")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Association of Clusterin Levels in Cerebrospinal Fluid with Synaptic Degeneration Across the Alzheimer’s Disease Continuum
Authors Wang J, Zhang X, Zhu B, Fu P
Received 26 July 2019
Accepted for publication 20 December 2019
Published 20 January 2020 Volume 2020:16 Pages 183—190
DOI https://doi.org/10.2147/NDT.S224877
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jun Chen
Jun Wang,* Xin Zhang,* Bihong Zhu, Pan Fu On the behalf of Alzheimer’s Disease Neuroimaging Initiative
Department of Neurology, Taizhou First People’s Hospital, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Pan Fu
Department of Neurology, Taizhou First People’s Hospital, 218 Hengjie Road, Huangyan District, Taizhou City, Zhejiang Province, People’s Republic of China
Email [email protected]
Purpose: Although emerging evidence has suggested that clusterin is involved in the pathogenesis of Alzheimer’s disease (AD), the association of clusterin with synaptic degeneration in living human is unclear. In the present study, we aimed to examine the association of CSF clusterin levels with synaptic degeneration in individuals with different severities of cognitive impairment.
Patients and Methods: In the present study, we compared levels of clusterin in CSF among individuals with normal cognition (NC), mild cognitive impairment (MCI), and AD. Further, linear regression models were performed to examine the association of CSF clusterin with neurogranin (NG, reflecting synaptic degeneration) with adjustment of several potential confounders.
Results: We found that CSF clusterin levels were positively correlated with NG in the NC and MCI groups, but not the AD group. In all subjects, linear regression models suggested that clusterin levels were positively associated with NG levels independent of age, gender, apolipoprotein E4 (APOE4) genotype, clinical diagnosis, and CSF Aβ 42 levels.
Conclusion: Our data indicated that clusterin was associated with CSF NG levels among older individuals with different severities of cognitive impairment.
Keywords: clusterin, neurogranin, synaptic degeneration, Alzheimer’s disease, mild cognitive impairment
Introduction
Clusterin, also known as apolipoprotein J, is involved in the pathogenesis of Alzheimer’s disease (AD).1 Previous studies showed that clusterin levels were significantly elevated in cerebrospinal fluid (CSF) and brain of AD patients, and clusterin levels in plasma were found to be related with disease severity and brain atrophy in AD patients.2,3 It has been suggested that clusterin could interact with β-amyloid (Aβ) and facilitate its clearance from brain.4,5 Further, it has also been reported that clusterin could interact with Aβ to form a stable complex6–9 and enhance its clearance from brain across the blood–brain barrier (BBB) via low-density lipoprotein receptor-related protein-1 (LRP1).6 However, the exact mechanisms by which clusterin contributes to the pathogenesis of AD remain elusive.
It has been reported that synaptic degeneration is an important mechanism underlying cognitive deficit in AD.10 Neurogranin (NG), a postsynaptic protein, has been reported to be significantly increased in the CSF of patients with mild cognitive impairment (MCI) and AD.11–13 In addition, CSF NG levels can predict progression of MCI to AD dementia.14 In animals, knockdown of NG inhibits long-term potentiation (LTP) and cognition,15 while upregulation enhances LTP and cognition.16 Given the potential roles of both clusterin and NG in AD pathogenesis, we hypothesized that clusterin may contribute to synaptic dysfunction, which leads to cognitive deficits in AD.
First, we compared CSF clusterin levels among individuals with normal cognition (NC), MCI, and AD. Second, to examine the relationship between CSF clusterin and NG levels, the Pearson correlation test was conducted in each diagnostic group. Finally, linear regression models were performed to examine the association of CSF clusterin with NG levels by controlling for age, gender, educational attainment, APOE4 genotype, clinical diagnosis, and CSF Aβ42 levels.
Patients and Methods
Alzheimer’s Disease Neuroimaging Initiative (ADNI)
Cross-sectional data used in the preparation of this paper were extracted from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database. The primary aim of ADNI has been to examine whether demographics, cognitive assessments, serial MRI, PET, blood, and CSF biomarkers could be integrated to predict the progression of MCI and AD. This study was approved by local ethical committees. All participants provided written informed consent. The official or affiliated names of the approving local ethics committee is available in the Supplementary document.
Participants
We selected subjects who met criteria for mild AD, MCI and NC and had CSF clusterin and NG samples.
Subjects with NC had a Mini-Mental State Examination (MMSE)17 score ranging from 24 to 30 and a Clinical Dementia Rating (CDR)18 score of 0. Individuals with MCI had a MMSE score ranging between 24 and 30, a CDR score of 0.5, an objective memory decline as evidenced by the Wechsler Memory Scale Logical memory II, and an absence of dementia. In the ADNI study, the type of MCI participants was amnestic MCI. Patients with mild AD fulfilled the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria for probable AD dementia19 and had a MMSE score ranging between 20 and 26 and a CDR score ranging between 0.5 and 1.
Measurement of Clusterin Levels in CSF
Clusterin levels in CSF were measured using Rules Based Medicine (Human Discovery MAP, v1.0),20 details of which can be found at the ADNI website (http://www.adni-info.org). The analyte was log-transformed for statistical analyses. Values are given as ug/mL. The average of the coefficient of variation (CV) of clusterin levels was 9.63%.
Measurement of NG Levels in CSF
NG levels in CSF were determined by electrochemiluminescence technology (Meso Scale Discovery, Maryland, USA) with NG7 (a monoclonal antibody specific for NG)21 as the coating antibody and polyclonal NG anti-rabbit (ab 23570, Upstate) as the detector antibody11. Values are given as pg/mL.
Measurement of CSF Aβ42, t-tau, and p-tau Levels
CSF Aβ42, t-tau, and p-tau levels were measured by the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX), details of which have been described elsewhere22 and can be found at the ADNI website (http://www.adni-info.org). Values are given as pg/mL.
Statistical Analysis
F-tests and χ2 tests were utilized to examine differences in demographics and clinical variables between the three diagnostic groups. The Pearson correlation analysis was applied to examine the relationship between clusterin and NG levels in CSF in each diagnostic group. Then, univariate analyses of predictors for CSF NG levels were conducted. Further, forward stepwise regression models were performed to examine the relationships between CSF clusterin and NG levels. The Akaike information criterion (AIC) was utilized to select the best model. All statistical analyses were conducted using R software (version 3.5.1). The level of statistical significance was set at P < 0.05.
Results
Demographic and Clinical Information on Study Participants
In the present study, there was a total of 294 individuals (84 individuals with NC, 143 individuals with MCI, and 67 patients with mild AD). There were significant differences in several variables between the three diagnostic groups (Table 1).
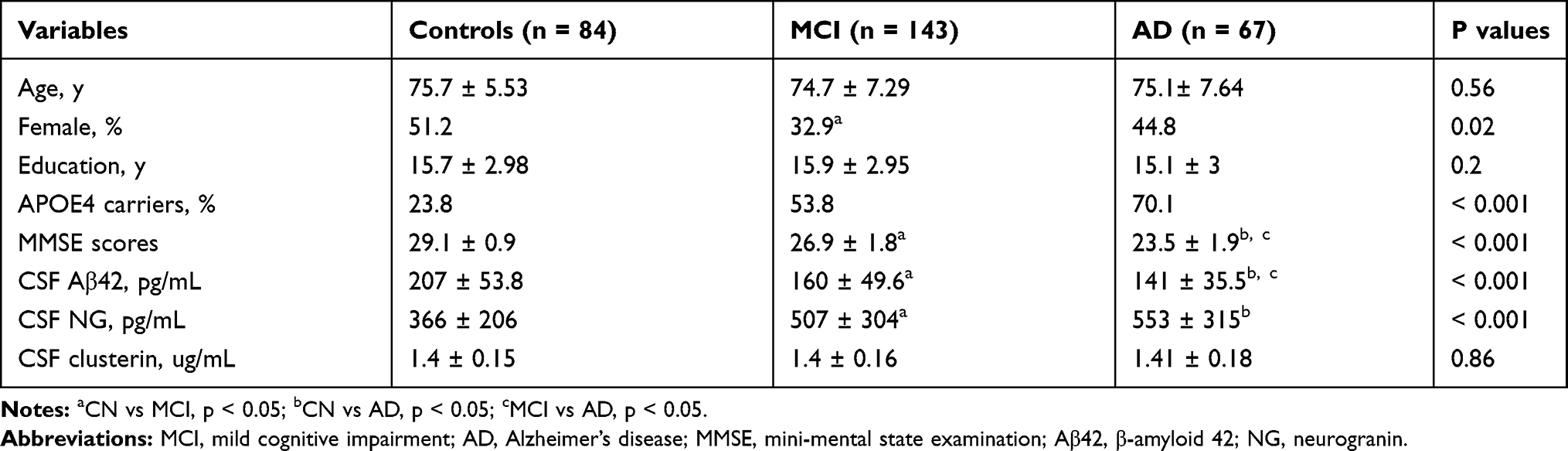 |
Table 1 Demographic and Clinical Characteristics and Protein Levels |
CSF Clusterin and NG Levels in the Three Diagnostic Groups
No significant difference was observed in CSF clusterin levels between the three diagnostic groups (Table 1 and Figure 1A). However, there was a significant difference in CSF NG levels between the three groups (Table 1 and Figure 1B).
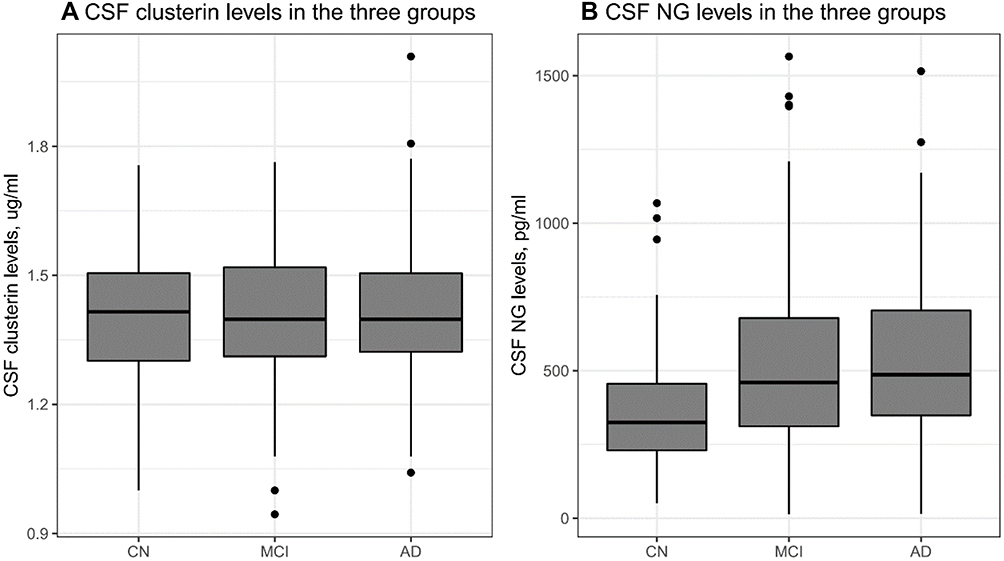 |
Figure 1 CSF clusterin and NG levels in the three diagnostic groups. (A) CSF clusterin levels were similar across subjects. (B) There was a significant difference in CSF NG levels across subjects. Abbreviations: CN, cognitively normal; MCI, mild cognitive impairment; AD, Alzheimer’s disease; NG, neurogranin. |
Associations of CSF Clusterin with CSF NG and AD Pathologies in All Subjects
To examine the associations of CSF clusterin with CSF NG and AD pathologies, the Pearson correlation tests were applied in the overall sample. As shown in Figure 2, we found that CSF clusterin levels were positively correlated with NG levels (r = 0.22, p < 0.001), t-tau (r= 0.29, p < 0.001) and p-tau (r = 0.12, p = 0.04), but not Aβ42 levels (r = 0.06, p = 0.288).
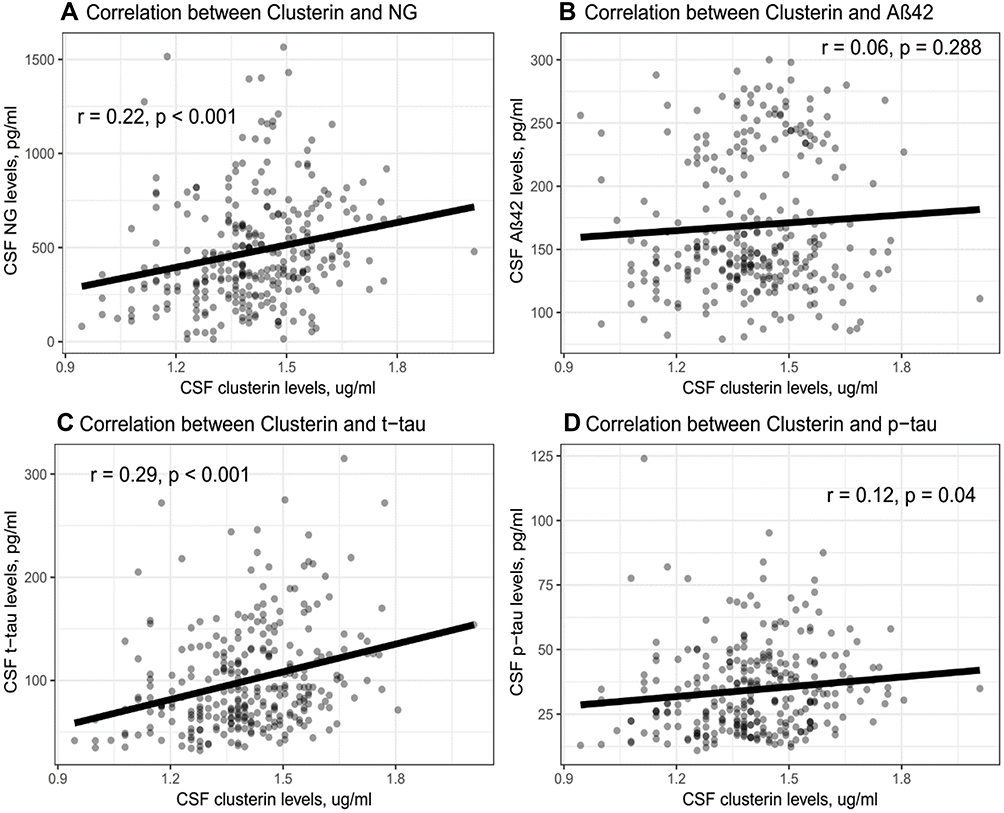 |
Figure 2 (A–D) Associations of CSF clusterin levels with NG levels and AD pathologies. We found that CSF clusterin levels were positively correlated with NG levels (r = 0.22, p < 0.001), t-tau (r= 0.29, p < 0.001) and p-tau (r = 0.12, p = 0.04), but not Aβ42 levels (r = 0.06, p = 0.288). Abbreviations: Aβ42, β-amyloid 42; NG, neurogranin. |
Correlations Between the Various Protein Levels and MMSE Scores in the Whole Sample
In order to examine the correlations between the various protein levels in CSF (clusterin, NG, and Aβ42) and MMSE scores in the whole sample, Spearman correlation tests were performed in the whole sample. We found that MMSE scores were associated with CSF NG (rho = −0.19, p = 0.001) and Aβ42 levels (rho = 0.35, p < 0.001), but not CSF clusterin levels (rho = 0.03, p = 0.66).
Correlations Between CSF Clusterin and NG Levels in Each Diagnostic Group
To examine whether the relationship between CSF clusterin and NG levels was modified by cognitive status, Pearson’s correlation tests were utilized to examine the correlation between CSF clusterin and NG levels in each diagnostic group. Interestingly, we found that CSF clusterin levels were associated with NG levels in the CN (r = 0.46, p < 0.001) and MCI (r = 0.23, p = 0.005) groups, but not the AD group (r = 0.046, p = 0.71; Figure 3).
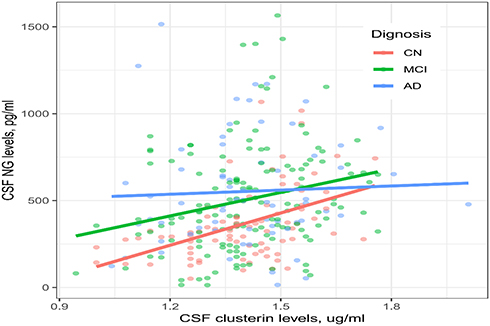 |
Figure 3 Correlation between CSF clusterin and NG levels in each diagnostic group. Abbreviations: CN, cognitively normal; MCI, mild cognitive impairment; AD, Alzheimer’s disease; NG, neurogranin. |
Association of CSF Clusterin with NG Levels
First, univariate analyses were conducted to systematically examine the relationships between each individual predictor and CSF NG levels. The results are demonstrated in Table 2. Second, to further examine the association of clusterin with NG levels in CSF, forward stepwise regression models were performed, and AIC was used to select the best fit. We found that CSF clusterin levels were associated with NG levels in CSF after adjusting for age, gender, APOE4 genotype, clinical diagnosis, and CSF Aβ42 levels (Table 3).
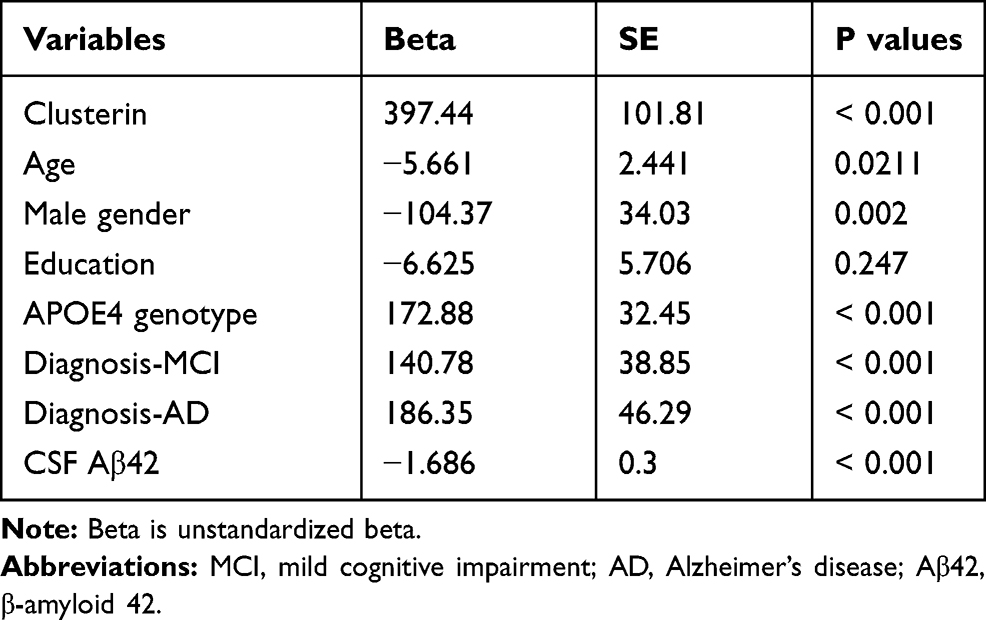 |
Table 2 Univariate Analysis of Predictors for CSF NG Levels |
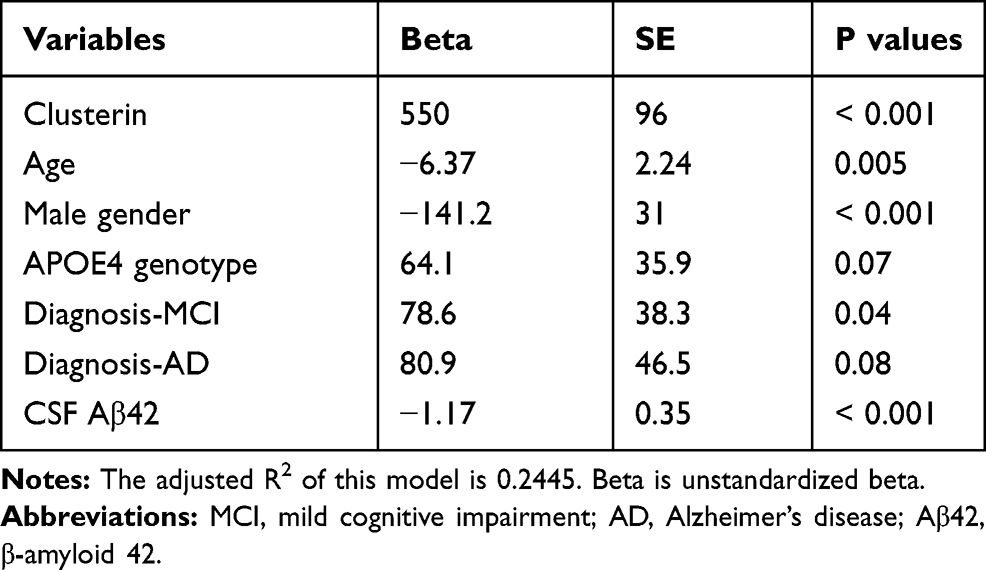 |
Table 3 Summary of Forward Stepwise Regression Model |
Discussion
To the best of our knowledge, this is the first study to report that CSF clusterin levels were positively associated with NG levels in CSF (reflecting synaptic degeneration) independent of age, gender, APOE4 genotype, clinical diagnosis, and CSF amyloid pathology. In addition, levels of clusterin in CSF were positively correlated with NG levels in the CN and MCI groups, but not the AD group. However, no significant difference was observed in CSF clusterin levels between the three diagnostic groups.
Although clusterin gene has been regarded as a strong genetic risk factor for late-onset AD,23,24 the mechanism by which clusterin contributes to the pathogenesis of AD remains unclear. We provided in vivo evidence that clusterin may impair synaptic function to increase the risk of AD. To the best of our knowledge, this is the first study to examine the relationship between CSF clusterin and NG levels in living human. In line with these findings, a preclinical study found that in an in vitro model of ischemia, exogenous administration of recombinant clusterin induced a substantial worsening of synaptic damage, suggesting a deleterious role of clusterin in synaptic structure and function.25 In clinical studies, structural magnetic resonance imaging (MRI) allows for the assessment of brain atrophy, including loss of synapses and neurons.26,27 A longitudinal prospective study showed a significant association of the clusterin *Aβ interaction with longitudinal entorhinal cortex atrophy in non-demented older adults,28 indicating that clusterin may accelerate amyloid deposition and induce Aβ-associated neuronal and synaptic degeneration in the early stage of AD.29–32 Increasing evidence has been suggested that β-amyloid-mediated synaptic loss is dependent on the upregulation of the NMDA receptor-mediated activity and cytoplasmic calcium ions, and this process contributes to dendritic spine loss.33–35 Recently, in non-demented older adults, Slot and colleagues found that CSF clusterin levels partially mediated the relationship between CSF ApoE levels and CSF tau levels. However, they did not find that CSF clusterin mediated the relationship between APOE4 genotype and CSF Aβ42 levels.36 In the present study, we found that CSF clusterin was associated with CSF NG levels after adjusting for CSF Aβ levels, indicating that this association may go beyond the effect of Aβ on synaptic function. However, further preclinical and clinical studies are needed to support this notion.
However, it is also likely that the upregulation of clusterin levels may represent a neuroprotective response. For instance, it has been reported that clusterin can reduce amyloid formation and provide significant cytoprotection.37 In addition, clusterin has been reported to enhance amyloid clearance from brain across the BBB via LRP2.6 Further, a prospective longitudinal study conducted in Rotterdam found that clusterin levels in plasma were associated with the prevalence and severity of AD, but not the incidence of AD during follow-up, suggesting that the upregulation of clusterin may be triggered by the neurodegenerative changes that occur in AD.2
Interestingly, we found that levels of clusterin in CSF were positively correlated with NG levels in the CN and MCI groups, but not the AD group. Our data highlighted a potential role of clusterin in the early stages of AD. However, further studies are warranted to examine the mechanism by which clusterin affects synaptic degeneration in the early stages of AD.
Several limitations should be noted. First, the cross-sectional design applied in the present study limits the ability to differentiate whether increased clusterin levels result from, cause, or are just correlated with synaptic degeneration among older individuals. Further preclinical and clinical studies are needed to examine the precise relationship between clusterin and synaptic degeneration. Second, CSF readout may not reflect what exactly happening in the brain. Therefore, recent advancements, such as multiplexed single synapse assay using SynTOF panel,38 and more direct assessments of biomarkers in interstitial brain tissue39 should be utilized in further studies. Third, the ADNI study represents a convenience sample of volunteers, which may reduce the power to generalize our findings to other populations. Further, most of the participants of the ADNI study are Caucasian and well educated, which may also limit the generalizability of our findings. However, the ADNI study is one of the most successful studies focusing on examining whether multiple biomarkers could be used to measure the progression of MCI and early AD dementia. More markers and more neurodegenerative diseases should be included in the ADNI study to further facilitate the biomarker discovery.
In conclusion, our data indicated that CSF clusterin was associated with CSF NG levels among older individuals with different severities of cognitive impairment.
Acknowledgments
Data collection and sharing for this project were funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Disclosure
The authors declare that they have no conflict of interest.
References
1. Harold D, Abraham R, Hollingworth P, Sims R, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088–1093. doi:10.1038/ng.440
2. Schrijvers EM, Koudstaal PJ, Hofman A, Breteler MM. Plasma clusterin and the risk of Alzheimer disease. JAMA. 2011;305(13):1322–1326. doi:10.1001/jama.2011.381
3. Thambisetty M, Simmons A, Velayudhan L, et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry. 2010;67(7):739–748. doi:10.1001/archgenpsychiatry.2010.78
4. Nelson AR, Sagare AP, Zlokovic BV. Role of clusterin in the brain vascular clearance of amyloid-beta. Proc Natl Acad Sci U S A. 2017;114(33):8681–8682. doi:10.1073/pnas.1711357114
5. Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell. 2015;163(5):1064–1078. doi:10.1016/j.cell.2015.10.067
6. Bell RD, Sagare AP, Friedman AE, et al. Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2007;27(5):909–918. doi:10.1038/sj.jcbfm.9600419
7. Zlokovic BV, Martel CL, Matsubara E, et al. Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci U S A. 1996;93(9):4229–4234. doi:10.1073/pnas.93.9.4229
8. Zlokovic BV. Cerebrovascular transport of Alzheimer’s amyloid beta and apolipoproteins J and E: possible anti-amyloidogenic role of the blood-brain barrier. Life Sci. 1996;59(18):1483–1497. doi:10.1016/0024-3205(96)00310-4
9. Zlokovic BV, Martel CL, Mackic JB, et al. Brain uptake of circulating apolipoproteins J and E complexed to Alzheimer’s amyloid beta. Biochem Biophys Res Commun. 1994;205(2):1431–1437. doi:10.1006/bbrc.1994.2825
10. Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science (New York, NY). 2002;298(5594):789–791. doi:10.1126/science.1074069
11. Kvartsberg H, Duits FH, Ingelsson M, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dement. 2015;11(10):1180–1190. doi:10.1016/j.jalz.2014.10.009
12. Thorsell A, Bjerke M, Gobom J, et al. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain Res. 2010;1362:13–22. doi:10.1016/j.brainres.2010.09.073
13. Sun X, Dong C, Levin B, et al. APOE epsilon4 carriers may undergo synaptic damage conferring risk of Alzheimer’s disease. Alzheimers Dement. 2016;12(11):1159–1166. doi:10.1016/j.jalz.2016.05.003
14. Tarawneh R, D’Angelo G, Crimmins D, et al. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer disease. JAMA Neurol. 2016;73(5):561–571. doi:10.1001/jamaneurol.2016.0086
15. Hayashi Y. Long-term potentiation: two pathways meet at neurogranin. EMBO J. 2009;28(19):2859–2860. doi:10.1038/emboj.2009.273
16. Zhong L, Cherry T, Bies CE, Florence MA, Gerges NZ. Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J. 2009;28(19):3027–3039. doi:10.1038/emboj.2009.236
17. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198. doi:10.1016/0022-3956(75)90026-6
18. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412. doi:10.1212/WNL.43.11.2412-a
19. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology. 1984;34(7):939–944. doi:10.1212/WNL.34.7.939
20. Mattsson N, Insel P, Nosheny R, et al. Effects of cerebrospinal fluid proteins on brain atrophy rates in cognitively healthy older adults. Neurobiol Aging. 2014;35(3):614–622. doi:10.1016/j.neurobiolaging.2013.08.027
21. De Vos A, Jacobs D, Struyfs H, et al. C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer’s disease. Alzheimers Dement. 2015;11(12):1461–1469. doi:10.1016/j.jalz.2015.05.012
22. Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65(4):403–413. doi:10.1002/(ISSN)1531-8249
23. Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088–1093. doi:10.1038/ng.440
24. Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–1099. doi:10.1038/ng.439
25. Hakkoum D, Imhof A, Vallet PG, et al. Clusterin increases post-ischemic damages in organotypic hippocampal slice cultures. J Neurochem. 2008;106(4):1791–1803. doi:10.1111/j.1471-4159.2008.05519.x
26. Freeman SH, Kandel R, Cruz L, et al. Preservation of neuronal number despite age-related cortical brain atrophy in elderly subjects without Alzheimer disease. J Neuropathol Exp Neurol. 2008;67(12):1205–1212. doi:10.1097/NEN.0b013e31818fc72f
27. Bobinski M, de Leon MJ, Wegiel J, et al. The histological validation of post mortem magnetic resonance imaging-determined hippocampal volume in Alzheimer’s disease. Neuroscience. 2000;95(3):721–725. doi:10.1016/S0306-4522(99)00476-5
28. Desikan RS, Thompson WK, Holland D, et al. The role of clusterin in amyloid-beta-associated neurodegeneration. JAMA Neurol. 2014;71(2):180–187. doi:10.1001/jamaneurol.2013.4560
29. Oh SB, Kim MS, Park S, et al. Clusterin contributes to early stage of Alzheimer’s disease pathogenesis. Brain Pathol. 2018;29(2):217–231.
30. Malkki H. Alzheimer disease: chaperone protein clusterin is involved in amyloid-beta-associated entorhinal atrophy in early AD. Nat Rev Neurol. 2014;10(2):60.
31. Thambisetty M, An Y, Kinsey A, et al. Plasma clusterin concentration is associated with longitudinal brain atrophy in mild cognitive impairment. NeuroImage. 2012;59(1):212–217. doi:10.1016/j.neuroimage.2011.07.056
32. Killick R, Ribe EM, Al-Shawi R, et al. Clusterin regulates beta-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway. Mol Psychiatry. 2014;19(1):88–98. doi:10.1038/mp.2012.163
33. Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13(7):812–818. doi:10.1038/nn.2583
34. Tu S, Okamoto S, Lipton SA, Xu H. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol Neurodegener. 2014;9:48. doi:10.1186/1750-1326-9-48
35. Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192(1):106–113. doi:10.1016/j.bbr.2008.02.016
36. Slot RER, Kester MI, Van Harten AC, et al. ApoE and clusterin CSF levels influence associations between APOE genotype and changes in CSF tau, but not CSF Abeta42, levels in non-demented elderly. Neurobiol Aging. 2019;79:101–109. doi:10.1016/j.neurobiolaging.2019.02.017
37. Yerbury JJ, Poon S, Meehan S, et al. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007;21(10):2312–2322. doi:10.1096/fj.06-7986com
38. Gajera CR, Fernandez R, Postupna N, et al. Mass synaptometry: high-dimensional multi parametric assay for single synapses. J Neurosci Methods. 2019;312:73–83. doi:10.1016/j.jneumeth.2018.11.008
39. Cirrito JR, Yamada KA, Finn MB, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48(6):913–922. doi:10.1016/j.neuron.2005.10.028
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.