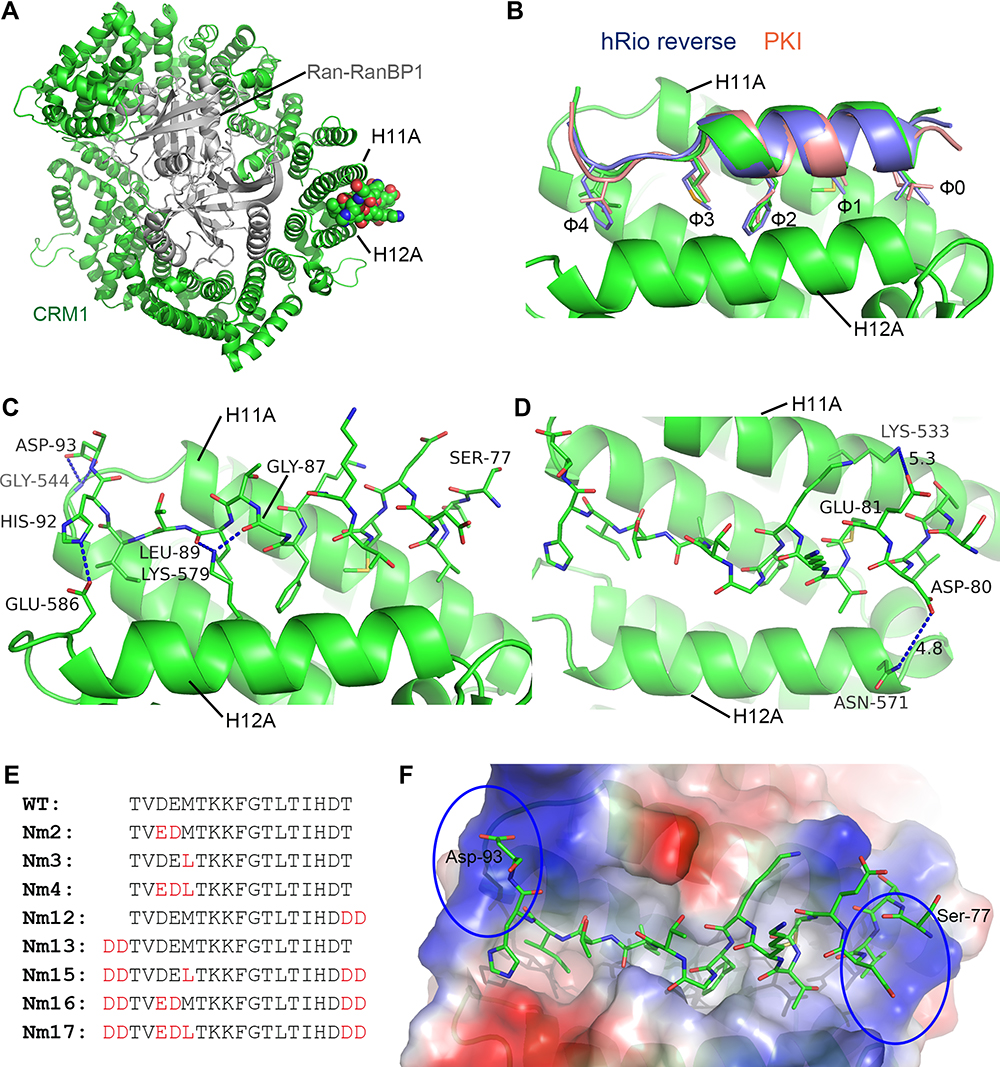 |
Figure 1 Crystal structure of NS2 NES in complex with CRM1/Ran/RanBP1 and the sequences of designed peptides. (A) The overall structure of NS2 NES in complex with CRM1/Ran/RanBP1. NS2 NES is shown as a ball and sticks representation. (B) Structural comparison of NS2 NES with that of PKI (pink, PDB 3NBY) and hRio reversed (blue, PDB 5DI9). (C) Hydrogen bonds formed between NS2 NES and yCRM1. (D) D80 and E81 contact distances with CRM1. The distances between opposite charges (labeled with dashes) could possibly be optimized by mutating to E80/D81. (E) List of mutations that were designed and discussed in the text. (F) The electrostatic surface potential map of CRM1. N and C terminuses of NS2 NES are surrounded by highly basic (circled) CRM1 surfaces. |
Structure-Guided Design of MVM Mutants
Closer examination of the structure shows that the distance between D80/E81MVM and K533/N571CRM1 is slightly sub-optimal (Figure 1D), and that switching to E80/D81 could optimize their interactions. We named this NS2 NES mutant as Nm2 (NS2 mutant 2, Figure 1E). Previously, Guttler et al reported that L is most preferred at the Φ1 position.40 Therefore M82L (Φ1) was designed and named Nm3. We also made a mutation combining Nm2 and Nm3 and named this construct as Nm4. Since the two ends of NS2 NES are adjacent to highly basic CRM1 electrostatic potential surfaces (Figure 1F), adding acidic residues on either end of NS2 NES may increase binding. Nm12 and Nm13 were designed by adding residues DD at C terminus and N terminus, respectively. Nm15-17 is similar to Nm2-4, except that they have DD at both termini. More mutants were designed than listed in Figure 1F (see Figure S1 for all of the other mutants).
CRM1-Binding Affinities of Designed Mutants
The binding potency of these peptides to CRM1 was first compared using a GST pull-down assay (Figure 2A). Since MVM WT NES already binds CRM1 tightly in the absence of Ran, incubation with competitor MBP-NESMVM was necessary in order to distinguish the increase in affinity. Indeed, either M82L or adding DD at the N or C terminus could increase the binding, comparing Nm3/Nm12/Nm13 with WT. Mutating 80DE81 to 80ED81 seemed to slightly decreased the binding between MVM and CRM1, judging by comparing Nm2 with WT, Nm3 with Nm4, and Nm15 with Nm17. Combining M82L with adding DD at both termini (Nm15) produced the greatest improvement of binding.
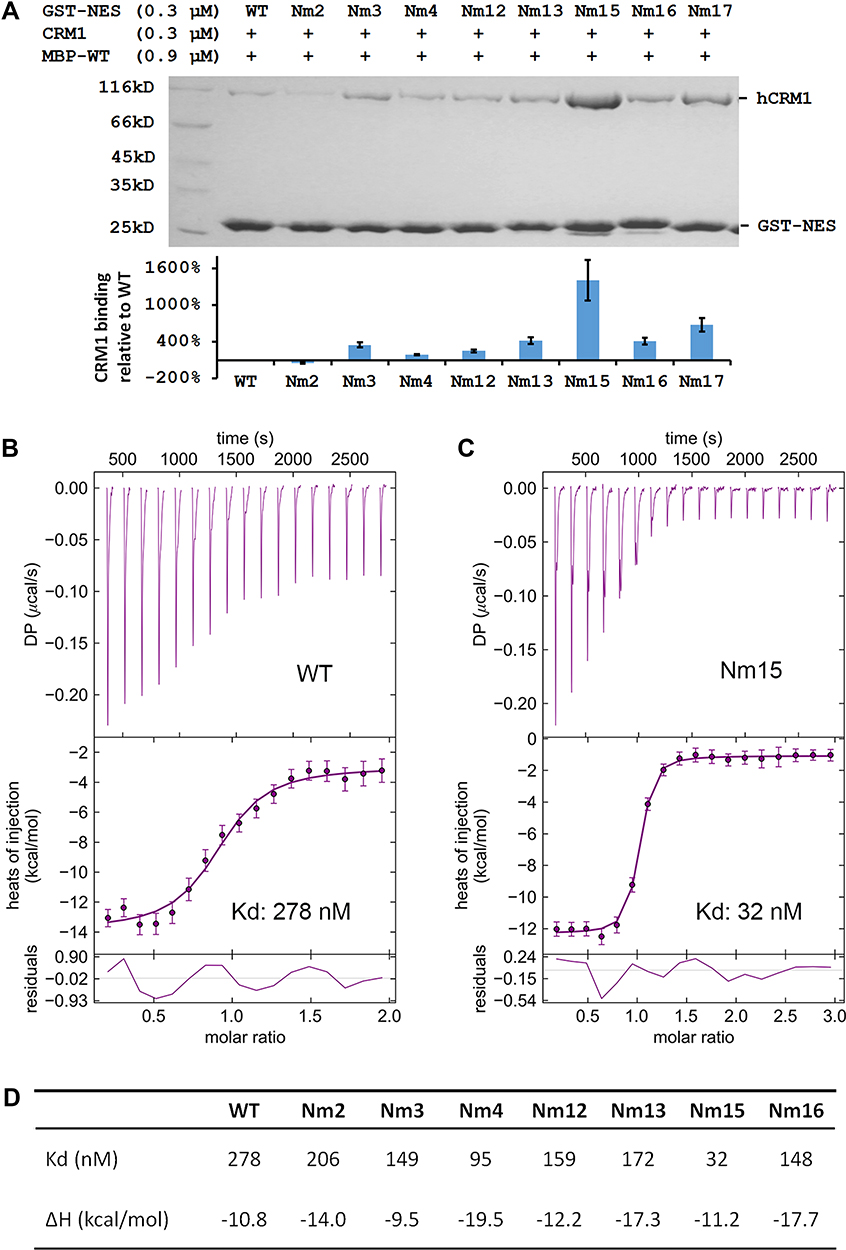 |
Figure 2 Comparing mutants’ affinities to CRM1 by pull-down and ITC experiments. (A) Top: pull-down analysis of different GST-MVM mutants in binding to CRM1 in the presence of competitor MBP-WT (MBP-tagged MVM WT NES). Bottom: quantification of CRM1 binding normalized by GST-NES bands and setting WT as 1. The error bars represent the standard deviation (SD) of three quantifications on the gel shown above. (B and C) ITC analysis of WT and Nm15 binding to CRM1. Error bars represent 68.3% confidence interval of measurements. (D) Affinity (Kd) and enthalpy change (ΔH) of different NES peptides binding to CRM1. Please refer to Figure S2 for isotherms and fitting for Nm2, Nm3, Nm4, Nm12, Nm13, and Nm16. |
These peptides’ affinities to CRM1 were further quantified by isothermal titration (ITC). The reported affinity of NS2 NES WT binding to CRM1 in the presence of Ran is beyond the measurement limit of ITC (10nM).38 In the absence of RanGTP, the affinity should decrease about 500 times41,42 to allow affinity measurement; however, that caused CRM1 precipitation and noisy isotherms in practice. We succeeded by adding a slight excess of RanGTP and yeast RanBP1 (yRanBP1) in the sample cell, which increased CRM1 stability probably by forming a loose inhibitory complex with CRM1.43 Under this condition, the WT peptide showed a 278 nM affinity to CRM1 (Figure 2B). Either M82L or adding DD at N or C terminus increased binding slightly, and altogether Nm15 displayed an affinity of 32 nM (9-fold higher than WT) (Figure 2C and D). Overall, ITC agreed well with pull-down, except that ITC showed a slight increase of binding affinity for 80DE81 to 80ED81 (Figure S2, Figure 2D).
Crystal Structures of Several Mutants in Complex with CRM1
We crystallized Nm2, Nm12, Nm13, and Nm15 in complex with CRM1/Ran/RanBP1 to visualize their mode of binding to CRM1. These structures were solved at 2.30–2.74 Å resolution, and the omit map electron densities are well defined (Figure S3). The crystal structure of Nm2 compared with WT shows that the dipole distances are indeed optimized: from 4.8 Å to 3.4 Å for D80E mutation, and from 5.3 Å to 4.4 Å for E81D mutation (Figures 1D and 3A). An extra electron density for D94 is observed in the crystal structure of Nm12, which forms electrostatic interaction with R543 of CRM1 (Figure 3B). Similarly, an extra electron density for D76 is observed in the crystal structure of Nm13, which forms electrostatic interaction with K525 of CRM1 (Figure 3C). These new interactions may enhance the binding between NES and CRM1. The crystal structure of Nm15 not only confirms what is observed with Nm12 and Nm14 but also shows the mutation of M82L, which may improve binding by interacting with more residues in the pocket (Figure 3D, S4). Altogether, these structures agree very well with our hypothesis, and with the observed higher affinities by pull-down and ITC.
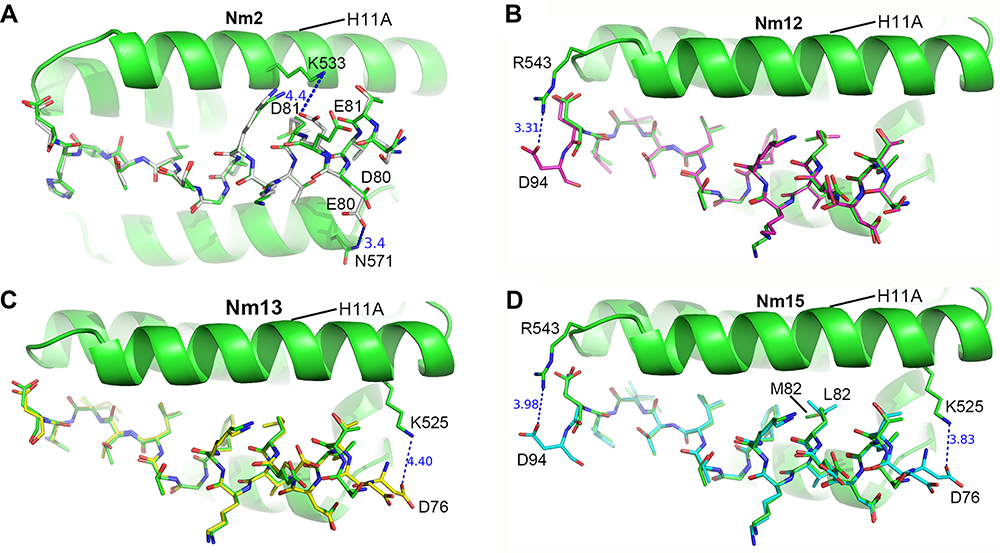 |
Figure 3 Crystal structures of MVM mutant peptides binding to CRM1. (A–D) the structure of Nm2 (grey), Nm12 (magenta), Nm13 (yellow), and Nm15 (cyan) stick structures with CRM1 shown as a cartoon. WT MVM is superimposed in each figure for comparison. Hydrogen bonds and electrostatic interactions are represented as dash lines. The angle of viewing is slightly adjusted for different figures to show the changes. |
Tighter Association with CRM1 in Cells
We next fused WT or Nm15 to the C terminus of GFP to allow easy detection by western and florescent imaging, and compared the their cellular localization and association with CRM1 in cells. Both GFP-WT and GFP-Nm15 were strongly co-localized with CRM1 in contrast to GFP in 293T cells (Figure 4A, top panel), suggesting that they formed a complex in cells. Additionally, a fraction of CRM1 was delocalized from the nucleus to the nuclear envelope for GFP-WT and GFP-Nm15 (Figure 4A, white arrows), but not for untransfected (yellow arrows) cells nor cells transfected with GFP-N1 (coding only GFP). Re-localization of CRM1 was possibly due to the formation of a tight complex with NS2 NESs, which stuck at the cargo dissociation stage at the cytoplasmic rim of the nuclear pore complex.22 Statistical analysis showed that the level of CRM1 co-localization was significantly stronger (p<0.0001) for GFP-Nm15 than GFP-WT (correlation coefficient 0.67 vs 0.36, Figure 4A, middle and bottom panel), most likely due to higher binding affinity with CRM1. Immunoprecipitation using anti-GFP or IgG showed that GFP-Nm15 was more associated with CRM1 than GFP-WT or GFP-N1 (Figure 4B), consistent with earlier results. We performed the above experiments in HeLa cells, and the results were similar (Figure 4C,D), suggesting that the stronger association with CRM1 for Nm15 is not cell-line specific.
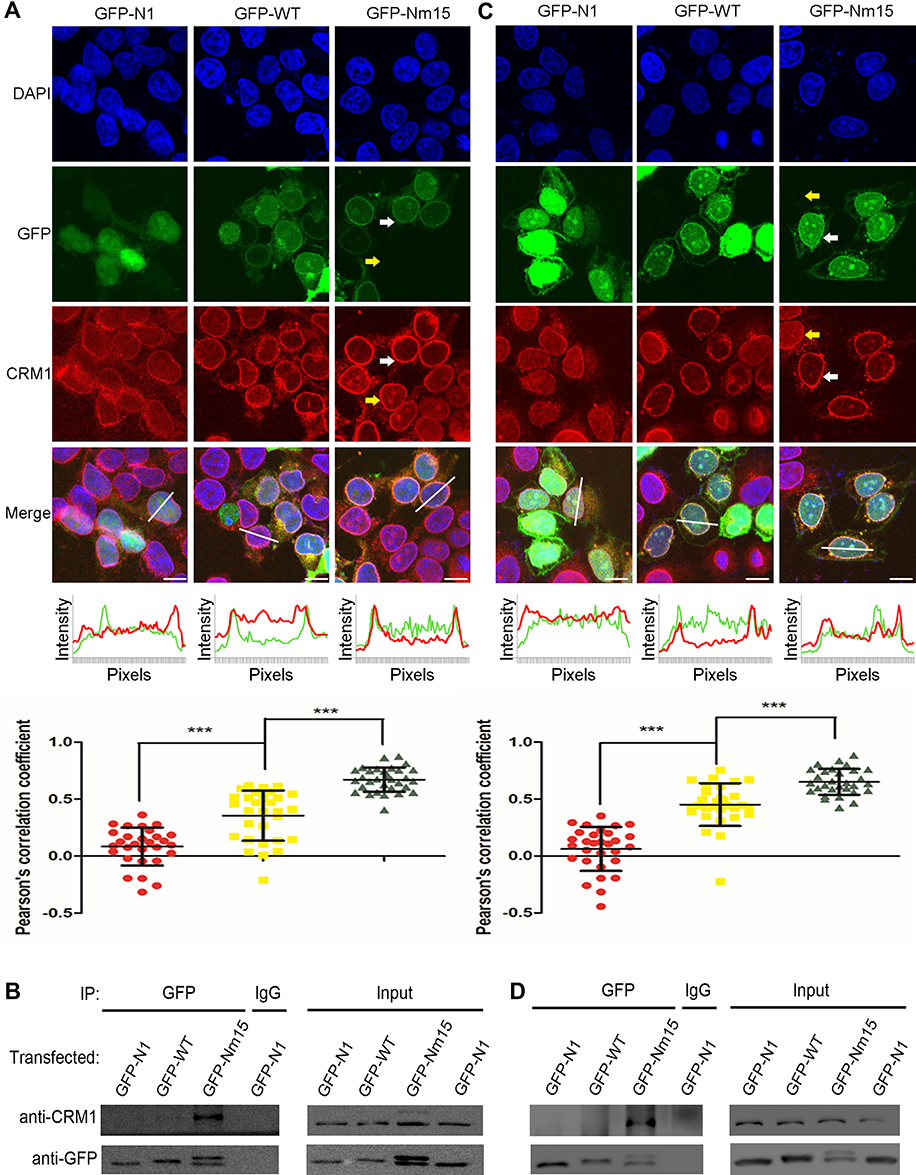 |
Figure 4 Stronger co-localization with CRM1 by Nm15 in 293T and HeLa cells. (A) Colocalization of CRM1 with GFP-N1, GFP-WT, or GFP-Nm15 in 293T cells. Confocal images in the top panel show the localization of endogenous CRM1 and transfected GFP proteins. White and yellow arrows represent transfected and untransfected cells, respectively. Scale bars represent 10 µm. The middle panel shows the GFP (green) and CRM1 (red) intensities of pixels for the co-responding white lines in the top panel. The bottom panel shows the dot plots for Pearson’s correlation coefficient calculated from at least 30 cells for each sample and the one-way ANOVA statistical analysis. Each dot represents a correlation coefficient calculated from one cell as done in the middle panel. *** denotes p<0.001. Error bars represent the standard deviation (SD) of each sample. (B) Immunoprecipitation to analyze cellular CRM1 binding strength. Transfected 293T cell lysates were immunoprecipitated using GFP or IgG antibodies and blotted for CRM1 and GFP. The bottom band is a proteolyzed fragment of full length GFP-Nm15 (the top band). (C and D) Performed the same as in (A and B), except using HeLa cells. |
Stronger Inhibition of Nuclear Export and Cell Growth
We then tested cellular inhibition of nuclear export by Nm15 in HeLa and A549 cells. As expected, KPT-330 relocated the mCherry-NES-NLS into the nucleus but not the GFP-N1 transfection (Figure 5A and B). GFP-WT transfection slightly increased the nuclear mCherry signal in both cells. In contrast, GFP-Nm15 markedly promoted nuclear localization of mCherry-NES-NLS, probably due to its higher affinity to CRM1. Similar results were observed in 293T (Figure S5), suggesting that GFP-Nm15 may function as a nuclear export inhibitor in a wide range of cell types.
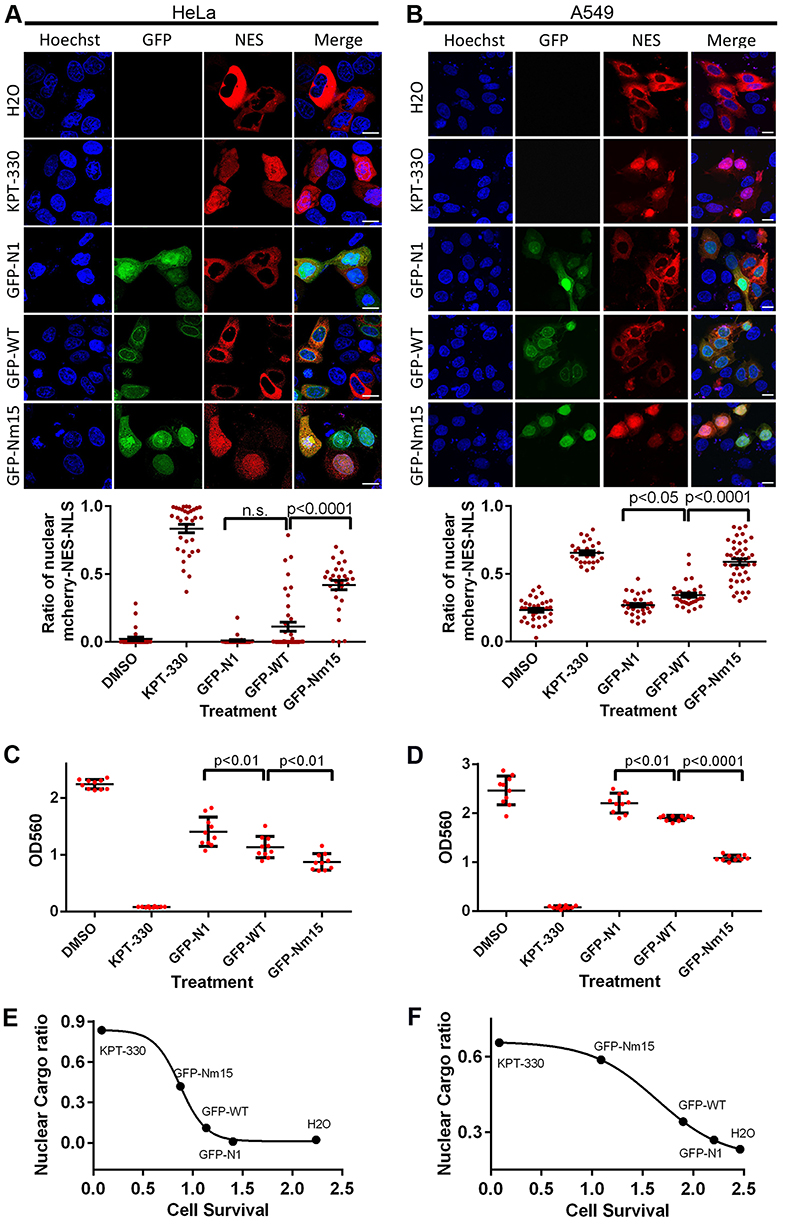 |
Figure 5 Nuclear export and cell survival inhibition by transfected GFP-Nm15. (A and B) Confocal images on nuclear export cargo (mCherry-NES-MBP-NLS, labeled as NES) transfected HeLa (A) or A549 (B) cells treated by KPT-330 or listed transfections. Bottom panels show the quantification and statistical analysis of nuclear cargo percentages on images collected in the top panels. Error bars represent the standard error of measurements. Scale bars represent 10 µm. (C and D) Cell survival and statistical analysis for HeLa (C) and A549 (D) cells under different transfection treatments. Error bars represent the standard deviation (SD) of each sample. (E and F) Negative correlations between the percent of nuclear cargo (Y-axis) and cell survival (X-axis) in HeLa (E) and A549 (F) cells. Average values from nuclear export (A and B) and cell survival (C and D) experiments were plotted and fitted using non-linear regression (four variables) in Graphpad software. |
Further, we showed that transfection of GFP-WT or GFP-Nm15 inhibited HeLa and A549 cell survival, with GFP-Nm15 notably stronger than GFP-WT (Figure 5C and D). KPT-330 exhibited the most potent growth inhibition to HeLa and A549 cells, consistent with its highest activity in nuclear export inhibition. When the nuclear cargo ratio and cell survival were plotted together, excellent correlations were observed: a lower ratio of nuclear cargo correlates with higher cell survival (Figure 5E and F). In other words, better nuclear export inhibition translates into better inhibition of cell growth. In the midst of our work, two high-affinity peptides of CRM1 were reported.38 Our pull-down, cellular nuclear export inhibition and cell survival inhibition assays showed that Nm15 was stronger than those peptides in binding to CRM1 and cell growth inhibition (Figure S6).
Further Optimization and Formation of the iDPP/DNA Nanocomplex
Direct addition of Nm15 peptide to cell culture medium was not effective in killing cancer cells at 100 µM concentration, even after different ways were employed to improve the net positive charge of the peptides, and/or to limit their cellular digestion (Figure S7). Meanwhile, we tried to further improve CRM1 binding affinity by virtually mutating each residue to all possible residues and calculating binding energy change through the Mutabind program,44 followed by testing some of mutants by pull-down. One of the new peptides, Nm42, consistently showed slightly tighter binding affinity than Nm15 (Figure 6A and B). ITC showed that Nm42 bound to CRM1 with a 22 nM affinity, which is 13-fold relative to WT (Figure 6C). The complex crystal structure shows that this peptide has substantially more hydrophobic interaction with CRM1 (Figure 6A, Figure S8), which might explain the much lower ΔH observed (water molecules that are squeezed out of the binding site could form more hydrogen bonds) (Figure 6C).
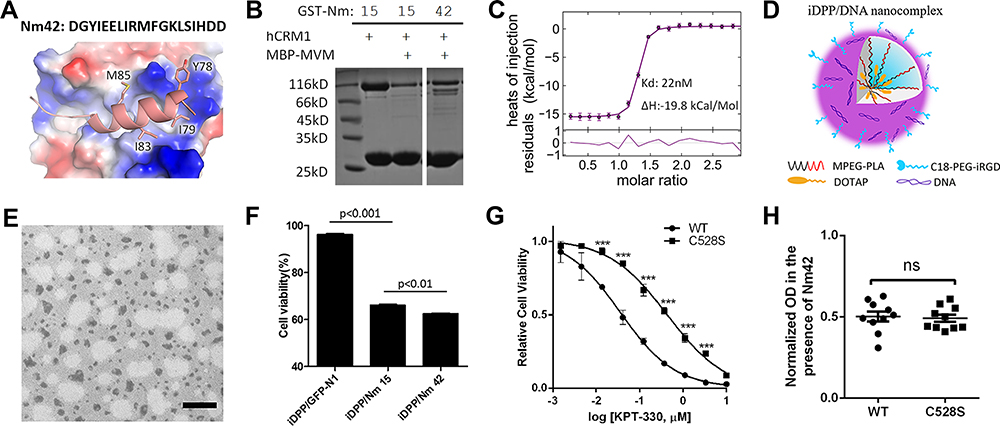 |
Figure 6 Design of Nm42 and nano-packaging of the Nm42 plasmid. (A) The sequence and crystal structure of Nm42 (cartoon) in complex with CRM1 (electrostatic surface potential map). The residues that increased hydrophobic interactions with CRM1 are shown as sticks. (B) SDS-PAGE of pull-down performed using Nm15 and Nm42 in the presence of competitor MBP-MVM WT NES (3 µM). (C) ITC analysis of Nm42 binding to CRM1, performed as in Figure 2. Error bars represent the 68.3% confidence interval of measurements. (D) Schematic illustration of iDPP/DNA nanocomplex. C18-PEG-iRGD, tumor-targeting peptide; DOTAP, cationic N-[1-(2, 3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium chloride; mPEG-PLA, block copolymer monomethoxy poly(ethylene glycol)-poly(d,l-lactide). (E) Morphology of iDPP/DNA nanocomplex determined by TEM. Scale bar, 200 nm. (F) Cell viability of B16-F10 cells after treated with different iDPP/DNA nanocomplexes (2 µg DNA). (G) HeLa stable cell lines (CRM1 WT or C528S) in the presence or absence of variable KPT-330 concentrations. Each concentration is performed with 6 repeats and normalized by the average reading of DMSO-treated samples. IC50s are 36 nM and 432 nM for WT and C528S mutant, respectively. Error bars represent the standard error of measurement. *** denotes p < 0.001. (H) Similar potency against WT and C528S HeLa cells by Nm42 plasmid transfection (2 µg). OD readings are normalized by non-transfected samples for both WT and C528S. |
Plasmids encoding Nm15 and Nm42 peptides were then incubated with the iDPP delivery system to form the iDPP/DNA nanocomplexes according to our previously reported method.30 As shown in Figure 6D, the iDPP/DNA nanocomplex was formed by electrostatically combining iDPP and plasmid DNA at a ratio of 25:1. The prepared iDPP/DNA nanocomplex exhibited a monodisperse and spherical appearance as seen by transmission electron microscopy (Figure 6E). The iDPP/DNA nanocomplex displayed an average diameter of 193 ± 5 nm with a PDI of 0.217, and a zeta potential of 2.12 ± 0.27 mV (Figure S9). Judging by the green fluorescence, there was a high transfection efficiency towards B16-F10 cells by iDPP/pEGFP-N1 (Figure S10). Further, MTT assay showed the lowest cell viability for the iDPP/Nm42 group (Figure 6F), indicating that it might inhibit B16-F10 cell growth in vivo. While KPT-330’s potency against the C528-mutated HeLa stable cell line was substantially reduced, Nm42 plasmid demonstrated similar anti-cancer potency on WT and C528S cells (Figure 6G and H). The IC50 for Nm42 plasmid was not measured since its concentration is not linearly correlated with cellular protein level nor with transfection efficiency.
Anti-Melanoma Activity and Safety Evaluation of iDPP/Nm42 Nanocomplex
To determine the targeting ability of iDPP/DNA nanocomplex in vivo, we used pGL-6 as a luciferase reporter gene. When iDPP/pGL-6 nanocomplex (125 µg iDPP/5 µg pGL-6) was administered through the tail vein, luciferase only accumulated in mouse tumor site (Figure 7A), demonstrating that iDPP had good targeting ability in vivo. Next, iDPP/Nm 42x5, iDPP/EM (empty vector) and NS (5% glucose) were used to study the anticancer ability in vivo. The growth curve of B16-F10 subcutaneous tumors under each treatment are displayed in Figure 7B, in which iDPP/Nm42x5 nanocomplex significantly inhibited the tumor growth when compared with NS (p = 0.0004) and iDPP/N1 (p = 0.022). Compared with the NS group (1.369 ± 0.362 g) and the iDPP/EM group (0.851 ± 0.131 g), there was a significant reduction of tumor size and weight of mice in iDPP/Nm 42x5 group (0.684 ± 0.104 g) (Figure 7C, Figure S11). During the treatment, the body weight of mice in each group did not significantly change (Figure 7D). All mice were sacrificed by cervical dislocation when the mice in the NS group became significantly weak. H&E staining analysis of vital organs, including heart, liver, spleen, lung, and kidney, showed that iDPP/Nm42 nanocomplex did not cause visible pathological changes (Figure 7E). The serum alanine aminotransferase (ALT), glutamate aminotransferase (AST), and creatinine (CREA) levels showed no significant difference from normal mice after iDPP/Nm42 treatment (Figure S12). These results suggested that iDPP/Nm42 nanocomplex was effective against melanoma cells and non-toxic to mice.
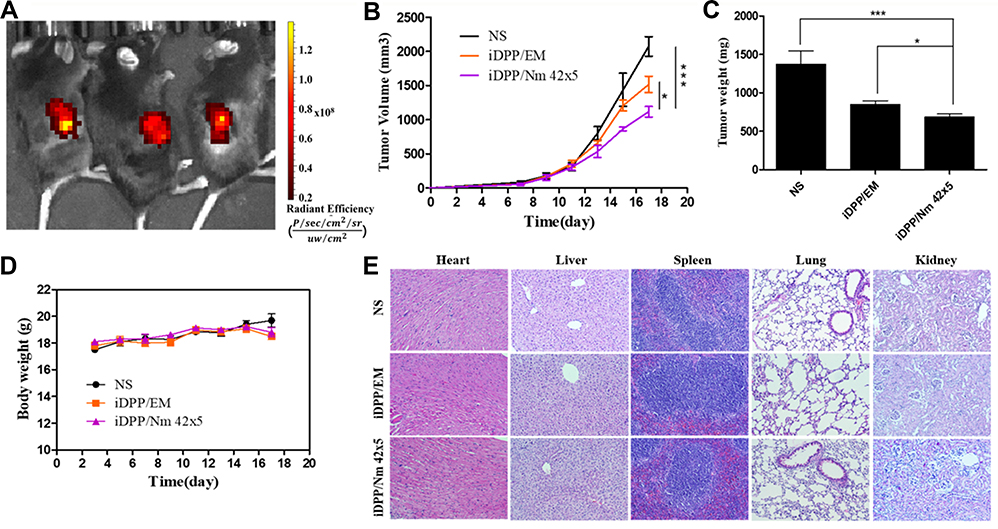 |
Figure 7 Anti-tumor activities and toxicity evaluation of iDPP/Nm42 nanocomplex on B16-F10 tumor in vivo. (A) In vivo imaging of luciferase activity, which indicated that iDPP/DNA nanocomplex possessed high tumor-targeting efficiency in vivo. (B) Tumor volumes (TV) of mice in each group, calculated according to the formula TV=1/2 × length × (width)2 (n = 5, mean ± SD). NS: treated with 5% glucose. EM, empty vector. (C) Weight of xenograft tumors of each treatment group (n = 5, mean ± SD). *p < 0.05 and ***p < 0.001. (D) Mouse body weight during the treatment. (E) No significant pathological changes from H&E staining of vital organ sections. Scale bar, 100 μm. Abbreviation: n.s., non-significant. |
Discussion
In this study, we improved the CRM1 binding affinity of a naturally existing NES and created several higher affinity NESs. The affinity between WT and CRM1 in the presence of Ran-yRanBP1 is 278 nM, which agrees well with previous literature.22,39,41,42 Under the same condition, Nm15 and Nm42 showed affinities of 32 nM and 22 nM, respectively. Since Ran-yRanBP1 is slightly inhibitory to NES binding,43 the real binary affinities might be slightly stronger than the measured values. In human cells, the environment on the cytoplasmic side of the nuclear pore complex (where the complex is stuck) is rich in RanBP1 and low in RanGTP.45 High affinity in the absence of RanGTP by Nm15 and Nm42 ensures CRM1 being sequestered and not recycled for further rounds of transport, possibly explaining their mechanism of action in cancer cells.
Our high-affinity NESs were shown to be useful to competitively inhibit CRM1 in molecular, cellular, and animal studies. Crystal structures showed that our designed peptides formed more electrostatic or hydrophobic interactions with CRM1. The increase in affinity was readily observable by pull-down, ITC, cellular co-localization with CRM1, nuclear export inhibition assay, and anti-cancer potency. The direct correlation between the level of nuclear export inhibition and the level of cell growth inhibition concurs with the importance of CRM1 in cancer. Though two other high affinity NESs were recently reported,38 their anti-tumor activities were never tested. Our designed peptides showed greater binding affinities and anti-cancer activities than those reported peptides (Figure S6).
The fact that non-covalent inhibitors of CRM1 were not discovered in different drug screen projects46,47 suggests that either non-covalent inhibitors may not bind to the NES groove sufficiently tightly to inhibit nuclear export of NES cargoes, or that covalent binding is essential for all CRM1 inhibitors. Since Nm42 does not form covalent bonds with CRM1 yet still inhibits cancer cells, it is clear that the covalent binding feature of CRM1 inhibitors is not essential, and that non-covalent CRM1 inhibitors should be pursued in future drug discovery.
C528T mutation has been found in majority of fungal CRM1, which provides resistance to the anti-fungal agent LMB.48 Likewise, treatment with KPT-330 may induce the evolution of C528 mutations. Compared to the covalent inhibitor KPT-330, we also showed that the peptide did not lose potency against CRM1 C528S-mutated cells. It should be noted that KPT-330’s IC50 towards CRM1-C528S HeLa cells was still very low (432 nM). This off-target effect might relate to KPT-330’s covalent binding nature and clinical toxicity. However, this type of off-target toxicity is likely not applicable to the peptide.
We showed for the first time that CRM1-targeted gene therapy against melanoma is feasible. As is well-known, peptides are limited in clinical application due to their high molecular weight, susceptibility to enzymatic decomposition, and low bioavailability. The nanotechnological delivery of peptide drugs often encounters problems such as low loading rates, insufficient encapsulation, and poor stability. In this work, we succeeded using a gene-therapy strategy to deliver the gene encoding the peptide of interest instead of the peptide itself into the target cells. The gene delivery system iDPP displayed a very high target site enrichment which enabled tumor-specific inhibition. Meanwhile, this delivery system does not contain positive charges, and did not induce obvious side effects following intravenous injection. In contrast to nano-packaging of peptides, the preparation of the DNA-nano complex is simple, requiring only mixing DNA and nanocarrier together. The current work provides technological support for CRM1-targeted peptide therapy against melanoma and other CRM1 overexpression diseases.
Using peptides to target a specific molecule’s structure is an important strategy in drug design.2 The approach to express peptides in cancer cells to inhibit the target protein is potentially applicable in cancer treatment. Previously, RNA interference was often used to achieve target protein inhibition.49 Proteins usually have several interaction surfaces, involved in different or related functions.2 RNA interference would inhibit all the interactions and functions of a protein. Our alternative strategy has the potential to selectively and desirably inhibit only one of the interactions, which might be beneficial in some cases. This work may enlighten future design of cancer gene therapy.
Conclusion
Through structure-guided protein engineering, we designed the first series of peptide-based non-covalent CRM1 inhibitors. Biochemical, cellular, and animal studies demonstrated that 1) non-covalent CRM1 inhibitors were effective to treat cancers, and 2) the potency of non-covalent inhibitors depended on their high CRM1-binding affinities. Superior to covalent CRM1 inhibitors, the peptide inhibitors were not sensitive to cysteine (528) mutation. Whether the non-covalent inhibitors are less toxic and/or more effective warrants further study. Our gene therapy approach offers a plausible strategy for structure-guided peptide therapy for cancers.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article and its supporting information files. Structure factor and atomic coordinates were deposited to Protein Data Bank (PDB) with accession codes 6A38, 6A3A, 6A3B, 6A3C, 6A3E, and 6KFT.
Acknowledgments
We thank the staff from BL17U1/BL18U/BL19U1 beamlines of Shanghai Synchrotron Radiation Facility (SSRF) for assistance during data collection. We acknowledge funding from NSFC (81502629 and 82073363) and 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYYC08007).
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
QS, YL, MS, MG, and MX have applied patents on the designed peptides in this study. The authors report no other conflicts of interest in this work.
References
1. Huang PS, Boyken SE, Baker D. The coming of age of de novo protein design. Nature. 2016;537(7620):320–327. doi:10.1038/nature19946
2. Zhang G, Andersen J, Gerona-Navarro G. Peptidomimetics targeting protein-protein interactions for therapeutic development. Protein Pept Lett. 2018;25(12):1076–1089. doi:10.2174/0929866525666181101100842
3. Spicer CD, Jumeaux C, Gupta B, Stevens MM. Peptide and protein nanoparticle conjugates: versatile platforms for biomedical applications. Chem Soc Rev. 2018;47(10):3574–3620.
4. Shin WH, Christoffer CW, Kihara D. In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods. 2017;131:22–32. doi:10.1016/j.ymeth.2017.08.006
5. Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278(5335):141–144. doi:10.1126/science.278.5335.141
6. Stade K, Ford CS, Guthrie C, Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 1997;90(6):1041–1050. doi:10.1016/S0092-8674(00)80370-0
7. Mathew C, Ghildyal R. CRM1 inhibitors for antiviral therapy. Front Microbiol. 2017;8:1171. doi:10.3389/fmicb.2017.01171
8. Ishizawa J, Kojima K, Hail N
9. Stelma T, Chi A, van der Watt PJ, Verrico A, Lavia P, Leaner VD. Targeting nuclear transporters in cancer: diagnostic, prognostic and therapeutic potential. IUBMB Life. 2016;68(4):268–280.
10. Kojima K, Kornblau SM, Ruvolo V, et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood. 2013;121(20):4166–4174. doi:10.1182/blood-2012-08-447581
11. Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83(8):1021–1032. doi:10.1016/j.bcp.2011.12.016
12. Khorashad JS, Eiring AM, Mason CC, et al. shRNA library screening identifies nucleocytoplasmic transport as a mediator of BCR-ABL1 kinase-independent resistance. Blood. 2015;125(11):1772–1781. doi:10.1182/blood-2014-08-588855
13. Sun Q, Chen X, Zhou Q, Burstein E, Yang S, Jia D. Inhibiting cancer cell hallmark features through nuclear export inhibition. Signal Transduction Targeted Ther. 2016;1(1):16010. doi:10.1038/sigtrans.2016.10
14. Kuruvilla J, Savona M, Baz R, et al. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood. 2017;129(24):3175–3183. doi:10.1182/blood-2016-11-750174
15. Hing ZA, Fung HY, Ranganathan P, et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia. 2016;30(12):2364–2372. doi:10.1038/leu.2016.136
16. Tai YT, Landesman Y, Acharya C, et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia. 2014;28(1):155–165. doi:10.1038/leu.2013.115
17. Newlands ES, Rustin GJ, Brampton MH. Phase I trial of elactocin. Br J Cancer. 1996;74(4):648–649. doi:10.1038/bjc.1996.415
18. Senapedis WT, Baloglu E, Landesman Y. Clinical translation of nuclear export inhibitors in cancer. Semin Cancer Biol. 2014;27:74–86. doi:10.1016/j.semcancer.2014.04.005
19. Sakakibara K, Saito N, Sato T, et al. CBS9106 is a novel reversible oral CRM1 inhibitor with CRM1 degrading activity. Blood. 2011;118(14):3922–3931. doi:10.1182/blood-2011-01-333138
20. Niu M, Chong Y, Han Y, Liu X. Novel reversible selective inhibitor of nuclear export shows that CRM1 is a target in colorectal cancer cells. Cancer Biol Ther. 2015;16(7):1110–1118. doi:10.1080/15384047.2015.1047569
21. XPO1. Inhibitor approved for multiple myeloma. Cancer Discov. 2019;9(9):1150–1151. doi:10.1158/2159-8290.CD-NB2019-085
22. Engelsma D, Valle N, Fish A, Salome N, Almendral JM, Fornerod M. A supraphysiological nuclear export signal is required for parvovirus nuclear export. Mol Biol Cell. 2008;19(6):2544–2552. doi:10.1091/mbc.e08-01-0009
23. Roy K, Kanwar RK, Kanwar JR. LNA aptamer based multi-modal, Fe3O4-saturated lactoferrin (Fe3O4-bLf) nanocarriers for triple positive (EpCAM, CD133, CD44) colon tumor targeting and NIR, MRI and CT imaging. Biomaterials. 2015;71:84–99. doi:10.1016/j.biomaterials.2015.07.055
24. Almeida JPM, Lin AY, Figueroa ER, Foster AE, Drezek RA. In vivo gold nanoparticle delivery of peptide vaccine induces anti-tumor immune response in prophylactic and therapeutic tumor models. Small. 2015;11(12):1453–1459. doi:10.1002/smll.201402179
25. Yu M, Wu J, Shi J, Farokhzad OC. Nanotechnology for protein delivery: overview and perspectives. J Control Release. 2016;240:24–37. doi:10.1016/j.jconrel.2015.10.012
26. Patil S, Vhora I, Amrutiya J, Lalani R, Misra A. Role of nanotechnology in delivery of protein and peptide drugs. Curr Pharm Des. 2015;21(29):4155–4173. doi:10.2174/1381612821666150901095722
27. Wang J, Hu X, Xiang D. Nanoparticle drug delivery systems: an excellent carrier for tumor peptide vaccines. Drug Deliv. 2018;25(1):1319–1327. doi:10.1080/10717544.2018.1477857
28. Garber K. Alnylam launches era of RNAi drugs. Nat Biotechnol. 2018;36(9):777–778. doi:10.1038/nbt0918-777
29. Morrison C. Landmark gene therapy poised for US approval. Nat Rev Drug Discov. 2017;16(11):739–741. doi:10.1038/nrd.2017.212
30. Luo L, Yang Y, Du T, et al. Targeted nanoparticle‐mediated gene therapy mimics oncolytic virus for effective melanoma treatment. Adv Funct Mater. 2018;201800173.
31. Fu SC, Fung HYJ, Cagatay T, Baumhardt J, Chook YM. Correlation of CRM1-NES affinity with nuclear export activity. Mol Biol Cell. 2018;29(17):2037–2044. doi:10.1091/mbc.E18-02-0096
32. Fung HY, Fu SC, Chook YM. Nuclear export receptor CRM1 recognizes diverse conformations in nuclear export signals. eLife. 2017;6. doi:10.7554/eLife.23961
33. Guttler T, Madl T, Neumann P, et al. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17(11):1367–1376. doi:10.1038/nsmb.1931
34. Monecke T, Haselbach D, Voss B, et al. Structural basis for cooperativity of CRM1 export complex formation. Proc Natl Acad Sci U S A. 2013;110(3):960–965. doi:10.1073/pnas.1215214110
35. Dong X, Biswas A, Suel KE, et al. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009;458(7242):1136–1141. doi:10.1038/nature07975
36. Li Y, Zhou J, Min S, et al. Distinct RanBP1 nuclear export and cargo dissociation mechanisms between fungi and animals. eLife. 2019;8:e41331. doi:10.7554/eLife.41331
37. Li M, Simonetti FL, Goncearenco A, Panchenko AR. MutaBind estimates and interprets the effects of sequence variants on protein-protein interactions. Nucleic Acids Res. 2016;44(W1):W494–501. doi:10.1093/nar/gkw374
38. Gorlich D, Seewald MJ, Ribbeck K. Characterization of Ran-driven cargo transport and the RanGTPase system by kinetic measurements and computer simulation. EMBO J. 2003;22(5):1088–1100. doi:10.1093/emboj/cdg113
39. Cautain B, de Pedro N, Murillo Garzon V, et al. High-content screening of natural products reveals novel nuclear export inhibitors. J Biomol Screen. 2014;19(1):57–65. doi:10.1177/1087057113501389
40. Kau TR, Schroeder F, Ramaswamy S, et al. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell. 2003;4(6):463–476. doi:10.1016/S1535-6108(03)00303-9
41. Yashiroda Y, Yoshida M. Nucleo-cytoplasmic transport of proteins as a target for therapeutic drugs. Curr Med Chem. 2003;10(9):741–748. doi:10.2174/0929867033457791
42. Shen J, Zhang W, Qi R, Mao ZW, Shen H. Engineering functional inorganic-organic hybrid systems: advances in siRNA therapeutics. Chem Soc Rev. 2018;47(6):1969–1995. doi:10.1039/C7CS00479F
43. Sun Q, Carrasco YP, Hu Y, et al. Nuclear export inhibition through covalent conjugation and hydrolysis of Leptomycin B by CRM1. Proc Natl Acad Sci U S A. 2013;110(4):1303–1308. doi:10.1073/pnas.1217203110
44. Scheuermann TH, Brautigam CA. High-precision, automated integration of multiple isothermal titration calorimetric thermograms: new features of NITPIC. Methods. 2015;76:87–98. doi:10.1016/j.ymeth.2014.11.024
45. Houtman JC, Brown PH, Bowden B, et al. Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. 2007;16(1):30–42. doi:10.1110/ps.062558507
46. Wang Z, Pan Q, Yang L, et al. Automatic crystal centring procedure at the SSRF macromolecular crystallography beamline. J Synchrotron Radiat. 2016;23(Pt 6):1323–1332. doi:10.1107/S160057751601451X
47. Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi:10.1107/S0907444996012255
48. Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Cryst. 2006;39:109–111. doi:10.1107/S0021889805038987
49. Sun Q, Yong X, Sun X, et al. Structural and functional insights into sorting nexin 5/6 interaction with bacterial effector IncE. Signal Transduction Targeted Ther. 2017;2:17030. doi:10.1038/sigtrans.2017.30
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.