")
Back to Journals » Journal of Inflammation Research » Volume 14
Aryl Hydrocarbon Receptor is Essential in the Control of Lung Club Cell Homeostasis
Authors Liu KY, Wang LT, Wang HC, Wang SN, Tseng LW, Chai CY, Chiou SS, Huang SK, Hsu SH
Received 5 October 2020
Accepted for publication 6 January 2021
Published 5 February 2021 Volume 2021:14 Pages 299—311
DOI https://doi.org/10.2147/JIR.S284800
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Kwei-Yan Liu,1 Li-Ting Wang,2 Hsueh-Chun Wang,3,4 Shen-Nien Wang,5– 7 Li-Wen Tseng,5 Chee-Yin Chai,8 Shyh-Shin Chiou,9,10 Shau-Ku Huang,1,11 Shih-Hsien Hsu5,12,13
1Department of Respirology & Allergy, Third Affiliated Hospital of Shenzhen University, Shenzhen, 518020, People’s Republic of China; 2Department of Life Science, National Taiwan Normal University, Taipei, Taiwan; 3Graduate Institute of Biomedical Sciences, China Medical University, Taichung, Taiwan; 4Department of Medical Research, China Medical University Hospital, China Medical University, Taichung, 40402, Taiwan; 5Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, 807, Taiwan; 6Division of General and Digestive Surgery, Department of Surgery, Kaohsiung Medical University Hospital; 7Department of Surgery, College of Medicine, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan; 8Department of Pathology, Faculty of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan; 9Division of Hematology-Oncology Department of Pediatrics, Kaohsiung Medical University Hospital Kaohsiung Medical University, Kaohsiung, Taiwan; 10Department of Pediatrics, Faculty of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, 807, Taiwan; 11National Institute of Environmental Health Sciences, National Health Research Institutes, Miaoli County, Taiwan; 12Department of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, 807, Taiwan; 13Research Center for Environmental Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
Correspondence: Shih-Hsien Hsu; Shau-Ku Huang Email [email protected]; [email protected]
Background: Club cells play an important role in maintaining lung homeostasis and aryl hydrocarbon receptor (AhR) is known to be important in xenobiotic metabolism, but its role in regulating club cells is currently unknown.
Methods: To this end, mice with club cell-specific AhR deficiency were generated and evaluated in a model of antigen (ovalbumin, OVA)-induced airway inflammation for the number of infiltrating inflammatory cells, the levels of cytokines and CC10 and Notch signaling by standard methods.
Results: After OVA sensitization and challenge, Scgb1a1-Cre; Ahrflox/flox mice showed aggravated levels of pulmonary inflammation with increased levels of inflammatory cells and cytokines 1 day after challenge as compared to those seen in their littermate controls, but in contrast to the littermate controls, no significant change in the levels of CC10 and SP-D was noted in Scgb1a1-Cre; Ahrflox/flox mice. Surprisingly, 7 days after the challenge, while, as expected, wild-type mice recovered from acute inflammation, significantly increased lymphocytic infiltration was noted in Scgb1a1-Cre; Ahrflox/flox mice, suggesting their defective mechanism of recovery. Mechanistically, this was due, in part, to the decreased Notch1 signaling and expression of its downstream gene, HES5, while AhR was shown to positively regulate Notch1 expression via its transactivating activity targeting the xenobiotic response element in the promoter region of Notch1 gene.
Conclusion: Under the condition of pulmonary inflammation, AhR is critical in controlling lung club cell homeostasis via targeting Notch1 signaling and the generation of anti-inflammatory mediators.
Keywords: AhR, club cells, CC10, Notch1, Hes5
Introduction
Lung epithelial cells are the first line of defense against external stressors, such as environmental pollutants, infectious agents, and allergens,1,2 and are known to be critical in maintaining lung homeostasis under stringent dynamic controls via, in part, the cell renewal, xenobiotic mechanism and secretion of various immune modulators.3 Dysregulation of these processes may have pathogenic consequences and lead to asthma, chronic obstructive pulmonary disease (COPD), and other lung diseases.4 Asthma is a common chronic airway inflammatory disease characterized by airway obstruction, bronchial hyper-responsiveness and airway inflammation,2 in which co-exposure to environmental pollutants and allergens potentiates and exacerbates allergic inflammation associated with Th2 cytokines, including IL-4, IL-33 and thymic stromal lymphopoietin (TSLP), and induce key characteristics of asthma. However, the underlying molecular mechanisms are currently not well established.
Among respiratory epithelial cell types,5 club cells are the most abundant cell populations and perform several protective functions. Club cells detoxify environmental toxins, produce surfactants, participate in mucociliary clearance along with ciliated cells, and maintain ciliated and non-ciliated cell populations, such as facultative progenitor cells.6 Furthermore, club cells are known to be important in the control of lung inflammation through their secretion of anti-inflammatory mediators, such as club cell 10kd protein (CC10) and surfactant D (SP-D),7–9 and have multiple functions that are fundamental to the health of conducting epithelium, but the precise regulatory mechanisms through which club cells control pulmonary homeostasis remain incompletely defined.
In the respiratory system, Notch signaling plays a critical role during lung development. Notch signaling is required for the pluripotent epithelial progenitors that give rise to lineage-restricted progenitors of the conducting airways in the developing lung. The regulation establishes epithelial club, ciliated and pulmonary neuroendocrine (NE) cell populations.10 It has been reported that Notch signaling is also required for the homeostasis of the adult lung under the lung injury by controlling stem cell maintenance and differentiation, cell proliferation and apoptosis. In allergic airway inflammation, Notch signaling has been focused on the regulation of T cells involved in activation of inflammatory response or resolution of the inflammation.11 However, the regulation of Notch signaling in epithelium, especially in club cells, has not been addressed.
Exposure to environmental pollutants, such as dioxins and polycyclic aromatic hydrocarbons (PAHs), is associated with various pulmonary diseases, including asthma, COPD, and lung cancer.10,11 Aryl hydrocarbon receptor (AhR) is a unique cellular chemical sensor and a ligand-activated transcription factor.12–15 Upon ligand binding, AhR is translocated to the nuclei and heterodimerize with the AhR translocator (Arnt), which subsequently transactivates the transcription of several detoxifying enzymes, such as cytochrome P4501A1 (CYP1A1) and cytochrome P4501B1 (CYP1B1).16 Exposure of epithelial cells to allergens and environmental AhR ligands, such as particulate matter (PM), 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) or Benzo(a)pyrene (B(a)P), exacerbates pulmonary inflammation. While accumulated evidence suggests that AhR in bronchial epithelial cells plays a role in the maintenance of lung homeostasis, the role of AhR in a specific lung epithelium, club cells, remains to be defined. In this study, we generated club cell-specific AhR-null mice and determined the role of AhR in lung homeostasis at the naïve state and under inflammatory conditions. Our data suggest that AhR is required for club cell homeostasis via, in part, targeting Notch 1 signaling and the generation of immune modulators, such as CC10 and SP-D.
Materials and Methods
Mice
All animal experiments were approved by the Kaohsiung Medical University–Institutional Animal Care and Use Committee (IACUC-106,054; Kaohsiung, Taiwan) and were in accordance with the guidelines (KMU Animal Care and Use Program for Center for Laboratory Animals, Kaohsiung Medical University, Taiwan) and regulations of the institution. Club cells specific AhR KO mice were generated by crossing Scgb1a1-Cre and Ahrflox/flox mice, which were from Rodent Model Resource Center and National Health Research Institutes. All mice were maintained in a specific pathogen-free facility.
Plasmids, Cell Lines, and Other Materials
Full-length AhR and GFP fused AhR constructs were generated as described previously. Briefly, AhR cDNA was subcloned into the Tet-on system (pAW4.puro, NCFPB, Taiwan) to express GFP-tagged AhR. A549 and NL-20 cell lines were purchased from American Type Culture Collection and maintained according to American Type Culture Collection protocols. Transfection was performed using a Lipofectamine transfection kit (GIBCO/BRL).
Luciferase Reporter Assays
The Notch1 promoter (between positions −1512 and +17 bp) was cloned from human placental genomic DNA and was used to construct a pGL3 luciferase reporter plasmid. The expression reporter constructs pSV40-Rluc and pGL3-Notch11Fire luciferase (Promega) were transfected into 2 × 105 A549 cells and the cells were harvested after 16 hrs for measurements of relative luciferase activities according to the manufacturer’s instructions. All data are expressed as the means ± standard deviations (SD) of at least three experiments.
RNA Extraction and Semi-Quantitative Real-Time PCR
NL-20 cells were collected from study subjects and extracted total RNA and synthesized cDNA were following the manufacturer’s instructions by using Trizol reagent (GIBCO/BRL) and SuperScript (Invitrogen, Carlsbad, CA), respectively. Notch1, Hes5, and cyp1B1 mRNA expression levels were determined using SYBR Green Quantitative RT-PCR kits (Invitrogen). All reactions and data analyses were performed according to the manufacturer’s instructions.
Western Blotting and Immunohistochemical Analyses
Western blotting and immunohistochemical (fluorescence) staining analyses were performed as described previously18,31 with the following primary antibodies: actin monoclonal (1:5000 dilution; Sigma–Aldrich), FITC-conjugated anti-goat IgG, rhodamine-conjugated anti-rabbit IgG, alkaline phosphatase-conjugated anti-rabbit IgG (Jackson ImmunoResearch Laboratories), AhR and CC10 goat polyclonal antibodies (Santa Cruz Biotechnology), Notch1 and Hes5 rabbit polyclonal antibodies (cell signaling technology), AhR monoclonal antibody (Enzo life Sciences), and GFP rabbit polyclonal antibody (Abcam). Percentages of CC10 and Notch1 or HES5 double-positive cells in total airway cells were counted on 3–6 random fields from three animals.
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed as described previously.32 All data are expressed as the means ± SD of at least three experiments. Notch1 promoter fragments were amplified using DRE1 Primer 1(5ʹ GAC CCGTTTGTGCTTTCTG 3ʹ) and DRE1 Primer 2 (5ʹ GACACGCTCCTCGGTCAC 3ʹ), and DRE2 Primer 1 (5ʹ GCAAATTTCAGTCGCCAGTT 3ʹ) and DRE2 Primer 2 (5ʹ GCGCCTGGGACTACTTCTC 3ʹ).
OVA-Induced Airway Inflammation in a Murine Model
OVA-induced asthma was generated as described previously with a minor modification.34 Briefly, mice were sensitized using intraperitoneal (i.p.) injections of OVA at 100 μg/mouse (grade V, Sigma, St. Louis, MO, USA) with 5 mg of aluminum hydroxide (Thermo Scientific, Rockford, MD, USA) in 200 μL of pyrogen-free phosphate-buffered saline (PBS, PH = 7.3). Mice were then challenged with 3% aerosolized OVA or PBS using an air-compressing nebulizer in a plexiglass chamber (403A, Yuyue, Danyang, Jiangsu, China) for 30 min over three successive days (days 11–13). Control mice were injected i.p. with equal volumes of PBS on day 1 and challenged with aerosolized PBS for 30 min over three successive days (days 11–13). Mice were sacrificed on day 14 for examinations of inflammatory infiltrates, cytokine production, and histology. The study of animals was approved by the Kaohsiung Medical University–Institutional Animal Care and Use Committee (IACUC-106,054; Kaohsiung, Taiwan).
Bronchoalveolar Lavage and Cell Count
Tracheae was cannulated and the lung was lavaged 2 times with 0.8 mL PBS. Each fluid was centrifuged and the supernatant was rapidly frozen at −80°C. The cells in BALFs were stained with PE-Cy7-anti-CD11c (N418; eBioscience), FITC-anti-I-Ad/I-Ed (M5/114.15.2; eBioscience), PE-anti-CCR3 (83,101; R&D Systems), APC-anti-CD3 (145–2C11; BD Biosciences, San Diego, California, USA) and anti-B220 (RA3–6B2; eBioscience) antibodies.33 The cell composition was examined by flow cytometry (LSR II; BD Biosciences).
Lung Pathology
The lung was fixed in 3.7% formaldehyde and embedded in paraffin. The tissue sections (3 μM) were stained by hematoxylin and eosin according to the routine histologic protocol and were observed by a light microscope (Axio Imager M1; Carl Zeiss, Oberkochen, Germany). To examine the inflammatory cell infiltration in the intraluminal, alveolar, peribronchial, and perivascular regions, cell counts were performed blindly based on five-point grading system for the following features: 0: normal, 1: few cells, 2: a ring of inflammatory cells 1 cell layer deep; 3: a ring of inflammatory cells 2–4 cells deep, 4: a ring of inflammatory cells of >4 cells deep as previously described.34 Eight fields were counted for each slide and the mean score was calculated from three animals.
Cytokine Measurements
BALF and serum were harvested as described in our previous report. IL-4, IL-33, TSLP, CC10 and SP-D levels in BALF supernatants were measured using sandwich ELIZA according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN, USA).
Statistical Analysis
Two groups comparisons were examined by Student’s t test, and more than 2 groups were 1-way ANOVA with Bonferroni post-test analysis using Prism 8 (GraphPad). Data are presented as means ± SEM or are expressed as percentages of controls ± SD. All statistical analyses were two tailed and differences were considered significant when P < 0.05.
Result
AhR Deficiency Impacts Lung Development with Reduced Levels of CC10 Expression
To investigate the importance of club cell-specific AhR signaling and its functional consequences. A cohort of conditional AhR-deficient mice were generated by crossing mice carrying an Ahr allele (Ahrflox/flox) in which the exon 2 was flanked by loxP sites with Scgb1a1-Cre mice with Cre recombinase expression being controlled by the CC10 (Scgb1a1) promoter sequences.17 Immunohistochemistry analysis of naïve mice with club cell-specific AhR deficiency (Scgb1a1-Cre; Ahrflox/flox) confirmed the absence of AhR expression in the bronchiolar (Figure 1A), while the expression of a club cell-specific marker, CC10, was seen, albeit at a lower level. The lung tissues of control and Scgb1a1-Cre; Ahrflox/flox mice confirmed the AhR knock-out efficiency (Supplemental Figure 1). It was also noted that while serial histological and immunohistochemical analyses of lungs from 7-week-old mice showed no significant difference in club cell numbers between Scgb1a1-Cre; Ahrflox/flox mice and their littermate controls (Figure 1B and C). Scgb1a1-Cre; Ahrflox/flox mice had smaller lungs than their Scgb1a1-Cre littermate controls during development (Supplemental Figure 2).
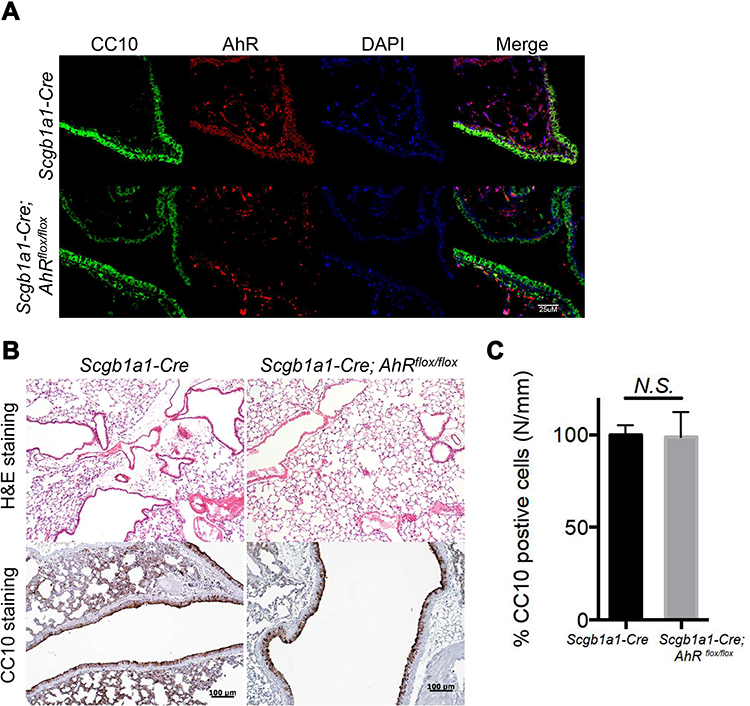 |
Figure 1 Mice with AhR-deficient club cells showed similar number of club cells to wild-type mice. (A) Lung sections from Scgblal-Cre or Scgb1a1-Cre; Ahrflox/flox mice were stained with DAPI (blue), anti-AhR antibody (red), and an anti-club Cell 10-kD protein (CC10) antibody (green). (B) Hematoxylin and eosin (H&E)- and CC10-stained sections are presented from control and club cell-specific AhR-deficient lungs, and quantification data were shown in (C). |
Club Cell-Specific AhR Deficiency Exacerbates Pulmonary Inflammation
Next, we investigated the role of AhR in club cells under an inflammatory condition, wherein an established mouse model of antigen (ovalbumin, OVA)-induced airway allergic inflammation (see Figure 2A for protocol) was analyzed. Results showed that following antigen challenge, significantly reduced numbers of CC10-positive club cells and decreased levels of CC10 and SP-D in the bronchoalveolar lavage fluids (BALFs) were noted in OVA-sensitized Scgb1a1-Cre; Ahrflox/flox mice as compared with those seen in their littermate controls (Figure 2B-D and Supplemental Figure 3, respectively), suggesting a potential defect in club cell regeneration after an inflammatory response.
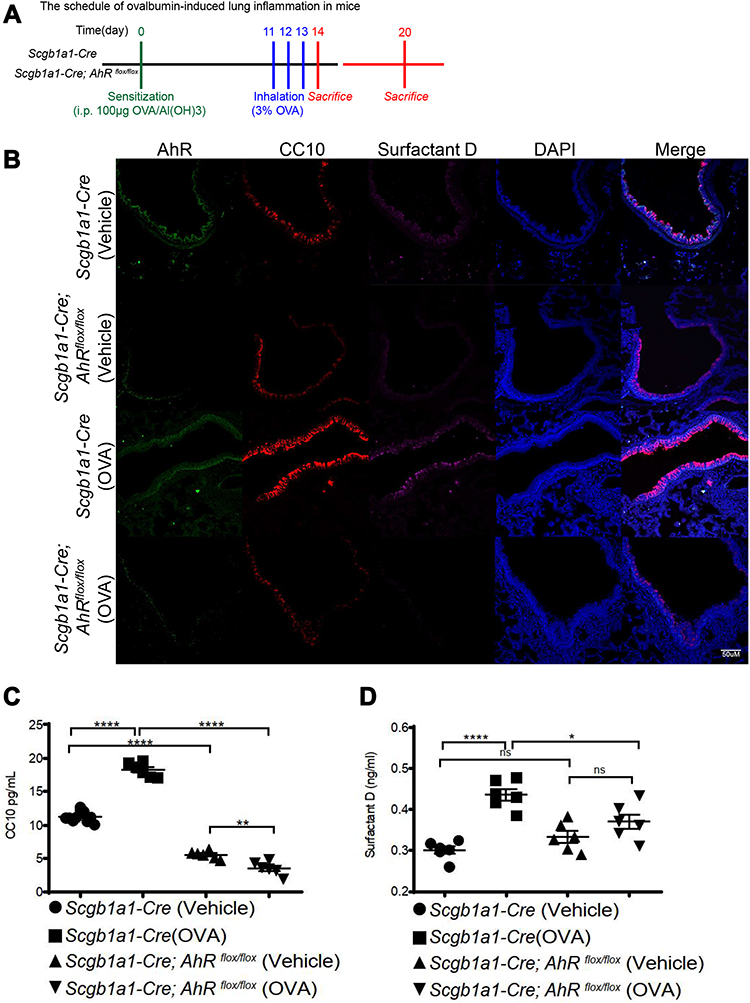 |
Figure 2 AhR deficiency resulted in reduced club cell-specific protein expression in OVA-sensitized and challenged mice. (A) Experimental protocol of Ag (OVA)-induced lung inflammation, in which mice were sacrificed one or 7 days after the last antigen challenge, and lung tissues and BALFs were collected for analysis; (B) lung sections from Scgblal-Cre or Scgb1a1-Cre; Ahrflox/flox mice with vehicle or OVA treatment were stained with DAPI (blue), anti-AhR antibody (green), anti-club Cell 10-kD protein (CC10) antibody (red), and anti-surfactant D (magenta). (C) CC10 and (D) surfactant D in bronchoalveolar lavage fluids were measured using ELISA; *P < 0.05, **P < 0.01 ****P < 0.0001 (One-Way ANOVA). |
Consequently, significantly enhanced levels of infiltrating inflammatory cells were observed in the lungs of Scgb1a1-Cre; Ahrflox/flox mice (Figure 3), and the numbers of total cells, eosinophils, lymphocytes and macrophages were significantly increased in BALFs (Figure 2B-F, respectively). Further, Ag-sensitization and challenge exacerbated the levels of Th2-associated cytokines, IL-4, IL-33 and TSLP (Figure 4A-C, respectively) in Scgb1a1-Cre; Ahrflox/flox mice as compared with those seen in OVA-challenged littermate controls, suggesting that loss of AhR in club cells enhanced the severity of OVA-induced airway inflammation. However, it was surprising to note that 7 days after the antigen challenge, the number of BALF lymphocytes was increased in Scgb1a1-Cre; Ahrflox/flox mice (Figure 5) as compared to those seen 1 day after the last challenge and as opposed to their littermate controls. In contrast, similar to those found in control mice, a significant reduction in the numbers of BALF total cells, eosinophils and neutrophils was seen 7 days after the antigen challenge (Figure 5A-C, respectively), although the degree of reduction in BALF total cells was not as dramatic as those seen in Scgb1a1-Cre; Ahrflox/flox mice. These results suggest that AhR in club cells may be consequential in the control of pulmonary recovery from the initial insult and the maintenance of lung homeostasis.
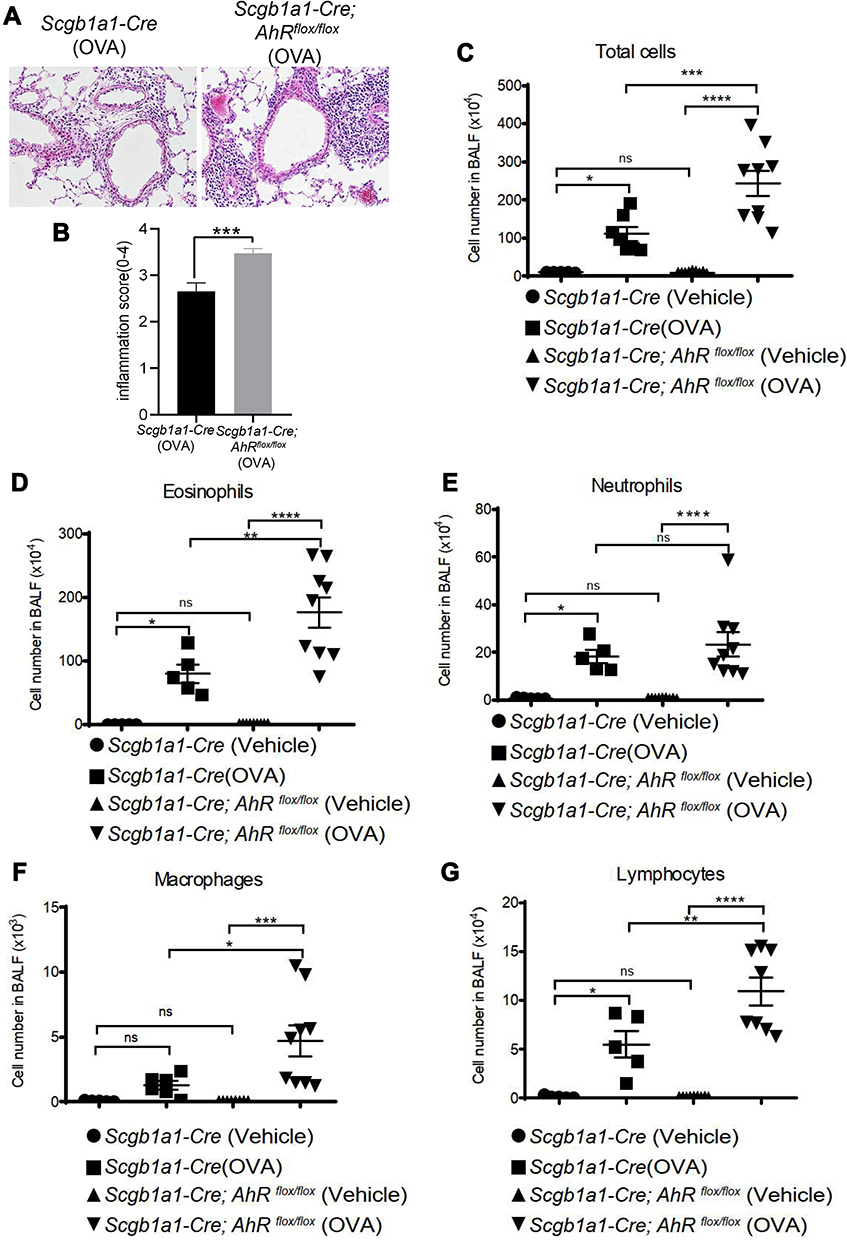 |
Figure 3 Club cell-specific AhR-null mice have enhanced severity of ovalbumin (OVA)-induced lung inflammation. (A) H&E-stained lung sections of antigen-sensitized and challenged mice; representative photomicrographs (200 ×) of lung sections are shown. (B) Inflammatory cell infiltration scores (mean ± SEM) were obtained from 3 animals). Cell subsets (C–G) in bronchoalveolar lavage fluids were determined using flow cytometry. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (t tests or One-Way ANOVA). |
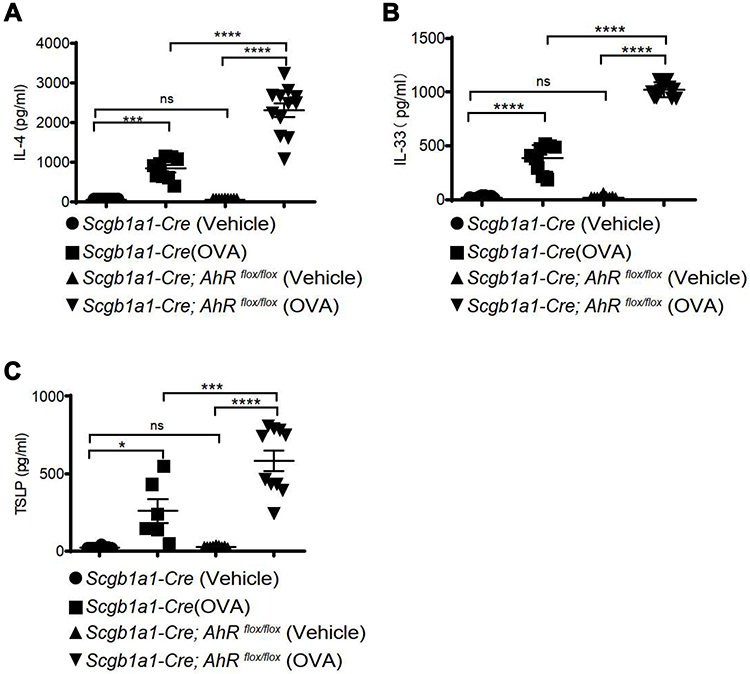 |
Figure 4 AhR deficiency resulted in reduced inflammatory cytokine expression in ovalbumin-sensitized and challenged mice. Levels of IL-4 (A), IL-33 (B), and thymic stromal lymphopoietin (TSLP), (C) in bronchoalveolar lavage fluids were measured using ELISA. *P < 0.05, ***P < 0.001, ****P < 0.0001 (One-Way ANOVA). |
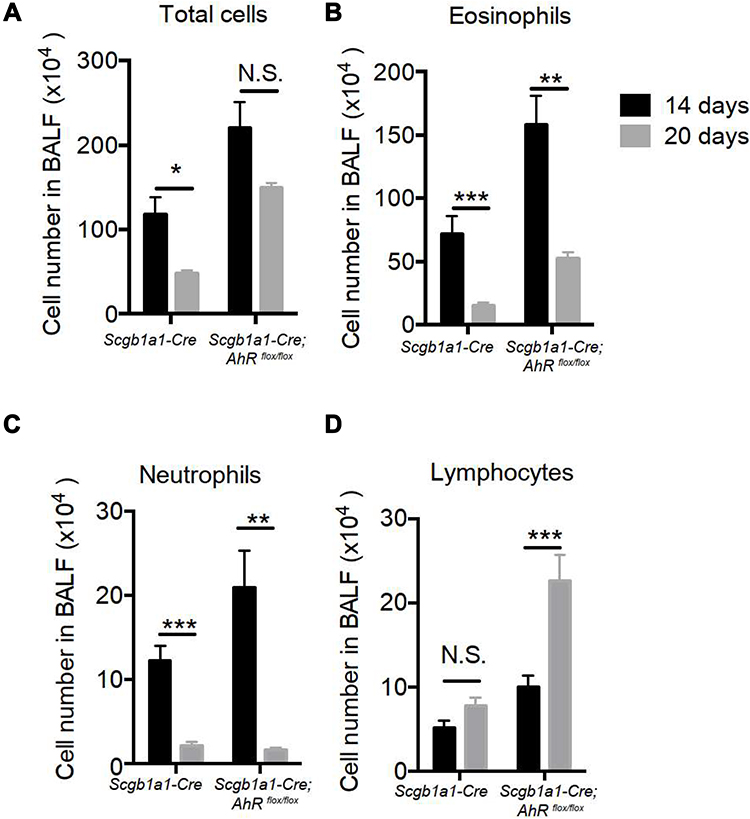 |
Figure 5 Club cell-specific AhR-null mice had prolonged ovalbumin-induced lung inflammation. (A) Total cell numbers, (B) eosinophils, (C) neutrophils and (D) lymphocytes, as determined by the use of flow cytometry. *P < 0.05, **P < 0.01, ***P < 0.001, (t tests). |
Club Cells with AhR Deficiency Fails to Activate Notch1 Signaling Under an Inflammatory Condition
It has been suggested that club cell progenitors participate in the repair of airway epithelia after pulmonary inflammation and injury, in which Notch1 signaling and its downstream target, HES5, is sequentially involved in cell regeneration.18 To examine the potential involvement of Notch 1 signaling in the renewal capacity of AhR-null club cells, we analyzed the expression of Notch 1 in lung tissues from OVA-sensitized and challenged Scgb1a1-Cre; Ahrflox/flox mice and control mice using immunofluorescence staining with relevant Abs. Results showed that Notch 1-positive cells were fewer in Scgb1a1-Cre; Ahrflox/flox mice after OVA sensitization and challenge (Figure 6A and C and Supplemental Figure 4), concomitant with a significantly lower level of Hes5 expression, as compared with those seen in the control lungs (Figure 6B and D). These data suggest that AhR is required for Notch1–Hes5 signaling and maintenance of club cell numbers under inflammatory condition.
 |
Figure 6 Expression levels of Notch1 signaling molecules are reduced in club cell-specific AhR-null mice. Lung sections were stained for Notch1 (red; (A)) or Hes5 (red; (B)) and CC10 (green) and DAPI (blue)), and the quantification data were showed in (C) and (D) (mean ± SEM scores were obtained from 3 animals), respectively. **P < 0.01, ***P < 0.001 (t tests). |
To establish the functional interaction of AhR and Notch 1 expression, we determined whether AhR activation promotes Notch1 expression in cultured lung club cells (NL20) following the treatment with an AhR ligand, Indeno[1,2,3-cd]pyrene (IP; a prominent PAH associated with air pollution19), or with ectopic AhR expression. Activation of AhR following treatments with its ligand led to time-dependent increases in the level of Notch1 transmembrane subunit (NTM) expression (Figure 7A). To confirm these results, we generated a doxycycline-inducible AhR expression lung epithelial cell line (Tet-On Systems). In these cells, induction of AhR overexpression enhanced Notch1 expression, as compared with that seen in mock control (Figure 7B). We also tested whether IP induced Notch1 expression via AhR in wild-type and AhR-null cells using RT-qPCR (Figure 7C) and Western blotting (Figure 7D). Results showed that as compared with those in vehicle control (methanol)-treated cells, the levels of expression for both NOTCH1 mRNA and protein expression were increased following IP treatment in wild-type cells, whereas AhR-null cells had decreased Notch1 expression levels. Also, the expression of Hes5 and an AhR target gene, CYP1B1, was similarly regulated in AhR-null cells treated with IP or vehicle control as compared to those found in wild-type control cells.
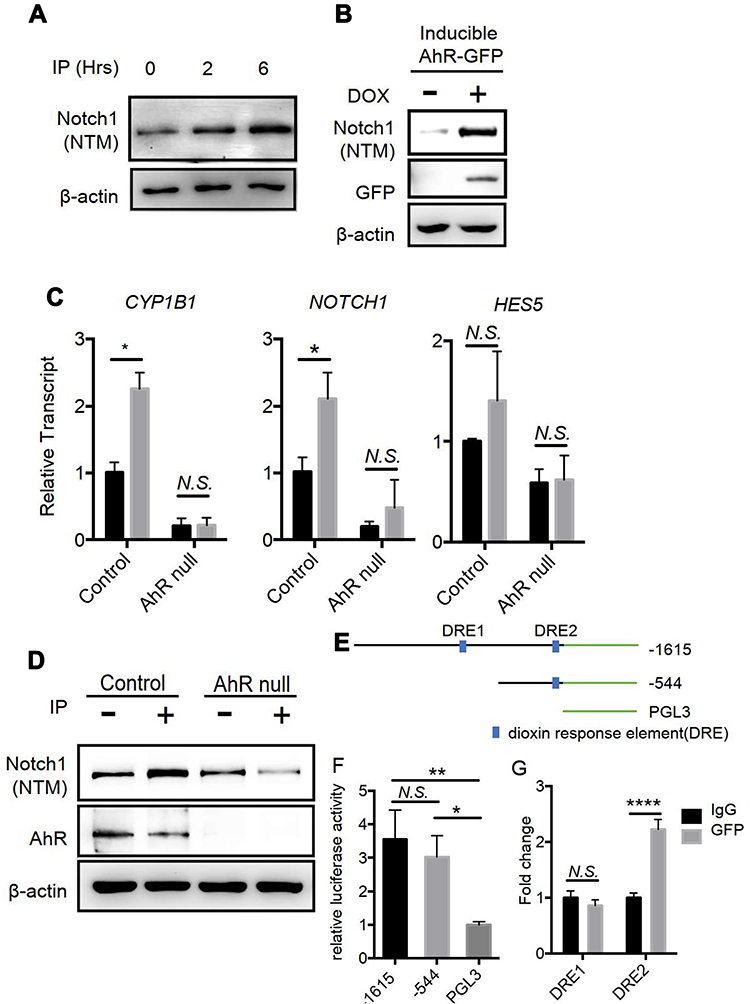 |
Figure 7 AhR is required for Notch1 mRNA and protein expression in Lung epithelial NL-20 cells; (A) NL-20 cells were treated with the AhR ligand Indeno[1,2,3-cd]pyrene (IP) at 1μM, and were examined in Western blotting analyses. Notch1 (NTM) is transmembrane/intracellular protein. (B) Doxycycline mediated ectopic AhR expression induces Notch1 in NL-20 cells. (C) IP-treated AhR-null cells showed lower expression levels of NOTCH1 and the downstream gene HES5 in NL-20 cells. (D) IP-treated (1μM) AhR-null cells had attenuated Notch1 (NTM) expression in NL-20 cells. (E) Serial deletion constructs of Notch1 promoters for promoter-reporter assays; (F) A549 cells were transfected with a series of deletion constructs of the Notch1 promoter and luciferase activities were determined. (G) A549 cells with ectopic AhR-GFP expression were analyzed in chromatin immunoprecipitation assays with IgG and GFP antibodies; *P < 0.05, **P < 0.01, ****P < 0.0001 (t tests or One-Way ANOVA). |
To confirm that Notch1 is a direct target of AhR, we performed luciferase reporter and chromatin immunoprecipitation assays. Initially, we screened the NOTCH1 promoter for AhR-binding consensus sequences and identified two putative dioxin response elements (DRE) at positions −734 to −726 (DRE1) and −119 to −111 (DRE2) relative to the transcriptional start site (Figure 7E). Subsequently, serial deletion constructs of NOTCH1 promoters were used to confirm and map the precise AhR-binding sites. The resultant reporter constructs encoding the 1.5-kbp NOTCH1 promoter produced 4-fold increases in luciferase activity compared with the empty vector. Moreover, constructs containing DRE2 only had similar increases in luciferase activity, whereas those containing DRE1 only did not (Figure 7F). Accordingly, chromatin immunoprecipitation analysis of the cells overexpressing AhR tagged with green fluorescent protein (GFP) indicated DNA enrichment at the DRE2 but not DRE1 site (Figure 7G). These results suggest that AhR binds the NOTCH1 promoter directly and regulates its transcription.
Discussion
The evidence provided herein supports the contention that AhR in club cells is a critical sensor that maintains a homeostatic state under the inflammatory condition in murine lungs. Under the condition of antigen-mediated lung inflammation, AhR deficiency in club cells failed to undergo cell renewal via, in part, dysregulated Notch1 and downstream Hes5 activation. As a corollary, regulation of Notch signaling has been demonstrated in multiple settings, although the detailed mechanism remains to be defined. For example, male infertility of AhR-null mice was associated with decreased Notch signaling and Hes1 expression, resulting in early maturation of spermatocytes and depletion of primary spermatids.20 In lungs, traffic-related particulate matter, including ultrafine particles and fine particles, stimulated AhR in alveolar macrophages, leading to induction of a Notch ligand, Jag1.21,22 Our experiments with club cells similarly showed that AhR is required for Notch1 and Hes5 expression, which might contribute to the maintenance of club cell homeostasis in pulmonary alveolar epithelium. It is not clear whether Notch1 activation is ligand-dependent or -independent under stress conditions. However, overexpression of AhR induced Notch1 expression, suggesting that AhR, at least in part, maintained Notch1 expression and downstream Hes5 expression in club cells.
It was noted that while no significant difference in club cell numbers was observed between Scgb1a1-Cre; Ahrflox/flox mice and their littermate controls, Scgb1a1-Cre; Ahrflox/flox mice had smaller lungs than their Scgb1a1-Cre littermate controls (Supplemental Figure 2). Hence, AhR in club cells may be associated with stem cell properties, epithelium cell differentiation, and lung development under aging and stress conditions. It is also worth noting that club cells are the progenitor cells of the trachea and the bronchiolar region, and identified by their resistance to naphthalene-induced injury.23 Also, studies have suggested that club cells have the ability to proliferate in response to injury,24 whereas club cell numbers are reduced in lung diseases, such as asthma.25 It is, therefore, likely that AhR is required for club cell homeostasis, particularly under the condition of antigen-induced lung inflammation.
In the lung microenvironment, club cells reportedly secrete various factors that are associated with defense mechanisms. Specifically, in an antigen-induced lung inflammation model, CC10 and SP-D were secreted by club cells and played a protective role from the Th2-associated allergic inflammatory responses.25–27 CC10 deficient mice showed increased numbers of eosinophils, neutrophils, and lymphocytes as compared with wild-type mice under condition of antigen-induced lung inflammation.8 In genetic analysis, CC10 polymorphisms are associated with low plasma CC10 concentrations and increased asthmatic episodes in asthma patients.28 Similarly, SP-D was reportedly to be induced by IL-4 and IL-13 as a part of a negative feedback circuit that represses allergic airway responses. In agreement, SP-D concentrations in bronchoalveolar lavage specimens were significantly decreased in severe asthma patients, as compared with those in healthy control subjects and patients with mild asthma.9,29,30 Therefore, club cell-specific secretory proteins likely play important roles in lung homeostasis during antigen-induced lung inflammation, and restoration of SP-D and CC10 protein concentrations using recombinant technologies has been used as therapeutic strategy for patients with antigen-induced lung inflammation.30 Our results suggest that exacerbated pulmonary inflammation in club cell-specific AhR-null mice reflects defective protective mechanisms of club cells. In fact, this is further supported by the finding that while acute airway inflammation appeared to have subsided 7 days after the antigen challenge in wild-type mice, a significant degree of lymphocytic inflammation selectively persisted and elevated over the 7-day observational period in Scgb1a1-Cre; Ahrflox/flox mice. The nature of this persistent inflammation due to the lack of AhR in club cells is currently unclear, but, nevertheless, this unique feature of AhR signaling in club cells may be instrumental in the control of respiratory homeostasis against toxic injury, inflammation and infection. Further work is clearly needed to delineate this seemingly fundamental defense mechanism.
Collectively, our findings demonstrated a novel mechanism of club cells and identified Notch1 activation as a critical outcome of AhR signaling. These data suggest that treatment options for pulmonary inflammatory disorders may be advanced by identifying cell functions in the lung microenvironment that promote Notch1 signaling and club cell homeostasis to balance pro- and anti-inflammatory cues, such as those relating to AhR. Future studies may determine the lung homeostatic contributions of the crosstalk between different cell types following AhR activation by endogenous or exogenous ligands.
Abbreviations
AhR, aryl hydrocarbon receptor; CC10, club cell 10 kd protein; OVA, ovalbumin; COPD, chronic obstructive pulmonary disease; PAHs, polycyclic aromatic hydrocarbons; CYP1A1, cytochrome P4501A1; CYP1B1, cytochrome P4501B1; SP-D, surfactant D; IP, Indeno[1,2,3-cd]pyrene; IL-4, interleukin-4; IL-33, interleukin-33; TSLP, thymic stromal lymphopoietin; DRE, dioxin response elements; NTM, Notch1 transmembrane subunit.
Acknowledgments
This work was supported by grants, in part, from National Health Research Institutes, Taiwan (EOPP10-014 and EOSP07-014), Ministry of Science and Technology, Taiwan (MOST 108-2320-B-039-058-MY3 to HCW, 106-2314-B-037-090-MY2, 107-2314-B-037-028-MY3, 107-2314-B-037-069-MY3, 109-2326-B-003-001-MY3), and China Medical University Hospital, Taiwan (DMR-108-126 to HCW), China Medical University, Taiwan (CMU109-N-08 to HCW), Kaohsiung Medical University Hospital, Taiwan (KMUH 106-6R33, KMUH107-7R46 and KMUH107-7R34), Kaohsiung Medical University, Taiwan (KMU-DK108012), The National Natural Science Fund (No. 31770984, 81929001, 81901634), Shenzhen Science and Technology Program (No.KQTD20170331145453160) and Shenzhen Nanshan District Pioneer Group Research Funds (No.LHTD20180007).
Disclosure
The authors declared they have no conflicts of interest for this work.
References
1. Xiao C, Puddicombe SM, Field S, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. 2011;128(3):e541–512. doi:10.1016/j.jaci.2011.05.038
2. Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med. 2012;18(5):684–692. doi:10.1038/nm.2737
3. Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. 2015;16(1):27–35. doi:10.1038/ni.3045
4. Lloyd CM, Marsland BJ. Lung homeostasis: influence of age, microbes, and the immune system. Immunity. 2017;46(4):549–561. doi:10.1016/j.immuni.2017.04.005
5. Franks TJ, Colby TV, Travis WD, et al. Resident cellular components of the human lung: current knowledge and goals for research on cell phenotyping and function. Proc Am Thorac Soc. 2008;5(7):763–766. doi:10.1513/pats.200803-025HR
6. Rokicki W, Rokicki M, Wojtacha J, Dzeljijli A. The role and importance of club cells (Clara cells) in the pathogenesis of some respiratory diseases. Kardiochir Torakochirurgia Pol. 2016;13:26–30. doi:10.5114/kitp.2016.58961
7. Mackay R-MA, Grainge CL, Lau LC, et al. Airway Surfactant Protein D deficiency in adults with severe asthma. Chest. 2016;149(5):1165–1172. doi:10.1016/j.chest.2015.11.012
8. Chen LC, Zhang Z, Myers AC, Huang SK. Cutting edge: altered pulmonary eosinophilic inflammation in mice deficient for Clara cell secretory 10-kDa protein. J Immunol. 2001;167(6):3025–3028. doi:10.4049/jimmunol.167.6.3025
9. Fakih D, Akiki Z, Junker K, et al. Surfactant protein D multimerization and gene polymorphism in COPD and asthma. Respirology. 2018;23(3):298–305. doi:10.1111/resp.13193
10. Morimoto M, Nishinakamura R, Saga Y, Kopan R. Different assemblies of Notch receptors coordinate the distribution of the major bronchial Clara, ciliated and neuroendocrine cells. Development. 2012;139(23):4365–4373. doi:10.1242/dev.083840
11. Huang MT, Chiu CJ, Chiang BL. Multi-faceted notch in allergic airway inflammation. Int J Mol Sci. 2019;20(14):3508. doi:10.3390/ijms20143508
12. Kim D, Chen Z, Zhou LF, Huang SX. Air pollutants and early origins of respiratory diseases. Chronic Dis Transl Med. 2018;4(2):75–94. doi:10.1016/j.cdtm.2018.03.003
13. Beamer CA, Shepherd DM. Role of the aryl hydrocarbon receptor (AhR) in lung inflammation. Semin Immunopathol. 2013;35(6):693–704. doi:10.1007/s00281-013-0391-7
14. Chang H, Chang LW, Cheng Y-H, et al. Preferential induction of CYP1A1 and CYP1B1 in CCSP-positive cells. Toxicol Sci. 2006;89(1):205–213. doi:10.1093/toxsci/kfj025
15. Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi:10.1146/annurev.pharmtox.43.100901.135828
16. Schmidt JV, Bradfield CA. Ah receptor signaling pathways. Annu Rev Cell Dev Biol. 1996;12(1):55–89. doi:10.1146/annurev.cellbio.12.1.55
17. Walisser JA, Glover E, Pande K, Liss AL, Bradfield CA. Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc Natl Acad Sci U S A. 2005;102(49):17858–17863. doi:10.1073/pnas.0504757102
18. Xing Y, Li A, Borok Z, Li C, Minoo P. NOTCH1 is required for regeneration of Clara cells during repair of airway injury. Stem Cells. 2012;30(5):946–955. doi:10.1002/stem.1059
19. Wong T-H, Lee C-L, Su -H-H, et al. A prominent air pollutant, Indeno[1,2,3-cd]pyrene, enhances allergic lung inflammation via aryl hydrocarbon receptor. Sci Rep. 2018;8(1):5198. doi:10.1038/s41598-018-23542-9
20. Huang B, Butler R, Miao Y, et al. Dysregulation of Notch and ERα signaling in AhR−/−male mice. Proc Natl Acad Sci U S A. 2016;113:11883–11888. doi:10.1073/pnas.1613269113
21. Xia M, Viera-Hutchins L, Garcia-Lloret M, et al. Vehicular exhaust particles promote allergic airway inflammation through an aryl hydrocarbon receptor-notch signaling cascade. J Allergy Clin Immunol. 2015;136(2):441–453. doi:10.1016/j.jaci.2015.02.014
22. Xia M, Harb H, Saffari A, Sioutas C, Chatila TA, Jagged A. 1-Notch 4 molecular switch mediates airway inflammation induced by ultrafine particles. J Allergy Clin Immunol. 2018;142(1243–1256):e1217. doi:10.1016/j.jaci.2018.03.009
23. Stripp BR, Maxson K, Mera R, Singh G. Plasticity of airway cell proliferation and gene expression after acute naphthalene injury.. Am J Physiol. 1995;269(6 Pt 1):L791–799. doi:10.1152/ajplung.1995.269.6.L791
24. Hiemstra PS, Bourdin A. Club cells, CC10 and self-control at the epithelial surface. Eur Respir J. 2014;44(4):831–832. doi:10.1183/09031936.00089214
25. Shijubo N, Itoh Y, Yamaguchi T, et al. Clara cell protein-positive epithelial cells are reduced in small airways of asthmatics. Am J Respir Crit Care Med. 1999;160(3):930–933. doi:10.1164/ajrccm.160.3.9803113
26. Hung C-H, Chen L-C, Zhang Z, et al. Regulation of TH2 responses by the pulmonary Clara cell secretory 10-kd protein. J Allergy Clin Immunol. 2004;114(3):664–670. doi:10.1016/j.jaci.2004.05.042
27. Xu Y-D, Cui J-M, Wang Y, et al. Proteomic analysis reveals the deregulation of inflammation-related proteins in acupuncture-treated rats with asthma onset. Evid Based Complement Alternat Med. 2012;2012:850512. doi:10.1155/2012/850512
28. Zhao G, Lin X, Zhou M, Zhao J. Association between CC10 +38A/G polymorphism and asthma risk: a meta-analysis. Pak J Med Sci. 2013;29(6):1439–1443. doi:10.12669/pjms.296.3724
29. Haczku A, Cao Y, Vass G, et al. IL-4 and IL-13 form a negative feedback circuit with surfactant protein-D in the allergic airway response. J Immunol. 2006;176(6):3557–3565. doi:10.4049/jimmunol.176.6.3557
30. Brandt EB, Mingler MK, Stevenson MD, et al. Surfactant protein D alters allergic lung responses in mice and human subjects. J Allergy Clin Immunol. 2008;121(5):e1142. doi:10.1016/j.jaci.2008.02.011
31. Li C, Hu L, Xiao J, et al. Wnt5a regulates Shh and Fgf10 signaling during lung development. Dev Biol. 2005;287(1):86–97. doi:10.1016/j.ydbio.2005.08.035
32. Wang L-T, Chiou -S-S, Chai C-Y, et al. Aryl hydrocarbon receptor regulates histone deacetylase 8 expression to repress tumor suppressive activity in hepatocellular carcinoma. Oncotarget. 2017;8(5):7489–7501. doi:10.18632/oncotarget.9841
33. van Rijt LS, Kuipers H, Vos N, et al. A rapid flow cytometric method for determining the cellular composition of bronchoalveolar lavage fluid cells in mouse models of asthma. J Immunol Methods. 2004;288(1–2):111–121. doi:10.1016/j.jim.2004.03.004
34. Kujur W, Gurram RK, Haleem N, Maurya SK, Agrewala JN. Caerulomycin A inhibits Th2 cell activity: a possible role in the management of asthma. Sci Rep. 2015;5(1):15396. doi:10.1038/srep15396
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.