Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
Association Between IL-17 and Chronic Obstructive Pulmonary Disease: A Systematic Review and Meta-Analysis
Authors Ma R, Su H, Jiao K, Liu J ![]()
Received 20 March 2023
Accepted for publication 24 July 2023
Published 2 August 2023 Volume 2023:18 Pages 1681—1690
DOI https://doi.org/10.2147/COPD.S412626
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Ru Ma,1– 3 Hongling Su,1– 3 Keping Jiao,1– 3 Jian Liu1,2
1The First Clinical Medical College of Lanzhou University, Lanzhou City, Gansu Province, People’s Republic of China; 2Lanzhou University, Lanzhou City, Gansu Province, People’s Republic of China; 3Gansu Provincial People’s Hospital, Lanzhou, Gansu Province, People’s Republic of China
Correspondence: Jian Liu, Department of Clinical Medicine, the First Clinical Medical College of Lanzhou University, No. 1, Donggang West Road, Chengguan District, Lanzhou City, Gansu Province, People’s Republic of China, Tel +86 136 0935 4197, Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory disease characterized by neutrophils airway infiltration. It is currently known that Interleukin-17 (IL-17) is an important pro-inflammatory factor. It can promote the accumulation of neutrophils and participate in the chronic inflammatory process of COPD. However, the value of IL-17 levels in the diagnosis and assessment of COPD remains controversial. In view of this, we conducted a systematic review and meta-analysis to assess its relevance.
Methods: We searched databases such as PubMed, Web of Science, Cochrane Library and Embase to extract original research.
Results: A total of 10 studies with 2268 participants were included in this meta-analysis. The results showed that the level of serum IL-17 in patients with stable COPD was significantly higher than that in healthy controls (standard mean difference SMD, 1.59, 95% CI 0.84– 2.34; p< 0.001). Compared with the stable COPD group, the serum IL-17 level in acute exacerbation (AECOPD) was significantly higher (SMD, 1.78, 95% CI 1.22– 2.33; p< 0.001). The level of IL-17 in sputum of COPD patients was also higher than that of healthy controls (SMD, 2.03, 95% CI 0.74– 3.31; p< 0.001).
Conclusion: Our results showed that IL-17 levels were elevated in serum and sputum in COPD patients compared with healthy controls, and IL-17 levels increased with disease progression. IL-17 serves as a potential biomarker to indicate the persistence of neutrophilic inflammation and exacerbation of COPD.
Keywords: chronic obstructive pulmonary disease, Interleukin-17, proinflammatory factor, meta-analysis
Introduction
Chronic obstructive pulmonary disease is a common, incurable, heterogeneous disease characterized by persistent airway inflammation and irreversible airflow limitation.1 About 3 million people worldwide die from the disease every year.2 COPD has been reported to be associated with multiple factors, including systemic and local inflammation, air pollution, and a sedentary lifestyle,3–5 Although the pathogenesis of COPD is unknown, it is well known that the chronic inflammatory response of the airways and lung parenchyma caused by cigarette smoke is the main cause of COPD.6,7 The toxic chemicals in cigarette smoke cause abnormal airway inflammation, which triggers the release of chemokines and promotes infiltration of neutrophils and other inflammatory cells into the airways. Accumulated neutrophils can produce and release a variety of pro-inflammatory mediators and enzymes, including neutrophil elastase (NE) and matrix metalloproteinases, which together contribute to the development of chronic bronchitis and emphysema.8–10
Interleukin-17 (IL-17) is considered to be one of the important pro-inflammatory factors involved in the persistent development of COPD airway inflammation. IL-17 is mainly secreted by helper T cells (Th) 17, and induces neutrophil activation by inducing chemokine to produce IL-17.11 Smokers with COPD showed higher levels of IL-17, p53 and plasminogen activator inhibitor-1 (PAI-1) than healthy smokers (HSs) and healthy controls (HCs).12 In experiments using bleomycin-induced inflammation in alveolar basal epithelial cells simulating in vitro inflammation, upregulation of IL-17 promoted alveolar basal epithelial cell motility and increased production of p53 and PAI-1. By raising p53 and PAI‐1, IL‐17 encourages neutrophil infiltration and lung damage. Alveolar epithelial cells can undergo apoptosis when exposed to both p53 and PAI‐1; however, PAI‐1 prevents neutrophil apoptosis and fibrinolysis in lung tissue. Additionally, IL‐17 stimulates the production of IL‐8, granulocyte‐colony stimulating factor (G‐CSF), and C–X–C motif chemokine ligand 2 (CXCL2), which draws in neutrophils and causes them to produce neutrophil elastase and myeloperoxidase, leading to the breakdown of the alveolar wall and the development of emphysema. Meanwhile, IL‐17 was also found to promote airway remodeling in COPD. COPD‐related lung structural remodeling can result in permanent airflow obstruction.13 In addition, cigarette smoke stimulation leads to increased IL-17 secretion in COPD patients, making COPD acutely exacerbated and thus contributing to disease progression.14
IL-17 promotes inflammatory response and participates in the pathological process of autoimmune diseases.15–17 Overproduction of IL-17 induces the expression of a large number of inflammatory factors, which may lead to conditions such as reduced tissue flexibility and tissue fibrosis.18 In rheumatoid arthritis (RA), IL-17 acts locally on synovial cells and osteoblasts, causing synovitis and joint destruction.19 In studies related to periodontitis it has been reported that neutrophil infiltration, triggering upregulation of IL17/Th17 responses, and Th17-driven mucosal inflammation lead to destruction of tooth-supporting bone.20 It has also been reported that the pro-inflammatory cascade controlled by the IL-23/IL-17 axis is thought to be the most important factor in the immunopathogenesis of psoriasis. il-23 plays a role in the differentiation and activation of Th 17 cells. In the presence of bacteria and fungi, IL-17 A induces chemokine expression in keratin-forming cells, induces the production of pro-inflammatory cytokines and leads to an immune response in the skin.21 However, the expression status and biological function of IL-17 in COPD are still unclear. To explore the relationship between IL-17 and COPD, we conducted a meta-analysis to assess the relationship between the two.
Methods
Data Source and Search Strategy
We systematically searched PubMed, Web of Science (WOS), Excerpted Medical Database (Embase), The Cochrane Library and review article reference lists without regard to publication date, status or language. The search terms were [“pulmonary disease, chronic obstructive” (MeSH Terms) or “chronic obstructive pulmonary disease” or “COPD” or “COAD” or “chronic obstructive airway disease” or “chronic obstructive lung disease” or “emphysema” or ‘chronic bronchitis’] and [“Interleukin-17”(MeSH Terms) or “IL-17”]. Only articles published in English were included. These search strategies can be found in the Supplementary Material and Table S1.
Eligibility Criteria
Eligible studies must meet all of the following criteria: describe the relationship between IL-17 and COPD; give specific concentrations of IL-17; provide control and COPD IL-17 levels; provide sufficient patient data to calculate standard mean Differences (SMD) and their 95% confidence intervals (CIs); COPD patients were diagnosed according to the criteria of the American Thoracic Association or the Global Chronic Obstructive Pulmonary Disease Initiative; included in the physical examination and laboratory tests without disease or abnormality, and healthy without symptoms of infection.
Exclusion Criteria
Conference abstracts, editorials, letters, reviews, meta-analyses; studies without a control group; Patients on nutritional support; patients with a history or diagnosis of respiratory disease other than asthma, allergy or COPD; patients with other immune system disorders were excluded, and animal experiments.
Data Extraction
Screening information was independently performed by two investigators from the original study. If relevant, the full article is retrieved. In these articles, references citing relevant reviews or original studies were also accessed to identify other eligible studies. Any disagreements between reviewers were resolved by a third researcher.
Quality Assessment
The following data information was extracted from the original included studies independently by two reviewers (DX and YY): Publication information (name of first author, year of publication), patient and control characteristics (country, sample size, mean age, sex, smoking status; extracted patient status, control status, and exposure assessment), and outcome variables (IL-17 concentration), and the predicted first second of forced expiration (FEV1). Any disagreements were resolved by consensus by a third reviewer. The Newcastle-Ottawa Scale (NOS) was used to assess the quality and bias of case control and cohort studies (Table S2).
The quality of included studies was independently assessed by two reviewers, YY and ZJ, using the Newcastle-Ottawa Quality Assessment Scale (NOS). NOS is a semi-quantitative scale consisting of three dimensions: selection (4 items), comparability (1 item), and degree of exposure (3 items), termed to assess case-control studies and cohort studies. A “star system” (range, 0–9) was developed for assessment. Studies with an overall score of ⩽3 were considered low-quality, 4–6 were considered moderate quality, and 7–9 were considered high-quality.
Statistical Analysis
The results of the meta-analysis were mainly to assess differences in IL-17 concentrations between healthy subjects, stable patients, and patients with acute exacerbations. The data we extract are all continuous. Forest plots for continuous data were constructed using standardized mean difference (SMD) and 95% confidence intervals (CIs). P< 0.05 was considered statistically significant. Because heterogeneity cannot be ignored, we used a random-effects model approach to calculate combined effect sizes and the I2 test to quantify heterogeneity between studies (I2< 25%, no heterogeneity; I2 between 25% and 50%, moderate heterogeneity; I2 between 50% and 75%, large heterogeneity; and I2> 75%, extreme heterogeneity). If there is significant heterogeneity, a sensitivity analysis is required. If the number of included studies exceeds 10, a funnel plot will be used to evaluate.22 Egger’s23 and Begg’s24 tests were also used to assess publication bias. All reported P-values are two-sided, and P<0.05 was considered statistically significant.
Results
Description of Included Studies
The literature search initially detected 2044 records, and 1530 records remained after screening out duplicate entries. After screening the titles and abstracts, 308 studies remained. Among these results, 298 studies that did not meet the inclusion criteria were excluded. Finally, 10 studies were included in this systematic review and meta-analysis, all of which were case-control studies (Figure 1).11,25–33 The population baseline characteristics and characteristics of the included studies are summarized in Table 1. All case-control studies were of moderate quality according to NOS scoring criteria.
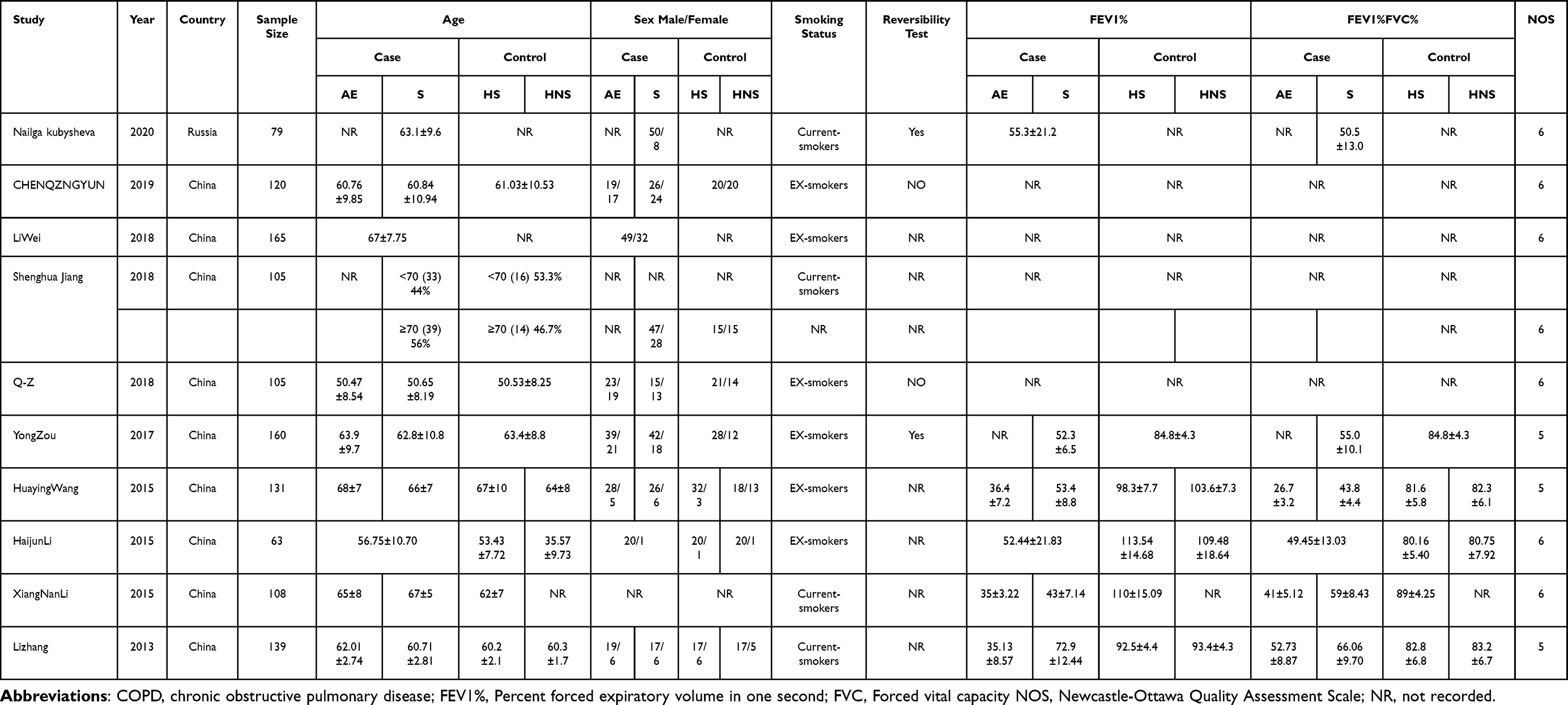 |
Table 1 Characterstics of the Included Studies |
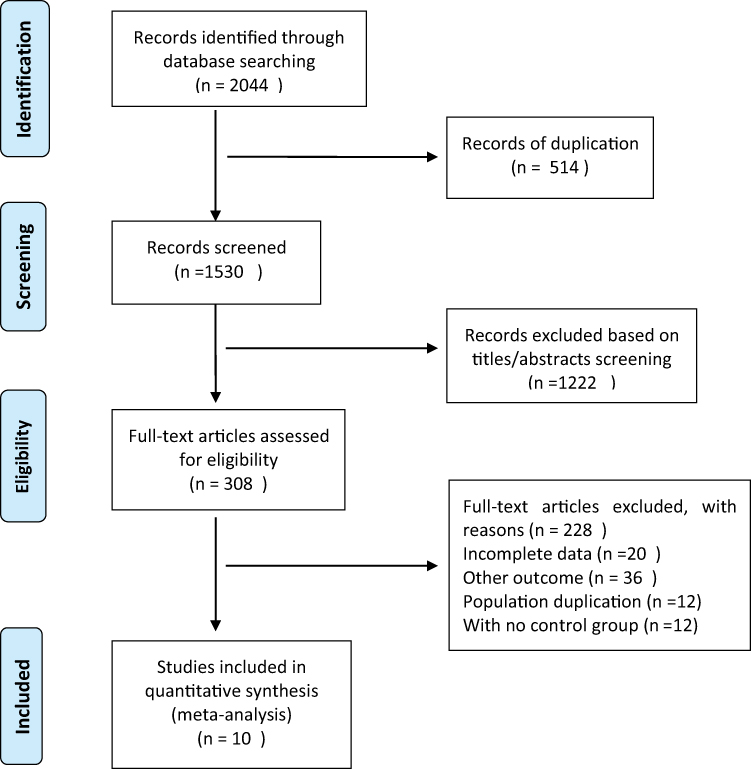 |
Figure 1 Study selection process: PRISMA flow diagram identifying studies included in the meta-analysis. Abbreviation: PRISMA, Preferred reporting Items for systematic reviews and Meta-analyses. Notes: PRISMA figure adapted from Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. Creative Commons.34 |
Serum IL-17 in Stable COPD Vs Control
A total of 811,25,26,29–33 out of 10 studies reported a correlation between serum IL-17 concentrations in stable COPD and controls. The forest plot showed that the serum IL-17 concentration in stable COPD patients was significantly higher than that in the control group (SMD: 1.77; 95% CI: 1.18–2.36; P<0.001; Figure 2). Due to the observed large heterogeneity (I2=91.04%; P<0.001), we performed a sensitivity analysis (Supplementary Figure 1), after excluding each study in turn from the pooled analysis, exclusion of any particular study did not affect the conclusions. In addition, the included studies were free of publication bias (Begg’s test z = 0.12, p = 0.902; Egger’s test P = 0.656). Supplementary Figure S2 shows Egger’s test plot. Supplementary Figure S3 shows Begg’s test plot.
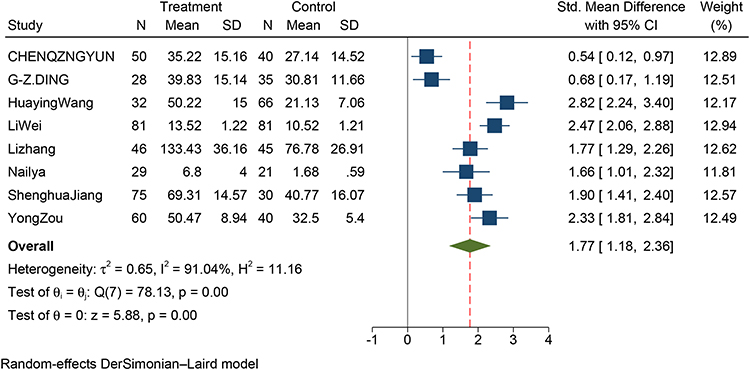 |
Figure 2 Forest plot of the serum IL-17 level between SCOPD and control patients. Abbreviations: SCOPD, stable chronic obstructive pulmonary disease; AECOPD, acute exacerbation chronic obstructive pulmonary disease; SMD, standard mean difference. |
Serum IL-17 in AECOPD Vs Stable COPD
Six studies reported the association of AECOPD with serum IL-17 concentrations in patients with stable COPD.25,26,29–31,33 We found that serum IL-17 concentrations were significantly higher in patients with AECOPD compared with patients with stable COPD (SMD: 1.78; 95% CI: 1.22–2.33; P<0.001; Figure 3).
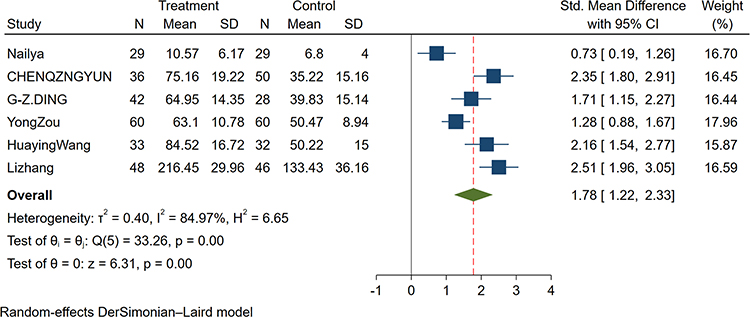 |
Figure 3 Forest plot of the serum IL-17 level between AECOPD and SCOPD patients. Abbreviations: SCOPD, stable chronic obstructive pulmonary disease; AECOPD, acute exacerbation chronic obstructive pulmonary disease; SMD, standard mean difference. |
Sputum IL-17 in Stable COPD Vs Control
A total of two studies reported the correlation of sputum IL-17 concentrations in COPD patients and controls.27,28 The study showed that the sputum IL-17 concentration of COPD patients was higher than that of the control group (SMD: 2.03; 95% CI: 0.74–3.31; P<0.001; Figure 4).
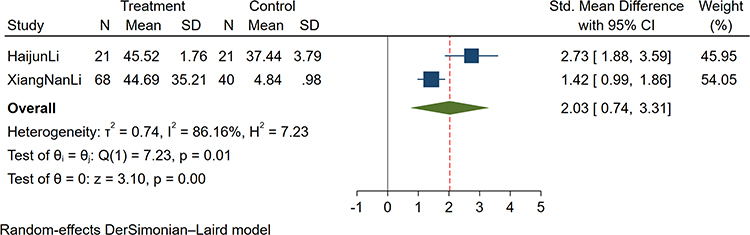 |
Figure 4 Forest plot of the sputum IL-17 level between COPD and control patients. Abbreviations: SCOPD, stable chronic obstructive pulmonary disease; AECOPD, acute exacerbation chronic obstructive pulmonary disease; SMD, standard mean difference. |
IL-17 and Lung Function
Five studies mentioned the association between IL-17 concentrations and lung function, but only two provided complete measurement data, so we could only perform a systematic review of them, not a meta-analysis (Table 1). Kubysheva et al26 reported that the concentration of IL-17 in blood was negatively correlated with FEV1% (r=−0.55, p<0.001). Chen et al31 found that FEV1/FVC and FEV1% in stable COPD group and AECOPD group were significantly lower than those in control group, p<0.05; FEV1/FVC and FEV1% in AECOPD group were significantly lower than those in stable COPD group. Sheng et al11 observed that serum IL-17 expression in stable COPD patients was negatively correlated with FEV1/FVC (p<0.001; regression equation: ^Y=−0.969–0.008X). Zou et al29 also found that serum IL-17 levels (r=−0.562, p<0.001) were significantly negatively correlated with predicted FEV1%. Li et al28 reported that sputum IL-17 was negatively correlated with predicted FEV1% (r=−0.522, p<0.001) and FEV1/FVC (r=−0.667, p<0.001). In conclusion, IL-17 concentrations were inversely correlated with lung function.
Discussion
There have been few meta-analyses of IL-17 concentrations and COPD in previous studies. The purpose of this meta-analysis was to evaluate the relationship between the pro-inflammatory factor IL-17 and COPD. A total of 10 studies were included, all of which were case-control studies. The comprehensive analysis results showed that the serum IL-17 concentration in stable COPD was significantly higher than that in the control group, and the serum IL-17 concentration in AECOPD was significantly higher than that in stable COPD; the sputum IL-17 concentration in COPD patients was significantly higher than that in the control group. Sensitivity analyses that excluded low-quality trials and studies that included only patients with special medical conditions did not alter these results. The research on the relationship between IL-17 and lung function has a small sample size and data, and needs to be further explored in the next work. Based on the above results, we can clarify the clinical significance of IL-17 expression, monitor the response of existing and new treatment strategies, and help doctors make accurate treatment decisions.
The etiology and pathogenesis of COPD have not been fully elucidated. Systemic and local chronic inflammation is recognized as the underlying cause.35,36 Adaptive immune processes are implicated in the pathogenesis of COPD. It has been hypothesized that susceptibility to COPD may arise by a shift from the non-specific innate response present in every smoker toward an adaptive immune response with features typical of autoimmune processes.6,37 An in vitro experiment investigated the process of tissue renewal and damage of IL-17 in bronchial and distal airway epithelial cells from COPD patients, further confirming the role of IL-17 in the induction of inflammatory gene expression in airway epithelial cells during COPD.38 It was also clarified that smoking habit can increase IL-17 expression in two regions in COPD patients. It has been observed that COPD and healthy smokers have increased numbers of IL-17 cells in the submucosa of the bronchi, where T cells may represent an important source of this cytokine in the presence of smoking habits.39 In addition, studies have shown that upregulation of Th17 may be associated with cigarette smoke in mouse lung tissue.40 Together with our findings, these data support a role for Th17 immunity in the pathogenesis of COPD, in relation to risk factors for smoking.
IL-17 is a pro-inflammatory cytokine secreted by Th17 cells after activation, which can accumulate neutrophils into the airways, aggravate the inflammatory response, and cause airway damage. At the same time, IL-17 is also the initiator of many inflammatory mediators.41 Studies have confirmed that IL-17 can promote the secretion of IL-6 and tumor necrosis factor alpfa (TNF-α), which in turn promotes airway fibrous connective tissue remodeling and smooth muscle proliferation, and is involved in COPD airway remodeling.15,33,42 Additional reports have confirmed that neutrophils mediate the formation of IL-1β and promote neutrophil recruitment to the airways. IL-1β can promote IL-17 expression in the lung by increasing the number of IL-17-producing T lymphocytes (αβ T cells and γδ T cells)43,44 and thus contribute to the progressive development of COPD.
Biomarkers are defined as the objective measurable characteristics indicative of normal biological processes, pathogenic processes and prognosis that predict pharmacological response to therapeutic interventions. The development of biomarkers that identifies the endotypes most likely to respond to targeted drug therapy is essential for precision medicine.45 Although there are many studies on serum biomarkers, so far they have not been validated, however, some studies on blood-based markers have shown promising results. IL-17 as a biomarker showed that serum IL-17 concentrations in patients with stable COPD were significantly higher than those in healthy controls, and Serum IL-17 concentrations in AECOPD were significantly higher than those in stable COPD. Roos et al analyzing IL-17 expression in lung tissue samples from patients with stable COPD found increased numbers of IL-17+ cells in COPD patients. This is consistent with our findings. They further demonstrated that mast cells are primary cells expressing IL-17A in advanced COPD.46 Therefore, IL-17A may contribute to the progression of advanced COPD.47 Another study showed that AECOPD patients had significantly higher serum IL-17 concentrations than stable COPD patients, which were lower than healthy controls.48 This differs from our results. These differences may be different from the types and doses of long-term medication COPD patients take, and the severity of COPD stages. In addition, IL-17 is mainly secreted by IL-17-producing T lymphocytes, including αβ T cells and γδ T cells, both of which are induced by IL-1β in COPD lung tissue and BLF43,44. It can be seen that COPD is caused by the interaction of various inflammatory mediators, resulting in systemic and local inflammatory responses. Our analysis also showed that COPD patients had significantly higher sputum IL-17 concentrations than healthy controls. Previous studies have found that the concentration of IL-17 in the bronchial mucosa and sputum of COPD patients is increased, and the number of IL-17+ cells is increased,39,49–51 This is consistent with our aggregated results. Furthermore, it has been reported that serum IL-17A concentrations in COPD patients were inversely correlated with predicted FEV1%.29,33 We also systematically reviewed the negative correlation between serum IL-17 and lung function in this article, and the results were consistent. The relationship between IL-17 and COPD is not limited to clinical studies. Shan et al used quantitative microcomputed tomography (CT) to show that Il-17a−/− mice exposed to cigarette smoke for 16 weeks did not increase lung volume and lung density, whereas Il-17a overexpression resulted in increased lung volume and lung density lower.52 Based on these findings, IL-17 may be a useful prognostic biological indicator in COPD. There are, however, conclusions that differ from ours, and there are so many influencing factors in COPD. As stated in the text, IL-17 interacts with other cytokines, so, we cannot rely on IL-17 alone to evaluate the development of the disease and its prognosis. There are many factors that influence the prognosis of COPD, such as age, lung function, comorbidities, medication use, frequency of hospitalisation, and smoking cessation. Therefore, we cannot limit ourselves to analysing IL-17 as a specific prognostic factor for disease. In future work we need to be more rigorous and comprehensive in our research.
Despite our use of an international standard for GOLD diagnosis and a robust case-control study design, there is significant heterogeneity in this study. The existence of heterogeneity may have the following reasons: First, although the included COPD patients were judged by GOLD criteria, the clinical variability of the patients was uncontrollable; Second, as mentioned above, the research subjects are different in the types and doses of therapeutic drugs. When analyzing subgroup data, the impact of drug intervention cannot be accurately assessed due to insufficient original data; Finally, IL-17 is an initiator of many inflammatory mediators that interact to exert a pro-inflammatory response, so other pro-inflammatory factors activated by IL-17 can also affect the expression of IL-17+ cell numbers. Therefore, we adopted a random-effects model to ensure the stability of the meta-analysis results.
It is worth noting that some of the limitations of this meta-analysis and systematic review itself will also have an impact on our evidence. First, due to the small sample size and incomplete data, the aggregated results are inaccurate, and some statistical results are of little significance; second, the data in this study are published studies, and publication bias cannot be avoided; finally, the few included studies were unable to assess publication bias.
Conclusion
COPD is a chronic inflammatory disease that causes high rates of disability and mortality worldwide. increased expression of IL 17 A contributes to the progression of COPD. Various studies based on COPD have found that the pro-inflammatory role of IL-17 in COPD is manifested in the following ways: First, IL-17 inhibits inflammation-associated mediators and cellular autophagy and promotes airway remodelling. Secondly, IL-17 increases CXCL 1, CXCL 2 and CXCR 2 by promoting the expression of p53 and PAI-1, thereby inducing neutrophil recruitment and triggering inflammation. Finally, IL-17 interacts with a variety of cytokines to trigger inflammation. Therefore, there is substantial evidence that T lymphocytes are increased in the lungs of COPD patients. But these findings also bring us many questions. How do T lymphocytes interact with other immune and non-immune cells in the body? What is the role of signalling pathways of immune cell differentiation in immune regulation and pathogenesis? Can targeted therapy achieve satisfactory results in the treatment of COPD?
It is well known that the treatment of COPD is constantly advancing. However, the most critical knowledge deficit remains our inability to predict the risk of active or former smokers developing progressive lung disease. Ideally, interventions should focus on smoking cessation, but smokers should also be assessed for evidence of autoreactive inflammation using non-invasive tools to provide prognostic information. Studies on targeted therapies are also emerging, but anti-IL-17 monoclonal antibodies have not shown significant results in the treatment of patients with severe COPD. Defining molecular COPD phenotypes and identifying molecular COPD phenotypes with IL-17 characteristics consistent with treatment targeting IL-17 signaling are important strategies for future treatment. There are still many challenges in the pathogenesis of cigarette smoke‐induced COPD, and we need to develop more effective animal models that realistically replicate the natural history, pathological features, and comorbidities of COPD in humans, as well as explore new treatment approaches.
Acknowledgments
The authors gratefully acknowledge the support of the Lanzhou University, the First Clinical Hospital of Lanzhou University, and all the authors who participated in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Science and Technology Projects of Gansu Province (grant number 18JR3RA344).
Disclosure
The authors declare that there is no conflict of interest.
References
1. Asia Pacific COPD Roundtable Group. Global initiative for chronic obstructive lung disease strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease: an Asia-Pacific perspective. Respirology. 2005;10(1):9–17. doi:10.1111/j.1440-1843.2005.00692.x
2. Christenson SA, Smith BM, Bafadhel M, et al. Chronic obstructive pulmonary disease. Lancet. 2022;399(10342):2227–2242. doi:10.1016/S0140-6736(22)00470-6
3. Cai C, Xu CQ, Jin HL, et al. Combined effects of chronic obstructive pulmonary disease and depression on spatial memory in old rats. Chin Med Sci J. 2018;33(4):260–266. doi:10.24920/003470
4. Yazdani R, Marefati H, Shahesmaeili A, et al. Effect of aerobic exercises on serum levels of apolipoprotein A1 and apolipoprotein B, and their ratio in patients with chronic obstructive pulmonary disease. Tanaffos. 2018;17(2):82–89.
5. Emami Ardestani M, Zaerin O. Role of serum Interleukin 6, albumin and C-reactive protein in COPD patients. Tanaffos. 2015;14(2):134–140.
6. Cosio MG, Majo J, Cosio MG. Inflammation of the airways and lung parenchyma in COPD: role of T cells. Chest. 2002;121(5 Suppl):160s–165s. doi:10.1378/chest.121.5_suppl.160S
7. Snider GL. Understanding inflammation in chronic obstructive pulmonary disease: the process begins. Am J Respir Crit Care Med. 2003;167(8):1045–1046. doi:10.1164/rccm.2302002
8. Profita M, Sala A, Bonanno A, et al. Chronic obstructive pulmonary disease and neutrophil infiltration: role of cigarette smoke and cyclooxygenase products. Am J Physiol Lung Cell Mol Physiol. 2010;298(2):L261–269. doi:10.1152/ajplung.90593.2008
9. Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2013;48(5):531–539. doi:10.1165/rcmb.2012-0492TR
10. Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):71–86. doi:10.1016/j.ccm.2013.10.004
11. Jiang S, Shan F, Zhang Y, et al. Increased serum IL-17 and decreased serum IL-10 and IL-35 levels correlate with the progression of COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:2483–2494. doi:10.2147/COPD.S167192
12. Gouda MM, Prabhu A, Bhandary YP. Curcumin alleviates IL-17A-mediated p53-PAI-1 expression in bleomycin-induced alveolar basal epithelial cells. J Cell Biochem. 2018;119(2):2222–2230. doi:10.1002/jcb.26384
13. Barnes PJ, Burney PG, Silverman EK, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers. 2015;1:15076. doi:10.1038/nrdp.2015.76
14. Ko FW, Chan KP, Hui DS, et al. Acute exacerbation of COPD. Respirology. 2016;21(7):1152–1165. doi:10.1111/resp.12780
15. Chang SH, Dong C. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. 2007;17(5):435–440. doi:10.1038/cr.2007.35
16. Iwakura Y, Nakae S, Saijo S, et al. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev. 2008;226:57–79. doi:10.1111/j.1600-065X.2008.00699.x
17. Collison LW, Pillai MR, Chaturvedi V, et al. Regulatory T cell suppression is potentiated by target T cells in a cell contact, IL-35- and IL-10-dependent manner. J Immunol. 2009;182(10):6121–6128. doi:10.4049/jimmunol.0803646
18. Lei Y, Wang K, Li X, et al. Cell-surface translocation of annexin A2 contributes to bleomycin-induced pulmonary fibrosis by mediating inflammatory response in mice. Clin Sci. 2019;133(7):789–804. doi:10.1042/CS20180687
19. Robert M, Miossec P. IL-17 in rheumatoid arthritis and precision medicine: from synovitis expression to circulating bioactive levels. Front Med. 2018;5:364. doi:10.3389/fmed.2018.00364
20. Kim TS, Silva LM, Theofilou VI, et al. Neutrophil extracellular traps and extracellular histones potentiate IL-17 inflammation in periodontitis. J Exp Med. 2023;220(9):e20221751. doi:10.1084/jem.20221751
21. Prieto-Pérez R, Solano-López G, Cabaleiro T, et al. The polymorphism rs763780 in the IL-17F gene is associated with response to biological drugs in patients with psoriasis. Pharmacogenomics. 2015;16(15):1723–1731. doi:10.2217/pgs.15.107
22. Lau J, Ioannidis JP, Terrin N, et al. The case of the misleading funnel plot. BMJ. 2006;333(7568):597–600. doi:10.1136/bmj.333.7568.597
23. Egger M, Davey Smith G, Schneider M, et al. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315(7109):629–634. doi:10.1136/bmj.315.7109.629
24. Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics. 1994;50(4):1088–1101. doi:10.2307/2533446
25. Ding GZ, Li WS. The expressions and significance of APN, D-D, IL-17 and hs-CRP in patients with acute exacerbation of chronic obstructive pulmonary disease. Eur Rev Med Pharmacol Sci. 2018;22(19):6463–6468. doi:10.26355/eurrev_201810_16059
26. Kubysheva N, Boldina M, Eliseeva T, et al. Relationship of serum levels of IL-17, IL-18, TNF-α, and lung function parameters in patients with COPD, asthma-COPD overlap, and bronchial asthma. Mediators Inflamm. 2020;2020:4652898. doi:10.1155/2020/4652898
27. Li XN, Pan X, Qiu D. Imbalances of Th17 and Treg cells and their respective cytokines in COPD patients by disease stage. Int J Clin Exp Med. 2014;7(12):5324–5329.
28. Li H, Liu Q, Jiang Y, et al. Disruption of th17/treg balance in the sputum of patients with chronic obstructive pulmonary disease. Am J Med Sci. 2015;349(5):392–397. doi:10.1097/MAJ.0000000000000447
29. Zou Y, Chen X, Liu J, et al. Serum IL-1β and IL-17 levels in patients with COPD: associations with clinical parameters. Int J Chron Obstruct Pulmon Dis. 2017;12:1247–1254. doi:10.2147/COPD.S131877
30. Wang H, Ying H, Wang S, et al. Imbalance of peripheral blood Th17 and Treg responses in patients with chronic obstructive pulmonary disease. Clin Respir J. 2015;9(3):330–341. doi:10.1111/crj.12147
31. Qingyun C, Fei C. Study on level changes and the significance of serum inflammatory medium IL-21,Il-6 and IL-17 in patients with chronic obstructive pulmonary disease. Acta Med Mediterr. 2019;35:2087–2091.
32. Wei L, Wang K, Ran Z, et al. Auxiliary diagnostic value of γδΤ cell, IL-17, and IFN-γ levels in peripheral blood and bronchoalveolar lavage fluid for lung cancer complicated with chronic obstructive pulmonary disease. Int J Clin Exp Med. 2018;11(7):7183–7191.
33. Zhang L, Cheng Z, Liu W, et al. Expression of interleukin (IL)-10, IL-17A and IL-22 in serum and sputum of stable chronic obstructive pulmonary disease patients. Int J Chron Obstruct Pulmon Dis. 2013;10(4):459–465. doi:10.3109/15412555.2013.770456
34. Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71
35. Tashkin DP, Wechsler ME. Role of eosinophils in airway inflammation of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2018;13:335–349. doi:10.2147/COPD.S152291
36. Arora S, Dev K, Agarwal B, et al. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223(4–5):383–396. doi:10.1016/j.imbio.2017.11.001
37. Lee SH, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13(5):567–569. doi:10.1038/nm1583
38. Montalbano AM, Riccobono L, Siena L, et al. Cigarette smoke affects IL-17A, IL-17F and IL-17 receptor expression in the lung tissue: ex vivo and in vitro studies. Cytokine. 2015;76(2):391–402. doi:10.1016/j.cyto.2015.07.013
39. Di Stefano A, Caramori G, Gnemmi I, et al. T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol. 2009;157(2):316–324. doi:10.1111/j.1365-2249.2009.03965.x
40. Duan MC, Zhong XN, Huang H, et al. Mechanisms and dynamics of Th17 cells in mice with cigarette smoke-induced emphysema. Zhonghua Yi Xue Za Zhi. 2011;26:1996–2000. Chinese.
41. Yu H, Rong F. Relationship between alveolar lavage fluid, serum IL-17 and pulmonary function in patients with chronic obstructive pulmonary disease. Mod Diagn Treat. 2017;2:26–28.
42. Ding Q, Liu GQ, Zeng YY, et al. Role of IL-17 in LPS-induced acute lung injury: an in vivo study. Oncotarget. 2017;8(55):93704–93711. doi:10.18632/oncotarget.21474
43. D’Hulst AII, Vermaelen KY, Brusselle GG, et al. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J. 2005;26(2):204–213. doi:10.1183/09031936.05.00095204
44. Warnatsch A, Ioannou M, Wang Q, et al. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349(6245):316–320. doi:10.1126/science.aaa8064
45. Stockley RA, Halpin DMG, Celli BR, et al. Chronic obstructive pulmonary disease biomarkers and their interpretation. Am J Respir Crit Care Med. 2019;199(10):1195–1204. doi:10.1164/rccm.201810-1860SO
46. Roos AB, Sandén C, Mori M, et al. IL-17A is elevated in end-stage chronic obstructive pulmonary disease and contributes to cigarette smoke-induced lymphoid neogenesis. Am J Respir Crit Care Med. 2015;191(11):1232–1241. doi:10.1164/rccm.201410-1861OC
47. Roos AB, Mori M, Gura HK, et al. Increased IL-17RA and IL-17RC in end-stage COPD and the contribution to mast cell secretion of FGF-2 and VEGF. Respir Res. 2017;18(1):48. doi:10.1186/s12931-017-0534-9
48. Jin Y, Wan Y, Chen G, et al. Treg/IL-17 ratio and Treg differentiation in patients with COPD. PLoS One. 2014;9(10):e111044. doi:10.1371/journal.pone.0111044
49. Chang Y, Nadigel J, Boulais N, et al. CD8 positive T cells express IL-17 in patients with chronic obstructive pulmonary disease. Respir Res. 2011;12(1):43. doi:10.1186/1465-9921-12-43
50. Doe C, Bafadhel M, Siddiqui S, et al. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest. 2010;138(5):1140–1147. doi:10.1378/chest.09-3058
51. Eustace A, Smyth LJC, Mitchell L, et al. Identification of cells expressing IL-17A and IL-17F in the lungs of patients with COPD. Chest. 2011;139(5):1089–1100. doi:10.1378/chest.10-0779
52. Chang Y, Al-Alwan L, Audusseau S, et al. Genetic deletion of IL-17A reduces cigarette smoke-induced inflammation and alveolar type II cell apoptosis. Am J Physiol Lung Cell Mol Physiol. 2014;306(2):L132–143. doi:10.1152/ajplung.00111.2013
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
High-Flow Nasal Cannula Oxygen Therapy versus Non-Invasive Ventilation for AECOPD Patients After Extubation: A Systematic Review and Meta-Analysis of Randomized Controlled Trials
Feng Z, Zhang L, Yu H, Su X, Shuai T, Zhu L, Chen D, Liu J
International Journal of Chronic Obstructive Pulmonary Disease 2022, 17:1987-1999
Published Date: 30 August 2022
Comparison of High-Flow Nasal Cannula with Conventional Oxygen Therapy in Patients with Hypercapnic Chronic Obstructive Pulmonary Disease: A Systematic Review and Meta-Analysis
Zhang L, Wang Y, Ye Y, Gao J, Zhu F, Min L
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:895-906
Published Date: 16 May 2023
Clinically Important Deterioration (CID) and Ageing in COPD: A Systematic Review and Meta-Regression Analysis According to PRISMA Statement
Manzetti GM, Ora J, Sepiacci A, Cazzola M, Rogliani P, Calzetta L
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:2225-2243
Published Date: 10 October 2023
Efficacy and Safety of Bisoprolol in Patients with Chronic Obstructive Pulmonary Disease: A Systematic Review and Meta-Analysis
Feng Z, Zhang L, Wang Y, Guo H, Liu J
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:3067-3083
Published Date: 23 December 2023
Comparison of the Application of Vibrating Mesh Nebulizer and Jet Nebulizer in Chronic Obstructive Pulmonary Disease: A Systematic Review and Meta‐analysis
Feng Z, Han Z, Wang Y, Guo H, Liu J
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:829-839
Published Date: 28 March 2024