Back to Journals » Journal of Inflammation Research » Volume 17
BAP31 Promotes Adhesion Between Endothelial Cells and Macrophages Through the NF-κB Signaling Pathway in Sepsis
Authors He J ![]() , Jing D, Zhao S, Duan M
, Jing D, Zhao S, Duan M ![]()
Received 14 November 2023
Accepted for publication 20 February 2024
Published 26 February 2024 Volume 2024:17 Pages 1267—1279
DOI https://doi.org/10.2147/JIR.S448091
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Jiawei He,* Danyang Jing,* Shen Zhao, Meili Duan
Department of Critical Care Medicine, Beijing Friendship Hospital, Capital Medical University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shen Zhao; Meili Duan, Department of Critical Care Medicine, Beijing Friendship Hospital, Capital Medical University, Betijing, 100050, People’s Republic of China, Tel +86-15960008819 ; +86-18811695963, Email [email protected]; [email protected]
Purpose: To investigate the role of B cell receptor associated protein 31 (BAP31) in the pathogenesis of sepsis.
Methods: Cecal ligation and puncture (CLP)-induced C57BL/6J mice, and LPS-challenged endothelial cells (HUVECs) were established to mimic a sepsis animal model and a sepsis cell model, respectively. Cre/LoxP and shRNA methods were used for BAP31 knockdown in vivo and in vitro respectively. Neutrophils/macrophages-endothelial cocultures were used to evaluate neutrophils or macrophages infiltration and adhesion to endothelial cells. Cox proportional hazards model was used to evaluate the survival time of mice. Western blotting (WB) and Quantitative real-time polymerase chain reaction (qRT-PCR) were used to detect toll-like receptor (TLR) signaling pathway, transforming growth factor β activated kinase 1 (TAK1) signaling pathway and phosphoinositide-3 kinases-protein kinase B (PI3K/AKT) signaling pathway.
Results: Deletion of BAP31 reduced CLP-induced mortality of mice, histological damage with less interstitial edema, and neutrophils and macrophages infiltration. IHC and IF showed that BAP31 knockdown significantly decreases the expressions of ICAM1 and VCAM1 both in vivo and in vitro. Coculture showed that LPS-induced neutrophils or macrophages adhesion to endothelial cells was significantly weakened in BAP31 knockdown cells. In addition, BAP31 knockdown of endothelial cells decreased the expression of CD80 and CD86 on the surface of macrophages as well as interleukin 1β (IL-1β) and tumor necrosis factor α (TNF-α) during sepsis. Mechanistically, LPS-induced the activation of TLR4, MyD88 and TRAF6, and the phosphorylation of TAK1, PI3K, AKT, IκBα and IKKα/β, resulting in activation of nuclear factor kappa B (NF-κB) p65 in endothelial cells. However, BAP31 knockdown significantly reversed the expressions of associated proteins.
Conclusion: BAP31 up-regulated the expressions of ICAM1 and VCAM1 in endothelial cells leading to sepsis-associated organ injury. This may be involved in activation of TLR signaling pathway, TAK1 pathway, and PI3K-AKT signaling pathway.
Keywords: BAP31, sepsis, macrophages, endothelial cells, NF-κB
Introduction
Sepsis triggered by an infective agent contributes to a systemic, deleterious inflammatory host response resulting in multi-organ dysfunction.1 Infiltration of immune cells and activation of inflammatory signaling cascades are critical in the pathogenesis of sepsis.2 Immune cells, such as neutrophils and mononuclear macrophages, contact with endothelial cells (EC) to fulfill subsequent adhesion and infiltration in the process of inflammation.3 Meanwhile, the intercellular cell adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1) on EC rapidly mediate immune cell recruitment.4 However, the cellular and molecular mechanisms to ensure rapid encounter and infiltration between immune cell and endothelial cell in sepsis is undefined.
Previous studies demonstrated that pathogen associated molecular pattern (PAMP), such as lipopolysaccharide (LPS), could identify surface receptors including Toll like receptor 4 (TLR4) on EC.5 Next, myeloid differentiation primary response 88 and tumor necrosis factor (TNF) receptor associated factor 6 (MyD88/TRAF6) binding-dependent TLR4 signaling triggered p65 acetylation or phosphorylation, resulting in activation of nuclear factor kappa B (NF-κB) signal pathway in the EC.6 Following translocation to the nucleus, p65 bound to the promoters of ICAM1 and VCAM1 inducing the latter transcription.7 NF-κB is a known targets of both TAK1 pathway and PI3K/AKT pathway which controls inflammation and apoptosis.8 However, either TAK1 pathway or PI3K/AKT pathway mediates LPS-induced TLR4/NF-kB pathway in EC remain to be elucidated.
TLR4 is one of the receptors for communication between immune cells and endothelial cells, which is an important part of the inflammation in sepsis.9 Recent research has been focus on B cell receptor associated protein 31 (BAP31), an endoplasmic reticulum associated protein, in the field of immunity and inflammation.10 BAP31 functions as a carrier protein to facilitate the transport of transmembrane proteins. For example, BAP31 can regulate cell surface adhesion molecules, such as cluster of differentiation 9 (CD9), lymphocyte function associated antigen 1 (LFA1), macrophage antigen 1 (MAC1), and CD81.11,12 However, it is unclear whether BAP31 could regulate TLR-mediated ICAM1 and VCAM1 activation on EC. Thus, the goal of this study was to investigate the effect of BAP31 on regulating the interaction between EC and immune cells via ICAM1 and VCAM1 during sepsis. Further investigation into the underlying mechanism was to show that this process involves TLR4/MyD88/TRAF6 signal, then either TAK1 or PI3K/AKT pathway, and ultimate NF-kB pathway.
Materials and Methods
Animals
In this study, 6 to 8 week old C57BL/6J male mice were used, in which the control group was operated by sham operation, and the experimental group was used to establish sepsis model with cecal ligation and puncture (CLP). Three mice in each group were assigned to extract blood tissue and other samples, and euthanasia was given 24 hours after the establishment of the model. Cre/LoxP system was used to construct BAP31 deletion mice in TIE2 targeted knockdown EC. The BAP31 floxed mice (BAP31flox/flox) were bred with transgenic C57BL/6 mice carrying a cre recombinase and TIE2 promotor. Then, the mice with a special deletion of BAP31 in EC were acquired by mating BAP31flox/flox TIE2 Cre (BAP31−/−) mice strain with BAP31flox/flox (BAP31+/+) mice strain. The genotype of the mice was analyzed by PCR of tail DNA using primers: BAP31 primers: sense 5′-AAGGGGAGCCAGGAATAGTGGTG-3′; antisense 5′-TCCTGGCAGTTTCCAGTAAGGGTAAC-3′; Cre primers: sense 5′-GCGGTCTGGCAGTAAAAACTATC-3′; antisense 5′-GTGAAACAGCATTGCTGTCACTT-3′. CD31 positive EC were extracted from mouse inferior vena cava by flow cytometry and verified by BAP31. All mice were fed in specific pathogen free facilities and received good artificial feeding and care.
Histopathology
The brain, lung, heart, liver and kidney of mice were stained with Hematoxylin and eosin (HE) for analysis under light microscope. Firstly, the tissue was embedded and fixed in paraffin, and then the tissue was sliced with a paraffin slicer with a thickness of about 5um. The cut tissue was fully dewaxed in xylene, and then soaked in 100%, 95%, 85% and 70% ethanol to achieve full hydration. The hydrated tissue samples were dehydrated in ethanol and xylene with hematoxylin and eosin stains that had been prepared in advance. Finally, neutral gum was used to seal the film to facilitate preservation and observation. Three independent pathologists take a quantitative score of tissue injury. According to the degree of parenchymal cell injury, the number of inflammatory cells infiltrated in the stroma and whether there was edema around the blood vessels, the tissue injury score was divided into 5 grades: 0: no obvious abnormality; 1: mild abnormality; 2: moderate abnormality; 3: severe abnormality; 4: extremely severe abnormality.
Immunohistochemistry
We also extracted the vascular tissue of mice to evaluate the expression of ICAM1 and VCAM1. Vascular tissue fixation and other steps are described earlier, and finally antigen repair is carried out in accordance with the instructions. The first antibody working solution of ICAM1 (ab171123; Abcam) or VCAM1 (ab134047; Abcam) was stored overnight at 4°C, and the corresponding second antibody working solution was stored at 37°C for 1 hour. After completing the above steps, seal the sheet with a clean cover slide so that it can be preserved and observed under an optical microscope.
Cell Culture
We extracted EC from inferior vena cava from BAP31 knockdown mice and purchased human umbilical vein EC (HUVEC) from American Type Culture Collection (ATCC). The two kinds of cells were cultured in ECM (CM-0122; Procell), which contained Ham’sF-12K, 0.1mg/mL heparin, 0.05mg/mL endothelial cell growth supplement (ECG), 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). We also divided HUVEC into the control group and the experimental group, in which the experimental group added LPS of 1ug/mL and the control group added corresponding dose of phosphate buffered solution (PBS) to observe the effect of LPS on cells. In addition, we also purchased neutrophils (CP-M150; Procell), Raw264.7 (CL-0190; Procell), THP-1 (CL-0233; Procell) and their corresponding media. PMA was added after the THP-1 cells density increased to 1 × 106 so that the concentration of PMA was 50 ng/mL. We also extracted a small number of neutrophils from the peripheral blood of healthy volunteers for the purpose of the experiment (P9402; Solarbio). For more information on volunteers, please refer to Supplementary Table S1. Since neutrophils are terminal cells and are not suitable for long-term culture, cells other than neutrophils are placed in a cell incubator containing 5%CO2 at 37°C.
Cell Transfection
Lentiviral particles containing shNC and shBAP31 were transfected into HUVEC and cultured in cell incubator for 24 hours. BAP31 shRNA sequence sense 5′- CCGGTACACGTTCAGAATGCGTTTGCTCGAGCAAACGCATTCTGAACGTGTATTTTTG-3′ and antisense 5′-AATTCAAAAATACACGTTCAGAATGCGTTTCTCGAGCAAACGCATTCTGAACGTGTA-3′. After screening according to the instructions, further amplification was carried out, and the efficiency of knockdown was evaluated by quantitative real time PCR (qRT-PCR) and Western blotting (WB).
Immunofluorescence
A suitable size cover slide is added to the cell culture plate to enable the cell to crawl to its surface. The cells were fixed with 4% paraformaldehyde at room temperature, and then the first antibody working solution added with ICAM1 (ab171123; Abcam) or VCAM1 (ab134047; Abcam) was stored overnight at 4°C, and the corresponding secondary antibody working solution was stored at 37°C for 1h. Finally, PBS was used to remove the excess working liquid so that it could be observed under fluorescence microscope.
Cell Co-Culture
Neutrophils, Raw264.7 and THP-1 were added to Hoechst staining solution according to the instructions, so that cells in normal stock state could be labeled. After that, neutrophils, Raw264.7 and THP-1 were co-cultured with corresponding EC, and the changes of adhesion were evaluated by observing the fluorescence-labeled cells under fluorescence microscope.
Enzyme Linked Immunosorbent Assay (ELISA)
We tested ICAM1 (ab174445; Abcam; ab100688; Abcam), VCAM1 (ab174445; Abcam; ab100750; Abcam), TnT (E-EL-M1801c; Elabscience), TnI (E-EL-M1203c; Elabscience), AST (ab263882; Abcam), ALT (ab282882; Abcam), CR (aMBS3805641; Biocompare), BUN (MBS2611085; Biocompare), IL-1β (PI301; Beyotime; PI305; Beyotime), TNF-α (PT512; Beyotime; PT518; Beyotime), IL-10 (PI528; Beyotime; PI522; Beyotime) and TGF-β (PT878; Beyotime; PT880; Beyotime) according to the instructions provided by the reagent manufacturer.
Quantitative Real Time PCR (qRT-PCR)
Total RNA was extracted from different samples using TRIzol reagent (15,596,026; Invitrogen). After isolation, RNA concentration and quality were determined. Two microgram of total RNA was reverse transcribed to single strand cDNA using universal RT-PCR Kit (RP1100; Solarbio). Quantitative real time PCR was conducted using SYBR Green PCR Mastermix (SR1110; Solarbio) following the manufacturer’s instructions. The amplification condition was at 95°C for 10min, followed by 40 cycles of amplification that 95°C for 10s, 58°C for 30s, 72°C for 30s, and then plate reading and melting 65–95°C with plate readings every 0.5°C. The relative change in gene expression was analyzed by the 2-ΔΔCt method from triplicate determinations using β-actin as a housekeeping gene. Sequences of primers are referenced in the Supplementary Table S2.
Western Blot Analysis
According to the manufacturer’s instructions, total protein, and nuclear proteins from scramble or shBAP31 EC were respectively extracted using protein extraction kit (P0028; Beyotime). Protein concentration was determined by a BCA protein assay kit (P0010; Beyotime). Equal quantities of protein were separated by 8–12% SDS-PAGE, transferred onto polyvinylidene difluoride (PVDF) membranes (FFP28; Beyotime). Subsequently, the membranes were blocked with 5% skim milk for 1h and probed with specific primary antibodies overnight at 4°C. After washing three times with TBST, the membranes were probed with secondary antibodies for 1h. All protein bands were visualized using ECL Western blotting reagents by chemiluminescence (P0018S; Beyotime). The grayscale analysis of each band was quantified using Image Lab software. Additionally, the primary antibodies for BAP31 (4043S; Cell Signaling Technology), ICAM1 (ab171123; Abcam), VCAM1 (ab134047; Abcam), p65 (8242; Cell Signaling Technology), p-p65 (3033; Cell Signaling Technology), TAK1 (5206; Cell Signaling Technology), p-TAK1 (9339; Cell Signaling Technology), IKKα (61,294; Cell Signaling Technology), IKKβ (8943; Cell Signaling Technology), p-IKKα/β (2078; Cell Signaling Technology), IκBα (4812; Cell Signaling Technology), p-IκBα (2859; Cell Signaling Technology), AKT (9272; Cell Signaling Technology), p-AKT (4060; Cell Signaling Technology), PI3K (4249; Cell Signaling Technology), p-PI3K (4228; Cell Signaling Technology; 13,857;Cell Signaling Technology), TLR4 (14,358; Cell Signaling Technology; 38,519; Cell Signaling Technology), MyD88 (4283; Cell Signaling Technology), TRAF6 (67,591; Cell Signaling Technology), Histone H3 (4499; Cell Signaling Technology), β-actin (93,473; Cell Signaling Technology). Corresponding secondary antibodies could be obtained from manufacturers. The repetitive WB images are referenced in the Supplementary Figure S1.
Flow Cytometric Assay
The collected EC were resuspended in 100μL 5% BSA and incubated for 10min on ice. These primary antibodies CD31 (ab7388; Abcam), F4/80 (ab6640; Abcam), CD80 (ab225674; Abcam; ab222702; Abcam), CD86 (ab239075; Abcam; ab242142; Abcam), CD163 (ab182422; Abcam) and CD206 (12–2069-42; Invitrogen) were used to specifically label target proteins on ice water bath at 4°C for 45min. After washing with PBS, cells were resuspended in 5% BSA and stained with conjugated secondary antibody on ice water bath at 4°C for 45min in dark. Then, cells were washed three times with PBS, centrifuged and collected. Finally, cells were resuspended by addition of 300μL PBS and loaded onto a flow cytometer.
Statistical Analysis
The statistical analyses were performed using GraphPad Prism 8.0.2 Software and ImageJ software. All data were acquired from three repeated experiments and presented as the mean ± SD. The unpaired two-tailed Student’s t-test was used for data analysis between two groups. Group differences were calculated using one way analysis of variance (ANOVA). P < 0.05 were assumed as the significance thresholds.
Results
Endothelial BAP31 Induced Multi-Organ Dysfunction in Sepsis
We used the Cre/LoxP system to knockdown BAP31 expression in C57BL/6J mice. Cecal ligation puncture (CLP) was used to establish sepsis in the mice, and mice in the Control group received a sham procedure. Blood samples and organs were harvested 24 h following CLP or the sham procedure to determine changes in the blood and the degree of organ damage (Figure 1A). BAP31 knockdown was successfully verified by Western blot and PCR in EC isolated from inferior vena cava (Figure 1B and C).
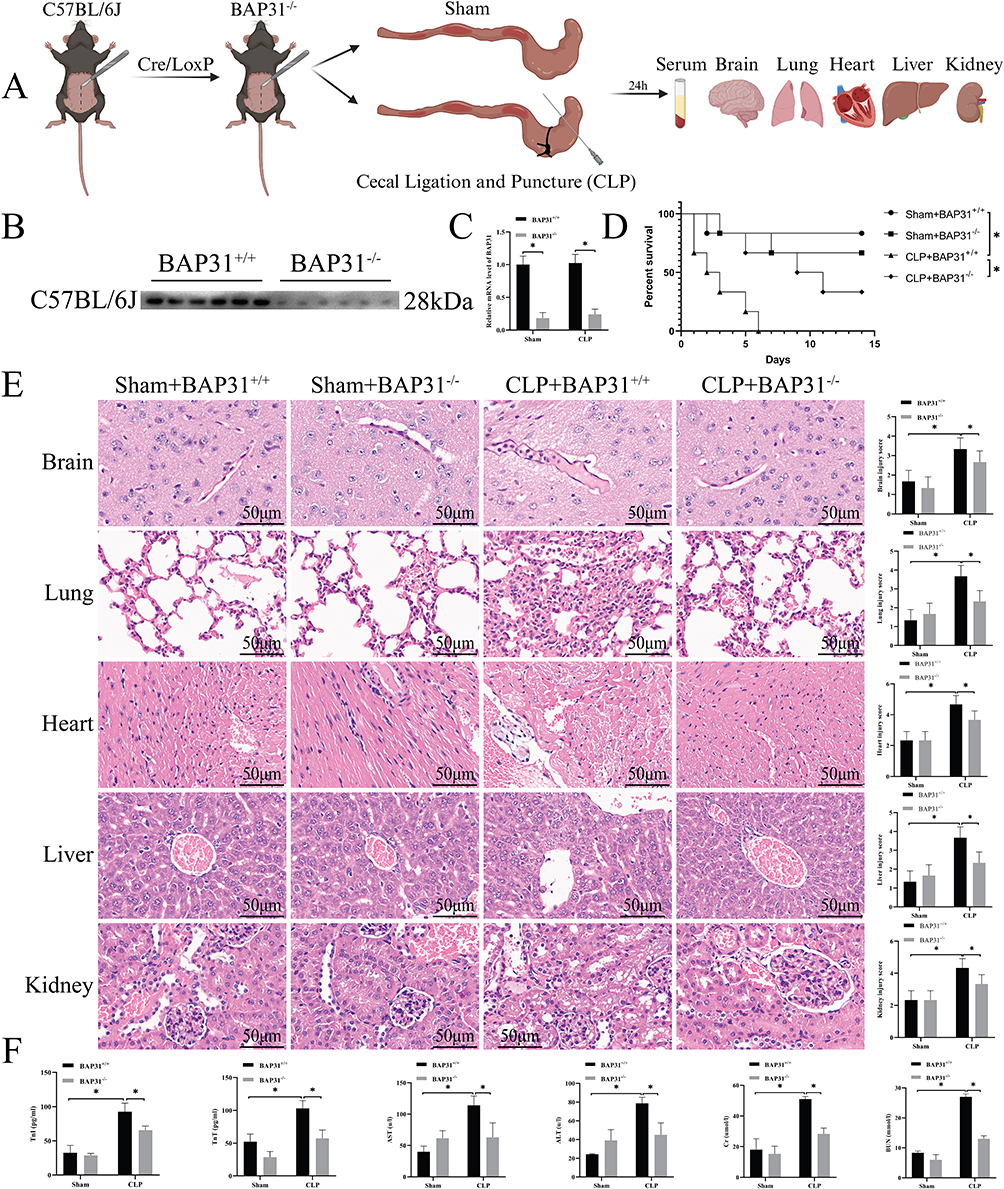 |
Figure 1 BAP31 knockout on animals. (A) Procedures for animal experiments. (B and C) Verification of BAP31 knockout. (D) The survival curve of animals. (E) The effect of BAP31 knockout on animal histopathology and its semi-quantitative score (Scale bars: 50μm). (F) The content of related substances in the blood of animals. Values were represented as mean ± SD, n=6-8 mice per group, *p < 0.05. Abbreviations: BAP31, B cell receptor associated protein 31; TnT, troponin T; TnI, troponin I; AST, aspartate aminotransferase; ALT, alanine aminotransferase; CR, creatinine; BUN, blood urea nitrogen. |
We used 6 mice to observe mortality in each group. All of the mice in the CLP+BAP31+/+ group died by day 6, while 5 (Sham+BAP31+/+), 4 (Sham+BAP31−/−), and 2 (CLP+BAP31−/−) mice remained by day 14 respectively. Compared to the Control groups, mortality at day 14 in the CLP groups was significantly increased. However, BAP31 knockdown improved the prognosis of sepsis (Figure 1D). HE staining showed more serious parenchymal cell injury with abundant inflammatory cell infiltration and tissue edema in an association of organs induced by CLP (Figure 1E). Consistent with HE staining, compared with Control, serum biomarkers related to organ damage including troponin T (TnT), troponin I (TnI), aspartate aminotransferase (AST), alanine aminotransferase (ALT), creatinine (CR), and blood urea nitrogen (BUN), greatly increased in CLP group (Figure 1F). However, BAP31 knockdown significantly attenuated sepsis-induced organ injury both in serology and in pathology. These results suggested that BAP31 may associate with sepsis induced multi-organ dysfunction, identifying BAP31 a target of organ protection.
BAP31 Promoted Adhesion of Neutrophils and Macrophages to EC
Previous studies reported that EC induced infiltration of inflammatory cells is one of the important pathogenetic mechanisms of sepsis.13 We further evaluated the effect of BAP31 on neutrophils or macrophage adhesion to EC. EC derived from the inferior vena cava of mice or HUVEC (Figure 2A) and then co-incubated with prelabeled neutrophils or macrophages. We found a significant increase in the adhesion of neutrophils and macrophages to sepsis-induced EC, whereas BAP31 knockdown weakened the adhesion capability (Figure 2B). Short hairpin RNA-mediated knockdown of BAP31 (shBAP31) was constructed in HUVEC and a non-specific shRNA (shNC) was used as the control. The in vitro sepsis model was established by incubating the HUVEC with LPS and PBS was used as the control and validation of BAP31 knockdown as shown (Figure 2C). We obtained results similar to those described above in the next cell experiments (Figure 2D). The result demonstrated that BAP31 could upregulate the adhesion of neutrophils and macrophages to EC during sepsis.
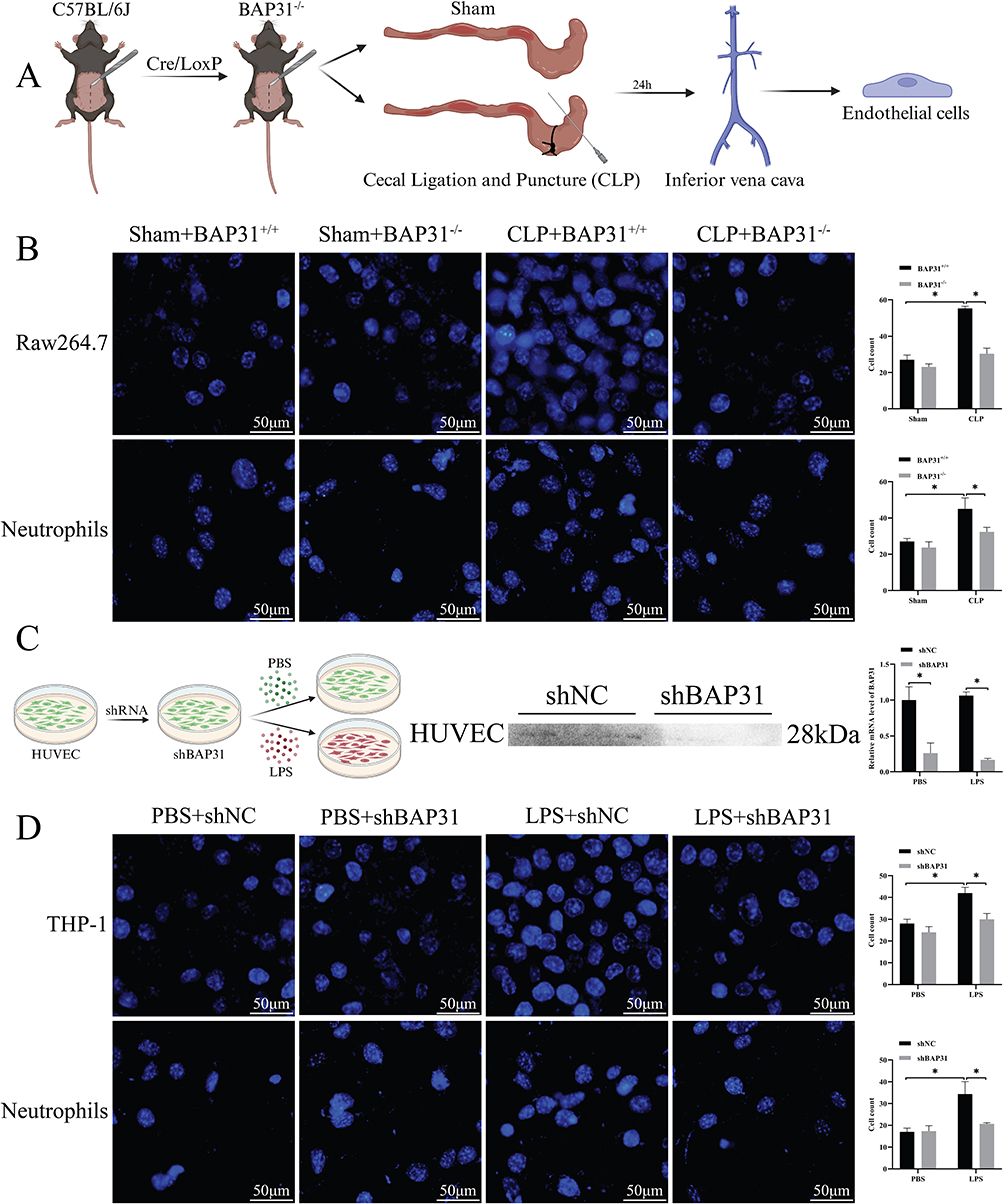 |
Figure 2 Effect of BAP31 knockout endothelial cells on the adhesion function of macrophages and neutrophils. (A) Endothelial cells were isolated from the inferior vena cava in animal experiments. (B) The co-culture results of animal-derived endothelial cells with corresponding macrophages and neutrophils and their semi-quantitative analysis (Scale bars: 50μm). (C) Flow chart of cell experiment and verification of BAP31 knockout. (D) The results of co-culture and semi-quantitative analysis of macrophages and neutrophils of HUVEC and its corresponding species (Scale bars: 50μm). Values were represented as mean ± SD, *p < 0.05. Abbreviation: HUVEC, human umbilical vein endothelial cells. |
BAP31 Regulated ICAM1 and VCAM1 Expression in EC Following LPS Stimulation
It has been demonstrated that ICAM1 and VCAM1 are involved in the interaction between endothelial and inflammatory cells.14 We further evaluated the effect of BAP31 on ICAM1 and VCAM1 expression in vitro. Compared to PBS, LPS stimulation increased ICAM1 and VCAM1 expression on the surface of the HUVEC. And BAP31 knockdown blocked the increase (Figure 3A and B). Further, the expression of ICAM1 and VCAM1 was detected in mouse tissues. We observed that the expression of ICAM1 and VCAM1 was significantly increased in the CLP group, whereas BAP31 knockdown reversed this effect (Figure 3C). The result further confirmed that BAP31 could regulate ICAM1 and VCAM1 in EC during sepsis.
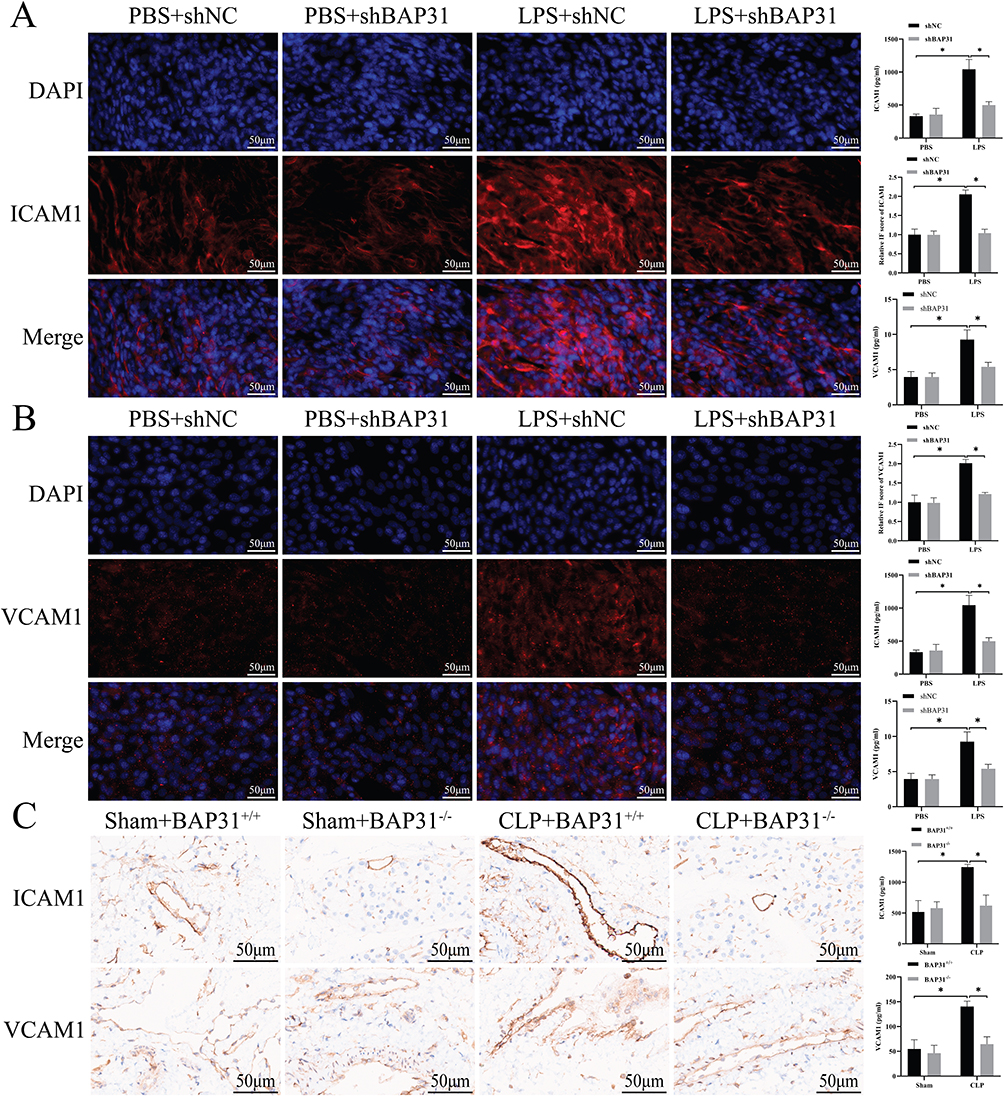 |
Figure 3 BAP31 knockout on HUVEC. (A) The effect of BAP31 knockout on ICAM1 expression in cells and its semi-quantitative analysis (Scale bars: 50μm). (B) The effect of BAP31 knockout on VCAM1 expression in cells and its semi-quantitative analysis (Scale bars: 50μm). (C) The effect of BAP31 knockout on ICAM1 and VCAM1 in animal tissues and its semi-quantitative score (Scale bars: 50μm). Values were represented as mean ± SD, *p < 0.05. Abbreviations: ICAM1, intercellular cell adhesion molecule 1; VCAM1, vascular cell adhesion molecule 1. |
BAP31 Induced Phenotypic Transformation of Macrophage Upon Interaction with EC
Previous studies demonstrated the phenotypic transformation of macrophages occurred in sepsis pathogenesis.15 Therefore, we investigated whether BAP31 knockdown in EC affects the phenotype of macrophages. EC also derived from the inferior vena cava of mice or HUVEC (Figure 2A). Surface markers of macrophages differentiation including CD80, CD86, CD163 and CD206, and inflammatory factors in supernatant including IL-1β, TNF-α, IL-10 and transforming growth factor-β (TGF-β) were detected by flow cytometer and by ELISA, respectively. With or without BAP31, there was no significant difference in the phenotype of macrophages between two Shams groups. Compared to Sham groups, the expression of CD80 and CD86 rather than CD163 and CD206 significantly increased in CLP groups. In addition, compared to Sham groups, IL-1β and TNF-α rather than IL-10 and TGF-β were significantly increased in CLP groups. However, BAP31 knockdown in sepsis-induced EC decreased the expression of CD80 and CD86 in macrophages as well as IL-1β and TNF-α in supernatant (Figure 4A and B). Similar results were observed in HUVEC (Figure 4C and D). This suggested that BAP31 could promote not only macrophages adhesion, but also differentiation to a certain extent, resulting in an enlarged inflammatory response.
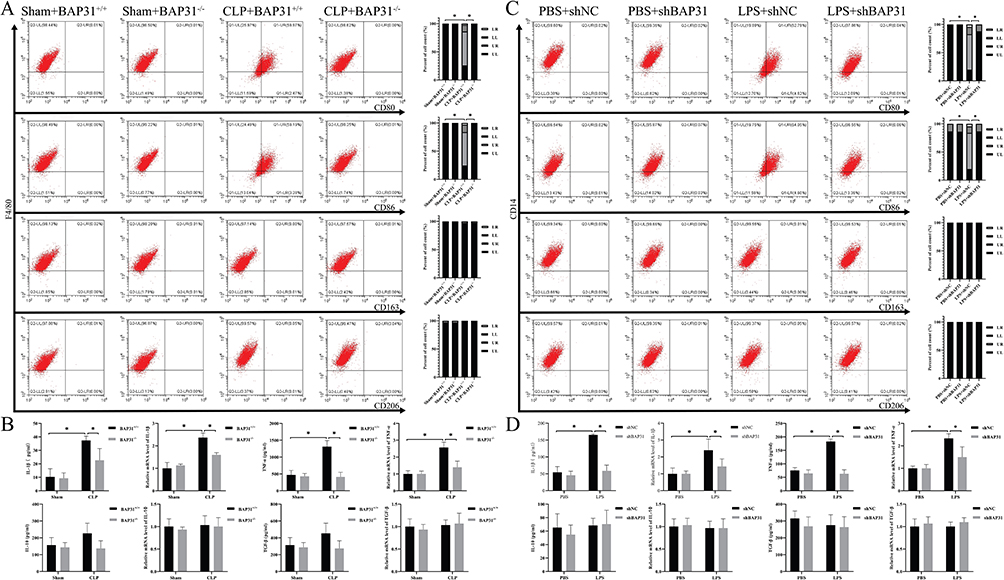 |
Figure 4 The effect of BAP31 knockout endothelial cells on the function of macrophages. (A) The effect of BAP31 knockout mouse endothelial cells on molecular proteins on the surface of macrophages. (B) The effect of BAP31 knockout mouse endothelial cells on the secretion of factors by macrophages. (C) The effect of BAP31 knockout HUVEC on molecular proteins on the surface of macrophages. (D) The effect of BAP31 knockout HUVEC on the secretion of factors by macrophages. Values were represented as mean ± SD, *p < 0.05. Abbreviations: IL-1β, interleukin 1β; TNF-α, tumor necrosis factor α; IL-10, interleukin 10; TGF-β, transforming growth factor β. |
BAP31 Increased the Expression of ICAM1 and VCAM1 on EC via TAK1 and PI3K-AKT Signaling Pathways
Previous studies have shown that BAP31 can affect TLR4-mediated intracellular signal transduction.7 Therefore, we explored the expression of TLR4 and its downstream pathways in EC both from murine inferior vena cava and HUVEC. Compared to Sham groups, CLP significantly increased the activation of MyD88 and TRAF6, which further affected the TAK1 pathway and PI3K-AKT pathway. However, BAP31 knockdown greatly reduced the activation of both pathways in sepsis-induced EC. Then, phosphorylation of IKKα, IKKβ and IκBα was significantly reduced in CLP plus BAP31 knockdown group, resulting in a decrease in phosphorylation and nuclear translocation of p65 (Figure 5A). Consistent results were observed in HUVEC (Figure 5B). This suggested that BAP31 in EC might regulate the expression of ICAM1 and VCAM1 by affecting both TAK1 pathway and PI3K-AKT pathway (Figure 5C).
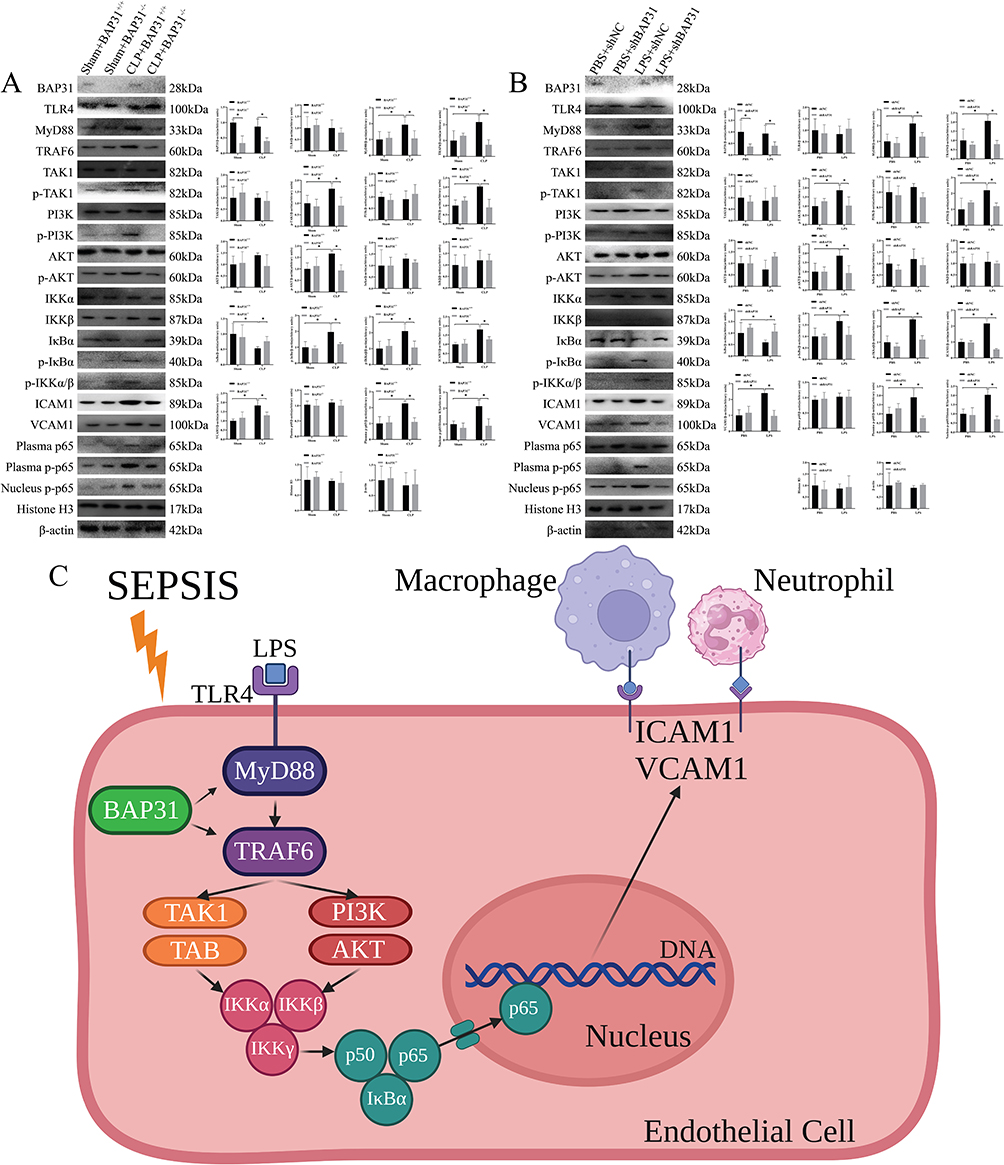 |
Figure 5 The effect of BAP31 on the signal pathway of endothelial cells. (A) The effect of BAP31 on the signal pathway of rat endothelial cells. (B) The effect of BAP31 on the signal pathway of HUVEC. (C) A pattern of the effects of BAP31 on TAK1 and PI3K-AKT signaling pathways. Values were represented as mean ± SD, *p < 0.05. Abbreviations: TLR4, toll-like receptor 4; MyD88, myeloid differentiation primary response 88; TRAF6, tumor necrosis factor receptor associated factor 6; TAK1, transforming growth factor β activated kinase 1; PI3K/AKT, phosphoinositide-3 kinases-protein kinase B. |
Discussion
Our results demonstrated that BAP31 up-regulated both ICAM1 and VCAM1 on the surface of EC, and thus aggravated sepsis-associated organ dysfunction. It may be due to a significantly strong adhesion caused by BAP31 between EC and neutrophils or macrophages. In terms of mechanism, BAP31 may activate MyD88/TRAF6 binding-dependent TLR4 signaling, and then both TAK1 and PI3K-AKT signal pathways. Subsequently, phosphorylation and nuclear translocation of p65 contributed to ICAM1 and VCAM1 expression on EC. Thus, BAP31 may be a new target for regulating the adhesion between EC and immune cells in the treatment of sepsis.
Sepsis is defined as a life-threatening organ dysfunction due to an imbalanced host immune response to infection. Timely diagnosis and sepsis care bundles have greatly improved outcomes in sepsis. However, it is still one of the leading causes of mortality worldwide. Consequently, increasing research focused on pathophysiology of sepsis at the molecular, cell, and organ level may contribute to recovery of immune homeostasis and better prognosis.
Previous studies have shown LPS is the major ligand for TLR4, initiating an immune inflammatory response during sepsis.16 Inhibition of TLR4 signaling rescued LPS-induced ICAM1 and VCAM1 expression levels in lung EC.17 However, mechanistic details of EC dysfunction remain poorly understood. NF-κΒ is essential for ICAM1 and VCAM1 activation.18 Furthermore, it is already known that PI3K-AKT and TAK1 complex signaling are identified as the canonical and the non-canonical pathway in NF-κΒ activation respectively.19 In this study, we firstly observed that LPS-induced MyD88/TRAF6 binding-dependent TLR4 signaling, both PI3K-AKT and TAK1 signaling, which in turn initiated the activation of NF-κB and ultimately induced ICAM1 and VCAM1 expression. Thus, the study might reasonably explain the TLR4 signal transduction pathway in LPS-induced EC dysfunction.
Adhesion and extravasation of immune cells is a key step during sepsis-induced organ dysfunction. BAP31 is an endoplasmic reticulum protein that is mainly involved in protein transport, degradation and cell adhesion.10 A recent study reported that myeloid-specific BAP31 knockdown attenuated the expression of adhesion molecules CD11b/CD18 and PSGL-1 exerting a protective effect on ALI.12 However, there is few research related to the effect of endothelial BAP31 on immune cells. Therefore, the study is designed to investigate whether EC-specific BAP31 depletion can regulate adhesion molecules such as ICAM1 and VCAM1, thus affecting the adhesion and infiltration of neutrophils and mononuclear macrophages. Our histopathological staining showed that ICAM1 and VCAM1 expression on the surface of EC in the CLP model or on LPS-induced HUVEC was significantly higher compared to the corresponding control groups. However, this effect was significantly weakened after BAP31 knockdown, which indicates that BAP31 could regulate ICAM1 and VCAM1 expression to a certain extent. Furthermore, BAP31 changes the infiltration of neutrophils and mononuclear macrophages in local tissues by regulating the expression of ICAM1 and VCAM1 on EC during LPS-induced sepsis. Eventually, BAP31 knockdown reduced the sepsis-related pathohistological injury, which may have been associated with the decreased expression of ICAM1 and VCAM1 on the surface of EC. These findings are partially inconsistent with a prior study, in which LPS induced down-regulation of BAP31 resulting in mitochondrial apoptosis.20 This might be due to a different concentration of LPS challenge or different animal model used. Therefore, more studies are needed to detect the additional effect of BAP31 on different inflammatory diseases.
Different phenotypes are the hallmarks of macrophages which represent two terminals of macrophage activation during inflammation.21 In this study, we wanted to understand whether BAP31 can affect the phenotype and function of macrophages upon binding to EC. As we know, M1 macrophages express surface molecular markers, such as CD80 and CD86, and secrete pro-inflammatory cytokines like IL-1β and TNF-α; while M2 macrophages express surface molecular markers, such as CD163 and CD206, and secrete cytokines, like IL-10 and TGF-β, which involved in anti-inflammatory reactions and tissue remodeling.22 Our results showed that BAP31 affected not only the adhesion, but also the phenotype and function of macrophages. Here, we observed that macrophages were significantly transformed to M1 following EC-specific BAP31 knockdown. A recent study referred to the effect of T cell specific BAP31 knockdown on macrophage polarization, which suggested that BAP31 had an immunoregulatory role on macrophage niche.23 However, further study is needed to investigate the association between macrophage polarization and the expression of ICAM1 or VCAM1 on EC.
Further, we studied the possible signaling pathways through which BAP31 regulates ICAM1 and VCAM1 expression. The activation of NFkB is a critical step in the pathogenesis of sepsis.24 TAK1 can induce the sequential activation of the classical NF-κB pathway. Previous studies have shown that TAK1 deficiency can lead to decreased NF-κB activation.25 Our study showed that the levels of phosphorylated TAK1 decreased following BAP31 knockdown in EC. Previous studies have shown that LPS can induce the phosphorylation of TAK1 through TLR4, which leads to the rapid activation of the IKK complex.26 This complex can further induce the phosphorylation of IκBα, and then promote NF-κB translocation into the nucleus, where NF-κB can rapidly bind to DNA resulting in increased transcription and translation of ICAM1 and VCAM1.27,28 Our study found that there is a close relationship between the IKK complex, IκBα, and p65 phosphorylation during sepsis. BAP31 knockdown in EC resulted in a significant decrease in the phosphorylation level of these factors. These results suggest that BAP31 may participate in the classical NF-κB pathway and regulate the occurrence and development of sepsis. In addition, previous studies have shown that PI3K-AKT is involved in the regulation of inflammatory pathways. The PI3K-AKT pathway can directly activate the IKK complex and the subsequent activation of NF-κB.29 Previous studies have shown that PI3K-AKT inhibitors can significantly downregulate NF-κB and reduce organ damage in sepsis.30 Our results showed that the phosphorylation levels of PI3K and AKT were also significantly decreased in EC following BAP31 knockdown. Therefore, this study suggested that BAP31 affected LPS-induced TLR4/NF-κB pathways by activation both PI3K-AKT signaling and TAK1 signaling pathway.
Several limitations of our experiments remain. First, we collected tissue specimens from mice 24 hours after surgery rather than longer. Because the inflammatory response in the early stage of sepsis and the recovery period may differ, our results may not be applicable to explore the pathophysiologic mechanisms during the recovery period of sepsis. Second, we did not detect additional cell surface adhesion molecules regulated by BAP31 on EC, which resulted in an amplified immune response during sepsis. It is also not clear whether BAP31 affect the interaction between EC and other immune cells, for example natural killer cell or dendritic cell. Of course, this in vitro and mouse model of sepsis differs in several crucial respects from a human infection, including the difference of infective route, infective dose, and specific immunity status. However, our results clearly demonstrated deletion of BAP31 significantly improved sepsis-associated organ injury as well as survival rate through attenuating endothelial ICAM1 and VCAM1 expression.
Conclusion
EC-specific BAP31 knockdown contributed to a reduced adhesion between EC and immune cells, attenuated tissue and organ injury, and a prolonged survival duration of in a mouse sepsis model. BAP31 may successively induce the activation of TLR4/MyD88/TRAF6 signaling, and then both PI3K-AKT and TAK1 signaling, and NF-κB p65, which eventually promote the expression of ICAM1 and VCAM1 on EC.
Institutional Review Board Statement
This experiment has been approved by the Experimental Animal and Ethics Committee of Beijing Friendship Hospital (No.22-2044; BFH20230320002/BFHHZS20230092) which following the GB/T 35892-2018 guidelines of laboratory animal welfare.
Data Sharing Statement
The raw data presented in this study will be made available by the authors, without undue reservation.
Acknowledgments
Graphical illustrations were created with Biorender.com.
Funding
This work was funded by grants from the Beijing Key Clinical Specialty Excellence Project.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315:801–810. doi:10.1001/jama.2016.0287
2. Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The pathogenesis of sepsis. Annu Rev Pathol. 2011;(6):19–48. doi:10.1146/annurev-pathol-011110-130327
3. Ziegelstein RC, Corda S, Pili R, et al. Initial contact and subsequent adhesion of human neutrophils or monocytes to human aortic endothelial cells releases an endothelial intracellular calcium store. Circ. 1994;(90):1899–1907. doi:10.1161/01.cir.90.4.1899
4. Boscacci RT, Pfeiffer F, Gollmer K, et al. Comprehensive analysis of lymph node stroma-expressed Ig superfamily members reveals redundant and nonredundant roles for ICAM-1, ICAM-2, and VCAM-1 in lymphocyte homing. Blood. 2010;(116):915–925. doi:10.1182/blood-2009-11-254334
5. Richard K, Piepenbrink KH, Shirey KA, et al. A mouse model of human TLR4 D299G/T399I SNPs reveals mechanisms of altered LPS and pathogen responses. J Exp Med. 2021;218. doi:10.1084/jem.20200675
6. Menden H, Xia S, Mabry SM, et al. Histone deacetylase 6 regulates endothelial MyD88-dependent canonical TLR signaling, lung inflammation, and alveolar remodeling in the developing lung. Am J Physiol Lung Cell Mol Physiol. 2019;(317):L332–l346. doi:10.1152/ajplung.00247.2018
7. Li G, Jiang X, Liang X, et al. BAP31 regulates the expression of ICAM-1/VCAM-1 via MyD88/NF-κB pathway in acute lung injury mice model. Life Sci. 2023;(313):121310. doi:10.1016/j.lfs.2022.121310
8. Yang HL, Thiyagarajan V, Shen PC, et al. Anti-EMT properties of CoQ0 attributed to PI3K/AKT/NFKB/MMP-9 signaling pathway through ROS-mediated apoptosis. J Exp Clin Cancer Res. 2019;(38):186. doi:10.1186/s13046-019-1196-x
9. Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 2021;(78):1233–1261. doi:10.1007/s00018-020-03656-y
10. Quistgaard EM. BAP31: physiological functions and roles in disease. Biochim. 2021;(186):105–129. doi:10.1016/j.biochi.2021.04.008
11. Stojanovic M, Germain M, Nguyen M, Shore GC. BAP31 and its caspase cleavage product regulate cell surface expression of tetraspanins and integrin-mediated cell survival. J Biol Chem. 2005;280(280):30018–30024. doi:10.1074/jbc.M501306200
12. Zen K, Utech M, Liu Y, Soto I, Nusrat A, Parkos CA. Association of BAP31 with CD11b/CD18. Potential role in intracellular trafficking of CD11b/CD18 in neutrophils. J Biol Chem. 2004;279(279):44924–44930. doi:10.1074/jbc.M402115200
13. Molema G, Zijlstra JG, van Meurs M, Kamps J. Renal microvascular endothelial cell responses in sepsis-induced acute kidney injury. Nat Rev Nephrol. 2022;(18):95–112. doi:10.1038/s41581-021-00489-1
14. Caprio M, Newfell BG, la Sala A, et al. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ Res. 2008;(102):1359–1367. doi:10.1161/circresaha.108.174235
15. Leaver SK, MacCallum NS, Pingle V, et al. Increased plasma thioredoxin levels in patients with sepsis: positive association with macrophage migration inhibitory factor. Intensive Care Med. 2010;(36):336–341. doi:10.1007/s00134-009-1640-z
16. Vasudevan SO, Russo AJ, Kumari P, Vanaja SK, Rathinam VA. A TLR4-independent critical role for CD14 in intracellular LPS sensing. Cell Rep. 2022;(39):110755. doi:10.1016/j.celrep.2022.110755
17. Obi AT, Andraska E, Kanthi Y, et al. Endotoxaemia-augmented murine venous thrombosis is dependent on TLR-4 and ICAM-1, and potentiated by neutropenia. J Thromb Haemost. 2017;(117):339–348. doi:10.1160/th16-03-0218
18. Hashmi SF, Rathore HA, Sattar MA, et al. Hydrogen sulphide treatment prevents renal ischemia-reperfusion injury by inhibiting the expression of ICAM-1 and NF-kB concentration in normotensive and hypertensive rats. Biomolecules. 2021;(11). doi:10.3390/biom11101549
19. Chang MC, Tsai YL, Chang HH, et al. IL-1β-induced MCP-1 expression and secretion of human dental pulp cells is related to TAK1, MEK/ERK, and PI3K/Akt signaling pathways. Arch Oral Biol. 2016;(61):16–22. doi:10.1016/j.archoralbio.2015.10.008
20. Zhang J, Wang L, Xie W, et al. Melatonin attenuates ER stress and mitochondrial damage in septic cardiomyopathy: a new mechanism involving BAP31 upregulation and MAPK-ERK pathway. J Cell Physiol. 2020;235(3):2847–2856. doi:10.1002/jcp.29190
21. Wang Z, Wang Z. The role of macrophages polarization in sepsis-induced acute lung injury. Front Immunol. 2023;(14):1209438. doi:10.3389/fimmu.2023.1209438
22. Lazarov T, Juarez-Carreño S, Cox N, Geissmann F. Physiology and diseases of tissue-resident macrophages. Nature. 2023;(618):698–707. doi:10.1038/s41586-023-06002-x
23. Zhao B, Sun L, Yuan Q, et al. BAP31 knockout in macrophages affects CD4(+)T cell activation through upregulation of MHC class II molecule. Int J Mol Sci. 2023;(24). doi:10.3390/ijms241713476
24. Adhikari A, Xu M, Chen ZJ. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene. 2007;(26):3214–3226. doi:10.1038/sj.onc.1210413
25. Xu YR, Lei CQ. TAK1-TABs complex: a central signalosome in inflammatory responses. Front Immunol. 2020;(11):608976. doi:10.3389/fimmu.2020.608976
26. Shi JH, Sun SC. Tumor necrosis factor receptor-associated factor regulation of nuclear factor κB and mitogen-activated protein kinase pathways. Front Immunol. 2018;(9):1849. doi:10.3389/fimmu.2018.01849
27. Zhong L, Simard MJ, Huot J. Endothelial microRNAs regulating the NF-κB pathway and cell adhesion molecules during inflammation. FASEB J. 2018;(32):4070–4084. doi:10.1096/fj.201701536R
28. Xiong Y, Qiu F, Piao W, Song C, Wahl LM, Medvedev AE. Endotoxin tolerance impairs IL-1 receptor-associated kinase (IRAK) 4 and TGF-beta-activated kinase 1 activation, K63-linked polyubiquitination and assembly of IRAK1, TNF receptor-associated factor 6, and IkappaB kinase gamma and increases A20 expression. J Biol Chem. 2011;(286):7905–7916. doi:10.1074/jbc.M110.182873
29. Tao Y, Yu S, Chao M, Wang Y, Xiong J, Lai H. SIRT4 suppresses the PI3K/Akt/NF‑κB signaling pathway and attenuates HUVEC injury induced by oxLDL. Mol Med Rep. 2019;(19):4973–4979. doi:10.3892/mmr.2019.10161
30. Song J, Chen D, Pan Y, et al. Discovery of a Novel MyD88 Inhibitor M20 and its protection against sepsis-mediated acute lung injury. Front Pharmacol. 2021;(12):775117. doi:10.3389/fphar.2021.775117.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
CD146+ Umbilical Cord Mesenchymal Stem Cells Exhibit High Immunomodulatory Activity and Therapeutic Efficacy in Septic Mice
Zhang L, Zhang X, Liu Y, Zhang W, Wu CT, Wang L
Journal of Inflammation Research 2023, 16:579-594
Published Date: 11 February 2023
Crosstalk Between H-Type Vascular Endothelial Cells and Macrophages: A Potential Regulator of Bone Homeostasis

Fan J, Xie Y, Liu D, Cui R, Zhang W, Shen M, Cao L
Journal of Inflammation Research 2025, 18:2743-2765
Published Date: 25 February 2025
Lung Tissue Extracellular Vesicles-Mediated Delivery of miR-128-3p as a Novel Mechanism of Acute Lung Inflammation
Deng W, Zhu X, Li H, Hu P, Qian K, Liu F
International Journal of Nanomedicine 2025, 20:4831-4848
Published Date: 15 April 2025
Co-Targeting Biomimetic Nanoparticles Alleviate Atherosclerosis by Inhibiting the Vicious Circle Between Inflammation and Lipids
Wu C, Li Y, Liu Y, Wang X, Yuan P, Xu M, Deng Y, Zhang Z, Li C, Zhou X
International Journal of Nanomedicine 2025, 20:15705-15721
Published Date: 25 December 2025
Macrophage Hypoxia Signaling Pathways and Their Roles in Sepsis
Yuan M, Tian L, Xu X
Journal of Inflammation Research 2026, 19:562619
Published Date: 10 February 2026