Back to Archived Journals » Journal of Neurorestoratology » Volume 4
Can mammalian vision be restored following optic nerve degeneration?
Authors Kuffler D
Received 30 March 2016
Accepted for publication 3 May 2016
Published 13 July 2016 Volume 2016:4 Pages 51—62
DOI https://doi.org/10.2147/JN.S109523
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Hongyun Huang
Damien P Kuffler
Institute of Neurobiology, School of Medicine, University of Puerto Rico, San Juan, Puerto Rico
Abstract: For most adult vertebrates, glaucoma, trauma, and tumors close to retinal ganglion cells (RGCs) result in their neuron death and no possibility of vision reestablishment. For more distant traumas, RGCs survive, but their axons do not regenerate into the distal nerve stump due to regeneration-inhibiting factors and absence of regeneration-promoting factors. The annual clinical incidence of blindness in the United States is 1:28 (4%) for persons >40 years, with the total number of blind people approaching 1.6 million. Thus, failure of optic nerves to regenerate is a significant problem. However, following transection of the optic nerve of adult amphibians and fish, the RGCs survive and their axons regenerate through the distal optic nerve stump and reestablish appropriate functional retinotopic connections and fully functional vision. This is because they lack factors that inhibit axon regeneration and possess factors that promote regeneration. The axon regeneration in lower vertebrates has led to extensive studies by using them as models in studies that attempt to understand the mechanisms by which axon regeneration is promoted, so that these mechanisms might be applied to higher vertebrates for restoring vision. Although many techniques have been tested, their successes have varied greatly from the recovery of light and dark perceptions to partial restoration of the optomotor response, depth perception, and circadian photoentrainment, thus demonstrating the feasibility of reconstructing central circuitry for vision after optic nerve damage in mature mammals. Thus, further research is required to induce the restoration of vision in higher vertebrates. This paper examines the causes of vision loss and techniques that promote transected optic nerve axons to regenerate and reestablish functional vision, with a focus on approaches that may have clinical applicability.
Keywords: vision, RGC, axon regeneration, optic nerve trauma, restoration of vision
Introduction
All forms of injury to an adult mammalian eye, including glaucoma, blunt or penetrating trauma, tumors, and optic nerve trauma, lead to irreversible loss of vision. Central nervous system (CNS) axons cannot regenerate, and there is no effective means of preventing optic nerve degeneration. Sports-related injuries, such as in baseball or boxing, frequently cause secondary glaucoma. However, following injury to the visual system of the embryonic mammal and adult fish and lower vertebrates, retinal ganglion cells (RGCs) proliferate, their axons regenerate, and vision can be restored. The major questions regarding this are why the mammalian CNS changes from being permissive to inhibiting neuron proliferation and axon regeneration, and what can be learned about the mechanisms that allow and promote axon regeneration in the fish and lower vertebrate CNS that may be applicable to the adult mammalian CNS to both allow and promote axon regeneration. Thus, extensive studies have been performed aimed at understanding which trophic factors and other bioactive molecules promote cell neuron survival and axonal regeneration, which might be applied to the adult mammalian CNS to allow for axon regeneration and recovery of neurological function. To date, no successful therapeutic interventions for restoring vision in the adult mammalian visual system have been found.
Loss of vision: causes and incidences
Glaucoma is a progressive optic neuropathy due to RGC degeneration and is one of the leading causes of irreversible blindness. Primary open-angle glaucoma (POAG) is the second most common cause of blindness in the United States,1 with approximately 2.5 million Americans having POAG, with approximately half being unaware of it.2 Untreated glaucoma is the leading cause of human blindness. Because there are virtually no symptoms or pain associated with increased eye or intraocular pressure (IOP), it is difficult to recognize its presence. And even with treatment, approximately 10% of people with glaucoma lose their vision.3 Its first indications are loss of peripheral vision, but this is typically not recognized because individuals compensate by using a less central portion of the retina for their vision. Thus, the physical deficits are not diagnosed until vision has been lost significantly.4 Although medication and/or surgery can slow down the progression of glaucoma-induced vision loss, the lost vision cannot be recovered. Therefore, the most important mechanism for preventing glaucoma-induced vision loss is early glaucoma diagnosis and treatment.
Glaucoma strikes at all ages although increasing age brings a higher risk with approximately one in 10,000 babies in the United States born with glaucoma.5 Worldwide, approximately 6% of children are blind because of glaucoma. However, open-angle glaucoma (OAG) is also a hereditary problem with the presence of glaucoma in one’s family increasing the risk of developing glaucoma four- to nine-fold over that of individuals with no hereditary glaucoma background.6
Among young adults, especially African-Americans, glaucoma is the leading cause of blindness,7 which is 6–8 times more common than in Caucasians.2 The American Hispanic population has the greatest risk of developing glaucoma more rapidly and at a younger age,8 while people of Asian descent appear to have no significant risks of developing glaucoma.9 Therefore, it is urgent to develop improved methods of screening and therapy for glaucoma.
Although glaucoma is often associated with elevated IOP, IOP elevation is not detected in a significant subset of glaucomas such as normal tension glaucoma (NTG). Moreover, in some glaucoma patients, significant IOP reduction does not prevent the progression of the disease. Thus, understanding IOP-independent mechanisms of RGC loss is important. Glutamate transporters are active during RGC degeneration, and the loss of glutamate transporters leads to the death of the RGCs due to glutamate neurotoxicity and oxidative stress. Mice deficient in the glutamate transporters GLutamate ASpartate Transporter (GLAST) and Excitatory Amino Acid Transporter 1 (EAAT1) show spontaneous RGC and optic nerve degeneration without elevated IOP. In GLAST-deficient mice, the glutathione level in Müller glia is decreased, and the administration of glutamate receptor blocker prevents RGC loss. In EAAC1-deficient mice, RGCs are more vulnerable to oxidative stress. Glutamate transporters are necessary to prevent excitotoxic retinal damage and to synthesize glutathione, a major cellular antioxidant and tripeptide of glutamate, cysteine, and glycine.10 Thus, glutamate transporter KO mice can be used as NTG models for the experiments of neuroprotection.
Various glaucoma etiologies based on genetic backgrounds
While considering the impact of optic disc topography (optic disc area, cup area, rim area, cup volume, rim volume, cup-to-disc area ratio, mean cup depth, maximum cup depth, cup shape measure, height variation contour, mean retinal nerve fiber layer thickness, and retinal nerve fiber layer cross-sectional area) in NTG and POAG of different patients, it is important to consider their genetic backgrounds and even variability seen for populations within various Southeast Asian countries. The optic disc parameters in patients with NTG and POAG in Malaysia differed from those in other Asian countries with the eyes for NTG patients having significantly larger optic disc areas and cup areas, respectively. A comparison of other parameters between the two groups revealed no significant differences.10 However, patients with moderate and severe NTG showed significantly deeper cups and larger disc and cup areas compared with moderate and severe POAG patients. Similar differences were seen in the studies of NTG and POAG in Korean patients.10 The deeper cups and larger disc and cup areas may serve as indicators of severity when comparing NTG with POAG.11,12
Optic nerve regeneration in lower vertebrates
In Meyer and Sperry,13 Sperry (1943, 1944) showed that in lower vertebrates, fish and frogs, precise vision can be reestablished following complete optic nerve transection and removal and replacement of the eye, even when reinserted after a 180° rotation. Thus, in lower vertebrates, RGC axons have an intrinsic ability to locate their appropriate central targets and reform retinotopic patterns.14 These experiments provide important information about retinal map formation. The reestablishment of a retinotopic map does not require a precise realignment of the two cut ends of the optic nerve, and the appropriate axon trajectories appear to be established by interactions between the regenerating axons and guidance cues along the visual pathway. Therefore, most of the research studies are aimed at promoting recovery of vision on adult mammalian animal models.
Inhibition of optic nerve axon regeneration
Although axons in the developing mammalian CNS can regenerate, the same axons in the developed mammalian CNS cannot regenerate.15 Severed RGC axons, such as the spinal cord long tracts, have no capacity to regenerate under normal physiological circumstances. This is due to the virtually ubiquitous presence of factors that inhibit axon regeneration, such as Nogo, chondroitin sulfate proteoglycan (CSPG), and myelin-associated glycoprotein (MAG).15 Similarly, although axons of embryonic RGCs can grow axons into the superior colliculus, RGC axons of postnatal animals fail to extend into CNS tissue, even when it is derived from the embryonic tectum.16 Thus, the failure to reestablish vision is a multifactorial phenomenon:
- Most RGCs die if they are axotomized close to the RGCs.17
- Adult RGCs lose their intrinsic potential to regenerate.18
- Axon lesions induce the upregulation of factors that inhibit axon regeneration.18
- Glial cells at the site of injury produce scars that inhibit axon regeneration.19
Thus, in this cellular environment, induction of vision following RGC axon injury requires maintaining RGC viability, blocking the upregulation and activity of regeneration-inhibiting factors, providing axon regeneration-promoting factors, and preventing scar tissue formation.
Changing cellular environment from one that inhibits to one that promotes axon regeneration
Axon regeneration in the CNS requires reactivating intrinsic growth state of the injured neurons and enabling growth in an inhibitory environment. CAST/Ei mouse adult dorsal root ganglion neurons extend axons more extensively on CNS myelin than the other strains, especially following a pre-injury. Injury-primed CAST/Ei neurons also regenerate markedly within the spinal cord and optic nerve more than those from C57BL/6 mice and show greater sprouting following ischemic stroke. The extent of growth of CAST/Ei neurons on myelin is genetically determined, and two whole-genome expression screens yield the Activin transcript Inhba as the most correlated with this ability. Inhibition of Activin signaling in CAST/Ei mice diminishes their CNS regenerative capacity, while its activation in C57BL/6 animals increases regeneration. Thus, mammalian CNS regeneration can take place, and modulation of part of the related molecular pathway contributes to this ability.20
The use of a length of vascularized peripheral nerve to the transected optic nerve results in enhancing the number of RGC axons that regenerate and the percent of myelinated RGC axons versus the number that regenerate and are myelinated when nonvascularized grafts are used.21
Although separate mechanisms can induce optic nerve regeneration singly, combining different therapies allows for synergistic actions that activate the intrinsic growth state of RGCs and enables them to regenerate their axons to the full length of the optic nerve, across the optic chiasm, and into the brain, where they establish synapses in appropriate target zones and restore limited visual responses.22,23 These treatments involve the induction of a limited inflammatory response in the eye, which increases the levels of oncomodulin and other growth factors, elevates intracellular Cyclic-adenosine-monophosphate (cAMP), and deletes the pten gene in RGCs. Although these approaches cannot be applied clinically, they indicate different approaches that may eventually be applied clinically.22
The optic nerve has been widely used to investigate factors that regulate axon regeneration in the mammalian CNS. Although RGCs show little capacity to regenerate their axons following optic nerve damage, studies show that some RGCs can regenerate axons through a segment of peripheral nerve grafted to the optic nerve.24 More recently, some degree of regeneration has been achieved through the optic nerve itself by factors associated with intraocular inflammation (oncomodulin) or by altering levels of particular transcription factors (Klf-4, -9, c-myc), cell-intrinsic suppressors of axon growth (PTEN, SOCS3), receptors to cell-extrinsic inhibitors of axon growth (Nogo receptor, LAR, PTP-sigma) or the intracellular signaling pathway activated by these receptors (RhoA). Regeneration partially restores the optomotor response, depth perception, and circadian photo-entrainment, demonstrating the feasibility of reconstructing central circuitry for vision after optic nerve damage in mature mammals.
In some cases, regenerating axons have been shown to innervate the appropriate central target areas and elicit postsynaptic responses. Many discoveries made in this system have been found to enhance axon regeneration after spinal cord injury. Thus, progress in optic nerve regeneration holds promise not only for visual restoration but also for improving outcome after injury to other parts of the mature CNS.25
Trauma-induced RGC apoptosis
RGC apoptosis occurs following optic nerve transection near the retina and after acute elevation of IOP that damages the retina.26 RGC apoptosis is also triggered by increased intra-retinal levels of glutamate27 with the consequent abnormal activation of N-methyl-d-aspartic acid (NMDA) and non-NMDA subtypes of glutamate receptors, leading to excitotoxic, glutamate-mediated, RGC death in the rat retina.28 The role of glutamate excitoxicity is demonstrated by the reduction in RGC apoptosis when glutamate release from injured RGCs is inhibited, inhibiting glutamate uptake, or blocking glutamate receptors on both injured and uninjured cells.28 RGC apoptosis can be prevented by injecting brain-derived neurotrophic factor (BDNF) into the eye at the time of the optic nerve lesion.29 Some growth factors induce the expression of β-tubulin mRNA in ganglion cells, which is important for axonal extension,30 and its effect appears independent of that of BDNF.31 Growth factors such as BDNF increase the phosphorylation of Akt, which may have potential therapeutic implications in the treatment of glutamate-related diseases.32 Autocamtide-2-related inhibitory peptide, a specific inhibitor of CaMKII, is also a neuroprotectant for RGC-5 cells against glutamate toxicity.33
BDNF regulates neural cell survival principally by activating TrkB receptors, and BDNF-TrkB signaling is important for RGC survival following optic nerve injury (ONI).34 Although the neuroprotective effect of exogenous BDNF is transient, glial cells act as mediators for neural cell survival, and TrkB expression in retinal glia suggesting its role in neuroprotection and that glial TrkB signaling plays an important role in the early stage of neural protection after traumatic injury.34
Glia are mediators of neuron survival and promotors of neural regeneration. For TrkB(GFAP) and TrkB(c-kit) knockout mice in which TrkB, a receptor for BDNF, is deleted in retinal glia or inner retinal neurons, the extent of glutamate-induced retinal degeneration is similar to that in the two mutant mice. For TrkB(GFAP) knockout mice, BDNF does not prevent photoreceptor degeneration and fails to stimulate Muller glial cell proliferation and expression of neural markers in the degenerating retina. Thus, BDNF signaling in glia has important roles in neural protection and regeneration, particularly in the conversion of Müller glia to photoreceptors.35
Scar tissue formation: inhibitory effects of astrocytes
Gliosis is the scar reaction around a CNS injury site and is mediated mainly by the proliferation of astrocytes.19 Trauma leads to reactive gliosis and failure of optic nerve axons to regenerate.36 Axon regeneration requires a second factor to overcome the inhibitory barrier produced by gliosis.37 Within the CNS, activated astrocytes form a dense plug of gliotic tissue in an attempt to wall off the CNS from pathogens and reestablish the blood–brain barrier.38 Thus, gliosis inhibits axon regeneration directly by presenting a physical barrier to regrowing axons and indirectly by the synthesis of regeneration-inhibiting molecules.39
Nogo
As stated earlier, the failure of axon to regenerate in the adult CNS is in part due to the presence of inhibitory molecules Nogo-A and MAG and repulsive guidance molecules Ephrins, Netrins, and Semaphorins.40 The long-term release of an antibody that blocks the regeneration-inhibiting activity of oligodendrocyte Nogo-A near a spinal cord lesion site allows approximately 10% of injured axons to regenerate into and within the CNS,41 while the intrinsic ability of adult mouse optic nerve RGCs to regenerate can be restored by blocking astrocyte-derived CSPGs or oligodendrocyte-derived Nogo receptor (NgR1) ligands.42
In zebrafish, Rtn4b, the homologue of mammalian Nogo-A/RTN4-A, is upregulated in axotomized RGCs and is primarily associated with the endoplasmic reticulum. Rtn4b serves as a neuron-intrinsic determinant required for axon regeneration in the zebrafish visual system. The lesion-induced upregulation of Rtn4b in fish correlates with an increase in endoplasmic reticulum, soma size, biosynthetic activity, and growth. This suggests that providing mammalian neurons with the same upregulation is required for the regeneration of RGC axons.43
Sema5A
RGC axons are also inhibited from regenerating within the optic nerve past postnatal day 4 due to the expression of sema5A expressed by optic nerve oligodendrocytes.44 Eliminating or neutralizing sema5A results in axon regeneration into and within the CNS,45 in which results in facilitating axon regeneration of lesioned optic nerve.
Promoting RGC survival
By postnatal day 4, RGC axons fail to regenerate within the optic nerve,36 which is correlated with the down regulation of Bcl-2 an anti-apoptotic gene36 required for neuronal survival during brain development46 while providing neuroprotection against axotomy-induced death.47 By inducing the overexpression of Bcl-2 axotomized, RGCs remain viable and their axons regenerate over long distances and innervate appropriate brain targets in postnatal mice of up to 2 weeks of age.36
Hypoxia-ischemia-induced neuronal death is an important pathophysiological process associated with ischemic stroke and represents a major challenge in preventing ischemic stroke. Exposing primary neurons to sodium dithionite and glucose deprivation (SDGD) mimics hypoxic-ischemic conditions. Exposing these neurons to N,N,N’,N’-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN), a specific Zn2+-chelating agent, significantly suppresses increased cell death, apoptosis, neuronal glutamate release into the culture medium, NR2B protein expression, and I K and decreased GluR2 protein expression and Na+ channel activity in primary cultured neurons exposed to SDGD. These results suggest that TPEN could inhibit SDGD-induced neuronal death by modulating apoptosis, glutamate signaling (via ligand-gated channels such as AMPA and NMDA receptors), and voltage-gated K+ and Na+ channels in neurons. Hence, Zn2+ chelation might be a promising approach for counteracting the neuronal loss caused by transient global ischemia. Moreover, TPEN could represent a potential cell-targeted therapy.48
The ability of fish RGC axons to regenerate requires the reexpression of the proteins reggie-1 and -2, but mammalian RGCs fail to upregulate reggie expression and regenerate axons after a lesion,49 but adult rat RGCs will regenerate following a lesion if reggie-1 expression is induced 14 days prior to ONI.49 Since RGCs grow long axons on CSPG and Nogo-A, this suggests that reggie-1 provides neurons the ability to override inhibitors of neurite growth.49
The inhibition of axon regeneration in mice by the rho pathway can be neutralized by the enzyme C3, allowing significant axon regeneration after optic nerve transection.50 The injection of cyclic adenosine monophosphate (cAMP) into the vitreous of mice a day after the crush injury also induces significant axon regeneration throughout an optic nerve crush site.50
Neuroprotection
Following ONI, neuroprotection of RGCs can be provided by recombinant human erythropoietin52 being injected into the vitreous cavity of the eye, resulting in the expression in the growth-associated protein 43, a marker for neuronal regeneration indicating that rhRPO promotes axon regeneration and functional restoration of the optic nerve.52 Axotomy-induced RGC death can be reduced by intraocular injection of molecules that inhibit the retinal microglia activity53 and the delivery of neurotrophic factors.54 An alternative technique involves applying growth factors directly onto the RGC axons.55
Fish: heat shock protein and insulin-like growth factor-1
Following ONI in fish, the mRNAs for heat shock protein 70 and insulin-like growth factor-1 rapidly increase in RGCs and act as neuroprotectors, which is also associated with a rapid increase in purpurin gene expression and the synthesis of purpurin in the photoreceptors. Purpurin promotes neurite sprouting, which is closely mimicked by retinoic acid and blocked by its inhibitor. Thus, purpurin appears to induce axon regeneration by acting as a retinol transporter to supply retinoic acid to damaged RGCs which in turn activates target genes, such as transglutaminase and neuronal nitric oxide (NO) synthase mRNAs, and retinoic acid genes.56
Neutrophils
ONI triggers the invasion into the eye and activation of neutrophils, the first responders of the innate immune system. The neutrophils express high levels of the atypical growth factor oncomodulin (Ocm). Immunodepletion of neutrophils diminishes Ocm levels without altering the levels of ciliary neurotrophic factor (CNTF), leukemia inhibitory factor, or interleukin-6, while suppressing the regeneration-promoting effects of inflammation. A peptide antagonist of Ocm also suppresses axon regeneration, indicating that neutrophils are a major source of Ocm that promotes axon regeneration in the CNS.57
NO
NO is a gaseous messenger molecule biosynthesized from l-arginine and molecular oxygen by NO synthase, and excess NO production plays a critical role in killing RGCs. However, NO also mediates neuroprotection via the signaling pathways, the NO/cyclic guanosine monophosphate pathway, and S-nitrosylation.58 However, a reduction in nitric oxide synthase or its production following ONI significantly increases RGC survival.59 RGCs can be protected after injury by reducing the production of N-nitro-l-arginase, nipradilol, and aminoguanidine, which reduce the production of NO or inhibit NO synthase.59
Neural progenitor cells
Müller glia may inhibit axon regeneration, but Müller glia produces trophic factors via BDNF–TrkB pathway, which may lead to RGC protection after ONI. In addition, this BDNF–TrkB pathway may stimulate the dedifferentiation of Müller glia into retinal neurons.
Human neural progenitors transplanted into the crushed optic nerve of adult rats are induced to differentiate into anterior specified neural progenitors by the administration of Noggin and retinoic acid. This treatment provides significant RGC neuroprotection while inducing axon regeneration.60
Müller glia are resident radial glia in the vertebrate retina. In response to mammalian retinal injury, the Müller glia lead to glial scar formation and prevent the replacement of lost neurons. However, adult zebrafish Müller glia act as tissue-specific stem cells that generate neurogenic progenitors that regenerate all retinal neurons after damage. The proliferative response depends on the regulation of transforming growth factor beta (TGFbeta) signaling by the corepressors Tgif1 and Six3b in the adult zebrafish retina. Thus, Tgif1 expression and regulation of TGFbeta signaling are implicated in the function of several types of stem cells and is necessary for neuron regeneration.61
Brimonidine
Brimonidine, an alpha2-adrenergic receptor agonist commonly used to lower IOP in glaucoma, exerts neuroprotective effects and brimonidine treatment promotes optic nerve regeneration by inducing the activation of Erk1/2 after ONI.62
Taxol
The local application of Taxol to the site of an ONI markedly enhances neurite extension of mature RGCs and PC12 cells by stabilizing microtubules and desensitized axons toward myelin and CSPG inhibition in vitro without reducing RhoA activation.63 Taxol treatment also significantly increases lens injury-mediated axon regeneration in vivo, delays glial scar formation, suppresses CSPG expression, and transiently reduces the infiltration of macrophages at the injury site.63 Thus, microtubule-stabilizing compounds such as Taxol may be candidates for the treatment of CNS injuries particularly when combined with interventions stimulating the intrinsic regenerative state of neurons.63
CSPG is expressed during development where it has a putative role in axon guidance by forming barriers to direct growth cones.64 In contrast with myelin, gliosis is present only around the site of injury, and regrowing axons can simply detour around the glial scar to grow within undisrupted CNS tissue.65 However, several neurotrophic factors induce axons to regenerate through scar tissue64 or prevent scar tissue formation by inhibiting collagen biosynthesis and basement membrane deposition.66 Similarly, although the axon outgrowth of dorsal root ganglion neurons is inhibited by CSPG, the axons can be made insensitive to this inhibition by exposure to a number of different neurotrophic factors.67
CNTF and fibroblast growth factor-2 but not BDNF
Neurotrophins, such as CNTF, BDNF, and fibroblast growth factor-2, support neuron survival and axonal outgrowth during development. However, following an optic nerve crush in the adult frog, Rana pipiens, CNTF more than doubles the axon regeneration rate, fibroblast growth factor -2 increases regeneration, while BDNF has little effect.68
EphB3 axon guidance, promoting axon regeneration, and synapse formation
EphB3 proteins govern axon pathway development, including retinal axon projections to CNS targets. Thus, during development, EphB proteins serve as axon guidance molecules for growth cones of RGC axons.69 Retinal growth cone responsiveness to EphB3 is regulated by co-impinging signals from other axon guidance molecules involving the SCG10-mediated regulation of the growth cone microtubule cytoskeleton.70 The loss of EphB3 does not affect the ability of injured RGC axons to elaborate complex terminal branching, suggesting that additional EphB3-independent mechanisms govern adult axon branching triggered by CNS damage.71
Recruited macrophages upregulate mRNA, encoding the axon regeneration guidance molecule EphB3, and express EphB proteins capable of binding Ephrin B molecules in vivo and in vitro.71 Injured adult RGC axons in turn expressed EphrinB3, a known receptor for EphB3, and RGC axons bind recombinant EphB3 protein injected into the optic nerve. Thus, optic nerve-derived EphB3 is involved in regulating adult RGC axon plasticity after ONI.71
Exposing optic nerve axons to sciatic nerve cells and macrophages activates the macrophages and thus induces regeneration of injured optic nerve axons.72 These results are encouraging, but “vision” requires the establishment of a vast number of connections to provide more information than evoked potentials in the tectum, and achieving this goal has been elusive.
Peripheral nerve grafts promoting axon regeneration into the CNS
Some axon regeneration guidance molecules can be reexpressed in the adult,73 and in vivo studies show that regenerating RGC axons can restore physiological function when one end of a length of peripheral nerve is inserted into the brain CNS and the other into the spinal cord. This results in RGC axons regenerating many centimeters through the nerve graft and into the spinal cord and superior colliculus where the regenerating CNS axons connect to central visual relays and restore light and dark perception vision sensitivity74 by the establishment of synapses with appropriate superior colliculus target cells74 when light-evoked synaptic activity is elicited when light is shined into the eye,75 to partial restoration of the optomotor response, depth perception, and circadian photoentrainment, thus demonstrating the feasibility of reconstructing central circuitry for vision after optic nerve damage in mature mammals.76 Another example is the reestablishment of appropriate RGC axon tectal connections evoking the pupil constriction reflex when a peripheral nerve graft is connected to the pretectal nucleus in rats,77 indicating the reestablishment of functional connection with higher visual centers.78 The transplanted Schwann cells appear to induce modifications of functional properties in neighboring optic nerve glial cells. This is likely due to their release of a number of axon regeneration-promoting neurotrophic factors, such as nerve growth factor (NGF), BDNF, neurotrophic factor (NT)-3, NT-4/5, and NT-6.79 The implanted Schwann cells also induce the invasion of macrophages into the optic nerve, which is accomplished by their release of oncomodulin, a calcium-binding protein, and potent macrophage-derived growth factor with high affinity in a cAMP-dependent manner.80 In vivo delivery of oncomodulin stimulates up to a sevenfold increase in the extension of optic nerve axons versus other trophic agents.80
Astrocytes, which usually form a glial scar, appear to guide the regenerating axons in cooperation with Schwann cells.75 There is also a decrease in the oligodendrocyte marker O4 and migration of ED-1-positive macrophages into the optic nerve stump.75 Accordingly, the ability of RGC axons to regenerate is not a simple phenomenon of axonal regeneration along the Schwann cell membrane, but results from direct and dynamic communication between the axon and the Schwann cells are accompanied by changes in the glial cells induced by the Schwann cells.75
Bone marrow stem cells
The application of stem cells, such as bone marrow mesenchymal stem cells (BM-MSCs), can be used to replace lost neurons, restore neural circuits, or act as paracrine-mediated therapies in which stem cell-derived trophic factors protect compromised endogenous retinal neurons from death and induce the growth of new connections. BM contains a population of pluripotent cells that can differentiate into a variety of cell lineages, including neural cells that promote significant optic nerve axon regeneration and remyelination following systemic delivery of BM-MSCs to a site of contused spinal cord and in stroke models.81 Non retinal-derived adult stem cells, in particular neural stem cells, and dental pulp stem cells, together with embryonic stem cells (ESC)/pluripotent stem cells (iPSC), can provide trophic support, repair and replacement of retinal neurons, retinal pigment epithelial cells, and glia in degenerative retinal diseases.81 Thus, pluripotent stem cells ESCs/iPSCs and endogenous retinal stem cells may replace lost photoreceptors and retinal pigment epithelial cells leading to restoration of vision in the diseased eye.81 The MSCs may be a useful source of paracrine factors that protect RGCs and stimulate regeneration of their axons into the injured optic nerve.
Pathway selectivity: restoration of functional retinotopic maps
The greatest challenge in restoring vision is to assure that optic nerve axons take the correct pathway at the optic chiasm and then form the appropriate synaptic connections in the lateral geniculate nucleus. In the peripheral nervous system, regenerating motor axons show the capacity to accurately select between a presented motor and a sensory nerve.82 This appropriate reinnervation of the distal nerve bundle significantly increases the accuracy of neurological recovery. The precision of the selection of the appropriate distal nerve pathway can be significantly enhanced by electrical stimulation.83 Appropriate optic nerve axon pathway selectivity and optic chiasm specificity of regenerating lesioned RGC axons take place,65 indicating that RGC axons can make the appropriate major crossed pathway selection, preferentially regenerating within the degenerating trajectories from their own eye, with minor uncrossed projections into both the optic tracts.84 Electrical stimulation of the optic nerve may enhance axon regeneration and improve the specificity pathway selection to distal neuronal targets.
Olfactory ensheathing cells
Olfactory ensheathing cells (OECs) are glial cells of the first cranial nerve of the nasal mucosa where they accompany and myelinate the axons of the olfactory neurons. OECs exhibit phenotypic characteristics of both astrocytes and Schwann cells and show plasticity. In vitro, OECs promote axonal growth, while in vivo they promote remyelination of damaged axons.85 These OECs can serve as a source of myelinating glia that remyelinate axons in demyelinating diseases such as multiple sclerosis, which allows the axons to survive and regenerate within the CNS.86 Thus, OECs can serve clinically as an alternative to Schwann cells for the treatment of spinal cord injury and for promoting injured spinal cord axons.87 Additional cells that can evoke myelination and induce axon regeneration in animal models of demyelinaton are progenitor cells derived from the subventricular zone of the brain and from BM that can form myelin when transplanted into demyelinated lesions in rodents.88 Furthermore, OECs cultured from the olfactory lamina propria or pieces of lamina propria from the olfactory mucosa transplanted into the transected spinal cord promote regeneration of spinal cord axons and partial recovery of movement of hind limbs.89 Thus, transplanting OECs stimulate axonal regeneration and sprouting, increase remyelination, confer neuroprotection, enhance neovascularization, and replace lost RGCs.
Clinically, following resection of a glial scar associated with a spinal cord injury, cultured OECs transplanted into a spinal cord lesion promote axonal regeneration, reorganize the glial scar, remyelinate axons, stimulate blood vessel formation, have phagocytic properties, and modulate the immune response.90 When OECs are similarly implanted into spinal cord stumps and the spinal cord gap is bridged with lengths of autologous sural nerve, one patient recovered partial voluntary movements of the lower extremities, an increase of the muscle mass in the left thigh, and partial recovery of superficial and deep sensation.91 The simultaneous treatment of injuring adult rat optic nerve with OEC under the nerve sheaths and injecting CNTF into the vitreous body reduces apoptosis and induces optic nerve repair and axon regeneration.92
Lens injury promotes optic nerve regeneration
Lens injury induces an inflammatory response, which causes macrophage invasion of the vitreous humor. The invading macrophages appear to release neurotrophic factors that induce regeneration of injured RGC axons.93 Lens injury also promotes the recovery of vision when the optic nerve is cut and the stumps are anastomosed. Thus, natural events induced by lens injury can induce some axons of the optic nerve to regenerate and restore functional activation of the visual cortex.94 Part of this function of an injured lens is the activation of retinal glia and their release of CNTF and leukemia inhibitory factor ,which provide potent protection of axotomized RGCs against apoptosis while also promoting axon regeneration in the injured optic nerve.95
Novel technique for promoting regeneration of injured optic nerve axons
The clinical “gold standard” surgical technique used for restoring neurological function of a peripheral nerve with a gap when the nerve stumps cannot be anastomosed is to bridge the gap with sensory (sural) nerve grafts. The concept is that the Schwann cells within the graft will release a cocktail of neurotrophic and the other factors will support the survival of axotomized neurons, induce their regeneration across the gap in association with the nerve graft, and promote axons to regenerate into the distal portion of the nerve, into which they normally do not regenerate. However, such grafts have significant limitations and are not very effective for nerve gaps >2 cm, for repairs performed >2 months post trauma, and in patients >25 years of age. Although motor nerve grafts are more effective, they are not used due to ethical reasons of sacrificing a motor function.96
A recent clinical study showed that bridging nerve with a length of sensory nerve graft within a collagen tube filled with autologous platelet-rich plasma (PRP) or even with a collagen tube containing only PRP induces axons to regenerate under conditions where other technique is effective: across nerve gaps up to 16 cm in length, for repairs performed >3 years post-trauma, and in patients up to 58 years of age.97 Figure 1 shows an example of an 8-cm long peripheral nerve gap repaired with a collagen tube filled with PRP.
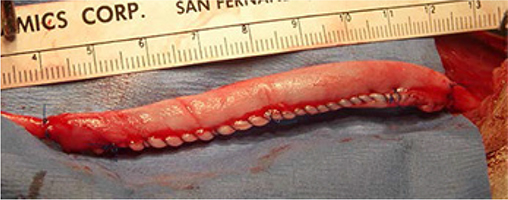 | Figure 1 An 8-cm long peipheral nerve gap bridged with a collagen tube filled with PRP. Note: Axons regenerated entirely across the gap, through the distal nerve, and reestablished good neurological function. Abbreviation: PRP, platelet-rich plasma. |
It is hypothesized that these techniques are effective because the PRP provides an abundance of axon regeneration-promoting factors that are taken up by the severed axons and transported to their soma where they trigger neurons to extend axons they would otherwise not extend, significantly increase the distance axons regenerate, and promote the axons to regenerate into the distal nerve stump, which is normally inhibitory to axon in regeneration. It is reasonable to assume that such techniques would be effective in promoting optic nerve regeneration.
The success in inducing large number of axons to regenerate long distances when a nerve graft within a collagen tube filled with PRP is used to bridge a peripheral nerve gap, suggests that this approach would induce similar axon regeneration by RGCs leading to the reestablishment of vision. Figure 2 presents the proposed technique.
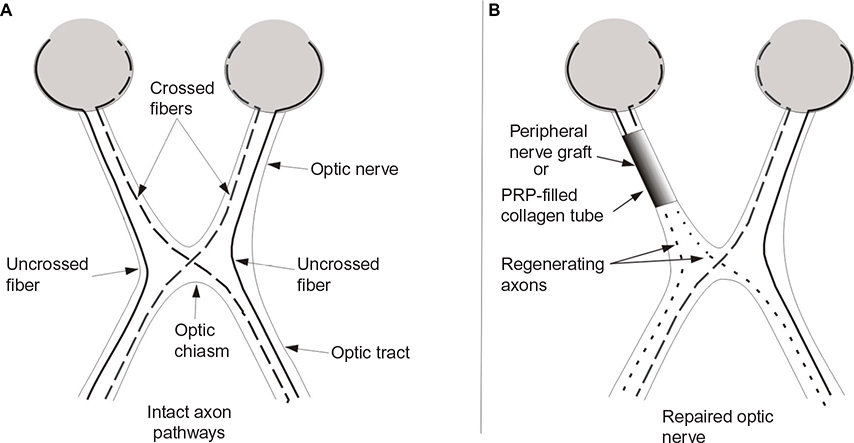 | Figure 2 Schematic representation of RGC axon pathways. Notes: (A) Intact RGC axon pathways. (B) Transected optic nerve stumps secured a collagen tube filled with autologous PRP to provide neurotrophic factors to promote RGC survival and axon regeneration and enhance specificity of target reinnervation. Abbreviations: PRP, platelet-rich plasma; RGC, retinal ganglion cell. |
The advantage of this technique is that the collagen tube prevents the invasion of fibroblasts that can form scar tissue that inhibits axon regeneration, while PRP provides the transected RGCs a host of neurotrophic and regeneration-promoting factors that can promote axon regeneration that wound not otherwise occur, enhance the extent of that regeneration, and block the factors in the distal optic nerve that inhibit axon regeneration, thereby creating a cellular environment that strongly promotes axon regeneration and restoration of functional vision.
Conclusion
Although in lower vertebrates RGCs survive optic nerve trauma and the optic nerve axons regenerate and vision can be restored, in higher vertebrates RGC survival and axon regeneration are poor to nonexistent. The underlying difference is that in higher vertebrates there is 1) an absence of factors to promote RGC survival, 2) a presence of factors that inhibit axon regeneration, and 3) an absence of factors to promote axon regeneration. Recovery of vision in higher vertebrates requires changing the cellular environment from one that inhibits to one that promotes axon regeneration. Although many mechanisms have been tested in higher vertebrates for their ability to restore vision, none has provided more than minimal success with the greatest being using peripheral nerve grafts. Although a peripheral nerve graft alone may not be sufficient for promoting optic nerve axon regeneration and restoring vision, bridging an optic nerve gap with a peripheral nerve graft within a collagen tube filled with PRP may create the cellular environment in which regeneration is successful in inducing neurological recovery.
Acknowledgments
No funding was involved in the production of this paper.
Disclosure
The authors declare no conflicts of interest in this work.
References
Tielsch JM, Katz J, Singh K, Quigley HA, Gottsch JD, Javitt J, Sommer A. A population-based evaluation of glaucoma screening: the Baltimore Eye Survey. Am J Epidemiol. 1991;134:1102–1110. | ||
Quigley HA, Vitale S. Models of open-angle glaucoma prevalence and incidence in the United States. Invest Ophthalmol Vis Sci. 1997;38:83–91. | ||
Quigley HA. Number of people with glaucoma worldwide. Br J Ophthalmol. 1996;80:389–393. | ||
Odberg T, Riise D. Early diagnosis of glaucoma. The value of successive stereophotography of the optic disc. Acta Ophthalmol (Copenh). 1985;63:257–263. | ||
Balashova LM, Saksonova EO, Zaitseva NS, Slepova OS, Teplinskaia LE, Il’nitskii VV, Grishin VL. [Role of immunological factors in peripheral vitreo-chorioretinal dystrophies and macular ruptures of the retina]. Vestn Oftalmol. 1995;111:16–18. | ||
Kato K, Sasaki N, Matsunaga S, Nishimura R, Ogawa H. Incidence of canine glaucoma with goniodysplasia in Japan : a retrospective study. J Vet Med Sci. 2006;68:853–858. | ||
Congdon N, O’Colmain B, Klaver CC, et al. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004;122:477–485. | ||
Wilson MR, Eezzuduemhoi DR. Ophthalmologic disorders in minority populations. Med Clin North Am. 2005;89:795–804. | ||
Cunningham ET Jr, Lietman TM, Whitcher JP. Blindness: a global priority for the twenty-first century. Bull World Health Organ. 2001;79(3):180. | ||
Harada T, Harada C, Nakamura K et al. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J Clin Invest. 2007;117:1763–1770. | ||
Adlina AR, Alisa-Victoria K, Shatriah I, Liza-Sharmini AT, Ahmad MS. Optic disc topography in Malay patients with normal-tension glaucoma and primary open-angle glaucoma. Clin Ophthalmol. 2014;8:2533–2539. | ||
Adlina AR, Shatriah I, Liza Sharmini AT, Ahmad MS. Optic disc topography of normal tension glaucoma patients in Malaysia. Med J Malaysia. 2013;68(4):338–342. | ||
Meyer RL, Sperry RW. Tests for neuroplasticity in the anuran retinotectal system. Exp Neurol. 1973;40(2):525–539. | ||
Fujisawa H, Tani N, Watanabe K, Ibata Y. Branching of regenerating retinal axons and preferential selection of appropriate branches for specific neuronal connection in the newt. Dev Biol. 1982;90(1):43–57. | ||
Vajda F, Jordi N, Dalkara D, et al. Cell type-specific Nogo-A gene ablation promotes axonal regeneration in the injured adult optic nerve. Cell Death Differ. 2015;22(2):323–335. | ||
Chen DF, Schneider GE, Martinou JC, Tonegawa S. Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature. 1997;385(6615):434–439. | ||
Aguayo AJ, Clarke DB, Jelsma TN, Kittlerova P, Friedman HC, Bray GM. Effects of neurotrophins on the survival and regrowth of injured retinal neurons. Ciba Found Symp. 1996;196:135–144; discussion 144–138. | ||
Thanos S, Thiel HJ. Mechanisms governing neuronal degeneration and axonal regeneration in the mature retinofugal system. J Cell Sci Suppl. 1991;15:125–134. | ||
Lefrancois T, Fages C, Peschanski M, Tardy M. Neuritic outgrowth associated with astroglial phenotypic changes induced by antisense glial fibrillary acidic protein (GFAP) mRNA in injured neuron-astrocyte cocultures. J Neurosci. 1997;17(11):4121–4128. | ||
Omura T, Omura K, Tedeschi A, et al. Robust axonal regeneration occurs in the injured CAST/Ei mouse CNS. Neuron. 2015;86(5):1215–1227. | ||
Hall SM, Kent AP. The response of regenerating peripheral neurites to a grafted optic nerve. J Neurocytol. 1987;16:317–331. | ||
de Lima S, Habboub G, Benowitz LI. Combinatorial therapy stimulates long-distance regeneration, target reinnervation, and partial recovery of vision after optic nerve injury in mice. Int Rev Neurobiol. 2012;106:153–172. | ||
Cen LP, Luo JM, Geng Y, Zhang M, Pang CP, Cui Q. Long-term survival and axonal regeneration of retinal ganglion cells after optic nerve transection and a peripheral nerve graft. Neuroreport. 2012;23:692–697. | ||
Benowitz LI, He Z, Goldberg JL. Reaching the brain: Advances in optic nerve regeneration. Exp Neurol. 2015;15:30141–30142. | ||
Benowitz LI, He Z, Goldberg JL. Reaching the brain: advances in optic nerve regeneration. Exp Neurol. Epub 2015 Dec 31. | ||
Nucci C, Tartaglione R, Rombola L, Morrone LA, Fazzi E, Bagetta G. Neurochemical evidence to implicate elevated glutamate in the mechanisms of high intraocular pressure (IOP)-induced retinal ganglion cell death in rat. Neurotoxicology. 2005;26(5):935–941. | ||
Li Y, Schlamp CL, Poulsen GL, Jackson MW, Griep AE, Nickells RW. p53 regulates apoptotic retinal ganglion cell death induced by N-methyl-D-aspartate. Mol Vis. 2002;8:341–350. | ||
Guo L, Salt TE, Maass A, Luong V, Moss SE, Fitzke FW, Cordeiro MF. Assessment of neuroprotective effects of glutamate modulation on glaucoma-related retinal ganglion cell apoptosis in vivo. Invest Ophthalmol Vis Sci. 2006;47(2):626–633. | ||
Weibel D, Kreutzberg GW, Schwab ME. Brain-derived neurotrophic factor (BDNF) prevents lesion-induced axonal die-back in young rat optic nerve. Brain Res. 1995;679(2):249–254. | ||
McKerracher L, Essagian C, Aguayo AJ. Marked increase in beta-tubulin mRNA expression during regeneration of axotomized retinal ganglion cells in adult mammals. J Neurosci. 1993;13(12):5294–5300. | ||
Fournier AE, Beer J, Arregui CO, Essagian C, Aguayo AJ, McKerracher L. Brain-derived neurotrophic factor modulates GAP-43 but not T alpha1 expression in injured retinal ganglion cells of adult rats. J Neurosci Res. 1997;47(6):561–572. | ||
Nakazawa T, Shimura M, Endo S, Takahashi H, Mori N, Tamai M. N-Methyl-D-aspartic acid suppresses Akt activity through protein phosphatase in retinal ganglion cells. Mol Vis. 2005;11:1173–1182. | ||
Fan W, Agarwal N, Kumar MD, Cooper NG. Retinal ganglion cell death and neuroprotection: Involvement of the CaMKIIalpha gene. Brain Res Mol Brain Res. 2005;139(2):306–316. | ||
Harada C, Azuchi Y, Noro T, Guo X, Kimura A, Namekata K, Harada T. TrkB Signaling in Retinal Glia Stimulates Neuroprotection after Optic Nerve Injury. Am J Ophthal. 2015;85:3238–3247. | ||
Harada C, Guo X, Namekata K et al. (2011). Glia- and neuron-specific functions of TrkB signalling during retinal degeneration and regeneration. Nat Commun. 2011;2:189. | ||
Cho KS, Yang L, Lu B, Feng Ma H, Huang X, Pekny M, Chen DF. Re-establishing the regenerative potential of central nervous system axons in postnatal mice. J Cell Sci. 2005;118(Pt 5):863–872. | ||
Johnson AR. Contact inhibition in the failure of mammalian CNS axonal regeneration. Bioessays. 1993;15(12):807–813. | ||
Mansour H, Asher R, Dahl D, Labkovsky B, Perides G, Bignami A. Permissive and non-permissive reactive astrocytes: immunofluorescence study with antibodies to the glial hyaluronate-binding protein. J Neurosci Res. 1990;25(3):300–311. | ||
Canning DR, Hoke A, Malemud CJ, Silver J. A potent inhibitor of neurite outgrowth that predominates in the extracellular matrix of reactive astrocytes. Int J Dev Neurosci. 1996;14(3):153–175. | ||
Joly S, Jordi N, Schwab ME, Pernet V. The Ephrin receptor EphA4 restricts axonal sprouting and enhances branching in the injured mouse optic nerve. Eur J Neurosci. 2014;40(7):3021–3031. | ||
Caroni P, Schwab ME. Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron. 1988;1(1):85–96. | ||
Wang X, Hasan O, Arzeno A, Benowitz LI, Cafferty WB, Strittmatter SM. Axonal regeneration induced by blockade of glial inhibitors coupled with activation of intrinsic neuronal growth pathways. Exp Neurol. 2012;237(1):55–69. | ||
Welte CS, Stuermer CA. Upregulation of the zebrafish Nogo-A homologue, Rtn4b, in retinal ganglion cells is functionally involved in axon regeneration. Neural Dev. 2015;10:6. | ||
Goldberg JL, Vargas ME, Wang JT, Mandemakers W, Oster SF, Sretavan DW, Barres BA. An oligodendrocyte lineage-specific semaphorin, Sema5A, inhibits axon growth by retinal ganglion cells. J Neurosci. 2004;24(21):4989–4999. | ||
Holmberg E, Nordstrom T, Gross M, Kluge B, Zhang SX, Doolen S. Simvastatin promotes neurite outgrowth in the presence of inhibitory molecules found in central nervous system injury. J Neurotrauma. 2006;23(9):1366–1378. | ||
Parsadanian AS, Cheng Y, Keller-Peck CR, Holtzman DM, Snider WD. Bcl-xL is an antiapoptotic regulator for postnatal CNS neurons. J Neurosci. 1998;18(3):1009–1019. | ||
Malik JM, Shevtsova Z, Bahr M, Kugler S. Long-term in vivo inhibition of CNS neurodegeneration by Bcl-XL gene transfer. Mol Ther. 2005;11(3):373–381. | ||
Zhang F, Ma XL, Wang YX, et al. TPEN, a specific Zn chelator, inhibits sodium dithionite and glucose deprivation (SDGD)-induced neuronal death by modulating apoptosis, glutamate signaling, and voltage-gated k and na channels. Cell Mol Neurobiol. Epub 2016 Mar 16. | ||
Koch JC, Solis GP, Bodrikov V, et al. Upregulation of reggie-1/flotillin-2 promotes axon regeneration in the rat optic nerve in vivo and neurite growth in vitro. Neurobiol Dis. 2013;51:168–176. | ||
Lehmann M, Fournier A, Selles-Navarro I, et al. Inactivation of rho signaling pathway promotes CNS axon regeneration. J Neurosci. 1999;19(17):7537–7547. | ||
Monsul NT, Geisendorfer AR, Han PJ, Banik R, Pease ME, Skolasky RL Jr, Hoffman PN. Intraocular injection of dibutyryl cyclic AMP promotes axon regeneration in rat optic nerve. Exp Neurol. 2004;186(2):124–133. | ||
Wang H, Liu ZL, Zhuang XT, Wang MF, Xu L. Neuroprotective effect of recombinant human erythropoietin on optic nerve injury in rats. Chin Med J (Engl). 2009;122(7):2008–2012. | ||
Thanos S, Mey J, Wild M. Treatment of the adult retina with microglia-suppressing factors retards axotomy-induced neuronal degradation and enhances axonal regeneration in vivo and in vitro. J Neurosci. 1993;13(2):455–466. | ||
Schuetz E, Rose K, Thanos S. Regeneration of ganglion cell axons into a peripheral nerve graft alters retinal expression of glial markers and decreases vulnerability to re-axotomy. Restor Neurol Neurosci. 2003;21(1–2):11–18. | ||
Li S, Hu B, Tay D, So KF, Yip HK. Intravitreal transplants of Schwann cells and fibroblasts promote the survival of axotomized retinal ganglion cells in rats. Brain Res. 2004;1029(1):56–64. | ||
Kato S, Matsukawa T, Koriyama Y, Sugitani K, Ogai K. A molecular mechanism of optic nerve regeneration in fish: the retinoid signaling pathway. Prog Retin Eye Res. 2013;37:13–30. | ||
Kurimoto T, Yin Y, Habboub G, et al. Neutrophils express oncomodulin and promote optic nerve regeneration. J Neurosci. 2013;33(37):14816–14824. | ||
Koriyama Y. Role of protein S-nitrosylation in central nervous system survival and regeneration. Yakugaku Zasshi. 2013;133(8):843–848. | ||
Manabe S, Gu Z, Lipton SA. Activation of matrix metalloproteinase-9 via neuronal nitric oxide synthase contributes to NMDA-induced retinal ganglion cell death. Invest Ophthalmol Vis Sci. 2005;46(12):4747–4753. | ||
Satarian L, Javan M, Kiani S, Hajikaram M, Mirnajafi-Zadeh J, Baharvand H. Engrafted human induced pluripotent stem cell-derived anterior specified neural progenitors protect the rat crushed optic nerve. PLoS One. 2013;8(8):e71855. | ||
Lenkowski JR, Qin Z, Sifuentes CJ, Thummel R, Soto CM, Moens CB, Raymond PA. Retinal regeneration in adult zebrafish requires regulation of TGFbeta signaling. Glia. 2013;61(10):1687–1697. | ||
Fujita Y, Sato A, Yamashita T. Brimonidine promotes axon growth after optic nerve injury through Erk phosphorylation. Cell Death Dis. 2013;4:e763. | ||
Sengottuvel V, Leibinger M, Pfreimer M, Andreadaki A, Fischer D. Taxol facilitates axon regeneration in the mature CNS. J Neurosci. 2011;31(7):2688–2699. | ||
Lu P, Jones LL, Tuszynski MH. Axon regeneration through scars and into sites of chronic spinal cord injury. Exp Neurol. 2007;203(1):8–21. | ||
MacLaren RE, Taylor JS. Chiasmatic specificity in the regenerating mammalian optic nerve. Exp Neurol. 1997;147(2):279–286. | ||
Klapka N, Hermanns S, Straten G, et al. Suppression of fibrous scarring in spinal cord injury of rat promotes long-distance regeneration of corticospinal tract axons, rescue of primary motoneurons in somatosensory cortex and significant functional recovery. Eur J Neurosci. 2005;22(12):3047–3058. | ||
Castro C, Kuffler DP. Membrane-bound CSPG mediates growth cone outgrowth and substrate specificity by Schwann cell contact with the DRG neuron cell body and not via growth cone contact. Exp Neurol. 2006;200(1):19–25. | ||
Vega-Melendez GS, Blagburn JM, Blanco RE. Ciliary neurotrophic factor and fibroblast growth factor increase the speed and number of regenerating axons after optic nerve injury in adult Rana pipiens. J Neurosci Res. 2014;92(1):13–23. | ||
Birgbauer E, Oster SF, Severin CG, Sretavan DW. Retinal axon growth cones respond to EphB extracellular domains as inhibitory axon guidance cues. Development. 2001;128(5):3041–3048. | ||
Tuszynski MH, Gabriel K, Gage FH, Suhr S, Meyer S, Rosetti A. Nerve growth factor delivery by gene transfer induces differential outgrowth of sensory, motor, and noradrenergic neurites after adult spinal cord injury. Exp Neurol. 1996;137(1):157–173. | ||
Liu X, Hawkes E, Ishimaru T, Tran T, Sretavan DW. EphB3: an endogenous mediator of adult axonal plasticity and regrowth after CNS injury. J Neurosci. 2006;26(12):3087–3101. | ||
Lazarov-Spiegler O, Solomon AS, Zeev-Brann AB, Hirschberg DL, Lavie V, Schwartz M. Transplantation of activated macrophages overcomes central nervous system regrowth failure. Faseb J. 1996;10(11):1296–1302. | ||
Bahr M, Bonhoeffer F. Perspectives on axonal regeneration in the mammalian CNS. Trends Neurosci. 1994;17(11):473–479. | ||
Fukuda Y, Watanabe M, Sawai H, Miyoshi T. Functional recovery of vision in regenerated optic nerve fibers. Vision Res. 1998;38(10):1545–1553. | ||
Dezawa M, Adachi-Usami E. Role of Schwann cells in retinal ganglion cell axon regeneration. Prog Retin Eye Res. 2000;19(2):171–204. | ||
de Lima S, Koriyama Y, Kurimoto T, et al. Full-length axon regeneration in the adult mouse optic nerve and partial recovery of simple visual behaviors. Proc Natl Acad Sci U S A. 2012;109(23):9149–9154. | ||
Thanos S. Adult retinofugal axons regenerating through peripheral nerve grafts can restore the light-induced pupilloconstriction reflex. Eur J Neurosci. 1992;4(8):691–699. | ||
Thanos S, Naskar R, Heiduschka P. Regenerating ganglion cell axons in the adult rat establish retinofugal topography and restore visual function. Exp Brain Res. 1997;114(3):483–491. | ||
Yin Q, Kemp GJ, Frostick SP. Neurotrophins, neurones and peripheral nerve regeneration. J Hand Surg [Br]. 1998;23(4):433–437. | ||
Yin Y, Henzl MT, Lorber B, et al. Oncomodulin is a macrophage-derived signal for axon regeneration in retinal ganglion cells. Nat Neurosci. 2006;9:843–852. | ||
Mead B, Berry M, Logan A, Scott RA, Leadbeater W, Scheven BA. Stem cell treatment of degenerative eye disease. Stem Cell Res. 2015;14(3):243–257. | ||
Brushart TM, Seiler WA 4th. Selective reinnervation of distal motor stumps by peripheral motor axons. Exp Neurol. 1987;97(2):289–300. | ||
Brushart TM, Jari R, Verge V, Rohde C, Gordon T. Electrical stimulation restores the specificity of sensory axon regeneration. Exp Neurol. 2005;194(1):221–229. | ||
Berry M, Carlile J, Hunter A, Tsang W, Rosenstiel P, Sievers J. Optic nerve regeneration after intravitreal peripheral nerve implants: trajectories of axons regrowing through the optic chiasm into the optic tracts. J Neurocytol. 1999;28(9):721–741. | ||
Pellitteri R, Cova L, Zaccheo D, Silani V, Bossolasco P. Phenotypic modulation and neuroprotective effects of olfactory ensheathing cells: a promising tool for cell therapy. Stem Cell Rev. 2015;12(2):224–234. | ||
Azimi Alamouti M, Bakhtiyari M, Moradi F, Mokhtari T, Hedayatpour A, Zafari F, Barbarestani M. Remyelination of the corpus callosum by olfactory ensheathing cell in an experimental model of multiple sclerosis. Acta Medica Iranica. 2015;53(9):533–539. | ||
Boyd JG, Skihar V, Kawaja M, Doucette R. Olfactory ensheathing cells: historical perspective and therapeutic potential. Anat Rec B New Anat. 2003;271(1):49–60. | ||
Kocsis JD, Akiyama Y, Lankford KL, Radtke C. Cell transplantation of peripheral-myelin-forming cells to repair the injured spinal cord. J Rehabil Res Dev. 2002;39(2):287–298. | ||
Lu J, Feron F, Ho SM, Mackay-Sim A, Waite PM. Transplantation of nasal olfactory tissue promotes partial recovery in paraplegic adult rats. Brain Res. 2001;889(1–2):344–357. | ||
Roet KC, Verhaagen J. Understanding the neural repair-promoting properties of olfactory ensheathing cells. Exp Neurol. 2014;261:594–609. | ||
Tabakow P, Raisman G, Fortuna W, et al. Functional regeneration of supraspinal connections in a patient with transected spinal cord following transplantation of bulbar olfactory ensheathing cells with peripheral nerve bridging. Cell Transplant. 2014;23(12):1631–1655. | ||
Yin DP, Liu L, Cao L. [Synergetic effects of ciliary neurotrophic factor and olfactory ensheathing cells on optic nerve reparation]. Zhonghua Yan Ke Za Zhi. 2013;49(11):1020–1028. | ||
Yin Y, Cui Q, Li Y, Irwin N, Fischer D, Harvey AR, Benowitz LI. Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci. 2003;23(6):2284–2293. | ||
Heiduschka P, Fischer D, Thanos S. Recovery of visual evoked potentials after regeneration of cut retinal ganglion cell axons within the ascending visual pathway in adult rats. Restor Neurol Neurosci. 2005;23(5–6):303–312. | ||
Heiduschka P, Renninger D, Fischer D, Muller A, Hofmeister S, Schraermeyer U. Lens injury has a protective effect on photoreceptors in the RCS rat. ISRN Ophthalmol. 2013;12:814814. | ||
Keirstead SA, Vidal-Sanz M, Rasminsky M, Aguayo AJ, Levesque M, So KF. Responses to light of retinal neurons regenerating axons into peripheral nerve grafts in the rat. Brain Res. 1985;359(1–2):402–406. | ||
Kuffler DP, Reyes O, Sosa IJ, Santiago-Figueroa J. Neurological recovery across a 12-cm-long ulnar nerve gap repaired 3.25 years post trauma: case report. Neurosurgery. 2011;69(6):E1321–E1326. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.