Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 20
Azithromycin Mitigates Tobacco Smoke-Induced Lung Senescence by Modulating the FOXO3A/CCND1 Signaling Pathway
Authors Chen Z, He Q, Yi X, Li T, Wei X ![]() , Chen Q, Ning R, Liang H, He Z
, Chen Q, Ning R, Liang H, He Z ![]()
Received 11 June 2025
Accepted for publication 29 October 2025
Published 11 November 2025 Volume 2025:20 Pages 3631—3645
DOI https://doi.org/10.2147/COPD.S534997
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jill Ohar
Zhangrong Chen,1,* Qiaoli He,1,* Xiaofei Yi,2 Tingting Li,3 Xuan Wei,4 Quanfang Chen,1 Ruiling Ning,5 Hanlin Liang,1 Zhiyi He1
1Department of Respiratory and Critical Medicine, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, 530021, People’s Republic of China; 2Department of Respiratory and Critical Care, Chengdu Third People’s Hospital, Chengdu, Sichuan, 610014, People’s Republic of China; 3Ningxia Medical University General Hospital, Ningxia, Yinchuan, 750004, People’s Republic of China; 4Department of Respiratory and Critical Care Medicine, The Fourth Affiliated Hospital of Guangxi Medical University, Liuzhou, Guangxi, 545005, People’s Republic of China; 5Department of Medical Oncology of Respiratory, Guangxi Medical University Cancer Hospital, Nanning, Guangxi, 530012, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhiyi He, Department of Respiratory and Critical Care Medicine, The First Affiliated Hospital of Guangxi Medical University, No. 6 Shuang Yong Road, Nanning, Guangxi, 530021, People’s Republic of China, Tel +86 187 7801 7698, Email [email protected]
Introduction: Accelerated lung aging is observed in chronic obstructive pulmonary disease (COPD), this study delved into the precise mechanisms through which azithromycin mitigated lung aging associated with COPD.
Methods: Employing network pharmacology, we predicted potential pathways through which azithromycin might affect COPD development. We collected lung tissues from non-smoking individuals, smokers with normal lung function, and smokers with COPD. COPD models were created by exposing mice to cigarette smoke (CS) for 24 weeks and stimulating human bronchial epithelial cells (BEAS-2B) with 0.2% cigarette smoke extract (CSE) for 24 hours. Azithromycin was then given to CS-exposed emphysema mice. BEAS-2B cells were pre-treated with azithromycin before being exposed to CSE and JY-2, a Forkhead box O3 (FOXO3A) inhibitor. Senescence-associated secretory phenotype (SASP) cytokines (interleukin-6, interleukin-8) in mouse BALF were quantified using ELISA. Markers associated with cellular aging (β-galactosidase activity, p53, and p21), FOXO3A, and Cyclin D1 (CCND1) were assessed via qPCR, Western blot, immunohistochemistry, and β-galactosidase staining.
Results: COPD patients who smoked showed increased pulmonary expression of CCND1, p53, and p21, with decreased FOXO3A in comparison with other groups. Similarly, CS-exposed mouse lung tissue exhibited reduced FOXO3A and elevated p53, p21, and CCND1, along with higher SASP secretion in BALF. Azithromycin treatment lowered SASP secretion and decreased CCND1, p53, and p21 expression, while increasing FOXO3A. In BEAS-2B cells, CSE and JY-2 stimulation raised senescence markers and CCND1 while lowering FOXO3A. Azithromycin preconditioning reduced p53, p21, and CCND1 expression and increased FOXO3A.
Conclusion: Azithromycin demonstrated anti-aging properties and modulated lung senescence in COPD via the FOXO3A/CCND1 pathway, presenting fresh insights for the treatment of COPD.
Keywords: azithromycin, COPD, cigarette smoke, senescence, FOXO3A/CCND1 pathway
Introduction
Chronic obstructive pulmonary disease (COPD) is a highly prevalent and persistent respiratory disorder, characterized by significant global morbidity and mortality. In 2019, COPD accounted for 3.3 million deaths worldwide, with 212.3 million prevalent cases and 16.2 million new cases reported,1 imposing a substantial economic burden globally.
Organismal aging is characterized by a key biological hallmark: the accumulation of senescent cells, which undergo permanent cell-cycle arrest in response to various damaging stimuli.2,3 This is accompanied by a widespread decline in stem-cell regenerative capacity across tissues.
The pathogenesis of COPD is intricately associated with accelerated senescence, which promotes the premature aging of lung tissues.4,5 Tobacco smoking remains the primary risk factor for COPD onset. Cigarette smoke extract (CSE) induces injury to bronchial and alveolar epithelial cells by exacerbating oxidative stress, amplifying inflammatory responses, and disrupting mitochondrial homeostasis.6 The consequent decline in lung function is a key indicator of COPD progression, and cigarette smoke exposuresignificantly accelerates this functional impairment.7 Furthermore, tobacco smoke acts as a potent accelerator of lung aging and COPD development.8 Smokers with COPD exhibit markedly elevated levels of aging biomarkers (e.g., p53, p21), components of the senescence-associated secretory phenotype (SASP) such as interleukin-6 (IL-6) and interleukin-8 (IL-8), and β-galactosidase activity compared to non-COPD individuals.9–11 Conversely, levels of key anti-aging factors, including Sirtuin 1 (SIRT1), Sirtuin 6 (SIRT6), and Forkhead box O3 (FOXO3A), are substantially reduced.12–14 These findings collectively indicate that lung tissues in COPD manifest an accelerated aging process.
Over the past few years, the anti-aging potential of macrolide antibiotics has garnered growing interest. Studies have shown that rapamycin attenuates pulmonary senescence in mouse models of COPD by inhibiting the mTOR signaling pathway.15 Our group has previously demonstrated that erythromycin alleviates oxidative-stress-induced cellular senescence in COPD by modulating the PI3K–mTOR axis.16 To date, both erythromycin and rapamycin have been proven to mitigate COPD-related lung aging. In contrast, azithromycin (AZM) has traditionally been recognized for its antimicrobial and immunomodulatory properties. However, emerging evidence suggests that azithromycin also possesses anti-aging activities. For example, studies have demonstrated that AZM down-regulates NADPH oxidase 4 (NOX4) in human lung fibroblasts, thereby suppressing fibroblast-to-myofibroblast differentiation and exerting an anti-fibrotic effect.17 This finding is highly relevant given that the progression of idiopathic pulmonary fibrosis is closely linked to accelerated cellular senescence and myofibroblast differentiation.18 Furthermore, AZM has been shown to selectively clear senescent fibroblasts with minimal impact on non-senescent cells.19 Despite these promising observations, the mechanisms underlying AZM’s actions in COPD-related senescence remain largely unexplored and warrant further investigation.
In this study, we predicted the targets of AZM for COPD using network pharmacology and identified key targets and pathways through bioinformatic analysis. These findings were then validated in clinical samples, animal models, and in vitro experiments. Furthermore, inhibitors targeting these key pathways were employed to explore the mechanism by which AZM alleviates cellular senescence in COPD. Our work aims to inform future therapeutic strategies for delaying lung aging in COPD.
Materials and Methods
Network Pharmacology Methods and the Targets Genes of COPD
Utilize the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) to look up the 3D structure of azithromycin, only to find that it is available solely in 2D. Save this in sdf format and then upload the sdf format file to the PharmMapper website (http://lilab-ecust.cn/pharmmapper/index.html). All pharmacophore targets were exported, and all target designations were standardized to their official nomenclature by referencing the UniProt database (https://www.uniprot.org/). By searching through OMIM (https://omim.org/), TTD (https://omim.org/), and Genecards databases (https://www.genecards.org/), using “chronic obstructive pulmonary disease” as the keyword, redundant targets were eliminated, and the disease-related targets from the three databases were summarized (3431 targets in total). Pharmaceutical and disease targets were submitted into Venny2.1 (https://bioinfogp.cnb.csic.es/tools/venny/) to identify intersection genes, followed by their visualization with a Venn diagram (Figure 1A).
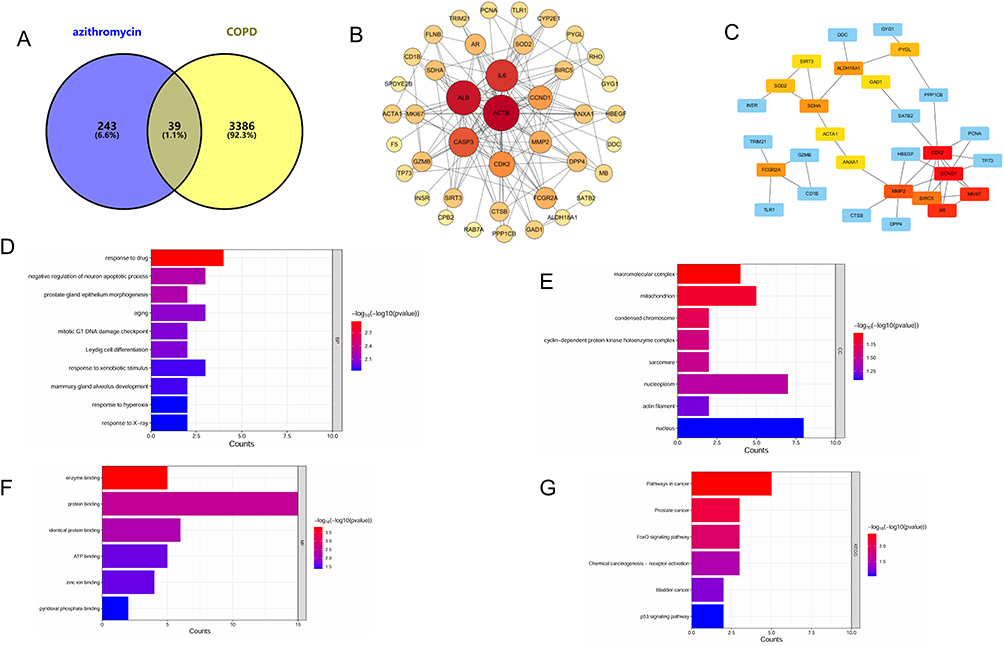 |
Figure 1 Network-pharmacology analysis of azithromycin binding interactions with COPD target genes.(A) Venn diagram showing the number of unique targets for azithromycin and COPD, respectively, and their overlapping shared genes.(B) A protein-protein interaction network depicting the interactions among the shared target genes.(C) A network map of the interactions among the key proteins. The 15 key proteins were subjected to functional enrichment analysis, including (D) Gene Ontology biological processes (GO-BP), (E) Gene Ontology cellular components (GO-CC), (F) Gene Ontology molecular functions (GO-MF), and (G) KEGG pathways. |
Import drug and disease targets into the STRING database (https://string-db.org/), discard targets that lack reciprocal interactions, and subsequently incorporate the interacting targets into Cytoscape to construct a PPI network. Meanwhile, the cytoHubba plugin within Cytoscape was employed to extract the 15 most critical proteins in the PPI network for visual analysis. Furthermore, these 15 pivotal proteins were submitted to the David database (https://david.ncifcrf.gov/) to conduct enrichment analyses for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG).
Patients and Samples
Specimens were gathered from 18 patients who underwent surgical treatment for lung cancer at the First Affiliated Hospital of Guangxi Medical University. The samples were taken from histologically normal-appearing lung tissue located >5 cm from the primary tumor. Patients were categorized into non-smoking normal individuals, smokers with normal lung function, and smokers with COPD according to smoking history and pulmonary function test outcomes, with similar age demographics across the groups. According to the GOLD criteria, patients with stable COPD exhibited an FEV1/FVC ratio of less than 70% following bronchodilator administration, and they had a definitive clinical diagnosis of COPD. Surgical patients had no severe lung or vital organ system lesions. All patients were informed, and all signed the informed consent form before the surgery. The study received ethical clearance from the Ethics Review Board at the First Affiliated Hospital of Guangxi Medical University (NO:2022-KT-guoji-039), and every aspect of the study adhered strictly to the principles set out in the Declaration of Helsinki. The clinical information of the patients is summarized in Table 1.
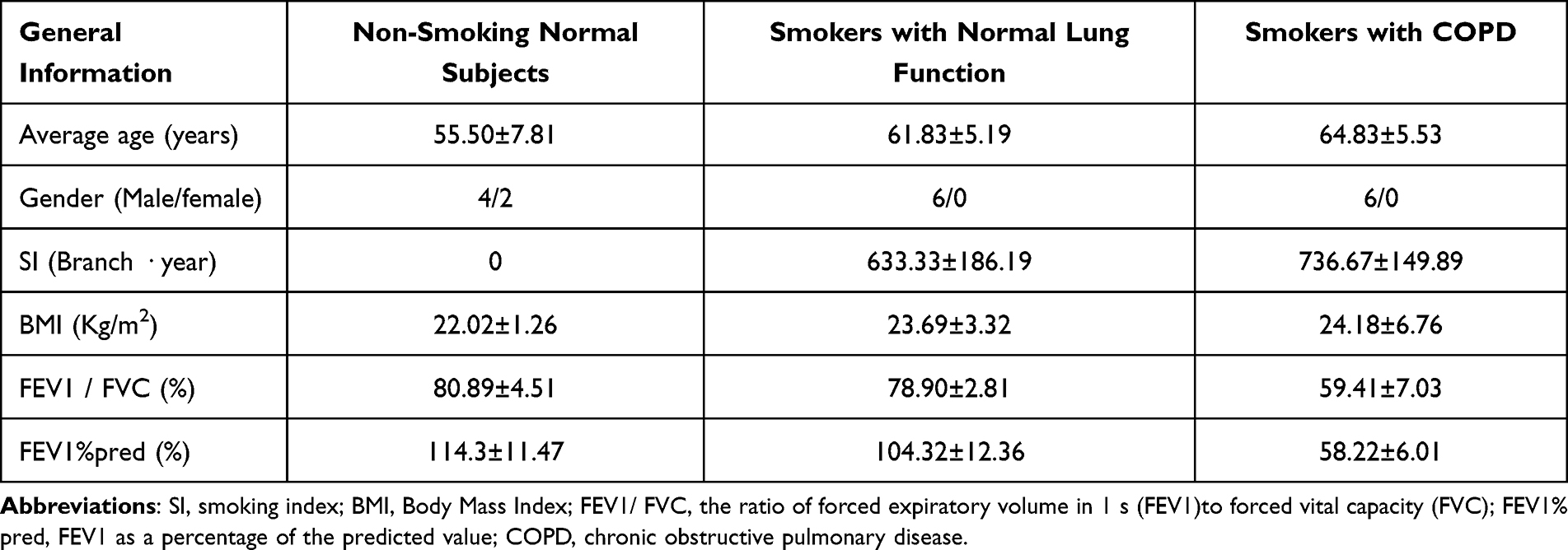 |
Table 1 Primary Clinical Information of Patients |
Animal Models of COPD
6- to 8-week-old C57BL/6J mice were randomly assigned to AIR, cigarette smoke exposure (CS), and azithromycin intervention (CS + AZM) groups. The AIR group was exposed to air for 24 weeks, while the CS and CS + AZM group underwent a 24-week smoke exposure period, as detailed in prior accounts.16,20 The CS + AZM group started gavage administration of azithromycin (Solarbio, Beijing, China) at 100mg/kg/day, the other groups received saline gavage. After modeling and anesthesia, lung tissue and bronchoalveolar lavage fluid (BALF) were extracted for further experiments. All animal experiments were approved by the Animal Ethics Committee of Guangxi Medical University (No.:202404008) and were conducted in strict compliance with the protocols outlined in the US National Institutes of Health (NIH) “Guide for the Care and Use of Laboratory Animals” and the relevant regulations of China’s “Implementation Rules for the Management of Laboratory Animals”.
Hematoxylin-Eosin (HE) Staining and Immunohistochemistry (IHC)
The mouse pulmonary tissue was placed in 4% paraformaldehyde, then processed for paraffin embedding, and sliced for HE and IHC. The slides were deparaffinized and progressively rehydrated with xylene and a series of ethanol solutions, then stained with HE and covered. Examined mouse lung tissue under a microscope for staining and pathology, and photographed it to calculate alveolar mean linear intercept (MLI).
The slides with fixed human and mouse lung tissue were deparaffinized and rehydrated using xylene and ethanol. Afterward, they were subjected to repair and inactivation processes, and were then incubated overnight at 4°C using antibodies against mouse FOXO3A (1:1000, proteintech66428-1-Ig, China), Cyclin D1 (CCND1) (1:1000, proteintech60186-1-Ig, China), p53 (1:3000, proteintech60283-2-Ig, China), and p21 (1:1000, proteintech10355-1-AP, China). After a night, the sectioned tissues underwent incubation with goat anti-mouse IgG secondary antibody alongside streptavidin, then stained using diaminobenzidine (Solarbio, DA1010, China) and hematoxylin (Biosharp, China). After acid alcohol differentiation, they were mounted and imaged for analysis.
Cell Culture and Treatment
BEAS-2B cells were obtained from China Typical Culture Preservation Center (CTCPC, China) and maintained in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum, under incubation conditions of 37°C and 5% CO2. They were exposed to CSE for 24h to model COPD in vitro. Cells were divided into Control, CSE, CSE+AZM, JY-2 and JY-2+AZM for experiments. The cells of the CSE + AZM and the JY-2 + AZM groups were initially incubated with 10 μg/mL AZM for 24 hours, then stimulated with 0.2% CSE and 40 μM JY-2 respectively for another 24 hours. The cells in the CSE group and the JY-2 group were subjected to intervention with 0.2% CSE and 40 μM JY-2 correspondingly for 24 hours.
Senescence-Associated β-Galactosidase (SA-β-Gal) Activity
To assess cellular senescence, we utilized a β-galactosidase staining protocol. Briefly, in a six-well plate, the cells were initially washed with phosphate buffered saline (PBS), subsequently fixed using β-galactosidase staining fixative for 15 minutes at ambient temperature, and finally cleansed again with PBS. The SA-β-gal staining mixture was applied and allowed to incubate in the dark at 37°C for an extended period overnight. The next day, staining was examined under a light microscope. This kit (Beyotime, Shanghai, China) utilizes X-Gal as a substrate, which turns senescent cells expressing β-galactosidase blue. The Image Pro Plus 6.0 analysis software was used to analyze the number of blue-stained cells.
Preparation of CSE
As previously described,21 a Zhenlong cigarette (manufacturer: China Tobacco Guangxi Industrial Co., Ltd., China; specifications: tar content 10 mg, nicotine content 0.8 mg per cigarette) was connected via a rubber tube to the inlet of a glass gas absorption tube containing 5 mL of PBS. The outlet of the absorption tube was then connected to a 50 mL syringe. After lighting the cigarette, the syringe was repeatedly pulled at a constant speed to draw smoke into the absorption tube until the cigarette burned to 0.5 cm from the filter. Smoke from five cigarettes was sequentially drawn into the same tube. The resulting solution was then filtered through a 0.22 μm sterile filter and adjusted to approximately pH 7.4 to obtain the CSE stock solution. To standardize the CSE quality, its absorbance (OD320) was measured at a wavelength of 320 nm using a spectrophotometer. If the value fell between 0.9 and 1.2, the CSE was considered qualified.
Western Blots
Add a mixture of RIPA lysis buffer (Solarbio, Beijing, China) and protease inhibitor (Solarbio, Beijing, China) to the cell or lung tissue samples at a volume ratio of 100:1. After lysis, cell samples were subjected to ice-based ultrasonication, while lung tissue samples were homogenized at 4°C. The lysate was then centrifuged to collect the supernatant, and the total protein concentration was determined using the BCA assay (Beyotime, Shanghai, China). Based on the measured protein concentration, an appropriate amount of protein supernatant was taken and mixed with 5× protein loading buffer (NCM, China) at a volume ratio of 1:4. The mixture was heated at 100°C for 10 minutes to denature the proteins for subsequent Western blot experiments.
Western blot analysis was performed to measure FOXO3A, CCND1, p53, and p21 protein expression levels across groups, with GAPDH as a control. Proteins were separated via SDS-PAGE and subsequently blotted onto a membrane. The membrane was first blocked with a 5% skim milk solution in TBST for 1 hour at ambient temperature, and then incubated with primary antibodies against FOXO3A, CCND1, p53, p21 (using the same antibodies as in the IHC procedure) and GAPDH (1:3000, affinity, America). The second day, the membrane was treated with a secondary antibody in TBST for 1 hour at room temperature, under dark conditions. It was then scanned on the LI-COR Odyssey dual-color infrared imaging system (LI-COR, USA), and band densities were quantified using ImageJ software (National Institutes of Health, USA).
Real-Time Fluorescence Quantitative PCR (RT-qPCR)
Total RNA from mouse lung tissue was extracted with the RNAkey kit (Seven Innovations, Beijing, China) and reverse-transcribed into cDNA using StarScript III All-in-one RT Mix (Genstar, Beijing, China). Amplification was performed with the StarScript III One-Step qRT-PCR SYBR Kit (Genstar, Beijing, China) on an ABI 7500 instrument (Applied Biosystems, USA). GAPDH served as the housekeeping gene, and Ct values were processed using the 2-ΔΔCT method. Primer sequences are in Table 2.
 |
Table 2 The PCR Primer Sequences of Mice |
Enzyme Linked Immunosorbent Assay (ELISA)
We used ELISA kits (mlbio, Shanghai, China) to measure IL6 and IL8 levels in mouse BALF. The optical density (OD) value was read at 450 nm with a microplate reader, then the sample levels were quantified by referencing a standard curve.
Cell Counting Kit-8 (CCK8) Assay
BEAS-2B cells, at a density of 5 × 10^3 cells per well, were plated in a 96-well plate and maintained in culture for a duration of 24 hours. Different concentrations of azithromycin and JY-2 were added and incubated for 12, 24, and 36 hours, respectively. The cells were cultivated with CCK8 solution (NCM, China) and absorbance was measured at a wavelength of 450 nm utilizing a microplate reader.
Statistical Analysis
Data are presented as mean ± SD. Comparisons among multiple groups were analyzed by one-way ANOVA followed by Tukey’s post-hoc test, whereas CCK-8 results were evaluated with two-way ANOVA and Tukey’s post-hoc test. Graphs were prepared in GraphPad Prism 8, and statistical analyses were performed with SPSS 23.0 A P-value of less than 0.05 was considered statistically significant.
Results
Protein-Protein Interaction Network Graph of Drug Targets and Disease Targets
We identified 39 common targets by intersecting the predicted targets of azithromycin with COPD-associated targets using a Venn diagram (Figure 1A). These targets were used for subsequent protein-protein interaction (PPI) network analysis (Figure 1B). In the resulting network, nodes are colored and sized to reflect their topological importance: red hues indicate a higher degree of clustering (closer to the network hub), and larger size represents greater significance. Key targets, including ACTB, ALB, CCND1, and CDK2, emerged as central hubs within the network, as indicated by their prominent size and color.
Key Protein Targets and Network Diagram
Following this topological analysis, we extracted the 15 most critical proteins from the PPI network based on their significance scores (Figure 1C): CCND1, CDK2, AR, MKI67, MMP2, BIRC5, SDHA, ALDH18A1, FCGR2A, SOD2, PYGL, ACTA1, ANXA1, SIRT3, and GAD1. In this representation, the darker the color, the higher the significance level. Notably, the top 5 targets in the network graph were CCND1, CDK2, AR, MKI67, and MMP2.
GO and KEGG Enrichment Analysis
Building upon the identification of these core targets, our functional enrichment analysis provided critical mechanistic insights: Gene Ontology-Biological Process (GO-BP) terms (Figure 1D) were significantly enriched in senescence-associated processes, DNA damage checkpoint signaling during the G1 phase of mitosis, and cellular response to drug stimuli; concurrently, GO-Cellular Component (GO-CC) analysis (Figure 1E) revealed a strong association with the cyclin-dependent kinase holoenzyme complex, while GO-Molecular Function (GO-MF) results (Figure 1F) highlighted prominent roles in enzyme binding and protein binding. Importantly, KEGG pathway analysis (Figure 1G) further contextualized these findings by demonstrating significant enrichment in the FOXO and p53 signaling pathways, which are master regulators of cell cycle arrest, and senescence, thus creating a coherent biological narrative that directly connects the molecular functions and cellular components to high-level pathways governing cellular aging and drug mechanism.
Based on the top five critical targets (eg, CCND1) identified from the PPI network via the cytoHubba plugin, along with GO enrichment results, we propose that azithromycin may alleviate COPD through mechanisms associated with aging. Further supported by KEGG enrichment analysis highlighting the FOXO signaling pathway, we hypothesize that azithromycin could modulate pulmonary aging in COPD via the FOXO/CCND1 axis. Notably, FOXO3A (a well-established longevity gene most strongly associated with aging in the FOXO family) has been consistently linked to pulmonary senescence in COPD, as evidenced by previous studies.14,22 In conclusion, integrating these network pharmacology results, we suggest that azithromycin may mitigate pulmonary aging in COPD by regulating the FOXO3A/CCND1 pathway.
Smokers with COPD Demonstrated an Accelerated Aging Phenomenon Within Their Pulmonary Tissue
To validate network pharmacology results and study the aging characteristics of lung tissue in smokers with COPD, we collected lung tissue samples over 5 cm distant from the tumor site. The general information of the patients was shown in the table Protein expression of CCND1, FOXO3A, p53, and p21 in three groups showed that in smokers with normal lung function, FOXO3A, CCND1 and senescence markers p53 and p21 levels were intermediate between non-smokers and smokers with COPD (Figure 2A and B, p<0.01), suggesting smoking accelerates lung aging, with more significant aging in smokers with COPD.
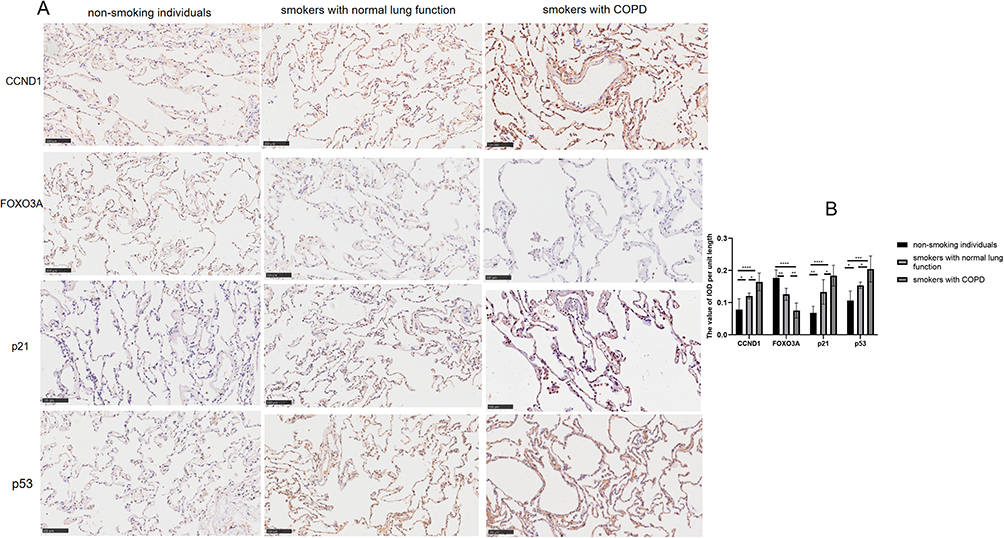 |
Figure 2 Smokers with COPD showed accelerated senescence in pulmonary tissue. (A) Immunohistochemical staining and (B) optical density measurement of CCND1, FOXO3A, p21, and p53 in the lung tissues from the three groups. Data were analyzed by one-way ANOVA followed by Tukey’s post hoc test (200x magnification; scale bar = 100 μm; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; n = 6 per group). |
Azithromycin Intervention Attenuated Lung Aging in Emphysema Mice Exposed to Smoke
To examine how azithromycin influences pulmonary aging in a mouse model of emphysema induced by CS, we created a CS-exposed emphysema model. MLI was measured to confirm the model’s success. The CS group showed increased alveolar volume and MLI compared to AIR group (Figure 3A and B, p<0.05), indicating emphysema. Azithromycin treatment significantly reduced MLI and improved emphysema.
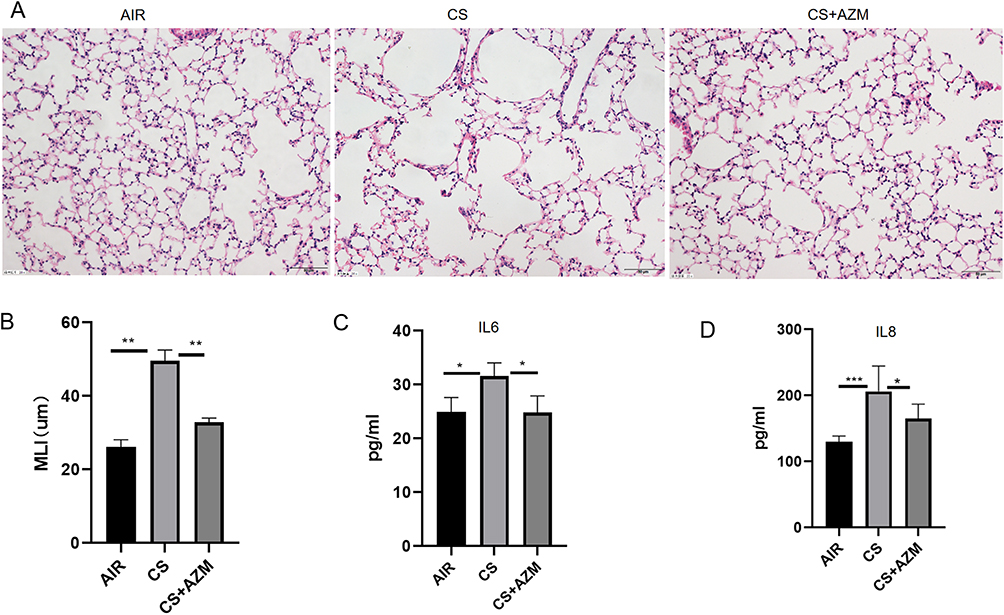 |
Figure 3 Azithromycin attenuated emphysema and reduced Senescence-associated secretory phenotype (SASP) secretion in mice exposed to cigarette smoke. Representative images of H&E-stained (A) lung tissue and the corresponding mean linear intercept (MLI) (B) quantification for each group (200x magnification; scale bar = 50 μm; n = 6). Levels of IL-6 (C) and IL-8 (D) in bronchoalveolar lavage fluid (BALF) (n ≥ 5). All data were analyzed by one-way ANOVA followed by Tukey’s post hoc test (*p < 0.05,**p < 0.01,***p < 0.001). |
The secretion levels of IL-6 and IL-8 were measured using ELISA. The results showed that, compared with the air control group, the secretion of IL-6 and IL-8 in BALF of mice in the CS exposure group was increased; after AZM treatment, the secretion of IL-6 and IL-8 was reduced (Figure 3C and D, p<0.05).
qRT-PCR results revealed that the mRNA levels of CCND1, p53, and p21 in the CS group were significantly higher than those in the air control group, while the mRNA expression of FOXO3A was decreased. After AZM intervention, the mRNA expression of FOXO3A increased, and the mRNA levels of CCND1, p53, and p21 significantly decreased (Figure 4A–D, p<0.05).
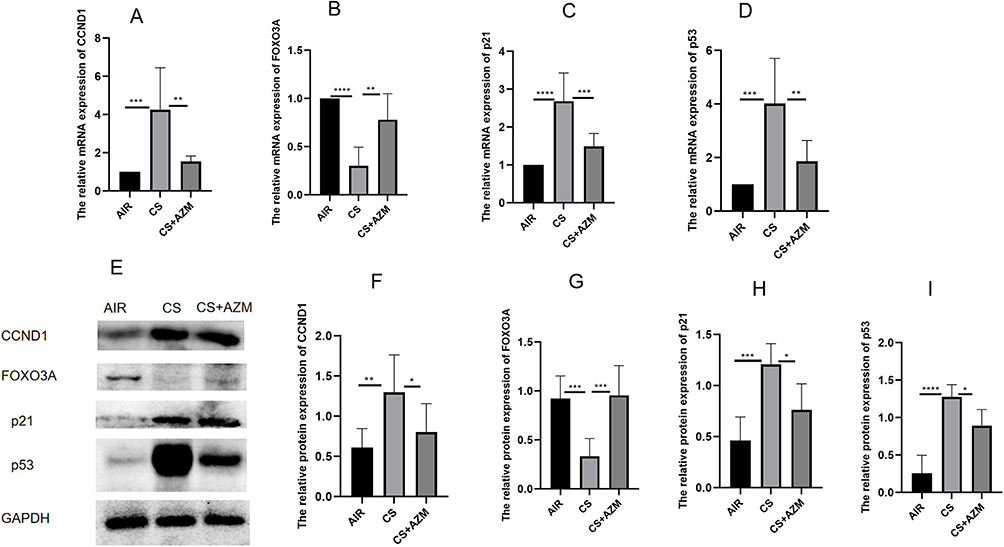 |
Figure 4 Azithromycin ameliorated tobacco smoke-induced pulmonary senescence in mice. mRNA expression levels of CCND1 (A), FOXO3A (B), p21 (C), and p53 (D) in lung tissues from the three groups were measured by qRT-PCR. Representative (E) Western blot and quantitative analyses of CCND1 (F), FOXO3A (G), p21 (H), and p53 (I) in lung tissues from the three mouse groups. All data were analyzed by one-way ANOVA followed by Tukey’s post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; n = 6 per group). |
Further detection by Western blotting (Figure 4E–I) and IHC (Figure 5A and B) of the protein expression of FOXO3A, CCND1, p53, and p21 showed that the expression of FOXO3A protein in the CS group was lower than that in the air control group, while the expressions of CCND1, p53, and p21 proteins were significantly increased. AZM treatment up-regulated FOXO3A protein expression and down-regulated the protein expressions of CCND1, p53, and p21 (p<0.05). These results indicate that AZM can alleviate pathological changes related to pulmonary aging in a mouse model of emphysema.
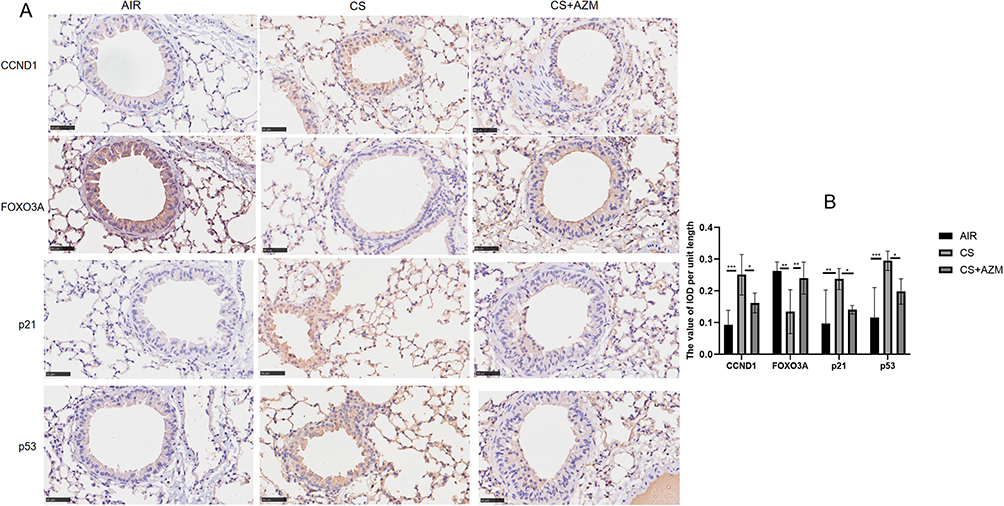 |
Figure 5 Azithromycin delayed the aging of pulmonary tissue in mice subjected to tobacco smoke exposure. (A) Immunohistochemical staining and (B) optical density measurement of CCND1, FOXO3A, p21, and p53 in the lung tissues across the three experimental groups (400x magnification; scale bar = 20 μm). Data were analyzed by one-way ANOVA followed by Tukey’s post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001; n = 6 per group). |
Azithromycin Exerted a Protective Effect Against CSE-Induced Senescence in BEAS-2B Cells by Regulating the FOXO3A/CCND1 Pathway
Based on the CCK-8 assay results (Figure 6A), the viability of cells treated with 10 μg/mL AZM for 36 hours was significantly lower than that of those treated for 24 hours (p < 0.01). Therefore, we selected the 24-hour treatment with 10 μg/mL AZM for subsequent experiments.
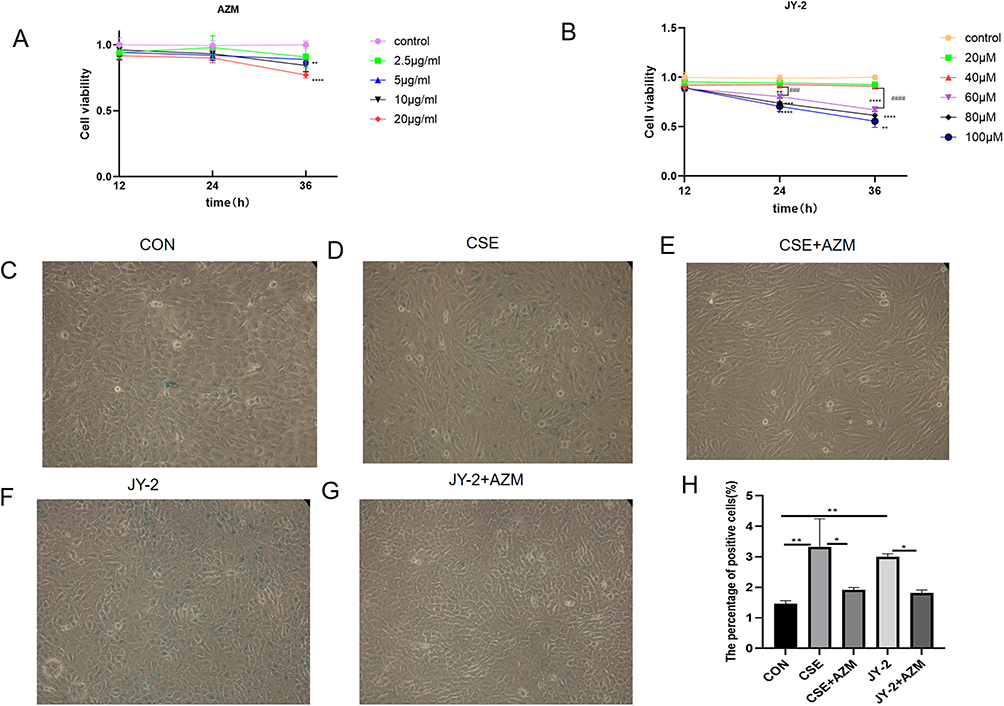 |
Figure 6 Azithromycin reduced the expression of senescence-associated β-galactosidase (SA-β-gal) in BEAS-2B cells induced by either CSE or JY-2. Dose-dependent effects of azithromycin and JY-2 on BEAS-2B cell viability. (A) Viability of BEAS-2B cells after treatment with increasing concentrations of azithromycin. (B) Viability after treatment with increasing concentrations of JY-2. SA-β-gal staining in BEAS-2B cells: representative images of the control (C), CSE-exposed (D), CSE+AZM-treated (E), JY-2-treated (F), and JY-2+AZM-treated (G) groups, with the corresponding percentage of SA-β-gal-positive cells (H) (200x magnification; n = 3 per group). Data in (A and B) were analyzed by two-way ANOVA followed by Tukey’s post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001,****p < 0.0001,###p < 0.001,####p < 0.0001). Data in (H) were analyzed by one-way ANOVA followed by Tukey’s post hoc test (*p < 0.05, **p < 0.01). |
As shown in Figure 6B, treatment with JY-2 at concentrations up to 40 μM for 12, 24, or 36 hours did not significantly affect cell viability (p > 0.05). No statistically significant difference in viability was observed between the 40 μM and 60 μM groups after 12 hours of exposure (p > 0.05). However, a highly significant decrease in cell viability emerged after 24 hours of treatment with 60 μM JY-2 compared to the 40 μM group (p < 0.001). Furthermore, at concentrations of 60 μM and above, cell survival rates significantly decreased across all treatment durations (12, 24, and 36 hours). Based on these findings, a 24-hour treatment with 40 μM JY-2 was selected for subsequent experiments.
BEAS-2B cells were pre-incubated with 10μg/mL azithromycin for 24 hours, then treated with 0.2% CSE (the concentrations, determined by CCK-8 cell viability assays, are unpublished data from our group) for another 24 hours to assess senescence and protein expression of FOXO3A, CCND1, p53 and p21. β-Galactosidase staining (Figure 6C–H) showed that, compared with controls, the CSE group had more blue cells, indicating elevated β-galactosidase activity. Additionally, Western blot analysis (Figure 7A–E) showed increased levels of CCND1, p53, and p21 proteins, whereas FOXO3A levels decreased. Azithromycin intervention reduced blue cell count and expression of CCND1, p53 and p21, and improved FOXO3A, suggesting a protective effect on senescent BEAS-2B cells.
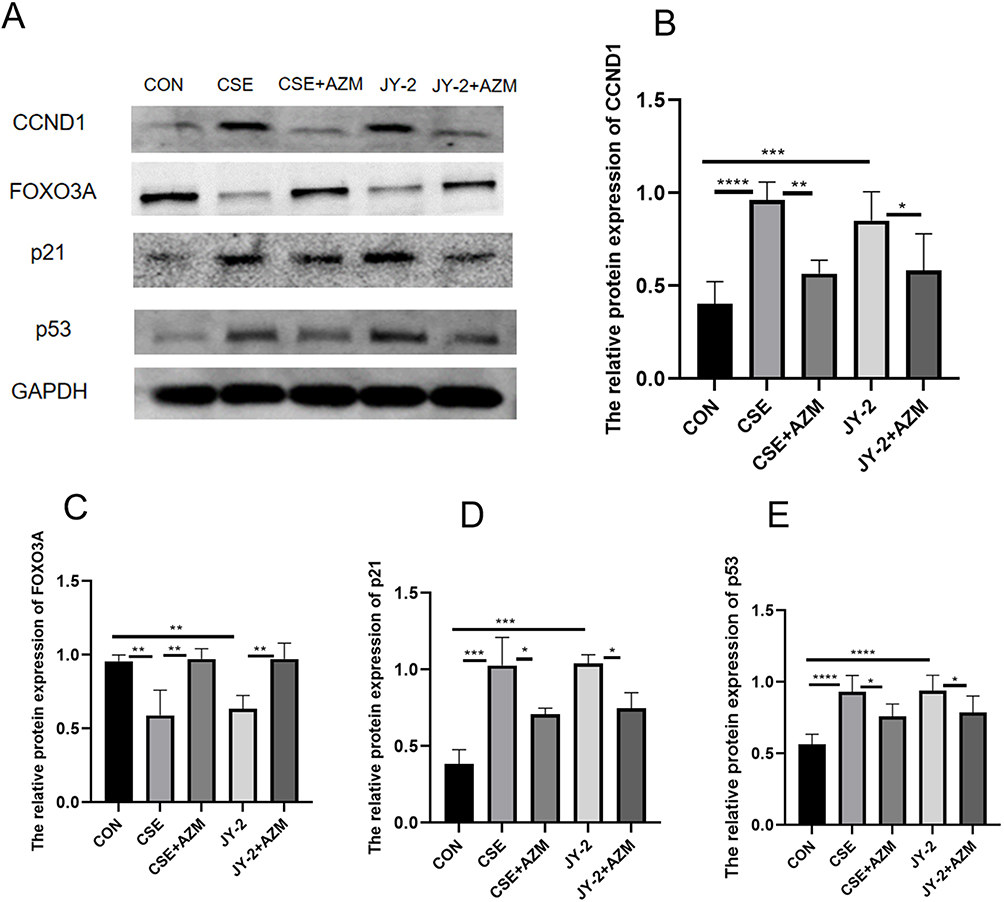 |
Figure 7 Azithromycin protected BEAS-2B cells from CSE-induced senescence by regulating the FOXO3A/CCND1 pathway. Representative (A) Western blot and quantitative analyses of CCND1 (B), FOXO3A (C), p21 (D), and p53 (E) in five groups of BEAS-2B cells. Data were analyzed by one-way ANOVA followed by Tukey’s post hoc test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; n ≥ 3 per group). |
To further investigate how azithromycin alleviates CSE-induced senescence in BEAS-2B cells, we sought to employ a FOXO3A agonist and/or inhibitor. Literature screening revealed no known FOXO3A agonist; JY-2 is the only reported inhibitor, exerting moderate suppression of FOXO3A activity.23 Using JY-2 (MCE, USA) as the inhibitor, we found that, compared with the control group, the JY-2 (40 μM) group displayed a comparable number of SA-β-gal-positive (blue) cells and similar expression trends for the senescence markers CCND1 and FOXO3A to the CSE group. The JY-2 + AZM group exhibited trends similar to those of the CSE + AZM group (Figure 7A–E). These data indicate that both CSE and JY-2 inhibit FOXO3A and accelerate senescence, whereas azithromycin can attenuate senescence induced by either stimulus, at least in part by modulating the FOXO3A/CCND1 pathway.
Discussion
COPD is a respiratory disease characterized by persistent respiratory symptoms and restricted airflow and is the third leading cause of death worldwide.1 After years of research, no drugs have been developed to cure COPD; they can only reduce the frequency of acute exacerbations and control symptoms. Thus, there is an urgent need to identify new targets or methods for treating COPD.
The lung function of normal individuals reaches its peak at about 25 years old, after which the first-second forced expiratory volume (FEV1), peak expiratory flow rate (PEFR) and forced vital capacity (FVC) gradually decrease as age advances and the absolute decline rate of male is greater than that of female.24 The decline in lung function is a hallmark characteristic of the progression of COPD. Our research found that smokers with COPD had lower FEV1%pred and FEV1/FVC compared to non-smoking individuals and smokers with normal lung function.
In patients suffering from COPD, the biological age was observed to be 13 to 23 years older than that of healthy individuals, and two characteristic biomarkers of aging, dehydroepiandrosterone (DHEA) and growth hormone (GH), were significantly reduced, suggesting an acceleration of aging in individuals with COPD.25 A study found26 that after three months of exposure to cigarette smoke, older (12-month) mice experienced accelerated lung inflammation leading to small airway remodeling, progression of emphysematous pathology, and increased lung compliance, whereas younger (2-month) mice remained free from COPD-associated features in the early stages of CS exposure. Cellular senescence may play a significant role in the pathogenesis of COPD.12,27 Cells enter a state of senescence when they stop dividing after reaching a limited number of replication cycles or in reaction to stress factors like oxidative stress,28 the triggering of the p53-p21 and p16-pRB pathways results in the cessation of the cell cycle and the onset of senescence.29 Numerous biomarkers are produced during the process of cellular senescence, characterized by elevated levels of p53, p21, and p16, as well as enhanced activity of SA-β-gal.30 Additionally, senescent cells are not static, they possess metabolic activity and secrete a range of inflammatory and chemotactic factors, including IL6, IL8, TNFα, etc.31 which is called the senescence-associated secretion phenotype (SASP).32 This study found that in mice with emphysema triggered by CS exposure, the expression of p53 and p21 proteins, as well as the levels of IL6, IL8, and MLI, were significantly increased, and their lung tissue showing signs of aging.
The FOXO3 family consists of FOXO3a and FOXO3b, wherein the FOXO3b gene encodes a protein that has no DNA-binding activity.33 FOXO3A is involved in oxidative stress, cell cycle arrest, DNA damage repair and apoptosis. FOXO3A is implicated in oxidative stress, cell cycle arrest, repairing DNA damage, and inducing apoptosis. FOXO3 is a principal gene linked to human longevity, and effectively managing FoxO3’s response to environmental stimuli could be vital for the prevention of aging and associated diseases.34 FOXO3A also plays a significant role in the accelerated aging of lungs in COPD. FOXO3A-driven PI3K/Akt signaling contributes to the rapid senescence of lung tissue in individuals with COPD.35 Furthermore, COPD patients and cigarette smoke-exposed mice showed significantly reduced levels of FOXO3A in their lung tissue, and the degree of emphysema and inflammatory response in the mice were markedly exacerbated, suggesting that FOXO3A deficiency was closely tied to the progression of COPD/emphysema.36 All of these indicated that FOXO3A was an important regulatory element in the aging trajectory of COPD.
Cyclin D1 (CCND1) is a gene linked to cancer, which promotes the cell cycle by creating complexes and boosting the activity of CDK4 or CDK6. Research had revealed37 that increased autophagy and decreased senescence in mammary epithelial cells of mice lacking CCND1. The regulation of cyclin D levels by the FOXO3 factor was achieved through transcriptional control,38 and the activated FOXO3A mutants can effectively suppress the promoter activity of CCND1 and CCND2, leading to a pause in cell division cycle. In cancer cells with CCND1 deletion, the absence of CCND1 triggers the accumulation of ROS, which subsequently activated FOXO3A, resulting in cellular senescence.39 These findings suggested a significant correlation between CCND1 and the process of cellular senescence.
Airway epithelial cells function as the initial shield against inhaled pathogens and foreign materials. In vitro cultivation of human bronchial epithelial cells from different age groups demonstrated a decline in the cells’ barrier capabilities with age. This, in turn, heightened vulnerability to inhalation-induced damages, including those from cigarette smoke, indicating that senescence is linked to impaired epithelial barrier function.40 Numerous detrimental substances found in cigarette smoke that had a damaging effect on the respiratory system. Cigarette use triggers oxidative stress in the airway epithelium, resulting in dysfunction of intercellular connections in airway epithelial cells.41 Oxidative stress served as a pivotal trigger for cellular senescence in COPD, directly activating the p53-p16INK4a pathway and damaging DNA, which leads to the activation of p21CIP1.28 The study showed that in smokers with COPD, there was a significant upregulation of p53 and p21, along with a notable decrease in FOXO3A expression. This indicated an accelerated rate of lung aging associated with COPD. Furthermore, the levels of CCND1 protein were significantly higher in smokers with COPD, which is consistent with previous research findings.42
The sirtuin family is a class of highly conserved histone deacetylases in the evolutionary process that regulate various intracellular functions, comprising seven members including SIRT1, SIRT2, and SIRT3. SIRT1 is closely associated with physiological processes such as cellular senescence, autophagy, and apoptosis in COPD.43 Reduced expression of SIRT1 leads to decreased expression of the telomere protection protein TPP1, thereby triggering telomere dysfunction, accelerating cellular senescence, and promoting the progression of COPD.44 FOXO3A is a downstream target molecule of SIRT1. Activation of SIRT1 enhances the expression of FOXO3, thereby delaying premature cellular senescence and preventing the occurrence of emphysema.45 Other studies have found that activation of SIRT1 can alleviate senescence in type II alveolar epithelial cells by deacetylating the FOXO3A/p53 signaling pathway, thus influencing the progression of COPD.14 SIRT3 is a deacetylase that regulates mitochondrial function. Its overexpression can mitigate mitochondrial oxidative stress and cellular damage, while its downregulation exacerbates airway inflammation, alveolar cavity enlargement, and mitochondrial damage in the airway epithelium of COPD rats.46 SIRT3 plays a key role in regulating mitochondrial function and cellular senescence. Its overexpression alleviates cadmium exposure-induced senescence in alveolar epithelial cells and COPD-like lung injury, whereas knockout of SIRT3 exacerbates these pathological changes.47 In summary, SIRT1 and SIRT3 are closely related to the process of cellular senescence in COPD. Under acute stress conditions, cathepsins can cleave SIRT1 in endothelial progenitor cells, leading to loss of function and thereby inducing cellular senescence.48 Azithromycin is a weak autophagy inducer that selectively acts on senescent cells, potentially promoting cell death by inducing autophagy,19 thereby exerting anti-aging effects. This study has not yet directly detected the expression levels of SIRT1/SIRT3, and future research will explore this direction in depth to more comprehensively elucidate the mechanism of action of azithromycin.
Previous work has focused almost exclusively on the antimicrobial and immunomodulatory properties of azithromycin, whereas its anti-senescent potential remains largely unexplored. To date, only Ozsvari19 et al have directly demonstrated that azithromycin can eliminate senescent fibroblasts. Using network pharmacology, we first predicted that azithromycin might ameliorate COPD-related senescence via the FOXO3A/CCND1 axis. This was corroborated in lung tissues from COPD patients, which exhibited reduced FOXO3A and elevated CCND1, p53 and p21—molecular signatures consistent with cellular senescence. In vivo experiments confirmed that azithromycin attenuates pulmonary senescence, and in vitro studies indicated that these effects are mediated, at least in part, through the FOXO3A/CCND1 pathway.
Collectively, our findings identify azithromycin as a promising new candidate for the treatment of senescence-driven lung diseases such as COPD and pulmonary fibrosis.
Limitations of this study include: the anti-aging effects of azithromycin have not yet been validated in COPD patients; only a single dose of azithromycin was used in the experiments, and its anti-aging mechanisms have not been sufficiently explored. In future studies, we will employ multiple doses to further investigate its anti-aging mechanisms and advance the research into clinical stages to evaluate its practical benefits in COPD patients.
Conclusion
In summary, this study reveals that azithromycin possesses therapeutic potential in alleviating COPD-related aging phenotypes by regulating the FOXO3A/CCND1 pathway, thereby providing a theoretical basis for its development as an anti-aging agent. However, how azithromycin precisely modulates this pathway and whether additional targets are involved remain to be elucidated. A deeper understanding of its anti-aging mechanisms will be crucial for future research on interventions aimed at delaying aging.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by the National Natural Science Foundation of China (82260012) and the self-raised fund scientific research project of the Health Commission of Guangxi Zhuang Autonomous Region (Z20210942). The funding agencies had no role in designing the study, conducting the experiment, analyzing the data, drafting the manuscript, or deciding on the publication of the paper.
Disclosure
The authors declare no competing interests in this work.
References
1. Korzh O, et al. GBD 2019 Chronic Respiratory Diseases Collaborators. Global burden of chronic respiratory diseases and risk factors, 1990-2019: an update from the Global Burden of Disease Study 2019. EClinicalMedicine. 2023;59:101936. doi:10.1016/j.eclinm.2023.101936
2. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi:10.1016/j.cell.2013.05.039
3. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482–496. doi:10.1038/nrm3823
4. Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135(1):173–180. doi:10.1378/chest.08-1419
5. Mercado N, Ito K, Barnes PJ. Accelerated ageing of the lung in COPD: new concepts. Thorax. 2015;70(5):482–489. doi:10.1136/thoraxjnl-2014-206084
6. Wang M, Zhang Y, Xu M, et al. Roles of TRPA1 and TRPV1 in cigarette smoke -induced airway epithelial cell injury model. Free Radic Biol Med. 2019;134:229–238. doi:10.1016/j.freeradbiomed.2019.01.004
7. Wang K, Liao Y, Li X, et al. Inhibition of neutrophil elastase prevents cigarette smoke exposure-induced formation of neutrophil extracellular traps and improves lung function in a mouse model of chronic obstructive pulmonary disease. Int Immunopharmacol. 2023;114:109537. doi:10.1016/j.intimp.2022.109537
8. Vij N, Chandramani-Shivalingappa P, Van Westphal C, Hole R, Bodas M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am J Physiol Cell Physiol. 2018;314(1):C73–c87. doi:10.1152/ajpcell.00110.2016
9. Wang W, Zhang S, Cui L, Chen Y, Xu X, Wu L. Bufei yishen formula inhibits the cell senescence in COPD by up-regulating the ZNF263 and Klotho expression. Int J Chron Obstruct Pulmon Dis. 2023;18:533–539. doi:10.2147/copd.S383295
10. Paschalaki K, Rossios C, Pericleous C, et al. Inhaled corticosteroids reduce senescence in endothelial progenitor cells from patients with COPD. Thorax. 2022;77(6):616–620. doi:10.1136/thoraxjnl-2020-216807
11. Cai Q, Chen S, Zhu Y, Li Z. Knockdown of GNL3L alleviates the progression of COPD Through inhibiting the ATM/p53 pathway. Int J Chron Obstruct Pulmon Dis. 2023;18:2645–2659. doi:10.2147/copd.S424431
12. Wu H, Ma H, Wang L, et al. Regulation of lung epithelial cell senescence in smoking-induced COPD/emphysema by microR-125a-5p via Sp1 mediation of SIRT1/HIF-1a. Int J Biol Sci. 2022;18(2):661–674. doi:10.7150/ijbs.65861
13. Takasaka N, Araya J, Hara H, et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J Immunol. 2014;192(3):958–968. doi:10.4049/jimmunol.1302341
14. Gu C, Li Y, Liu J, et al. LncRNA‑mediated SIRT1/FoxO3a and SIRT1/p53 signaling pathways regulate type II alveolar epithelial cell senescence in patients with chronic obstructive pulmonary disease. Mol Med Rep. 2017;15(5):3129–3134. doi:10.3892/mmr.2017.6367
15. Houssaini A, Breau M, Kebe K, et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight. 2018;3(3). doi:10.1172/jci.insight.93203
16. Xiaofei Y, Tingting L, Xuan W, Zhiyi H. Erythromycin attenuates oxidative stress-induced cellular senescence via the PI3K-mTOR signaling pathway in chronic obstructive pulmonary disease. Front Pharmacol. 2022;13:1043474. doi:10.3389/fphar.2022.1043474
17. Tsubouchi K, Araya J, Minagawa S, et al. Azithromycin attenuates myofibroblast differentiation and lung fibrosis development through proteasomal degradation of NOX4. Autophagy. 2017;13(8):1420–1434. doi:10.1080/15548627.2017.1328348
18. Araya J, Kojima J, Takasaka N, et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2013;304(1):L56–69. doi:10.1152/ajplung.00213.2012
19. Ozsvari B, Nuttall JR, Sotgia F, Lisanti MP. Azithromycin and Roxithromycin define a new family of “senolytic” drugs that target senescent human fibroblasts. Aging. 2018;10(11):3294–3307. doi:10.18632/aging.101633
20. Pei G, Guo L, Liang S, et al. Long-term erythromycin treatment alters the airway and gut microbiota: data from chronic obstructive pulmonary disease patients and mice with emphysema. Respiration. 2024;103(8):461–479. doi:10.1159/000538911
21. Wei Y, Li Q, He K, et al. Mechanism of cigarette smoke in promoting small airway remodeling in mice via STAT 3 / PINK 1-Parkin / EMT. Free Radic Biol Med. 2024;224:447–456. doi:10.1016/j.freeradbiomed.2024.08.036
22. Zeng Q, Zeng J. Inhibition of miR-494-3p alleviates oxidative stress-induced cell senescence and inflammation in the primary epithelial cells of COPD patients. Int Immunopharmacol. 2021;92:107044. doi:10.1016/j.intimp.2020.107044
23. Choi HE, Kim Y, Lee HJ, Cheon HG. Novel FoxO1 inhibitor, JY-2, ameliorates palmitic acid-induced lipotoxicity and gluconeogenesis in a murine model. Eur J Pharmacol. 2021;899:174011. doi:10.1016/j.ejphar.2021.174011
24. Thomas ET, Guppy M, Straus SE, Bell KJL, Glasziou P. Rate of normal lung function decline in ageing adults: a systematic review of prospective cohort studies. BMJ Open. 2019;9(6):e028150. doi:10.1136/bmjopen-2018-028150
25. Karametos I, Tsiboli P, Togousidis I, Hatzoglou C, Giamouzis G, Gourgoulianis KI. Chronic obstructive pulmonary disease as a main factor of premature aging. Int J Environ Res Public Health. 2019;16(4). doi:10.3390/ijerph16040540
26. John-Schuster G, Günter S, Hager K, Conlon TM, Eickelberg O, Yildirim A. Inflammaging increases susceptibility to cigarette smoke-induced COPD. Oncotarget. 2016;7(21):30068–30083. doi:10.18632/oncotarget.4027
27. Tao L, Lu X, Fu Z, et al. Tong Sai granule improves AECOPD via regulation of MAPK-SIRT1-NF-κB pathway and cellular senescence alleviation. J Ethnopharmacol. 2023;314:116622. doi:10.1016/j.jep.2023.116622
28. Devulder JV. Unveiling mechanisms of lung aging in COPD: a promising target for therapeutics development. Chin Med J Pulm Crit Care Med. 2024;2(3):133–141. doi:10.1016/j.pccm.2024.08.007
29. Huang W, Hickson LJ, Eirin A, Kirkland JL, Lerman LO. Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol. 2022;18(10):611–627. doi:10.1038/s41581-022-00601-z
30. Silwal P, Nguyen-Thai AM, Mohammad HA, et al. Cellular senescence in intervertebral disc aging and degeneration: molecular mechanisms and potential therapeutic opportunities. Biomolecules. 2023;13(4). doi:10.3390/biom13040686
31. Kandhaya-Pillai R, Yang X, Tchkonia T, Martin GM, Kirkland JL, Oshima J. TNF-α/IFN-γ synergy amplifies senescence-associated inflammation and SARS-CoV-2 receptor expression via hyper-activated JAK/STAT1. Aging Cell. 2022;21(6):e13646. doi:10.1111/acel.13646
32. Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol. 2020;16(5):263–275. doi:10.1038/s41574-020-0335-y
33. Santo EE, Paik J. A splice junction-targeted CRISPR approach (spJCRISPR) reveals human FOXO3B to be a protein-coding gene. Gene. 2018;673:95–101. doi:10.1016/j.gene.2018.06.048
34. Morris BJ, Willcox DC, Donlon TA, Willcox BJ. FOXO3: a major gene for human longevity--A mini-review. Gerontology. 2015;61(6):515–525. doi:10.1159/000375235
35. Yuan YM, Luo L, Guo Z, Yang M, Lin YF, Luo C. Smoking, aging, and expression of proteins related to the FOXO3 signaling pathway in lung tissues. Genet Mol Res. 2015;14(3):8547–8554. doi:10.4238/2015.July.31.2
36. Hwang JW, Rajendrasozhan S, Yao H, et al. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J Immunol. 2011;187(2):987–998. doi:10.4049/jimmunol.1001861
37. Brown NE, Jeselsohn R, Bihani T, et al. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res. 2012;72(24):6477–6489. doi:10.1158/0008-5472.Can-11-4139
38. Schmidt M, Fernandez de Mattos S, van der Horst A, et al. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol. 2002;22(22):7842–7852. doi:10.1128/mcb.22.22.7842-7852.2002
39. Laphanuwat P, Likasitwatanakul P, Sittithumcharee G, et al. Cyclin D1 depletion interferes with oxidative balance and promotes cancer cell senescence. J Cell Sci. 2018;131(12). doi:10.1242/jcs.214726
40. de Vries M, Nwozor KO, Muizer K, et al. The relation between age and airway epithelial barrier function. Respir Res. 2022;23(1):43. doi:10.1186/s12931-022-01961-7
41. Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: role of cigarette smoke exposure. Am J Respir Cell Mol Biol. 2018;58(2):157–169. doi:10.1165/rcmb.2017-0200TR
42. Yang C, Li L, Guo J, et al. Up-regulation of Pim-3 in chronic obstructive pulmonary disease (COPD) patients and its potential therapeutic role in COPD rat modeling. Pathol Res Pract. 2017;213(4):322–326. doi:10.1016/j.prp.2017.01.018
43. Li S, Huang Q, He B. SIRT1 as a potential therapeutic target for chronic obstructive pulmonary disease. Lung. 2023;201(2):201–215. doi:10.1007/s00408-023-00607-9
44. Ahmad T, Sundar IK, Tormos AM, et al. Shelterin telomere protection protein 1 reduction causes telomere attrition and cellular senescence via sirtuin 1 deacetylase in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2017;56(1):38–49. doi:10.1165/rcmb.2016-0198OC
45. Yao H, Chung S, Hwang JW, et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest. 2012;122(6):2032–2045. doi:10.1172/jci60132
46. Zhang M, Zhang Y, Roth M, et al. Sirtuin 3 inhibits airway epithelial mitochondrial oxidative stress in cigarette smoke-induced COPD. Oxid Med Cell Longev. 2020;2020:7582980. doi:10.1155/2020/7582980
47. Peng K, Yao YX, Lu X, et al. Mitochondrial dysfunction-associated alveolar epithelial senescence is involved in CdCl(2)-induced COPD-like lung injury. J Hazard Mater. 2024;476:135103. doi:10.1016/j.jhazmat.2024.135103
48. Chen J, Xavier S, Moskowitz-Kassai E, et al. Cathepsin cleavage of sirtuin 1 in endothelial progenitor cells mediates stress-induced premature senescence. Am J Pathol. 2012;180(3):973–983. doi:10.1016/j.ajpath.2011.11.033
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Identification of Small Airway Epithelium-Related Hub Genes in Chronic Obstructive Pulmonary Disease
Lin L, Lin G, Chen X, Lin H, Lin Q, Zeng Y, Xu Y
International Journal of Chronic Obstructive Pulmonary Disease 2022, 17:3001-3015
Published Date: 30 November 2022
Role of Digoxin in Preventing Cigarette Smoke-Induced COPD via HIF-1α Inhibition in a Mouse Model
Zhang K, Zhou F, Zhu C, Yuan L, Li D, Wang J, Lu W
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:1665-1678
Published Date: 23 May 2025
Chronic Exposure to Cigarette Smoke Induces Pyroptosis in Pulmonary Epithelial Cells via EGR1/USP44/TRAF6 Axis in COPD
Cao C, Zhong Z, Wu B, Zou B, Jia H, Xu J, Xiao G, Xia S
Journal of Inflammation Research 2025, 18:16619-16635
Published Date: 27 November 2025