Back to Journals » Journal of Inflammation Research » Volume 14
Bacterial Lipopolysaccharide Augmented Malignant Transformation and Promoted the Stemness in Prostate Cancer Epithelial Cells
Authors Tang S ![]() , Lian X, Cheng H, Guo J, Ni D, Huang C, Gu X, Meng H, Jiang J, Li X
, Lian X, Cheng H, Guo J, Ni D, Huang C, Gu X, Meng H, Jiang J, Li X ![]()
Received 9 August 2021
Accepted for publication 15 October 2021
Published 9 November 2021 Volume 2021:14 Pages 5849—5862
DOI https://doi.org/10.2147/JIR.S332943
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Sijie Tang,1,2 Xueqi Lian,1 Huiying Cheng,1 Jiaqian Guo,1 Daguang Ni,1 Can Huang,1 Xiang Gu,2 Hong Meng,3 Jiajia Jiang,1 Xiaohua Li1,4,5
1The Aoyang Cancer Institute, Affiliated Aoyang Hospital of Jiangsu University, Zhangjiagang, Suzhou, 215600, People’s Republic of China; 2Department of Urology, the Affiliated Aoyang Hospital of Jiangsu University, Zhangjiagang, Suzhou, 215600, People’s Republic of China; 3Perinatology Research Branch, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Detroit, MI, 48201, USA; 4The Laboratory of Clinical Genomics, Hefei KingMed Diagnostics Ltd, Hefei, 230088, People’s Republic of China; 5National Center for Gene Testing Technology Application & Demonstration (Anhui), Hefei, 230088, People’s Republic of China
Correspondence: Xiaohua Li; Jiajia Jiang
The Aoyang Cancer Institute, Affiliated Aoyang Hospital of Jiangsu University, 279 Jingang Blvd., Zhangjiagang, Suzhou, 215600, People’s Republic of China
Email [email protected]; [email protected]
Purpose: To study bacterial lipopolysaccharide (LPS)-induced cancer stem-like transformation and to investigate the inhibitory effect of Trichostatin A (TSA) on the malignant transformation through targeting p-Stat3 signaling.
Methods: 2D, 3D, and serum-free suspension culture system were used to study LPS-induced malignant transformation in series malignant grade of prostate cancer (PCa) epithelial cells. Flow cytometry assay and RT-PCR were utilized to evaluate the CD44+CD133+ stem cell population, the expression of inflammatory cytokines and series tumor stemness biomarkers. Meanwhile, Western blot was used to analyze the alteration of cell signaling associated-molecules by treatment with TSA, an original antifungal antibiotic and a panel inhibitor of histone deacetylase.
Results: Our study found that LPS promoted the migration, invasion and stem-like tumoroshpere forming in multiple PCa cell lines including DU145, PC3, 22RV1, LNCaP. LPS also enriched CD44+CD133+ stem cell population and increased the expression of series tumor stemness biomarkers (e.g., CD44, CD133, SOX-2, α-intergrin, Nestin, etc.). TSA was found to prevent tumor cell migration, invasion and tumorosphere forming in DU145 and PC3 cells with increasing tumor suppressive Maspin and reducing both phosphorylation of Stat3 (p-Stat3) and pro-oncogene c-Myc expression in LPS-treated DU145 cells. Furthermore, blocking Stat3 signaling pathway by treatment with TSA and/or small molecule compound Stattic of an p-Stat3 inhibitor effectively abrogated LPS-induced tumorosphere forming with decrease of IL-6, IL-8 and stemness biomarkers CD44, SOX-2 expression.
Conclusion: Our data demonstrated that the inflammatory agent of bacterial LPS augmented malignant transformation and promoted the cancerous stemness in PCa epithelial cells. TSA could prevent, at least in part, the LPS-induced malignant transformation by targeting p-Stat3/c-Myc signaling pathway and reducing inflammatory IL-6, IL-8. In addition, the assay of LPS-induced tumorosphere forming could serve as a simple and an easy handling method for targeting cancer stem cells drug screening in vitro in clinical practice.
Keywords: tumorosphere forming, prostate cancer, bacterial lipopolysaccharide, epithelial malignant transformation, cancer stemness
Introduction
It is well known that the occurrence of epithelial-to-mesenchymal transition (EMT) was a key event for tumor progression. This step helps malignant tumor cells acquire a more motile and invasive phenotype. Meanwhile, demonstrated by developing and utilizing the mammosphere forming assay, the process of EMT was directly linked to the gain of epithelial stem cell properties (e.g., self-renewal).1 Later on, this stem-like sphere forming assay was tested successfully in variant cancer tissue and cells. For example, by utilizing this assay, CagA oncoprotein from Helicobacter pylori was studied to unveils cancer stem cell (CSC)-like properties by induction of EMT-like changes in gastric epithelial cells,2 and TGF-β1 was also demonstrated to induce CSC in human cholangiocarcinoma cells.3 Recently, four spherical cancer models were developed and described.4 Among these, the tumorosphere forming assay was widely accepted to analyze both the clonogenic growth ability and self-renewal ability of CSC.5,6 Meanwhile, the method was also used to enrich CSCs from bulk cancer cells for screening potential anti-CSC and anti-cancer agents.4,7 Taken together, EMT was shown to be closely correlated with cancer stem cells, where cells undergoing EMT acquire properties of stem cells. Thus, the accumulated evidences indicated that EMT may be a key therapeutic target in prostate cancer.
Decades ago, microbial infections-induced abscess was found to be associated with prostate carcinogenesis.8,9 Later on, bacteria were found to invade the prostate gland/tissue through nastiness sexual intercourse, urinary infection and even leaky gut, etc. to form chronic inflammation, which further induced inflammation-mediated prostatic epithelial malignant transformation and carcinogenesis due to the constant exposure to microbial infection.10,11 However, the inside molecular mechanism was not clear. Lipopolysaccharide (LPS) is the outer membrane major component of Gram-negative bacteria. As the bacterial lipoglycans and an endotoxin, LPS recognizes cell membrane molecules of LBP (LPS-binding protein), CD14 (mCD14, sCD14), MD-2 (myloid differentiation protein-2), TLRs (Toll-like receptors), SR (scavenger receptor), etc. and activates the cell signaling transduction.12–14 And LPS was broadly reported to stimulate inflammatory responses to induce epithelial cell to undergo malignant transformation and to promote carcinogenesis both in vitro and in vivo. For instance, LPS was documented to enhance the migration and invasion of prostate cancer (PCa) cells,15–18 to promote EMT,15,19 to activate EMT-related signaling pathways such as Stat3 pathway.15,20,21 Moreover, TSA has been reported to reverse EMT in PCa PC3 cells.22 Importantly, LPS was reported to augment cancer stemness in esophageal squamous cell cancer,23 squamous cell carcinoma of the head and neck24 and colorectal cancer.25 Although chronic prostatic inflammation induced by bacterial infection may play a crucial role in PCa etiology and progression,26 and even LPS-induced prostatic epithelial carcinogenesis was reported,27,28 its underlying cellular and molecular mechanisms of LPS-induced cancer stemness has not been well studied experimentally and illustrated theoretically. In this study, to explore the possibility of LPS-associated chronic inflammation-induced prostate intraepithelial neoplasia (PIN), malignant stem cell formation and PCa initiation, LPS-induced EMT and stem-like tumorosphere formation was studied in multiple PCa cell lines. Then, an antifungal antibiotic and histone deacetylase (HDAC) inhibitor Trichostatin A (TSA) was tested to prevent the LPS-induced malignant transformation and stem-like tumorosphere formation.
Materials and Methods
Cell Culture and Reagents
Normal immortalized nonmalignant human prostate epithelial RWPE-1 cell was obtained from Beijing Beina Chuanglian Biotechnology Institute (Beijing, China). The cells were cultured and maintained in keratinocyte medium with keratinocyte growth supplement and 1% penicillin/streptomycin (ScienCell Research Laboratories, USA). PCa cell lines DU145, PC3, 22RV1, LNCaP were purchased from the cell bank of Chinese Academy of Sciences (Shanghai, China). DU145 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, ThermoFish Scientific, China) containing 10% FBS (Biological Industries, Israel), 1% penicillin/streptomycin according to the manufacturer’s description. PC3 cells were maintained in F-12 medium (Gibco, ThermoFish Scientific, China) and LNCaP, 22RV1 cells were maintained in RPMI 1640 medium with 10% FBS and the same additives as above. All cells were cultured in a humidified incubator with 6% CO2 at 37°C and were tested for free of mycoplasma contamination periodically with LookOut Mycoplasma PCR Detection Kit from Sigma-Aldrich (St Louis, MO, USA). LPS was purchased from Sigma-Aldrich (St. Louis, MO, USA) and TSA was obtained from Selleckchem (Houston, TX, USA).
Wound Healing Assay
Cells were seeded in 6-well plate (5 × 105 cells/2 mL/well) and cultured in complete growth medium. A wounded line at the bottom of the monolayer surface was scratched using a 200 μL sterile pipette tip when cells grew at about 80% confluence. After removing cellular debris by soft washing with phosphate-buffered saline (PBS), cells were cultured in serum-free medium containing the indicated concentration of TSA or LPS. At the indicated time, the wound monolayer cell gap was photographed at least three fields. The gap area was measured by ImageJ software, and the data were analyzed with the formula 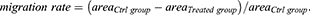
Transwell Invasion Assay
Cells were pre-treated with TSA followed by treatment with or without LPS for 24 hrs and prepared for a transwell assay. Briefly, cells were suspended with serum-free medium and were seeded on a Boyden chamber insert of 24-well plates pre-coated with matrigel (Corning, USA). The lower chambers were filled with medium containing 10% FBS and the cells were cultured for 24 hrs. At the end of the culture, the upper chamber was taken out and rinsed two times with PBS followed by fixation with cold methanol (Sinopharm Chemical Regent Co., Ltd, China) for 15 min at room temperature (RT). Then, a cotton swab moistened with the medium was inserted into the top of the matrix permeable support to rub the area gently. After repeating the above process with a new cotton swab, the permeable support was rinsed three times with PBS. The cells attached to the lower side of the support filter surface were stained with 0.1% crystal violet (Solarbio, China) for 15 min. The number of cells attached to the lower surface of the filter was counted and photographed.
Suspension Cell Culture and Tumorosphere Forming Assay
Cells were pretreated with or without TSA for 2 hrs followed by adding LPS for total 8 hrs, then harvested and washed with PBS. The cells were re-seeded into 6-well ultra-low attachment plates (Corning, USA) at 8×104 cells/well and cultured in 3 milliliters of serum-free DMEM/F12 medium supplemented with 1 × B27 (Gibco, ThermoFish Scientific, China), 0.4% BSA (Sigma-Aldrich, St. Louis, MO, USA), 20 ng/mL epidermal growth factor (EGF), 10 ng/mL basic fibroblast growth factor (bFGF) and 20 ng/mL leukemia inhibitory factor (LIF) (all purchased from PeproTech, Rocky Hill, NJ, USA). Cells were cultured in suspension condition for at least 2 weeks to obtain tumorospheres, and the medium was replaced every 3–4 days. The tumorospheres should be solid and fused together with a round structure and the size at least 50 μm. Then, the formed tumorospheres were photographed and counted for nine fields of a view.
3D Cell Culture
VitroGel 3D-RGD (TheWell Bioscience, USA) was used to culture tumorosphere cells by following the manufacturer’s instruction and our previous publication.29 Briefly, the tumorospheres were collected and singularized by trypsin-EDTA, then the cell suspension was gently mixed with the diluted VitroGel and the hydrogel mixture was transferred into a 6-well plate. After a soft gel was formed, an additional culture medium was carefully added to cover the hydrogel for incubating, and the cover medium was changed every 48 hrs. The cell clones were photographed under a microscope. Then, the hydrogel was fixed and a nucleus staining solution was added. The stained cells were observed and counted under a fluorescence microscope.
Western Blot (WB) Assay
Briefly, the cellular protein was extracted by utilizing RIPA buffer (Nobleryder, Beijing, China) containing with 1 mM PMSF (Phenylmethanesulfonyl fluoride) and quantified by using BCA kit (ThermoFisher Scientific, China). After the sample denaturing by boiling and resolving by running SDS–PAGE, the sample protein was transferred onto a polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA) for immune blotting assay. Firstly, the membrane was blocked with 5% milk for 40 min at RT followed by incubating with the primary antibodies at predetermined concentration overnight at 4°C. Then, the membrane was incubated with HRP-conjugated secondary antibodies (Abcam, Cambridge, MA, USA) for 1 hr at RT. At the end, the expression of protein was detected by enhanced chemiluminescence system (ECL) with autoradiography using Tanon 5500 (Shanghai, China). The primary antibodies against phosphorylated STAT3 (p-Stat3), total Stat3 and NF-κB p65 (D14E12) XP® Rabbit mAb were purchased from Cell Signaling Technology (Boston, MA, USA). The primary antibodies against Erk1 (pT202/pY204) + Erk2 (pT185/pY187) (p-Erk), Erk1 + Erk2 (Erk), c-jun (phospho S63) (p-c-jun), c-jun and c-Myc were purchased from Abcam (Cambridge, UK). The anti-Maspin monoclonal antibody was purchased from BD PharMingen (San Diego, CA, USA). The McAb against GAPDH purchased from Good HERE (Hangzhou, China) was used for equal loading and endogenous control.
Flow Cytometry Assay
The tumorospheres were collected and singularized by trypsin-EDTA. The single-cell suspensions in PBS with 0.5% BSA were incubated with primary antibodies of mouse anti-CD44 McAb (Abcam, USA) and rabbit anti-CD133 McAb (Abcam, USA) at 4°C for 30 min, isotype IgG control was also set up. Then, the cells were washed and incubated with second antibodies of goat anti-mouse IgG H&L (Alexa Fluor 488) and anti-rabbit IgG H&L (Alexa Fluor 647). Finally, the labeled cells were washed and analyzed by fluorescence-activated cell sorting (FACS) for CD44+CD133+ cells (Beckman Coulter, DxFLEX, USA).
Isolation of RNA and Quantitative RT-PCR
The RiboPure kit (Life Technologies, USA) was used to extract total cellular RNA followed by reverse transcription to generate cDNA using High Capacity cDNA Reverse Transcription Kit (ThermoFisher, China). Then the gene expression level was analyzed by quantitative real-time RT-PCR (qRT-PCR) utilizing POWER SYBR Green Master Mix (Applied Biosystems, USA). The following oligonucleotide primers were obtained from GENEWIZ (Suzhou, China). Human cytokeratin 8 (CK8), forward 5ʹ CTCCTCACCAAGAAGCAG 3ʹ and reverse 5ʹ CCTGATGGACATGGTAGAG 3ʹ; human CK18, forward 5ʹ ACTTTGCCATCCACTATCCGG 3ʹ and reverse 5ʹ ATCTTGGTGATGCCTTGGACA 3ʹ; human vimentin, forward 5ʹ AGGAAATGGCTCGTCACCTTCGTGAATA 3ʹ and reverse 5ʹ GGAGTGTCGGTTGTTAAGAACTAGAGCT 3ʹ; human α-intergrin, forward 5ʹ CAGGTCTGTTGGTCAATCTCAC 3ʹ and reverse 5ʹ TCCATATCGTGGAATGCTGAAG 3ʹ; human CD44, forward 5ʹ AGAAGGTGTGGGCAGAAGAAAA 3ʹ and reverse 5ʹ CATTCTGCAGGTTCCTTGTCT 3ʹ; human Nestin, forward 5ʹ CGTTGGAACAGAGGTTGGAG 3ʹ and reverse 5ʹ TCCTGAAAGCTGAGGGAAG 3ʹ; human Oct-4, forward 5ʹ GACAACAATGAAAATCTTCAGGAGA 3ʹ and reverse 5ʹ CTGGCGCCGGTTACAGAACCA 3ʹ; human Nanog, forward 5ʹ TGCCTCACACGGAGACTGTC 3ʹ and reverse 5ʹ TGCTATTCTTCGGCCAGTTG 3ʹ; human BCRP, forward 5ʹ CAGGTCTGTTGGTCAATCTCAC and reverse 5ʹ TCCATATCGTGGAATGCTGAAG; human Sox2, forward 5ʹ CACCTACAGCATGTCCTACTC 3ʹ and reverse 5ʹ CATGCTGTTTCTTACTCTCCTC 3ʹ; human GAPDH, forward 5ʹ ATCACCATCTTCCAGGAGCGA 3ʹ and reverse 5ʹ GCCAGTGAGCTTCCCGTTCA 3ʹ. PCR was initiated at 50°C for 2 min, then 95°C for 10 min and followed by a total of 40 cycles reaction of 95°C for 15 s, 60°C for 60 s in ABI 7500 systems (Applied Biosystems, USA). The relative gene expression was calculated according to 2− Δ(ΔCt) method and normalized to the endogenous housekeeping gene GAPDH.
Results
LPS Induced Epithelial-to-Mesenchymal Transition and Tumorosphere Formation
As a bacterial component and endotoxin, LPS was documented to be the most common inflammatory stimuli. To determine the capability of LPS on promoting epithelial cells transformation, we first performed a wound healing assay by treating DU145, PC3, RWPE-1 cells with LPS. The result showed that, treated with LPS at variant concentration from 0 to 0.02 μg/mL for 24 hrs, the gap area of wounded monolayer cells was healed in a dose-dependent manner (Figure 1A and C). Then, DU145 cells were treated with LPS at 0.01 μg/mL for up to 48 hrs, the gap area was healed in a time-dependent fashion (Figure 1B and D). The similar data were also obtained by using PC3 and RWPE-1 (Supplementary Figure 1 and 2). The result from transwell assay showed that, compared with the no treatment control group, treatment with LPS (0.01 μg/mL) alone increased the invaded cell numbers of DU145, PC3 and RWPE-1 significantly (Figure 1F and G). To further assess the LPS-induced cell transformation, the cells were treated with LPS (0.01 µg/mL) for the indicated time, and the expression of EMT-associated molecules CK8, CK18 and vimentin were evaluated using RT-PCR. The result showed that the ratio of both CK18/vimentin and CK8/vimentin were decreased (Figure 1E). Similar results were also obtained in PC3 cells (Supplementary Figure 1E and 2E). Interestingly, the occurring time of the ratio decrease in RWPE-1 cell was later than it in PCa cells after treatment. Thus, the above data indicate that LPS promoted prostatic epithelial cells to undergo EMT with an increase in migration and invasion.
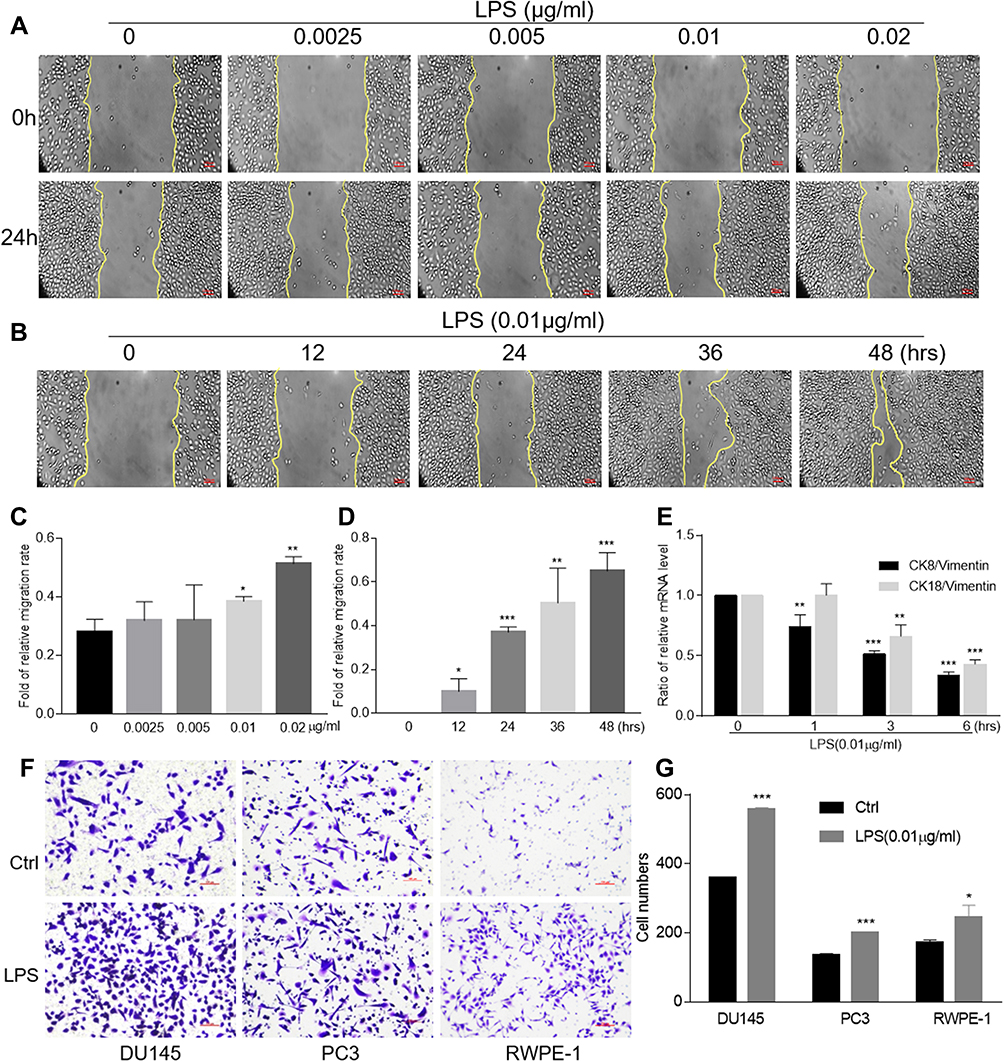 |
Figure 1 LPS-induced migration, invasion and epithelial-to-mesenchymal transition. The wound healing assays of DU145 cells with various concentrations of LPS stimulation for 24 hrs were conducted (A) and the data were presented as an average of three independent experiments (C). The wound healing assays of DU145 cells with LPS stimulation (0.01 μg/mL) at indicated period of time were conducted (B) and the data were presented as an average of three independent experiments (D). The expression of CK8, CK18 and vimentin in DU145 cells treated with LPS at 0.01 μg/mL for indicated period of time was evaluated using qRT-PCR, and the ratio of CK8/vimentin or CK18/vimentin was presented (E). The invasion assays of DU145, PC3 and RWPE-1 cells with LPS treatment at 0.01 μg/mL were conducted (F) and the data were presented as an average of three independent experiments (G). (By comparing with control group, *p<0.05; **p<0.01; ***p<0.001). |
To evaluate the LPS-induced self-renewal ability of malignant cells, DU145 and RWPE-1 cells were pre-treated with LPS for 6 hrs followed by culturing in suspension condition to form stem cell-like spheres. The result showed that both DU145 and RWPE-1 formed spheres after suspension culture, but the spheres derived from DU145 were bigger than those from RWPE-1 (Figure 2A). And treatment with LPS (0.01 μg/mL) significantly increased both the size and numbers of spheres derived from DU145 in comparison with the no-LPS treatment group (Figure 2A and B). We also found that the phenomena of LPS-induced spheres formation were common in other PCa cells (Figure 2B). To further determine its potency of clonal formation and propagation, the cells composed in the above LPS-induced tumorospheres were harvested, singularized and re-cultured in 2D and 3D culture condition. The results also showed that there were significantly higher numbers of cells in the LPS-induced tumorospheres than in the control group (Figure 2C and D, p<0.001). Taken together, the results demonstrated that LPS induced PCa epithelial cells undergoing EMT with an enhance of migration and invasion, a decrease in the ratio of CK8/vimentin and CK18/vimentin, and an increase in sphere formation.
 |
Figure 2 LPS-induced tumorosphere formation. (A) DU145 and RWPE-1 cells were pre-treated with indicated concentration of LPS followed by spheres forming assay. The formed spheres were photographed (10 ×). (B) Prostate RWPE-1, 22RV-1, LNCaP, PC3 and DU145 cells were treated with LPS (0.01 μg/mL) respectively and then were cultured in suspension condition for tumorosphere forming assay. (C) The DU145 tumorosphere cells were singularized by trypsin-EDTA treatment, and were re-seeded and cultured in 2D monolayer condition with complete growth medium. The cell number was counted at day 7. (D) The DU145 tumorosphere cells were singularized by trypsin-EDTA treatment and were cultured in 3D culture condition. The cell number was counted. (By comparing with control group, *p<0.05; **p<0.01; ***p<0.001). |
LPS Augmented Prostate Cancer Stemness
To investigate the stemness of above LPS-induced tumorosphere cells, flow cytometry assay was conducted to sort the defined stem cells with double positive of CD44+CD133+ expression in singularized DU145 tumorosphere cells. The data indicated that treatment with LPS at different concentrations increased the number of CD44+CD133+ stem cells (11.67%, 14.93%) in comparison with the no-LPS treatment control (5.86%) significantly (Figure 3A). Furthermore, RT-PCR was utilized to profile genes expression of cancer stem cell-associated biomarkers in DU145 tumorosphere cells. And the results also showed that LPS stimulated a series of cancer stem cell-associated biomarker transcription (e.g., α-intergrin, CD44, Nestin, OCT-4, Nanog, BCRP) and increased their mRNA levels (Figure 3B). Similar results were also obtained in PC3 and RWPE-1 cells (Figure 3A and B). Thus, these data demonstrated that treatment with bacterial LPS augmented cancer stemness features in PCa epithelial cells.
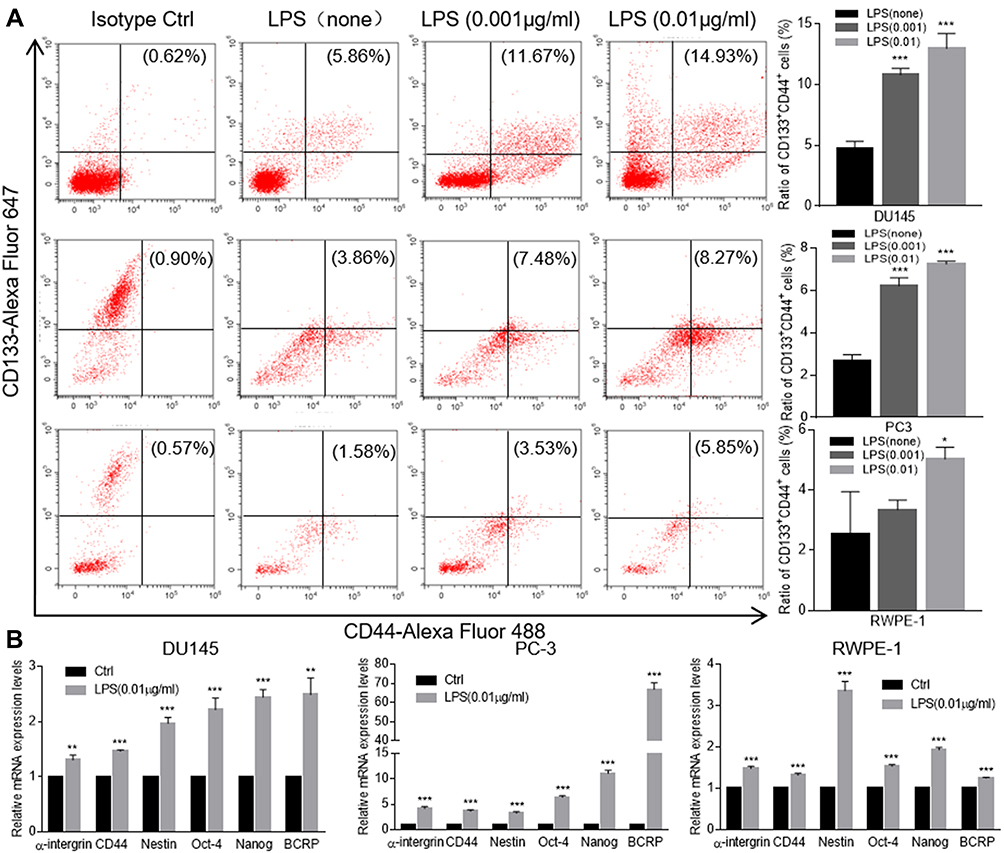 |
Figure 3 LPS promoted prostate cancer stemness. (A) The FACS assay was conducted to detect the CD44+CD133+ populations in LPS-induced DU145, PC3, RWPE-1 tumorospheres. A representative FACS result was presented (left panel). And the above FACS data were also presented as an average of three repeats (right figure). (B) The LPS-induced tumorospheres from DU145, PC3, RWPE-1 cells were harvested and total cellular RNA was extracted. The relative expression of cancer stem cell-associated genes of α-intergrin, CD44, Nestin, OCT-4, Nanog and BCRP were determined with qRT-PCR and normalized with GAPDH. (by comparing with control group, *p<0.05; **p<0.01; ***p<0.001). |
TSA Restrained LPS-Induced DU145 Cancer Cells Undergoing Migration, Invasion and Tumorosphere Formation
As originally found to be an antifungal antibiotic and later to be proved as a classic panel HDACs inhibitor,30,31 TSA was well documented to possess anti-tumor activities. To study the effect of TSA on epithelial transformation, DU145 cells were treated with different doses of TSA with or without LPS (0.01 μg/mL) for 24 hrs followed by a wound healing assay. The result showed that treatment with LPS alone healed the wounded monolayer cells by narrowing the wounded gap area more rapidly in comparison with the no treatment control group. Meanwhile, treatment with different doses of TSA alone showed a larger gap area than it in no treatment control group. Importantly, the results from combination treatment of TSA with LPS showed an increase in the area of the gap in a dose-dependent manner in comparison with LPS treatment alone control (Figure 4A and B). Similar results were also obtained by utilizing PC3 (Supplementary Figure 3A and C) and RWPE-1 cells (Supplementary Figure 3B and D). This data indicated that TSA appeared to inhibit both spontaneously occurred and LPS-induced tumor cell migration and retarded the wound healing process.
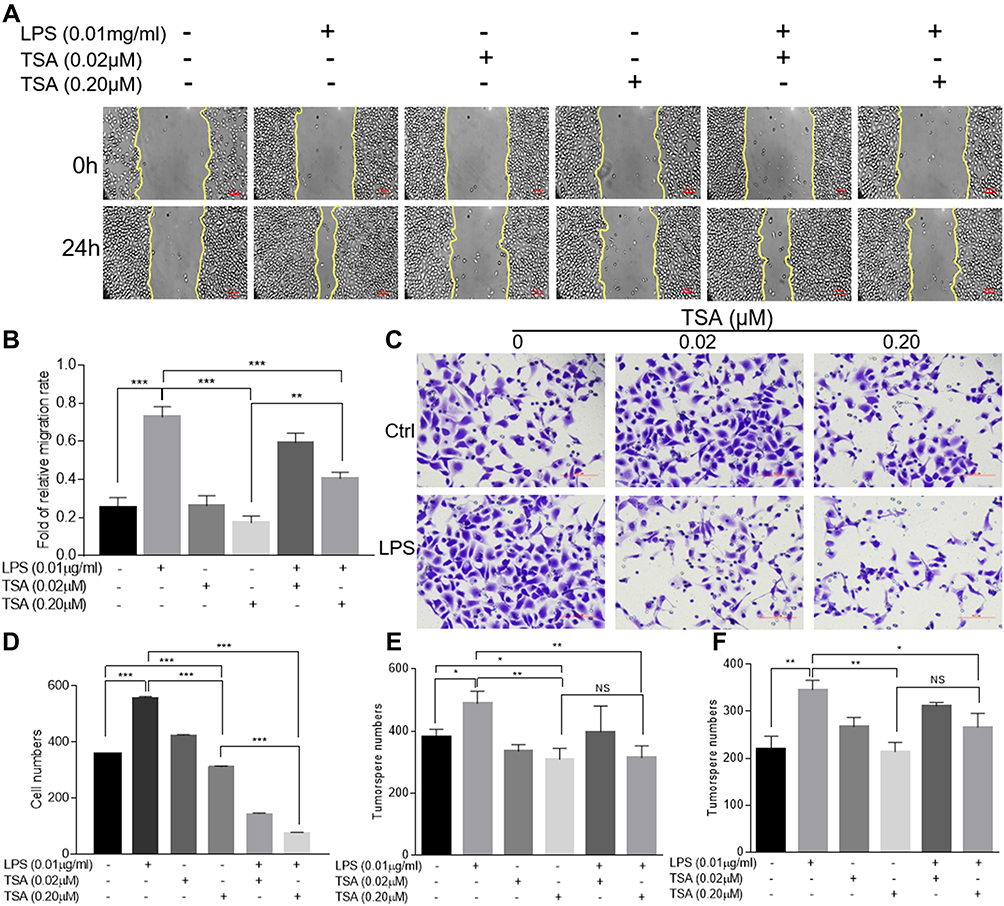 |
Figure 4 TSA restrained LPS-induced migration, invasion and tumorosphere formation. DU145 cells were treated with LPS at 0.01 μg/mL and/or indicated concentration of TSA for 24 hrs and the wound healing assays were conducted. The representative results were photographed (A), and the data were presented as an average of three independent experiments (B). DU145 cells pre-treated with TSA and LPS for 24 hrs and the invasion assay was conducted. The invaded cells with crystal violet staining were photographed and counted. A representative result (C) and an average of three independent repeats (D) were presented. The PCa cells were pre-treated with LPS (0.01 μg/mL) and/or indicated concentration of TSA. The tumorosphere forming assay was conducted. The formed tumorosphere number from DU145 (E) and PC3 (F) were counted and presented as an average of three independent repeats. (*p<0.05; **p<0.01; ***p<0.001). Abbreviation: NS, not significant. |
Next, a transwell assay was conducted to investigate the effects of TSA on invasion activity of LPS-treated DU145 cancer cells. The result showed that, in addition to increasing the invaded cell numbers significantly by LPS treatment alone (p>0.001), treatment with TSA alone from lower to higher concentration also tended to reduce the invaded cell numbers, typically at higher concentration of TSA (0.2 μM, p<0.05, Figure 4C and D). Notably, pretreatment with TSA significantly decreased the invaded cancer cell numbers of LPS-treated groups in comparison with other groups (Figure 4C and D, p<0.001). Similar results were also obtained by utilizing PC3 (Supplementary Figure 4A and B) and RWPE-1 cells (Supplementary Figure 4C and D). The data demonstrated that TSA restrained LPS-induced cancer cells invasion.
To evaluate the effect of TSA on cancer stem-like tumorosphere formation, DU145 and PC3 cells were pre-treated with TSA for 2 hrs prior to adding LPS for another 6 hrs. The cells were then cultured in suspension condition for tumorosphere forming assay. The result showed that, in compared with no treatment control group, treatment with LPS alone increased both DU145 and PC3 tumorosphere numbers significantly, but treatments with TSA at two doses alone reduced the tumorosphere numbers of DU145 cell, and the effects of TSA on tumorospher formation in PC3 cell were also trending toward inhibitory along with the increase in the concentration. Meanwhile, treatment with TSA decreased the LPS-induced tumorosphere number significantly. Notably, a higher dose of TSA (0.2 µM) showed more inhibitory effect on tumorosphere formation than a lower dose (0.02 µM) (Figure 4E and F). Thus, it could be concluded that treatment with TSA prevented prostate stem-like tumorosphere formation.
TSA Inhibited LPS-Induced Stat3 Phosphorylation and c-Myc Expression
Abnormal activation of cellular signaling could occur on a broad spectrum of human cells and tissues in response to bacterial LPS stimulation. To test whether the above suppressive effects of TSA on LPS-induced EMT and stem-like tumorosphere formation were through preventing LPS-activated abnormal cell signaling transduction, DU145 cells were treated with LPS with or without TSA, and the expression of cell signaling-related molecules were analyzed by WB. The result showed that, in line with others report,15,16,32 treatment with LPS alone increased the level of both c-Myc and p-Stat3. And treatment with TSA at 0.2 µM significantly reduced LPS-induced the level of p-Stat3, p-Erk, p-c-jun and decreased c-Myc expression (Figure 5A). It was also noticed that lower dose of TSA (0.02 µM) increased c-Myc expression, and higher dose of TSA (0.2 µM) increased NF-κB (p65) level (Figure 5A). Meanwhile, increasing tumor suppressive Maspin level and reducing IL-6 and IL-8 expression were also observed in DU145 cell after TSA treatment (Figure 5A and E).
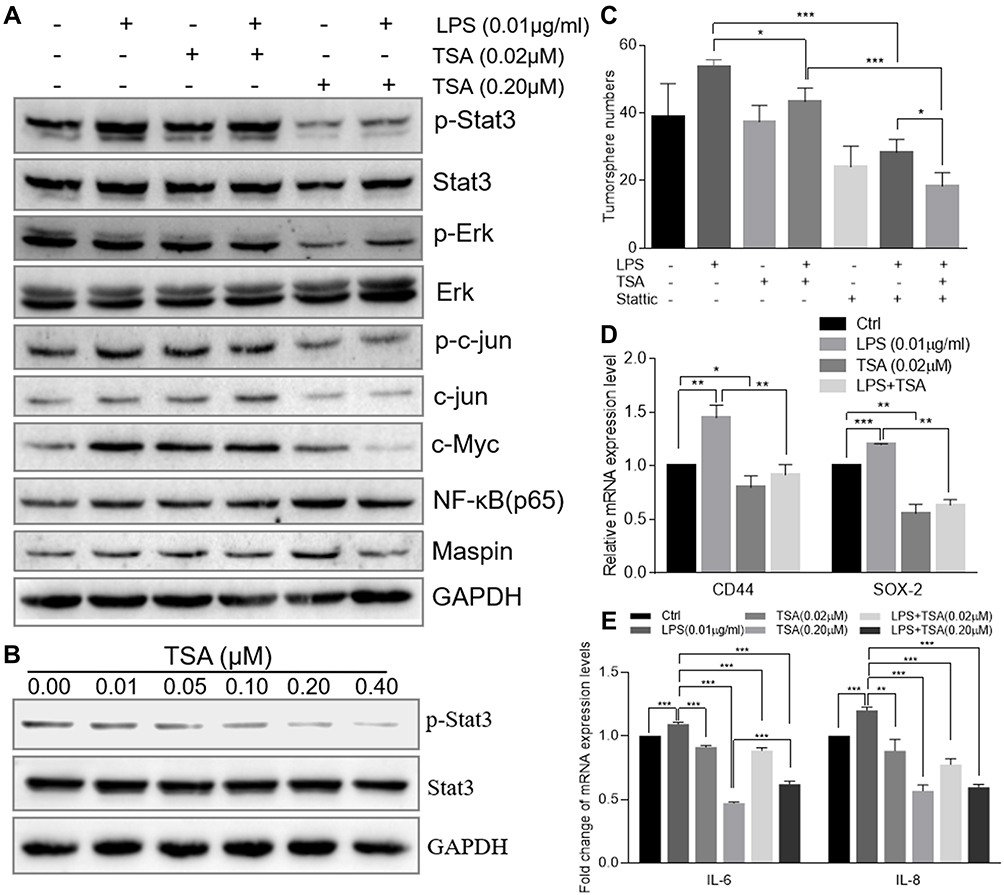 |
Figure 5 TSA prevented Stat3 phosphorylation and reduced LPS-induced cancer stem-like tumorosphere formation. (A) The total cellular protein of DU145 cell treated with LPS at 0.01 µg/mL and/or TSA was extracted by RIPA lysis buffer. The protein level of p-Stat3, Stat3 and GAPDH were evaluated by Western blot. (B) The total cellular protein of DU145 cell treated with variant concentrations of TSA was extracted by RIPA lysis buffer and the protein level of p-Stat3, Stat3 and GAPDH were evaluated by Western blot. (C) The DU145 cell was treated with TSA (0.02 µM) and/or Stattic (2 µM) and/or LPS (0.01 µM) followed by the tumorosphere forming assay. The formatted tumorospheres were counted and presented. (D) After LPS and/or TSA treatment, the relative expression of cancer stem cell-associated genes CD44 and SOX-2 in DU145 tumorosphere cells was determined with qRT-PCR and normalized with internal control GAPDH. (E) DU145 cancer cells were treated with TSA and /or LPS (0.01 µM) for 48 hrs and total cellular RNA was extracted. The relative expression of IL-6, IL-8 genes were determined with qRT-PCR and normalized with internal control GAPDH. (*p<0.05; **p<0.01; ***p<0.001). |
Stat3 signaling pathway played a crucial role in chronic inflammation-inducedStat3 phosphorylation. The result showed that TSA indeed inhibited Stat3 phosphorylation in a dose-dependent manner (Figure 5B).
Stattic is a Stat3 phosphorylation-mediatedStat3 phosphorylation on tumorosphere formation, DU145 cells were pre-cultured with TSA and/or Stattic for 2 hrs followed by addition of LPS and cultured continually for another 6 hrs. The cells were then harvested and re-cultured in suspension condition for tumorosphere forming assay. The result showed that treatment with a single agent of TSA or Stattic alone decreased LPS-induced stem-like tumorosphere numbers significantly. And combination treatment with TSA and Stattic further decreased the tumorosphere numbers despite no complete elimination (Figure 5C). Furthermore, the result from qRT-PCR analysis also showed that treatment with TSA inhibited both spontaneous-occurring and LPS-induced stemness biomarker CD44 and SOX-2 expression significantly in tumorosphere cells (Figure 5DStat3 phosphorylation was indispensable, at least in part, for TSA-restrained LPS-induced EMT and cancer stem-like tumorosphere formation.
Discussion
Knowing from the acute inflammation, which is a short-term response that usually results in healing, the chronic inflammation is defined as a prolonged, dysregulated and maladaptive response to inflammatory agents or factors. The accumulated evidence also showed that the core of chronic inflammation-induced cell malignant transformation lies in chronic and continuous stimulation with a low-dose of inflammatory agent or factors. Such persistent inflammation is associated with many chronic human conditions and diseases, including cancer development and autoimmune diseases.33,34 PCa is the third-leading cause of cancer-related men’s death due to the high rate of forming castration-resistant PCa with the most aggressive and malignant progress.35 The inflammatory environment derived from primary and/or adaptive pathogenic events has been demonstrated to play a critical role for prostate carcinogenesis.36,37 Meanwhile, the chronic inflammation-induced malignant transformation is usually accompanied by abnormal epigenetic modification with up regulation of oncogenes expression and/or down regulation of tumor suppressor level. Thus, the strategy of targeting both abnormal epigenetic modulators and inflammatory environments offers a novel approach for PCa treatment clinically. However, application of targeting epigenetic regulators (epi-drugs) affected global changes of cellular phenotypes and gene expression due to alteration of a gene chromatin environment.38,39 In fact, as an antifungal antibiotic and a classical HDAC inhibitor, epi-drug TSA showed a broad anti-tumor activity in vitro and in vivo.40–42 Recently, TSA was reported to reverse the EMT and decrease PCa tumorigenicity.22,41,43 Our studies also showed that inhibition of HDAC activity by TSA or other HDAC inhibitors reversed EMT and attenuated the processing of cell invasion and migration.29,44,45 However, the opposite effects of TSA on promoting cell transformation and proliferation were also observed. For instance, PCa cells developed resistance to TSA.46 TSA induced pleiotropic effects on PCa cells including increase of c-Myc and NF-κB expression.47,48 Therefore, the biological activity of TSA on EMT needs to be further clarified. Currently, the implication of epi-drugs still shows promising results in cancer clinics.49–51
In this study, we attempted to test LPS-induced malignant transformation in prostate epithelial cells by utilizing multiple tools including stem-like tumorosphere forming assay. Indeed, treatment with lower dose of LPS (less than 0.02 μg/mL), rather than with higher dose of LPS, promoted the epithelial cell undergoing migration, invasion with down regulation of CK8/vimentin and CK18/vimentin (Figure 1). Presumably, a lower concentration of LPS induced chronic inflammation and was responsible for the induction of EMT with alteration of biological activity (migration, invasion) and molecular phenotype (CK8/vimentin, CK18/vimentin), but a higher dose of LPS stimulated acute inflammatory reaction. Notably, the lower dose of LPS also increased stem-like tumorosphere formation in multiple prostatic epithelial cell lines (Figure 2A and B) with increase of clonogenic growth in 2D and 3D culture condition (Figure 2C and D). Furthermore, treatment with LPS enriched double positive CD44+CD133+ stem cells in the tumorospheres with augment of stem cell biomarkers expression in PCa DU145 cell (Figure 3). Although the LPS-induced tumorosphere cells were not tested to generate tumor in vivo in current study, this matter of fact was already demonstrated in our previous study and other's investigation.4,52
It was also aware that LPS alone induced fewer spheres with small size in normal RPEW-1 cells (Figure 2), this may be due to a short period of LPS treatment time or un-optimized treatment strategy. Further investigation found that treatment with TSA alone increased tumor suppressive Maspin expression, and TSA prevented LPS-induced stem-likeStat3 phosphorylation, down-regulating IL-6, IL-8, oncogene c-Myc and stem cell biomarker CD44 expression, which may contribute to abrogate LPS-induced malignant transformation and cancer stem cells formation (Figure 5). Although TSA showed inhibitory trend on tumor cells invasion along with an increase in the concentration, a lower dose of TSA alone (0.02 μM) was observed to increase cancer cells invasion (Figure 4B) and c-Myc expression, and a higher dose of TSA (0.2 μM) augmented NF-κB expression (Figure 5A). This data suggests that various effects of TSA on cell biology could occur in response to the implemented concentration and/or cell type.53,54 In addition, the phenomena also reflected the likely reason for the failure of multiple HDAC inhibitors in clinical trials.38,55 Thus, in terms of a certain disease with the individual pathologic tissue/cell types, a careful pharmacodynamic evaluation should be carried out by clinician prior to implementation of individual HDAC inhibitors clinically to avoid the side effect. Meanwhile, appropriate combination of epi-drugs (e.g., TSA) with other agent to maximize its therapeutic effect with minimizing the side effects would be an ideal strategy in a clinical practice.
Studies from basic and clinical investigation showed that, possessing the capacity to self-renew, cancer stem cells (CSCs) exist in a tumor with a very small population and were believed to cause the heterogeneous lineages of cancer cells that comprise the tumor. And been different from cancer cells which in general display higher intrinsic or spontaneous morphological, phenotypic, and functional plasticity compared to fully differentiated normal cells, CSC perhaps mainly undergo one-way maturation by developing into tumor progenitors and even into differentiated tumor cells.56,57 Thus, with its highly tumorigenic feature, CSC was evidenced and showed to be possibly responsible for the initiation and progression of cancer and for the invasive and metastatic disease. Thus, CSC was considered as the root of the cancer disease.58,59 Prostate CSC was reported to have a CD44+/alpha2beta1hi/CD133+ phenotype with approximately 0.1% population of tumor cells, which possess a significant capacity for self-renewal.59 Data from molecular profiling showed that PCa cells with EMT phenotype displayed stem-like cell features of increased expression of Sox2, Nanog, Oct4, Lin28B and/or Notch1, but decreased expression of miR-200 and/or let-7 family. These CSC-like cells also displayed an enhanced both sphere (prostasphere)-forming and clonogenic ability and tumorigenecity.60 Further study also found that inflammatory cytokine IL-6 was sufficient to convert none stem cancer cells into CSCs (CD44+/CD133+) and to sphere-forming CSCs in a prostate PC3 cell.57 Experimental evidence also showed that eradicating PCa stem cells improved the prognosis for patients with advanced stages of the disease. Thus, targeting CSC has been considered as a promising treatment strategy in PCa clinic practice.61,62
The origin and progress of malignant tumors were featured with EMT and unrestricted cell proliferation/tumor growth. The somatic mutation-induced malignant transformation is the most popular and acceptable theory for cancer origin since its being enunciated in 1914. In addition, multiple factors were also reported to induce epithelial malignant transformation, including those molecules generated from inflammatory tissues and cells. Although both external stimuli of pro-inflammatory molecules, and internal inflammatory cells infiltrated into tumor stroma were reported to play critical role in the process of malignant transformation and tumorigenesis, the theory of “chronic inflammation-induced cancer” has being suspected with constant debating due to lack of solid and direct evidence. Meanwhile, the cancer stem cell hypothesis was also reported to be associated with tumorigenesis, malignant progress and therapeutic resistance. And individual pro-inflammatory cytokines (e.g., IL-6) was able to convert non-stem cancer cells to cancer stem cells.57 Thus, considering our results, it could be speculated that, in a chronic inflammatory microenvironment, a persistent stimulation by bacterial agent and/or inflammatory factors could induce epithelial malignant transformation, which is responsible, at least in part, for cancer stem cells generation and prostatic carcinogenesis. Moreover, the attention also needs to be paid that the presence of bacterial agent and/or inflammatory factors (e.g., LPS) not only impact the epithelial cells but also affect the immune cells and other stroma compounds existing in the microenvironment, which may produce a cross interaction among the different components, make it more complicate for the understanding disease progresses, and put more challenge for the clinical treatment.
Conclusion
Our data demonstrated that the inflammatory agent of bacterial LPS augmented malignant transformation and promoted cancer stemness in PCa epithelial cells. TSA could prevent the LPS-induced malignant transformation by targeting p-Stat3/c-Myc signaling pathway and reducing inflammatory IL-6, IL-8. Thus, appropriate management and treatment of bacterial infection are crucial for preventing the chronic infectious inflammation-induced prostate epithelial malignant transformation and cancer origination.
Abbreviations
LPS, lipopolysaccharide; TSA, trichostatin A; EMT, epithelial-mesenchymal transformation; HDAC, histone deacetylase; CSC, cancer stem cell.
Data Sharing Statement
The data and materials used or analyzed in the current study are available from the corresponding author upon reasonable request.
Consent for Publication
Part of the preliminary data was presented as an abstract at CUA 2017 Annual Meeting (Hefei, China) and AACR 2019 Annual Meeting (Atlanta, USA).
Author Contributions
Most of the data were generated by ST and Lian X. HC, JG, and DN helped to conduct the experiments and generate the data. XG provided the clinic information and consultation for drug treatment. HM helped to design the experiment and discussed the data. JJ conducted a part of the study with flow cytometry data generation, managed the research and collected the raw data for finalizing the project. Li X designed the project, directed the research, evaluated the data and finalized the manuscript. All authors participated in data discussion, read and approved the final manuscript. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by National Natural Science Foundation of China (81572741) and Wu Jieping Medical Foundation (320.6750.14120) to Li X, by Zhangjiagang Science and Technology support program (ZKS2023, ZKS2047), and by Jiangsu University Clinical Medicine Science and Technology Development Foundation (JLY2021099). The funding sponsors had neither role in the study design, collection, analysis or interpretation of data, nor in writing the report.
Disclosure
The authors declare no potential conflicts of interest in this work.
References
1. Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi:10.1016/j.cell.2008.03.027
2. Bessede E, Staedel C, Acuna Amador LA, et al. Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene. 2014;33(32):4123–4131. doi:10.1038/onc.2013.380
3. Shuang ZY, Wu WC, Xu J, et al. Transforming growth factor-beta1-induced epithelial-mesenchymal transition generates ALDH-positive cells with stem cell properties in cholangiocarcinoma. Cancer Lett. 2014;354(2):320–328. doi:10.1016/j.canlet.2014.08.030
4. Weiswald LB, Bellet D, Dangles-Marie V. Spherical cancer models in tumor biology. Neoplasia. 2015;17(1):1–15. doi:10.1016/j.neo.2014.12.004
5. Tsunekuni K, Konno M, Haraguchi N, et al. CD44/CD133-positive colorectal cancer stem cells are sensitive to trifluridine exposure. Sci Rep. 2019;9(1):14861. doi:10.1038/s41598-019-50968-6
6. Weissenrieder JS, Reed JL, Green MV, et al. The dopamine D2 receptor contributes to the spheroid formation behavior of U87 glioblastoma cells. Pharmacology. 2020;105(1–2):19–27. doi:10.1159/000502562
7. Lee CH, Yu CC, Wang BY, Chang WW. Tumorsphere as an effective in vitro platform for screening anti-cancer stem cell drugs. Oncotarget. 2016;7(2):1215–1226. doi:10.18632/oncotarget.6261
8. Ellner HJ, Miller GC. Prostatic abscess simulating carcinoma. Northwest Med. 1964;63:174–175.
9. Jameson RM. Prostatic abscess and carcinoma of the prostate. Br J Urol. 1968;40(3):288–292. doi:10.1111/j.1464-410X.1968.tb09889.x
10. Ohadian Moghadam S, Momeni SA. Human microbiome and prostate cancer development: current insights into the prevention and treatment. Front Med. 2021;15(1):11–32.
11. Bruggemann H, Al-Zeer MA. Bacterial signatures and their inflammatory potentials associated with prostate cancer. APMIS. 2020;128(2):80–91. doi:10.1111/apm.13021
12. Cario E, Rosenberg IM, Brandwein SL, Beck PL, Reinecker HC, Podolsky DK. Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing Toll-like receptors. J Immunol. 2000;164(2):966–972. doi:10.4049/jimmunol.164.2.966
13. Li H, Li Y, Liu D, Liu J. LPS promotes epithelial-mesenchymal transition and activation of TLR4/JNK signaling. Tumour Biol. 2014;35(10):10429–10435. doi:10.1007/s13277-014-2347-5
14. Li XY, Yang X, Zhao QD, et al. Lipopolysaccharide promotes tumorigenicity of hepatic progenitor cells by promoting proliferation and blocking normal differentiation. Cancer Lett. 2017;386:35–46. doi:10.1016/j.canlet.2016.10.044
15. Tian QX, Zhang ZH, Ye QL, et al. Melatonin inhibits migration and invasion in LPS-stimulated and -unstimulated prostate cancer cells through blocking multiple EMT-relative pathways. J Inflamm Res. 2021;14:2253–2265. doi:10.2147/JIR.S305450
16. Xing WY, Zhang ZH, Xu S, et al. Calcitriol inhibits lipopolysaccharide-induced proliferation, migration and invasion of prostate cancer cells through suppressing STAT3 signal activation. Int Immunopharmacol. 2020;82:106346. doi:10.1016/j.intimp.2020.106346
17. Nam HW, Bae J, Kim YW, et al. Anti-cancer effects of RAW 264.7 cells on prostate cancer PC-3 cells. Ann Clin Lab Sci. 2020;50(6):739–746.
18. Lee CF, Dang A, Hernandez E, et al. Activation of sphingosine kinase by lipopolysaccharide promotes prostate cancer cell invasion and metastasis via SphK1/S1PR4/matriptase. Oncogene. 2019;38(28):5580–5598. doi:10.1038/s41388-019-0833-3
19. Li J, Chong T, Wang Z, et al. A novel anticancer effect of resveratrol: reversal of epithelial mesenchymal transition in prostate cancer cells. Mol Med Rep. 2014;10(4):1717–1724. doi:10.3892/mmr.2014.2417
20. Huo SF, Shang WL, Yu M, et al. STEAP1 facilitates metastasis and epithelial-mesenchymal transition of lung adenocarcinoma via the JAK2/STAT3 signaling pathway. Biosci Rep. 2020;40(6). doi:10.1042/BSR20193169.
21. Huang R, Wang S, Wang N, et al. CCL5 derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating beta-catenin/STAT3 signaling. Cell Death Dis. 2020;11(4):234. doi:10.1038/s41419-020-2435-y
22. Wang X, Xu J, Wang H, et al. Trichostatin A, a histone deacetylase inhibitor, reverses epithelial-mesenchymal transition in colorectal cancer SW480 and prostate cancer PC3 cells. Biochem Biophys Res Commun. 2015;456(1):320–326. doi:10.1016/j.bbrc.2014.11.079
23. Xu F, Liu Z, Liu R, et al. Epigenetic induction of tumor stemness via the lipopolysaccharide-TET3-HOXB2 signaling axis in esophageal squamous cell carcinoma. Cell Commun Signal. 2020;18(1):17. doi:10.1186/s12964-020-0510-8
24. Huang CF, Chen L, Li YC, et al. NLRP3 inflammasome activation promotes inflammation-induced carcinogenesis in head and neck squamous cell carcinoma. J Exp Clin Cancer Res. 2017;36(1):116. doi:10.1186/s13046-017-0589-y
25. Fan F, Wang R, Boulbes DR, et al. Macrophage conditioned medium promotes colorectal cancer stem cell phenotype via the hedgehog signaling pathway. PLoS One. 2018;13(1):e0190070. doi:10.1371/journal.pone.0190070
26. Ou T, Lilly M, Jiang W. The pathologic role of Toll-Like receptor 4 in prostate cancer. Front Immunol. 2018;9:1188. doi:10.3389/fimmu.2018.01188
27. Kundu SD, Lee C, Billips BK, et al. The toll-like receptor pathway: a novel mechanism of infection-induced carcinogenesis of prostate epithelial cells. Prostate. 2008;68(2):223–229. doi:10.1002/pros.20710
28. Omabe M, Omabe K, Okwuegbu M, Grace O, Okoro DU. Exposure of prostate to lipopolysaccharide and hypoxia potentiates neoplastic behavior and risk for prostate carcinogenesis in vivo. Int Sch Res Notices. 2014;2014:420429.
29. Bernardo MM, Meng Y, Lockett J, et al. Maspin reprograms the gene expression profile of prostate carcinoma cells for differentiation. Genes Cancer. 2011;2(11):1009–1022. doi:10.1177/1947601912440170
30. Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265(28):17174–17179. doi:10.1016/S0021-9258(17)44885-X
31. Li X, Chen BD. Histone deacetylase inhibitor M344 inhibits cell proliferation and induces apoptosis in human THP-1 leukemia cells. Am J Biomed Sci. 2009;1(4):352–363. doi:10.5099/aj090400352
32. Liao L, Zhang XD, Li J, et al. Pioglitazone attenuates lipopolysaccharide-induced depression-like behaviors, modulates NF-kappaB/IL-6/STAT3, CREB/BDNF pathways and central serotonergic neurotransmission in mice. Int Immunopharmacol. 2017;49:178–186. doi:10.1016/j.intimp.2017.05.036
33. Weiss U. Inflammation. Nature. 2008;454(7203):427. doi:10.1038/454427a
34. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi:10.1038/nature07205
35. iegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30.
36. De Nunzio C, Kramer G, Marberger M, et al. The controversial relationship between benign prostatic hyperplasia and prostate cancer: the role of inflammation. Eur Urol. 2011;60(1):106–117. doi:10.1016/j.eururo.2011.03.055
37. Sciarra A, Gentilucci A, Salciccia S, et al. Prognostic value of inflammation in prostate cancer progression and response to therapeutic: a critical review. J Inflamm. 2016;13:35. doi:10.1186/s12950-016-0143-2
38. Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours–lessons from the past. Nat Rev Clin Oncol. 2013;10(5):256–266. doi:10.1038/nrclinonc.2013.42
39. Zhang T, Pilko A, Wollman R. Loci specific epigenetic drug sensitivity. Nucleic Acids Res. 2020;48:4797–4810.
40. De Souza C, Chatterji BP. HDAC inhibitors as novel anti-cancer therapeutics. Recent Pat Anticancer Drug Discov. 2015;10(2):145–162. doi:10.2174/1574892810666150317144511
41. Zhang H, Zhao X, Liu H, Jin H, Trichostatin JY. A inhibits proliferation of PC3 prostate cancer cells by disrupting the EGFR pathway. Oncol Lett. 2019;18(1):687–693.
42. Liu X, Zhang Y, Gao H, et al. Induction of an MLKL mediated non-canonical necroptosis through reactive oxygen species by tanshinol A in lung cancer cells. Biochem Pharmacol. 2020;171:113684. doi:10.1016/j.bcp.2019.113684
43. Zhou R, Yang Y, Park SY, et al. p300/CBP-associated factor promotes autophagic degradation of delta-catenin through acetylation and decreases prostate cancer tumorigenicity. Sci Rep. 2019;9(1):3351. doi:10.1038/s41598-019-40238-w
44. Dzinic SH, Bernardo MM, Li X, et al. An essential role of maspin in embryogenesis and tumor suppression. Cancer Res. 2017;77(4):886–896. doi:10.1158/0008-5472.CAN-16-2219
45. Tang S, Lian X, Jiang J, et al. Tumor suppressive maspin-sensitized prostate cancer to drug treatment through negative regulating androgen receptor expression. Front Cell Dev Biol. 2020;8:573820. doi:10.3389/fcell.2020.573820
46. Taghiyev AF, Guseva NV, Sturm MT, Rokhlin OW, Cohen MB. Trichostatin A (TSA) sensitizes the human prostatic cancer cell line DU145 to death receptor ligands treatment. Cancer Biol Ther. 2005;4(4):382–390. doi:10.4161/cbt.4.4.1615
47. Kong D, Ahmad A, Bao B, Li Y, Banerjee S, Sarkar FH. Histone deacetylase inhibitors induce epithelial-to-mesenchymal transition in prostate cancer cells. PLoS One. 2012;7(9):e45045. doi:10.1371/journal.pone.0045045
48. Han RF, Li K, Yang ZS, Chen ZG, Yang WC. Trichostatin A induces mesenchymal-like morphological change and gene expression but inhibits migration and colony formation in human cancer cells. Mol Med Rep. 2014;10(6):3211–3216. doi:10.3892/mmr.2014.2594
49. Benedetti R, Conte M, Iside C, Altucci L. Epigenetic-based therapy: from single- to multi-target approaches. Int J Biochem Cell Biol. 2015;69:121–131. doi:10.1016/j.biocel.2015.10.016
50. Duan YT, Sangani CB, Liu W, Soni KV, Yao Y. New promises to cure cancer and other genetic diseases/disorders: epi-drugs through epigenetics. Curr Top Med Chem. 2019;19(12):972–994. doi:10.2174/1568026619666190603094439
51. Tomaselli D, Lucidi A, Rotili D, Mai A. Epigenetic polypharmacology: a new frontier for epi-drug discovery. Med Res Rev. 2020;40(1):190–244. doi:10.1002/med.21600
52. Bernardo MM, Kaplun A, Dzinic SH, et al. Maspin expression in prostate tumor cells averts stemness and stratifies drug sensitivity. Cancer Res. 2015;75(18):3970–3979. doi:10.1158/0008-5472.CAN-15-0234
53. Wawruszak A, Kalafut J, Okon E, et al. Histone deacetylase inhibitors and phenotypical transformation of cancer cells. Cancers (Basel). 2019;11(2):148. doi:10.3390/cancers11020148
54. Huang K, Liu Y, Gu C, Liu D, Zhao B. Trichostatin A augments esophageal squamous cell carcinoma cells migration by inducing acetylation of RelA at K310 leading epithelia-mesenchymal transition. Anticancer Drugs. 2020;31(6):567–574. doi:10.1097/CAD.0000000000000927
55. McClure JJ, Li X, Chou CJ. Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv Cancer Res. 2018;138:183–211.
56. Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012;22(3):457–472. doi:10.1038/cr.2012.13
57. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011;108(4):1397–1402. doi:10.1073/pnas.1018898108
58. Konrad CV, Murali R, Varghese BA, Nair R. The role of cancer stem cells in tumor heterogeneity and resistance to therapy. Can J Physiol Pharmacol. 2017;95(1):1–15. doi:10.1139/cjpp-2016-0079
59. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946–10951. doi:10.1158/0008-5472.CAN-05-2018
60. Kong D, Banerjee S, Ahmad A, et al. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS One. 2010;5(8):e12445. doi:10.1371/journal.pone.0012445
61. Jaworska D, Krol W, Szliszka E. Prostate cancer stem cells: research advances. Int J Mol Sci. 2015;16(11):27433–27449. doi:10.3390/ijms161126036
62. Yun EJ, Zhou J, Lin CJ, et al. Targeting cancer stem cells in castration-resistant prostate cancer. Clin Cancer Res. 2016;22(3):670–679. doi:10.1158/1078-0432.CCR-15-0190
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.