Back to Journals » Clinical Epidemiology » Volume 13
Assessing the Associations of Growth Differentiation Factor 15 with Rheumatic Diseases Using Genetic Data
Authors Ye D ![]() , Liu B, He Z, Huang L, Qian Y, Shao K, Wen C, Mao Y
, Liu B, He Z, Huang L, Qian Y, Shao K, Wen C, Mao Y ![]()
Received 2 February 2021
Accepted for publication 11 March 2021
Published 23 March 2021 Volume 2021:13 Pages 245—252
DOI https://doi.org/10.2147/CLEP.S305024
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Vera Ehrenstein
Ding Ye,1 Bin Liu,1 Zhixing He,2 Lin Huang,2 Yu Qian,1 Keding Shao,2 Chengping Wen,2 Yingying Mao1
1Department of Epidemiology and Biostatistics, Zhejiang Chinese Medical University School of Public Health, Hangzhou, Zhejiang, 310053, People’s Republic of China; 2Institute of Basic Research in Clinical Medicine, Zhejiang Chinese Medical University School of Basic Medical Sciences, Hangzhou, Zhejiang, 310053, People’s Republic of China
Correspondence: Yingying Mao
Department of Epidemiology and Biostatistics, Zhejiang Chinese Medical University School of Public Health, Hangzhou, Zhejiang, 310053, People’s Republic of China
Tel +86-571-8661-3536
Email [email protected]
Chengping Wen
Institute of Basic Research in Clinical Medicine, Zhejiang Chinese Medical University School of Basic Medical Sciences, Hangzhou, Zhejiang, 310053, People’s Republic of China
Tel +86-571-8663-3377
Email [email protected]
Objective: To investigate the potential causal associations of circulating levels of growth differentiation factor 15 (GDF-15) with the risk of systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and inflammatory bowel disease (IBD) using a Mendelian randomization (MR) design.
Methods: A genome-wide association study (GWAS) of GDF-15 among 5,440 individuals of European ancestry was used to identify genetic instruments. Summary statistics of SLE, RA and IBD were obtained from publicly available GWASs. We conducted an MR study using the inverse-variance weighted (IVW) method, supplemented with simple-median and weighted-median methods. Cochran Q test and MR-Egger regression were used to detect potential heterogeneity and directional pleiotropy. Odds ratios (ORs) with 95% confidence intervals (95% CIs) were calculated.
Results: We found that genetically predicted high circulating GDF-15 levels were associated with a decreased risk of SLE (OR 0.80, 95% CI 0.68– 0.92 by IVW), with similar results in sensitivity analyses. In replication analysis using summary data from another SLE GWAS, the results were consistent (OR 0.82, 95% CI 0.71– 0.93 by IVW). Moreover, no evidence of heterogeneity or pleiotropy was detected. However, genetically determined circulating levels of GDF-15 were not associated with risk of RA or IBD in the primary analysis and subsequent sensitivity analyses.
Conclusions: Our study suggested an inverse association between circulating GDF-15 levels and risk of SLE, and further studies are warranted to elucidate the underlying biological mechanisms. There was limited evidence supporting a causal association of circulating GDF-15 levels with risk of RA and IBD.
Keywords: growth differentiation factor 15, systemic lupus erythematosus, rheumatoid arthritis, inflammatory bowel disease, Mendelian randomization, single nucleotide polymorphism
Plain Language Summary
Growth differentiation factor 15 (GDF-15), is a divergent TGF-β family member, which was originally identified to play an important role in cell growth and differentiation. Recently, GDF-15 has been found to coordinate tolerance to inflammatory damage, and can be induced by inflammatory stimuli in both immune cells and parenchymal cells. Observationally, circulating levels of GDF-15 were found higher in patients with rheumatic diseases. However, this association could be possibly driven by reverse causation, confounding or selection bias. In this study, we performed a Mendelian randomization analysis using summary statistics from of rheumatoid arthritis, systemic lupus erythematosus and inflammatory bowel disease from publicly available genome-wide association studies. We found suggestive evidence for a protective effect of circulating GDF-15 on the risk of systemic lupus erythematosus, with similar results from sensitivity analysis, which would be useful for further elucidation of the underlying mechanism. However, no casual association between circulating GDF-15 levels and the risk of rheumatoid arthritis or inflammatory bowel disease was found.
Introduction
Rheumatic diseases, such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and inflammatory bowel disease (IBD) are major challenges for global health.1 These diseases have common pathogenesis of an immune-mediated attack on the body’s own organs, and both environmental and genetic risk factors contribute to the etiology.2
Studies have demonstrated that a variety of cytokines are altered in patients with rheumatic diseases, and the concentrations of certain cytokines also correlate with disease activity and severity, such as transforming growth factor β (TGF-β), interleukin-4 (IL-4) and tumor necrosis factor α (TNF-α).3 Growth differentiation factor 15 (GDF-15), also known as NSAID-activated gene 1 (NAG-1) and macrophage inhibitory cytokine 1 (MIC-1), is a divergent TGF-β family member, which has been identified to play a pivotal role in cell growth and differentiation.4 Recently, GDF-15 was reported to be induced by inflammatory stimuli in both immune cells and parenchymal cells, and coordinate tolerance to inflammatory damage.5
Moreover, GDF-15 has been reported to be involved in the etiology and pathogenesis of numerous diseases, including cardiovascular events,6 diabetes mellitus7 and cancers.8–10 Despite the unclear underlying biological mechanisms, the role of GDF-15 in inflammation has already become apparent.11 A case-control study has shown that serum levels of GDF15 were higher in patients with lupus and autoimmune hepatitis, as compared to healthy controls.5 In addition, higher serum levels of GDF-15 were observed in patients with RA, and the levels correlated with disease severity independently of classic disease markers.11–13 In addition, the expression level of GDF-15 was higher in the lesions of the patients with Crohn’s disease than those in the normal intestinal tissues.14 However, these observed associations could be possibly driven by reverse causation, confounding or selection bias inherent in conventional observational studies.
Mendelian randomization (MR) is a type of instrumental variable (IV) analysis that utilizes genetic variants, such as single nucleotide polymorphisms (SNPs), to infer the causality of the exposure with respect to disease.15 As genetic variants cannot be modified by disease status, reverse causation can be avoided in MR analyses. Moreover, as genetic variants are presumed to be randomly distributed in the general population according to Mendel’s laws of inheritance, MR analyses can minimize the risk of confounding bias.16 Thus, we used the MR approach to investigate whether genetically predicted circulating GDF-15 levels were associated with the risk of SLE, RA and IBD.
Methods
As the current study was based on published studies and public databases, no additional ethics approval or informed consent was required. The overall design of this study is displayed in Figure 1.
 |
Figure 1 Schematic diagram of the Mendelian randomization (MR) assumptions underpinning an MR analysis of the association between circulating GDF-15 levels and risk of rheumatic diseases. |
Data Sources
Summary statistics of SLE were obtained from a previously published meta-analysis of genome-wide association studies (GWASs).17 The genetic association data consisted of 4,036 SLE patients and 6,959 healthy controls, covering a total of 644,674 markers. As replication analysis, we utilized summary statistics of another SLE GWAS which included 1,311 SLE patients and 3,340 healthy controls.18 There was no overlap of the study population involved in the two datasets. For RA, a GWAS meta-analysis including 14,361 RA cases and 43,923 controls of European descent from 18 studies was used, with a total of 9,700,598 genotyped SNPs.19 Summary-level data for IBD was extracted from a meta-analysis of GWASs with 25,042 IBD cases and 34,915 controls of European ancestry, covering a total of 9,619,016 SNPs.20 Genetic data of inflammatory cytokines including TNF-α, IFN-γ, IL-1β, IL- 4, IL-6, IL-10, IL-12 and IL-17 was obtained from a GWAS enrolling 8,293 individuals of European ancestry.21 Detailed information about the studies and datasets used in the present study is shown in Supplementary Table 1.
Selection of GDF-15 Associated SNPs
The genetic predictors of circulating GDF-15 were extracted from a GWAS pooling four cohorts of individuals of European descent.22 Briefly, a total of 5,440 individuals with a mean age of 62 years (range: 14–94 years) were included, and 53% (range: 47.0–56.0%) of the participants were women. The concentrations of GDF-15 in serum or plasma were measured using immunoassays. Detailed genotyping and imputation procedures have been described previously.23 In the meta-analysis of all cohorts in the discovery and replication samples combined, eight SNPs achieved genome-wide significance level (P value < 5×10−8). In the current study, we selected three independent SNPs (rs888663, rs749451, and rs1054564) as IVs (Cheung et al, 2019), among which two SNPs were in PGPEP1 gene region and one was in GDF15 region (Supplementary Table 2). The pairwise linkage disequilibrium (in terms of r2) of these three SNPs was estimated to be 0.299 (rs888663 – rs749451), 0.136 (rs1054564 – rs749451), and 0.051 (rs888663 – rs1054564) by using the LDlinkR (https://ldlink.nci.nih.gov/) based on CEU populations.
We calculated F-statistics to quantify the strength of the selected instruments.24 The variance explained by the three SNPs was estimated according to minor allele frequency (MAF) and beta value (β), using the equation of 2 × MAF × (1-MAF) ×β2.25 The smallest effect detected by the sample size to provide >80% statistical power at an alpha level of 5% was computed using the online mRnd power tool (https://shiny.cnsgenomics.com/mRnd/). We checked whether the three selected SNPs were associated with other traits by using the GWAS Catalog (https://www.ebi.ac.uk/gwas/, accessed on February 1, 2020). We also checked whether these three SNPs were associated with any inflammatory cytokines, since multivariable MR analysis was limited by the relatively small number of instrumental SNPs in this study.
Statistical Analysis
We applied a two-sample MR analysis to assess the potential causal association of circulating GDF-15 levels with risk of SLE, RA and IBD using the MendelianRandomization package in R software (version 3.6.2).
The primary analyses were conducted using the inverse-variance weighted (IVW) method. The IVW method obtains an effect estimate of each SNP on circulating GDF-15 levels and rheumatic diseases (SLE, RA and IBD) to calculate Wald estimate.26 The standard error of the Wald estimate was calculated by the delta method.27 IVW uses an approach analogous to meta-analysis to combine these Wald estimates and provides a consistent estimate of the causal effect of circulating GDF-15 on risk of SLE, RA and IBD, when each genetic variant satisfies the assumptions of an IV.28 Cochran ’s Q test was used to test whether the estimate of the causal effect was consistent across each individual SNP.29 A fixed-effects model was applied when there was no heterogeneity, otherwise, the random-effects model was used to provide more conservative estimates.30
To assess the robustness of the results, we also performed sensitivity analyses. MR-Egger regression was used to identify potential directional pleiotropy, and simple-median and weighted-median methods were used to adjust for potential pleiotropic effects.31 The MR-Egger regression is disposed to regression dilution bias, and the average horizontal pleiotropic effect across all genetic variants can be interpreted by the intercept term.32 The simple- and weighted-median methods are more robust to violations of the standard IV assumptions when pleiotropic or invalid instruments are included. The weighted-median method provides consistent estimates of the causal effects if more than half of the weight is derived from valid IVs. In addition, we performed sensitivity analyses excluding rs749451, which had an r2 of 0.136 and 0.299 with the other two SNPs. The fluctuation of the results before and after removing the SNP reflects the stability of the association.
Results
Validity of the Selected SNPs
The detailed information about the three SNPs used as IVs and their effect estimates on circulating GDF-15 levels is presented in Supplementary Table 2.
The F statistics that quantify the strength of IVs ranged from 129.76 to 158.97, well above the threshold of F>10 typically recommended for MR analyses (Supplementary Table 3). It was estimated that 7.94% of the variance of circulating GDF-15 level was explained by the three SNPs. Based on the sample size of RA and IBD GWASs, there was >80% power to detect an association of circulating GDF-15 with the outcomes at an effect size of 1.10 for deleterious effect or 0.91 for protective effect. The statistical power was lower for SLE, but we used two independent datasets in discovery and validation stage, respectively. We checked whether the SNPs used as IVs were associated with other traits, and no potentially pleiotropic SNPs were found by using the GWAS Catalog. In addition, none of the individual SNPs were associated with any inflammatory cytokines (Supplementary Table 4). The included SNPs basically fulfill the assumptions of being valid IVs.
Association Between Circulating GDF-15 and Risk of SLE
Fixed-effects IVW method showed strong evidence of a potential causal association between genetically predicted circulating GDF-15 levels and risk of SLE (OR: 0.80, 95% CI 0.68–0.92). There was no evidence of directional pleiotropy assessed by the MR-Egger intercept. Similarly, we found suggestive evidence of a protective effect of circulating GDF-15 on the risk of SLE using the simple-median (OR: 0.80, 95% CI 0.65–0.97) and weighted-median methods (OR: 0.79, 95% CI 0.66–0.96). Sensitivity analysis suggested an inverse association between genetically predicted circulating GDF-15 and risk of SLE after removal of rs749451 (OR 0.80, 95% CI 0.66–0.95). Sing-SNP analysis showed that rs1054564 was associated with a decreased risk of SLE (OR 0.70, 95% CI 0.54–0.90; Supplementary Figure 1). Consistently, the results of replication analysis using an independent SLE GWAS suggested a potential protective effect of circulating GDF-15 on risk of SLE (Figure 2). Therefore, the consistent inverse association between GDF-15 status and risk of SLE in consequent sensitivity analyses, strengthening the conclusions.
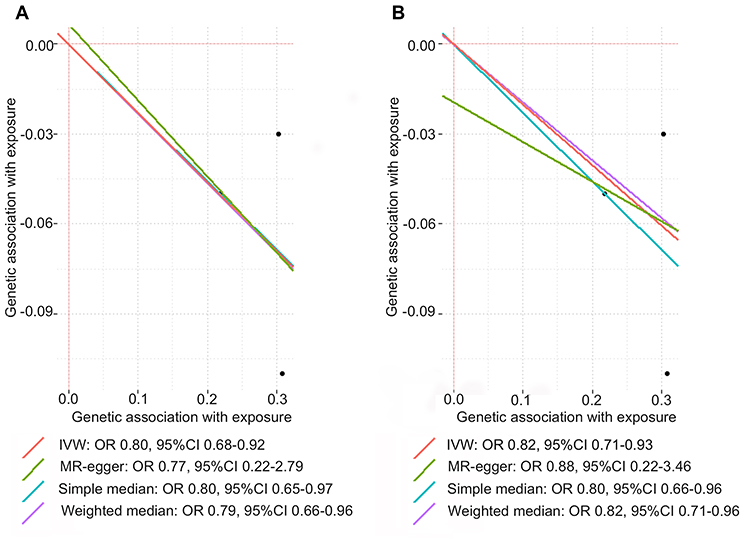 |
Figure 2 MR estimates for the association between circulating GDF-15 levels and risk of SLE in main analysis (A) and replication analysis (B). The single SNP effects on the circulating GDF-15 levels were plotted against the single SNP effects on the risk of SLE and the estimated regression lines of the multi-SNP analyses were added. The effect size was expressed as β value in the scatter plot. |
Association Between Circulating GDF-15 and Risk of RA
As shown in Table 1, there was no evidence of causal association between circulating GDF-15 level and risk of RA by fixed-effects IVW method (OR: 1.11, 95% CI 0.99–1.23). Likewise, the simple-median (OR: 1.13, 95% CI 0.98–1.30) and weighted-median methods (OR: 1.14, 95% CI 0.96–1.34) produced similar effect estimates. There was no evidence of heterogeneity as assessed by Cochran’s Q test and no indication for directional pleiotropy effects by the MR-Egger intercept. Thus, the overall conclusion of no association between GDF-15 and RA was unlikely to be severely influenced by bias.
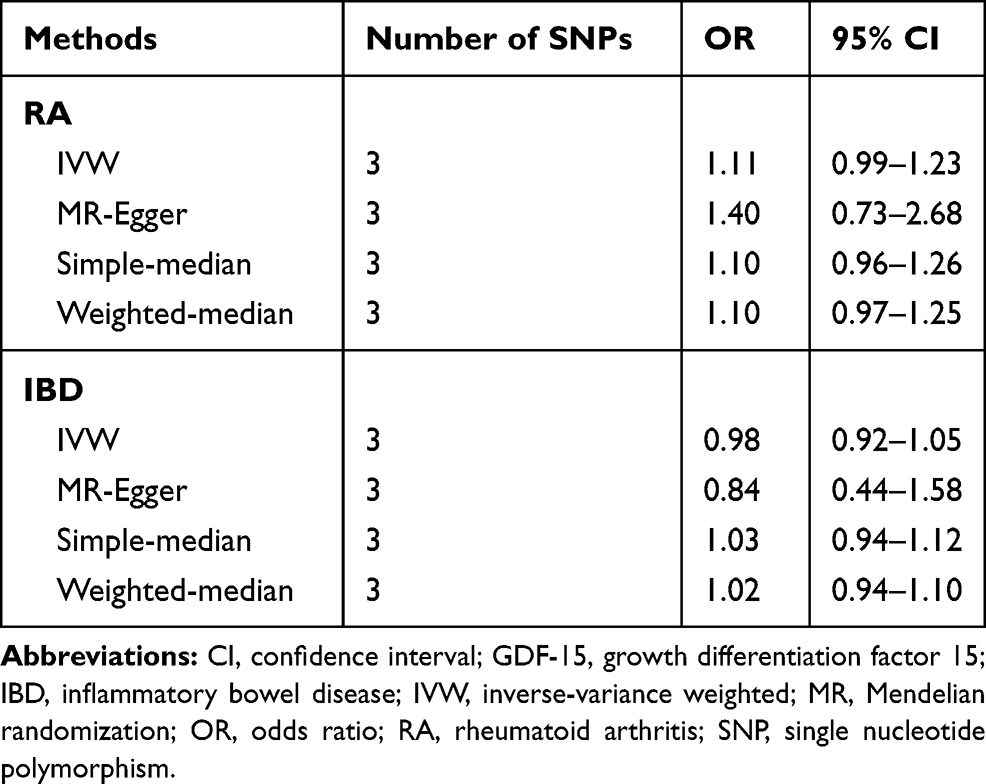 |
Table 1 MR Estimates of the Causal Effect of Circulating GDF-15 on the Risk of RA and IBD |
Association Between Circulating GDF-15 and Risk of IBD
No convincing association was found between circulating GDF-15 and the risk of IBD (OR: 0.98, 95% CI 0.92–1.05) by fixed-effects IVW method. The intercept from the MR-Egger regression analysis did not suggest the presence of directional pleiotropy. In addition, genetically determined higher circulating GDF-15 levels were not associated with risk of IBD using the simple-median (OR: 1.03, 95% CI 0.94–1.12) and weighted-median (OR: 1.02, 95% CI 0.94–1.10) methods (Table 1). The small differences in estimates and CI widths for the effects of the different MR methods might be explained by chance and, possibly, differential measurement errors, rather than indicating distinct estimates.
Discussion
In the present study, we found potential causal evidence of a protective effect of circulating GDF-15 on the risk of SLE; however, we did not find evidence of a causal association between circulating GDF-15 and the risk of RA or IBD. These results provide rationale for using GDF-15 status as a promising target for SLE prevention.
Previous observational studies found that circulating GDF-15 level was positively correlated with rheumatic diseases, however, the increased GDF-15 level could also be a symptom of disease.33 The principal response of the immune system is migration of macrophages and production of inflammatory cytokines to contain physiological balance and stabilization. The expression of GDF-15 is generally low in resting macrophages, but may be dramatically increased following an adaptive stress response to inflammation.34 An in vitro study has demonstrated that exogenous administration of GDF-15 well after the initial inflammatory phase of each disease model was sufficient to improve survival rates.5 Thus, the observational results may be explained by the frustrated attempt of GDF-15 to bring the underlying inflammatory process under control.35 Mechanistically, GDF-15 was found to promote tissue protection from inflammatory damage, primarily cardiac, via regulation of triglyceride metabolism.4 Consistently, GDF15 has been reported to exert a cardioprotective effect in inflammation-driven states.36 Despite the fact that the major function of GDF-15 in the pathogenesis of SLE is not certain, the findings of this MR analysis would be useful for further elucidation of the underlying mechanism.
In MR analysis, there are three assumptions that need to be satisfied. The first assumption is that the genetic variants used as IVs are truly predictive of the exposure.16 We used three independent SNPs associated with circulating levels of GDF-15 at genome-wide significance level. The SNP rs888663 is related to the expression level of GDF-15 in multiple types of tissues in the GTEx database,37 including whole blood (P=1.9×10−6), and the meta-analysis P value reached 5.9×10−16. Functionally, HaploReg from the ENCODE Project38 showed that rs888663 is located at the DNase hypersensitivity peak that highly enriched with H3K27ac. Notably, rs1054564 is located in the 3ʹ UTR of GDF-15, which may be a functional SNP. rs1054564 and rs888663 are the SNPs primarily driving the observed associations between the genes on chromosome 19 and circulating GDF-15 concentration.22 For rs749451, it was reported to be associated with PGPEP1 expression, a gene coding for pyroglutamyl peptidase I, residing on chromosome 9p13.11 abutting GDF-15.39
The second assumption is that the genetic variants are not associated with measured or unmeasured confounders that influence both exposure and outcome. As genotypes are presumed to be randomly allocated at conception, covariates are deemed as randomly distributed with respect to genotypes. Considering inflammatory cytokines might be confounders in the present MR study,21 we checked the association of the three SNPs used as IVs with TNF-α, IFN-γ, IL-1β, IL- 4, IL-6, IL-10, IL-12 and IL-17, and found none of the individual SNPs was associated with any of the inflammatory cytokines.
The third assumption is that the genetic variants affect outcome through their effects on exposure only and not through any alternative causal pathways. We examined the association between the SNPs used as IVs and their potential secondary traits by using the GWAS Catalog, and no other traits have been found for these SNPs. In general, the three SNPs are strong and independent genetic predictors of GDF-15. Since MR studies are susceptible to bias from pleiotropy, we further performed a series of sensitivity analysis to eliminate the influence of potential pleiotropy. Consistently, there was no evidence of potential pleiotropy found in MR-egger regression. Results from simple-median and weighted-median methods were also consistent with the primary analysis, and the replication analysis provided additional confidence of these findings.
This study has several limitations. First, the instrumental variables are located in or near GDF-15 and PGPEP1. Thus, our study needs confirmation when more genetic instruments for GDF-15 from different gene regions are identified. Though no associations between instrumental variables and other traits were found from the GWAS catalog, we cannot expel the possibility that pleiotropic effects may affect our results. Second, the summary-level data for our MR analysis was from participants of European ancestry. Therefore, our findings may be limited when extrapolated to other study populations with different ethnicities. Nevertheless, the MR estimates could be confounded by population stratification. Using genetic studies for both exposure and outcome among European ancestry could minimize such bias. Third, limited to the summary-level data in our MR analyses, we cannot rule out the possibility of a nonlinear causal relationship of circulating GDF-15 with the risk of SLE, RA and IBD. Further MR analysis with individual-level data and a longitudinal design would be ideal to fully evaluate the potential dose–response relationship. Fourth, we hypothesized that GDF-15 might be influenced by health status, ie, reverse causation. Limited to the access to genetic summary statistics for GDF-15, we were unable to examine the possibility using a bi-directional MR design. Finally, the MR analysis of SLE was based on summary statistics with relatively small sample sizes. The potential protective effect of GDF-15 on the risk of SLE should be explored further in larger samples.
In conclusion, this is the first MR study to evaluate the potential causal effect of circulating GDF-15 on SLE, RA and IBD. Our study suggested that genetically predicted circulating GDF-15 was associated with a reduced risk of SLE. Further studies are warranted to elucidate the underlying biological mechanism. However, we did not find associations of circulating GDF-15 with the risk of RA or IBD.
Abbreviations
CI, confidence interval; eQTL, expression quantitative trait loci; GDF-15, growth differentiation factor 15; GWAS, genome-wide association study; IL-4, interleukin-4; IVW, inverse-variance weighted; IBD, inflammatory bowel disease; LD, linkage disequilibrium; MAF, minor allele frequency; MIC-1, macrophage inhibitory cytokine 1; MR, Mendelian randomization; NAG-1, NSAID-activated gene 1; OR, odds ratio; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SNP, single nucleotide polymorphism; TNF-α, tumor necrosis factor α; TGF-β, transforming growth factor β.
Data Sharing Statement
The web source of summary data for this study can be found in Supplementary Table 1.
Ethics Approval
We adopted an MR approach by using publicly accessible summary-level statistics of published GWASs. Therefore, no additional ethical approval was required.
Acknowledgments
We thank the investigators of the genome-wide association studies for sharing their work. This work was supported by grants from the National Natural Science Foundation of China (81973663 and 81703864), the National Key R&D Program of China (2018YFC1705500), Talent Project of Zhejiang Association for Science and Technology (2018YCGC003), and Zhejiang Chinese Medical University Foundation (2020ZG16 and KC201905). The funders had no role in the study design, data analysis, interpretation of data, or preparation of the manuscript. We also thank Ying Guo for the substantial contributions to revising the manuscript.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Rose NR. Prediction and prevention of autoimmune disease in the 21st Century: a review and preview. Am J Epidemiol. 2016;183(5):403–406. doi:10.1093/aje/kwv292
2. Smith DA, Germolec DR. Introduction to immunology and autoimmunity. Environ Health Perspect. 1999;107(Suppl 5):661–665. doi:10.1289/ehp.99107s5661
3. Wahren-Herlenius M, Dorner T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet. 2013;382(9894):819–831. doi:10.1016/S0140-6736(13)60954-X
4. Bootcov MR, Bauskin AR, Valenzuela SM, et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci USA. 1997;94(21):11514–11519. doi:10.1073/pnas.94.21.11514
5. Luan HH, Wang A, Hilliard BK, et al. GDF15 is an inflammation-induced central mediator of tissue tolerance. Cell. 2019;178(5):1231–1244. doi:10.1016/j.cell.2019.07.033
6. Brown DA, Breit SN, Buring J, et al. Concentration in plasma of macrophage inhibitory cytokine-1 and risk of cardiovascular events in women: a nested case-control study. Lancet. 2002;359(9324):2159–2163. doi:10.1016/S0140-6736(02)09093-1
7. Dostalova I, Roubicek T, Bartlova M, et al. Increased serum concentrations of macrophage inhibitory cytokine-1 in patients with obesity and type 2 diabetes mellitus: the influence of very low calorie diet. Eur J Endocrinol. 2009;161:397–404. doi:10.1530/EJE-09-0417
8. Li C, Wang X, Casal I, et al. Growth differentiation factor 15 is a promising diagnostic and prognostic biomarker in colorectal cancer. J Cell Mol Med. 2016;20(8):1420–1426. doi:10.1111/jcmm.12830
9. Wang X, Li Y, Tian H, et al. Macrophage inhibitory cytokine 1 (MIC-1/GDF15) as a novel diagnostic serum biomarker in pancreatic ductal adenocarcinoma. BMC Cancer. 2014;14:578. doi:10.1186/1471-2407-14-578
10. Brown DA, Lindmark F, Stattin P, et al. Macrophage inhibitory cytokine 1: a new prognostic marker in prostate cancer. Clin Cancer Res. 2009;15(21):6658–6664. doi:10.1158/1078-0432.CCR-08-3126
11. Unsicker K, Spittau B, Krieglstein K. The multiple facets of the TGF-beta family cytokine growth/differentiation factor-15/macrophage inhibitory cytokine-1. Cytokine Growth Factor Rev. 2013;24:373–384. doi:10.1016/j.cytogfr.2013.05.003
12. Brown DA, Moore J, Johnen H, et al. Serum macrophage inhibitory cytokine 1 in rheumatoid arthritis: a potential marker of erosive joint destruction. Arthritis Rheum. 2007;56(3):753–764. doi:10.1002/art.22410
13. Tanrikulu O, Sariyildiz MA, Batmaz I, et al. Serum GDF-15 level in rheumatoid arthritis: relationship with disease activity and subclinical atherosclerosis. Acta Reumatol Port. 2017;42:66–72.
14. Choi HJ, Do KH, Park JH, et al. Early epithelial restitution by nonsteroidal anti-inflammatory drug-activated gene 1 counteracts intestinal ulcerative injuries. J Immunol. 2016;197:1415–1424. doi:10.4049/jimmunol.1501784
15. Burgess S, Small DS, Thompson SG. A review of instrumental variable estimators for Mendelian randomization. Stat Methods Med Res. 2017;26(5):2333–2355. doi:10.1177/0962280215597579
16. Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG, Consortium E-I. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. 2015;30(7):543–552. doi:10.1007/s10654-015-0011-z
17. Bentham J, Morris DL, Graham DSC, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015;47(12):1457–1464. doi:10.1038/ng.3434
18. Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med. 2008;358:900–909. doi:10.1056/NEJMoa0707865
19. Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi:10.1038/nature12873
20. de Lange KM, Moutsianas L, Lee JC, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017;49:256–261. doi:10.1038/ng.3760
21. Ahola-Olli AV, Wurtz P, Havulinna AS, et al. Genome-wide association study identifies 27 loci influencing concentrations of circulating cytokines and growth factors. Am J Hum Genet. 2017;100:40–50. doi:10.1016/j.ajhg.2016.11.007
22. Jiang J, Thalamuthu A, Ho JE, et al. A meta-analysis of genome-wide association studies of growth differentiation factor-15 concentration in blood. Front Genet. 2018;9:97. doi:10.3389/fgene.2018.00097
23. Cheung CL, Tan K, Au P, Li G, Cheung B. Evaluation of GDF15 as a therapeutic target of cardiometabolic diseases in human: a Mendelian randomization study. EBioMedicine. 2019;41:85–90. doi:10.1016/j.ebiom.2019.02.021
24. Palmer TM, Lawlor DA, Harbord RM, et al. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res. 2012;21:223–242. doi:10.1177/0962280210394459
25. Park JH, Wacholder S, Gail MH, et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42(7):570–575. doi:10.1038/ng.610
26. Palmer TM, Sterne JA, Harbord RM, et al. Instrumental variable estimation of causal risk ratios and causal odds ratios in Mendelian randomization analyses. Am J Epidemiol. 2011;173(12):1392–1403. doi:10.1093/aje/kwr026
27. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–665. doi:10.1002/gepi.21758
28. Pierce BL, Burgess S. Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol. 2013;178(7):1177–1184. doi:10.1093/aje/kwt084
29. Greco MF, Minelli C, Sheehan NA, Thompson JR. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med. 2015;34:2926–2940. doi:10.1002/sim.6522
30. Greenland S. Quantitative methods in the review of epidemiologic literature. Epidemiol Rev. 1987;9:1–30. doi:10.1093/oxfordjournals.epirev.a036298
31. Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology. 2017;28(1):30–42. doi:10.1097/EDE.0000000000000559
32. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi:10.1093/ije/dyv080
33. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–698. doi:10.1038/s41588-018-0099-7
34. Mimeault M, Batra SK. Divergent molecular mechanisms underlying the pleiotropic functions of macrophage inhibitory cytokine-1 in cancer. J Cell Physiol. 2010;224(3):626–635. doi:10.1002/jcp.22196
35. Breit SN, Johnen H, Cook AD, et al. The TGF-beta superfamily cytokine, MIC-1/GDF15: a pleotrophic cytokine with roles in inflammation, cancer and metabolism. Growth Factors. 2011;29(5):187–195. doi:10.3109/08977194.2011.607137
36. Ago T, Sadoshima J. GDF15, a cardioprotective TGF-beta superfamily protein. Circ Res. 2006;98(3):294–297. doi:10.1161/01.RES.0000207919.83894.9d
37. GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–585. doi:10.1038/ng.2653
38. Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44(D1):D877–81. doi:10.1093/nar/gkv1340
39. Ho JE, Mahajan A, Chen MH, et al. Clinical and genetic correlates of growth differentiation factor 15 in the community. Clin Chem. 2012;58:1582–1591. doi:10.1373/clinchem.2012.190322
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.