Back to Journals » Journal of Inflammation Research » Volume 19
Metformin Attenuates Osteoarthritis Progression by Modulating Mitochondrial Dynamics via Activation of the AMPK/Drp1 Pathway
Authors Yang Y, Zhou H, Yang J, Li F, Hu Q, Jia Y ![]() , Huang W
, Huang W ![]() , Qin L, Zhou Y
, Qin L, Zhou Y ![]()
Received 30 May 2025
Accepted for publication 22 December 2025
Published 2 February 2026 Volume 2026:19 540176
DOI https://doi.org/10.2147/JIR.S540176
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ujjwol Risal
Yaji Yang,1– 4,* Haotian Zhou,2– 4,* Jianye Yang,2– 5 Feilong Li,2– 4,6 Qianshui Hu,2– 4 Ying Jia,2– 4 Wei Huang,2– 4 Leilei Qin,2– 4 Yu Zhou1– 4
1Orthopedic Hospital, Chongqing University of Chinese Medicine, Chongqing, 400012, People’s Republic of China; 2Department of Orthopaedic Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China; 3Chongqing Municipal Health Commission Key Laboratory of Musculoskeletal Regeneration and Translational Medicine, Chongqing Medical University, Chongqing, 400016, People’s Republic of China; 4Orthopaedic Research Laboratory, Chongqing Medical University, Chongqing, 400016, People’s Republic of China; 5Orthopedics Department, Chongqing university Fuling hospital, Chongqing, 408107, People’s Republic of China; 6Department of Orthopaedics, The People’s Hospital of Dazu, Chongqing, 402360, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Leilei Qin, Department of Orthopaedic Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China, Email [email protected] Yu Zhou, Orthopedic Hospital, Chonqqing University of Chinese Medicine, Chongqing, 400012, People’s Republic of China, Email [email protected]
Background: Osteoarthritis (OA) is a chronic degenerative disease primarily characterized by articular cartilage degradation and chondrocyte dysfunction. Mitochondrial impairment and oxidative stress in chondrocytes are pivotal contributors to OA pathogenesis. Emerging evidence suggests that metformin, beyond its role in glucose regulation, exhibits antioxidative and anti-inflammatory properties via activation of AMP-activated protein kinase (AMPK). Nonetheless, how metformin regulates mitochondrial dynamics and autophagy in OA remains to be fully elucidated.
Methods: A mouse anterior cruciate ligament transection (ACLT) model and an IL-1β-induced oxidative stress model in human chondrocytes were established. Following metformin administration, a comprehensive assessment was conducted using histological staining, immunohistochemistry, Western blotting, flow cytometry, confocal microscopy, and AMPK siRNA transfection to evaluate the effects of metformin on mitochondrial function, autophagic activity, and oxidative stress in chondrocytes.
Results: Metformin markedly improved articular cartilage architecture in ACLT mice and enhanced the stability of the cartilage matrix. It activated AMPK signaling in chondrocytes while suppressing Dynamin-related protein 1 (Drp1) phosphorylation at Ser637, thereby promoting mitochondrial fission and mitophagy. By reducing reactive oxygen species accumulation, restoring mitochondrial membrane potential, and inhibiting NOD-like receptor thermal protein domain associated protein 3 inflammasome activation, metformin effectively mitigated oxidative stress in chondrocytes. AMPK siRNA experiments further demonstrated that the AMPK/Drp1 axis is pivotal for metformin-induced mitochondrial protection and promotion of chondrocyte proliferation.
Conclusion: This study demonstrates that metformin delays osteoarthritis progression by activating the AMPK/Drp1 pathway to modulate mitochondrial fission and mitophagy, attenuate oxidative stress, and restore chondrocyte function. These findings provide novel mechanistic insight into the therapeutic potential of metformin in osteoarthritis and highlight mitochondrial dynamics as a promising target for future OA interventions.
Keywords: AMPK, Drp1, metformin, mitochondrial fission, mitophagy, osteoarthritis
Introduction
Osteoarthritis (OA) is a chronic degenerative joint disorder primarily characterized by the progressive breakdown of articular cartilage. Pathological features include degradation of the cartilage matrix, chondrocyte apoptosis, and structural deterioration of the cartilage, ultimately leading to joint dysfunction.1 As aging progresses, chondrocyte metabolic activity diminishes, and mitochondrial dysfunction becomes increasingly pronounced. Especially in OA, mitochondrial dysfunction has become one of the key factors in OA progression.2 Studies have demonstrated that chondrocyte mitochondrial dysfunction in OA patients is accompanied by elevated reactive oxygen species (ROS) generation and diminished cellular antioxidant capacity, contributing to increased apoptosis and progressive cartilage degradation.3
Mitophagy is an important intracellular mechanism for selectively removing damaged mitochondria and is essential for maintaining mitochondrial homeostasis and normal cellular function.4 Under physiological conditions, mitophagy promptly eliminates damaged mitochondria, thereby preventing excessive ROS accumulation and protecting cells from oxidative stress–induced injury.5 Notably, mitochondria-derived ROS are not only a primary driver of oxidative stress but also activate the NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome, thereby triggering pro-inflammatory responses and pyroptosis—key contributors to OA progression.6 Therefore, preserving proper mitophagic activity and ensuring the timely removal of dysfunctional mitochondria are vital for preventing ROS accumulation and delaying oxidative damage and senescence in chondrocytes.7 Mitochondrial fission is a prerequisite for the initiation of mitophagy, in which dynamin-related protein 1 (Drp1) serves as a key regulatory protein essential for maintaining mitochondrial dynamics.8 Drp1 activity is modulated by its phosphorylation status, which governs its recruitment to mitochondrial membranes and its ability to induce fission, ultimately facilitating the fragmentation and autophagic clearance of damaged mitochondria.9 Phosphorylation at Ser616 promotes Drp1 activation and oligomerization, facilitating its rapid recruitment to the outer mitochondrial membrane and enhancing mitochondrial fission. In contrast, phosphorylation at Ser637 maintains Drp1 in an inactive cytosolic conformation, reducing its affinity for mitochondrial receptors and thereby restricting the occurrence of fission.10 Under pathological conditions such as OA, impaired Drp1 function disrupts mitophagy and promotes excessive ROS accumulation, thereby exacerbating oxidative stress, inflammatory responses, and cartilage tissue damage.11 Consequently, therapeutic targeting of Drp1 has emerged as a promising strategy for osteoarthritis management.
In this pathological context, AMP-activated protein kinase (AMPK) functions as a central energy sensor and plays a pivotal role in cellular adaptation.12 AMPK is activated under conditions of energy deficiency or oxidative stress and exerts protective effects through multiple pathways. First, activation of AMPK stimulates the nuclear factor erythroid 2–related factor 2 signaling pathway, suppresses ROS production, mitigates endoplasmic reticulum stress, and prevents NLRP3 inflammasome activation.13 Secondly, AMPK promotes autophagy, including the induction of mitophagy, thereby facilitating the removal of damaged mitochondria, limiting ROS accumulation, and preserving cellular homeostasis. Current studies have shown that AMPK may regulate mitochondrial fission and autophagy by modulating the phosphorylation state of Drp1.14 However, the precise molecular mechanisms underlying this hypothesis remain incompletely understood. On one hand, studies suggest that AMPK may promote mitophagy by indirectly modulating Drp1 activity through inhibition of the mammalian target of rapamycin (mTOR) pathway or activation of Unc-51-like kinase 1 (ULK1).15,16 On the other hand, other studies propose that AMPK-mediated regulation of Drp1 may involve upstream activators such as sirtuin 1.17 However, the means of activating AMPK under pathological conditions, and how AMPK modulates Drp1 to attenuate OA progression, remain to be fully clarified.
Metformin, a well-established anti-diabetic agent, has recently been shown to exert diverse non-glycemic biological effects, including anti-inflammatory, antioxidant, and anti-senescent activities.18 Multiple studies have demonstrated that metformin activates AMPK, thereby initiating various downstream pathways that reduce ROS production, suppress inflammation and apoptosis, and collectively exert protective effects on chondrocytes.19–21 These effects have shown beneficial impact on the pathophysiology of OA, offering novel therapeutic perspectives for OA management. Therefore, under osteoarthritic conditions, activation of AMPK is crucial for delaying chondrocyte degeneration and preserving cartilage homeostasis. However, the therapeutic efficacy of metformin in OA remains to be confirmed through additional preclinical studies and clinical trials.
Although current evidence suggests that metformin modulates OA pathogenesis via activation of AMPK, the underlying molecular mechanisms remain incompletely understood. It remains to be clarified whether metformin attenuates OA progression by modulating Drp1–mediated mitochondrial fission and mitophagy through AMPK activation. Therefore, this study aims to elucidate the novel mechanism by which metformin mitigates oxidative stress in chondrocytes, focusing on AMPK-dependent regulation of Drp1-mediated mitochondrial fission, through both in vitro and in vivo models. These findings may provide a theoretical foundation for the clinical use of metformin in osteoarthritis and potentially pave the way for novel therapeutic strategies.
Materials and Methods
Ethical Statement
All animal experiments were conducted using 6- to 8-week-old male C57BL/6 mice purchased from Beijing Weida Biotechnology Co., Ltd. Experimental procedures complied with the Guide for the Care and Use of Laboratory Animals and were approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (Approval No. 2021–526). All surgical interventions were performed under sodium pentobarbital anesthesia, and every effort was made to minimize animal suffering.
Establishment of the OA Mouse Model
The OA model was established in C57BL/6 mice using the anterior cruciate ligament transection (ACLT) procedure, following previously described methods.22 A total of eighteen mice were randomized into three groups (sham, ACLT, and ACLT + metformin; n = 6 per group). All surgical and postoperative procedures were performed under anesthesia. Repeated experiments under the same experimental settings were conducted to obtain the final sample size and ensure robustness of the in vivo findings. The sham group served as control, in which the joint cavity was opened under anesthesia and then sutured without ligament transection. In the ACLT group, the anterior cruciate ligament was transected under anesthesia, followed by layered closure of the surgical site. In the ACLT + metformin group, mice received oral metformin (200 mg/kg/day) following ACLT surgery, based on a human-equivalent dose (approximately 1000 mg/day) as reported previously.20 After six weeks of treatment, all mice were euthanized, and knee joint specimens were harvested for subsequent analyses.
Antibodies and Reagents
The antibodies used in this study included: anti-AMPK (CST, #2532); anti-phospho-AMPK (anti-p-AMPK, CST, #2535); anti-NLRP3 (Beyotime, AF2155); anti-collagen II (GenTex, GTX20300); anti-GAPDH (CST, #97166); anti-DRP1 (CST, #8570); anti-phospho-DRP1 (CST, # 20990); anti-Fis1 (CST, #32525); anti-P62 (CST, #39749); anti-LC3B (CST, #2775); anti-PINK1 (CST, #6946); anti-Parkin (CST, #2132); and anti-COX IV (Proteintech, 11242-1-AP). The reagents used included: metformin (MedChemExpress, MCE, HY-B0627); Mdivi-1 (MCE, HY-15886); interleukin-1β (IL-1β; PeproTech, AF-211-11B-500UG); JC-1 Mitochondrial Membrane Potential Assay Kit (Beyotime, C1071S); ROS Assay Kit (Beyotime, S0033M); MitoTracker Red (Beyotime, C1049B); LysoTracker Green (Invitrogen, L7526); BeyoClick™ EdU Cell Proliferation Kit with Alexa Fluor 488 (Beyotime, C0071S); and ATP Assay Kit (Beyotime, S0026).
Chondrocyte Culture
Human chondrocytes (Cat. ZQY063) were purchased from Zhongqiao Xinzhou Biotechnology Co., Ltd. (ZQXZBIO, Shanghai, China). Cells were cultured in Ham’s F-12K medium (Gibco, 21127030) supplemented with 10% fetal bovine serum (FBS; Gibco, A5670701) and 1% penicillin-streptomycin (Gibco, 15070063). Cells were maintained in a humidified incubator with 5% CO2 at 37°C. Based on previous studies, chondrocytes were treated with interleukin-1β (IL-1β; 10 ng/mL) for 24 hours to establish an in vitro osteoarthritis (OA) chondrocyte model.23 Subsequently, chondrocytes were exposed to metformin for 6 hours prior to downstream analyses.21
siRNA Transfection
Cells were seeded in culture plates at an appropriate density 24 hours prior to transfection to achieve 70–80% confluency at the time of transfection. Scramble siRNA and AMPK siRNA (GenePharma, Shanghai, China) were each diluted in Opti-MEM, while Lipofectamine RNAiMAX (Invitrogen) was separately diluted in Opti-MEM. The two solutions were combined at the recommended ratio and incubated at room temperature for 20 minutes to form siRNA–lipid complexes. Prior to transfection, the culture medium was removed and cells were gently washed with PBS, followed by the addition of serum- and antibiotic-free Ham’s F-12K medium. The siRNA–lipid complexes were then added to each well, gently mixed, and incubated at 37°C in a 5% CO2 incubator. Six hours post-transfection, the medium was replaced with complete medium containing 10% FBS to minimize cytotoxicity associated with the transfection reagents. Forty-eight hours after transfection, protein expression was analyzed by Western blotting.
Western Blotting
Primary articular chondrocytes were isolated from mouse knee joints as previously described.24 Briefly, the distal femur and proximal tibia were exposed under sterile conditions, and the articular cartilage was carefully shaved from the joint surface. Collected cartilage tissues were digested with 0.2% collagenase II at 37°C for 2–3 hours to obtain a single-cell suspension. Cells were washed, pelleted, and immediately used for protein extraction without further passaging.
As previously described, human chondrocytes were pre-seeded in culture dishes and either co-transfected with siRNA or treated with the indicated reagents as appropriate. Cells were harvested and used lysis buffer (Beyotime, P0013) supplemented with protease and phosphatase inhibitors (Beyotime, P1046). Lysates were centrifuged at 4°C, and the supernatants were collected for further analysis. Protein samples were separated via SDS-polyacrylamide gel electrophoresis (SDS-PAGE; Yamei, PG213) and transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% non-fat milk in TBST (Yamei, TF103) for 1 hour at room temperature, and then incubated overnight at 4°C with primary antibodies specific to the target proteins. Membranes were subsequently washed with TBST and incubated with HRP-conjugated secondary antibodies (anti-mouse or anti-rabbit IgG) at room temperature for 1 hour. Protein bands were visualized using an ECL detection kit (Thermo Scientific, 34580) and captured with the ChemiDoc MP Imaging System (Bio-Rad, USA). Band intensity was quantified using ImageJ software.
Flow Cytometry
Intracellular ROS levels were measured using the fluorescent probe DCFH-DA in ROS Assay Kit. According to the manufacturer’s instructions, DCFH-DA stock solution was diluted in culture medium to a final concentration of 10 μM and incubated with cells for 20 minutes at 37°C in the dark. Fluorescence was subsequently detected by flow cytometry using an excitation wavelength of 488 nm.
JC-1 staining was performed to assess mitochondrial membrane potential. The JC-1 stock solution was diluted in complete culture medium to a final concentration of 2 μM and incubated with cells at 37°C for 20 minutes, followed by PBS resuspension. The proportion of green fluorescent-positive cells (510/527 nm) was analyzed using the BD FACSVerse system (BD Biosciences, USA).
Confocal Laser Scanning Microscopy (CLSM)
Mitophagy was evaluated based on the colocalization of mitochondria and lysosomes using confocal laser scanning microscopy. Pretreated chondrocytes were incubated with 20 nM MitoTracker Red (Beyotime, C1049B) for 30 minutes at 37°C in the dark, followed by 2–3 washes with PBS. Subsequently, cells were incubated with 50 nM LysoTracker Green (Invitrogen, L7526) for 5 minutes, followed by PBS washing. After staining nuclei with Hoechst 33342 (Beyotime, C1011) for 30 minutes, cells were transferred to phenol red–free complete medium and imaged using a confocal laser scanning microscope (Leica TCS SP8, Germany).
Immunohistochemistry Analysis
As described above, mice were euthanized 6 weeks post-treatment. Knee joints were fixed in 4% paraformaldehyde overnight, decalcified in 0.5 M EDTA (pH 7.4) for 4 weeks, and subsequently embedded in paraffin. Immunohistochemical staining was performed on 5-μm-thick coronal sections. Antigen retrieval was performed by heating sections at 95°C for 15 minutes. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide, followed by permeabilization with 0.5% Triton X-100 and blocking of non-specific binding using an avidin-biotin blocking kit. After blocking with 10% normal goat serum for 1 hour, sections were incubated with primary antibodies against collagen II and P-AMPK. After overnight incubation at 4°C, sections were treated with biotinylated goat anti-rabbit secondary antibody for 30 minutes at room temperature, followed by signal amplification using the VECTASTAIN Elite ABC Kit (Vector Laboratories, USA). Immunoreactive signals were visualized using the ImmPACT DAB peroxidase substrate (Vector Laboratories, USA), captured with the Leica Aperio VERSA 8 system (Leica, Germany), and quantified using ImageJ software.21
Hematoxylin–Eosin (H&E) and Safranin O–Fast Green Staining Analysis
Paraffin-embedded mouse knee joints were sectioned at 5 μm and stained with either H&E (Beyotime, C0105S) or Safranin O–Fast Green (Beyotime, C0621S) following fixation in 4% paraformaldehyde. After coverslip sealing, stained sections were imaged using the Leica Aperio VERSA 8 system (Leica, Germany) and used for subsequent histological analysis.22
EdU-Azide 488 Staining Assay
According to the manufacturer’s instructions, chondrocytes pre-seeded in 20 mm dishes and cultured overnight were treated as indicated, then washed 2–3 times with PBS. A 2× EdU working solution (20 μM) was prepared according to the manufacturer’s protocol and mixed with complete medium at a 1:1 ratio, followed by incubation for 5 hours. After incubation, cells were washed three times with PBS. Cells were fixed for 15 minutes and then permeabilized twice with permeabilization buffer, each for 3–5 minutes. The Click reaction solution was prepared by mixing 4.3 mL Click Reaction Buffer, 200 μL CuSO4, 10 μL Azide 488, and 500 μL Click Additive Solution to obtain a final volume of 5 mL. Each dish received 0.5 mL of Click reaction solution and was incubated in the dark for 30 minutes to allow Azide 488 to label incorporated EdU. After PBS washing, nuclei were stained with Hoechst (Beyotime, C1011) for 15 minutes in the dark. Cells were then imaged using a CLSM (Leica TCS SP8, Leica, Germany), and the percentage of positive cell was quantified using ImageJ software. EdU–Azide 488 (excitation/emission: 495/519 nm); Hoechst (excitation/emission: 346/460 nm).
ATP Assay
Cellular ATP levels in chondrocytes from each treatment group were measured according to the manufacturer’s instructions. Cells were lysed on ice using lysis buffer, and the lysates were centrifuged at 12,000 × g for 5 minutes at 4°C to obtain the supernatants. The ATP detection reagent was diluted 1:9 with assay buffer to prepare the working solution, which was then added to the samples. Luminescence was measured using a microplate reader (BioTek Synergy H1, USA).
Statistical Analysis
To ensure data reproducibility and reliability, all experiments were performed in triplicate using three independent biological replicates. Quantitative results are expressed as the mean ± standard deviation (SD), derived from the combined analysis of three independent experiments. Statistical significance among multiple groups was assessed using one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc multiple comparisons test. Data analysis and visualization were performed using GraphPad Prism 8.0. A p-value of <0.05 was considered statistically significant.
Results
Metformin Activates AMPK to Attenuate OA Progression in a Mouse ACLT Model
To examine the effect of metformin on OA progression, mice underwent ACLT surgery and received oral metformin treatment. At 6 weeks post-surgery, mice were euthanized, and knee joints were collected for histological and immunohistochemical evaluation. H&E staining revealed that in the sham group, the cartilage surface was smooth and continuous, with a thick cartilage layer, intact cell morphology, and uniformly distributed extracellular matrix (ECM). In contrast, the ACLT group displayed pronounced cartilage erosion, disrupted architecture, and heterogeneous staining of chondrocytes and ECM. These degenerative changes were markedly attenuated in the metformin-treated group (Figure 1A). Furthermore, Safranin O–Fast Green staining showed that the ACLT group exhibited severe cartilage erosion, thinning, and reduced staining intensity, whereas the ACLT + metformin group showed cartilage morphology similar to that of the sham group. These findings were supported by Mankin scoring (Figure 1B). These results indicate that metformin can limit OA progression in ACLT-induced mouse models.
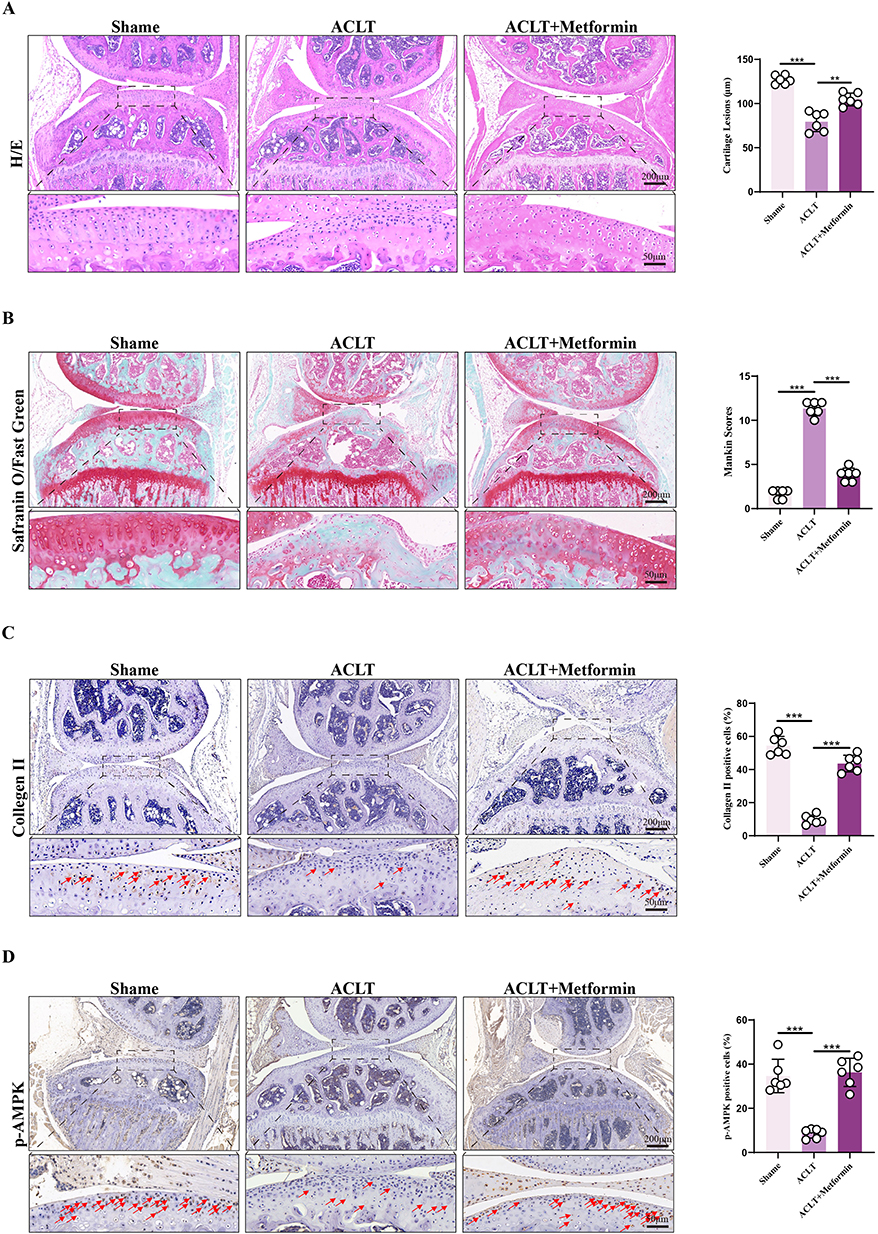 |
Figure 1 Histological assessment. (A) Hematoxylin and eosin (H&E) staining was performed on knee joint sections from each treatment group, and cartilage thickness was quantitatively analyzed (n = 6). (B) Safranin O/Fast Green staining was used to evaluate cartilage matrix integrity, and total Mankin scores were quantified for each group (n = 6). (C) Immunohistochemical analysis of Collagen II expression in knee joint sections, with quantification of Collagen II–positive chondrocytes (n = 6). Red arrows indicate chondrocytes showing positive immunoreactivity for Collagen II. (D) Immunohistochemical analysis of phosphorylated AMPK (p-AMPK) expression in knee joint sections, with quantification of p-AMPK–positive cells (n = 6). Red arrows indicate chondrocytes showing positive immunoreactivity for p-AMPK. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post-hoc multiple comparisons test: ns, P > 0.05; ** P < 0.01; *** P < 0.001. Data are presented as mean ± SD from at six independent experiments. |
Collagen II is the primary structural component of the articular cartilage matrix, and its expression serves as a marker of chondrocyte functional status. To assess the impact of metformin on cartilage matrix synthesis, immunohistochemical staining for Collagen II was performed. The results showed that Collagen II expression was significantly preserved in both the sham and ACLT + metformin groups compared to the ACLT group (Figure 1C). Given that AMPK is a key metabolic sensor involved in OA progression and metformin is a well-known AMPK activator, we next evaluated p-AMPK levels in knee cartilage tissues of all groups. At 6 weeks after ACLT, p-AMPK expression was markedly reduced in the ACLT group but significantly upregulated in the metformin-treated group (Figure 1D).
In conclusion, these results suggest that metformin activates AMPK signaling and enhances Collagen II expression, thereby inhibiting cartilage matrix degradation, maintaining chondrocyte function and ECM integrity, and delaying OA progression. However, the precise molecular mechanisms remain to be elucidated.
Metformin Alleviates Mitochondrial Dysfunction and Preserves Chondrocyte Homeostasis
In vitro, chondrocytes were stimulated with IL-1β to mimic the oxidative and inflammatory microenvironment of OA, followed by treatment with varying concentrations of metformin to assess its effects. Western blot results showed that IL-1β inhibited AMPK activation, while metformin restored AMPK signaling. Further analysis demonstrated a dose-dependent increase in p-AMPK levels following metformin treatment, with higher doses significantly enhancing AMPK phosphorylation despite persistent IL-1β exposure (Figure 2A and B). Meanwhile, metformin significantly inhibited IL-1β-induced NLRP3 inflammasome expression in a dose-dependent manner. As inflammasome activity was suppressed, Collagen II expression gradually recovered in chondrocytes, suggesting that metformin exerts both anti-inflammatory and cartilage-reparative effects (Figure 2A, C and D).
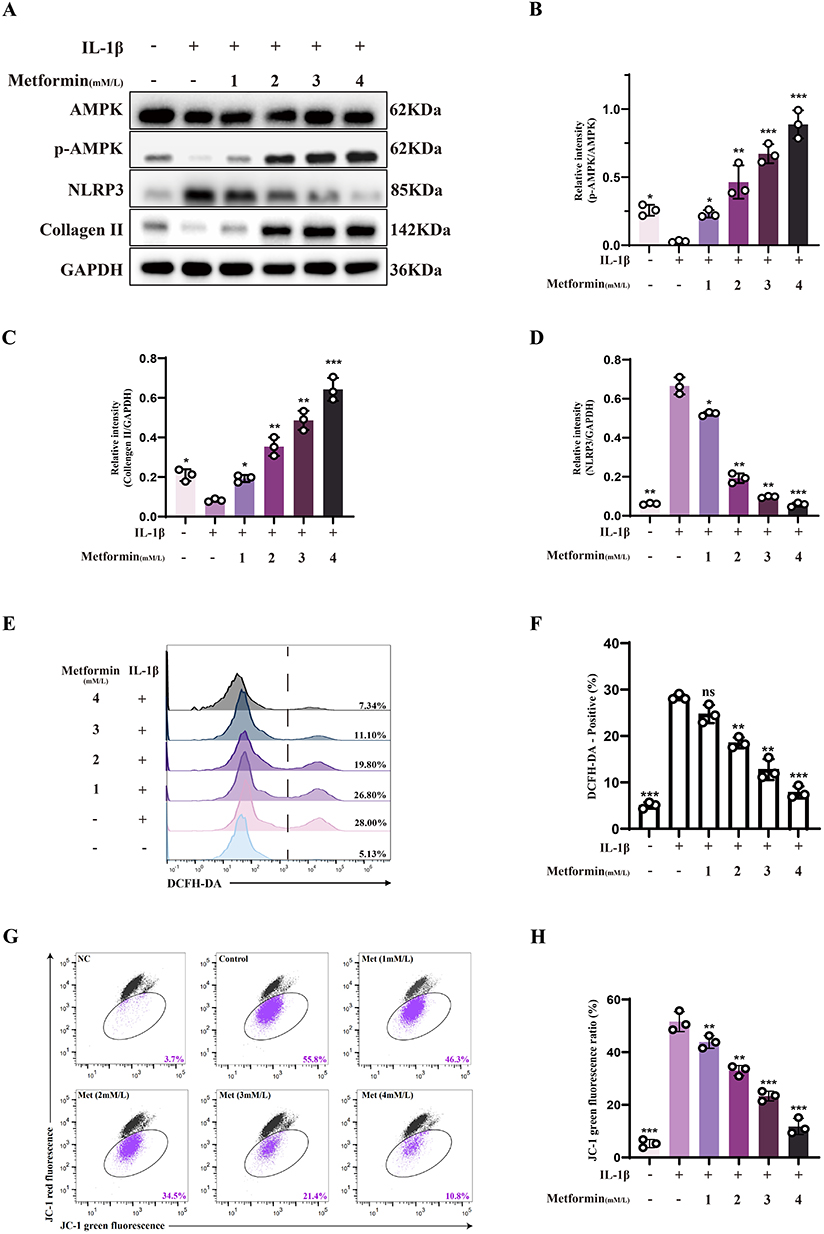 |
Figure 2 Metformin alleviates mitochondrial dysfunction and preserves chondrocyte homeostasis. Chondrocytes were treated with or without IL-1β (10 ng/mL) for 24 h, followed by metformin treatment at various concentrations (1, 2, 3, or 4 mM) for 6 h. (A) Western blot analysis of AMPK, phosphorylated AMPK (p-AMPK), NLRP3, and Collagen II expression in each group. (B–D) Densitometric quantification of p-AMPK/AMPK ratio, as well as relative expression levels of Collagen II and NLRP3 normalized to GAPDH. (E and F) Intracellular ROS levels were detected using the DCFH-DA fluorescent probe and analyzed by flow cytometry. The percentage of DCFH-DA–positive cells was quantified. (G and H) Mitochondrial membrane potential was assessed using JC-1 staining and flow cytometry. Red fluorescence indicates JC-1 aggregates representing intact membrane potential; green fluorescence indicates JC-1 monomers reflecting membrane depolarization. The percentage of JC-1 monomer–positive cells was quantified. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post-hoc multiple comparisons test: ns, P > 0.05; * P < 0.05; ** P < 0.01; *** P < 0.001. The data represent the mean ± SD from at three independent experiments. |
Mitochondrial dysfunction is a hallmark of OA-associated chondrocytes, and mitochondrial membrane potential (Δψm) serves as an essential indicator of mitochondrial integrity. Mitochondrial dysfunction is frequently accompanied by excessive production of ROS, which disrupts redox homeostasis and promotes chondrocyte apoptosis. ROS levels were measured using fluorescence probes and flow cytometry. Results showed that metformin reduced IL-1β-induced ROS accumulation, alleviating oxidative stress (Figure 2E and F). JC-1 staining revealed that metformin preserved mitochondrial Δψm in a concentration-dependent manner, indicating its role in maintaining mitochondrial function. As metformin concentration increased, the proportion of cells with disrupted membrane potential decreased, suggesting protection of mitochondrial function and structure under IL-1β stress (Figure 2G and H).
In conclusion, these results suggest that metformin activates AMPK, inhibits NLRP3 expression, reduces ROS, and stabilizes mitochondrial membrane potential, thus improving chondrocyte function. However, the precise molecular mechanisms through which metformin regulates mitochondrial homeostasis and exerts anti-inflammatory effects require further investigation.
Metformin Promotes Mitochondrial Fission to Preserve Mitochondrial Function
Drp1 is a central regulator of mitochondrial fission, and its phosphorylation at distinct serine residues determines whether fission is activated or suppressed. Specifically, phosphorylation of Drp1 at serine 637 (Ser637) inhibits its translocation to mitochondria and suppresses mitochondrial fission.25 Mitochondrial fission protein 1 (Fis-1), localized to the outer mitochondrial membrane, facilitates Drp1 recruitment and promotes mitochondrial fission and subsequent mitophagy when upregulated.26 Western blot showed that IL-1β stimulation increased p-Drp1 (Ser637) levels and decreased Fis-1 expression, indicating impaired mitochondrial fission under oxidative stress. Treatment with the Drp1 inhibitor Mdivi-1 further aggravated this suppression. Metformin reversed these effects by reducing p-Drp1 (Ser637), restoring Fis-1 expression, and alleviating Mdivi-1–induced fission inhibition, suggesting a positive role in mitochondrial dynamics (Figure 3A–C).
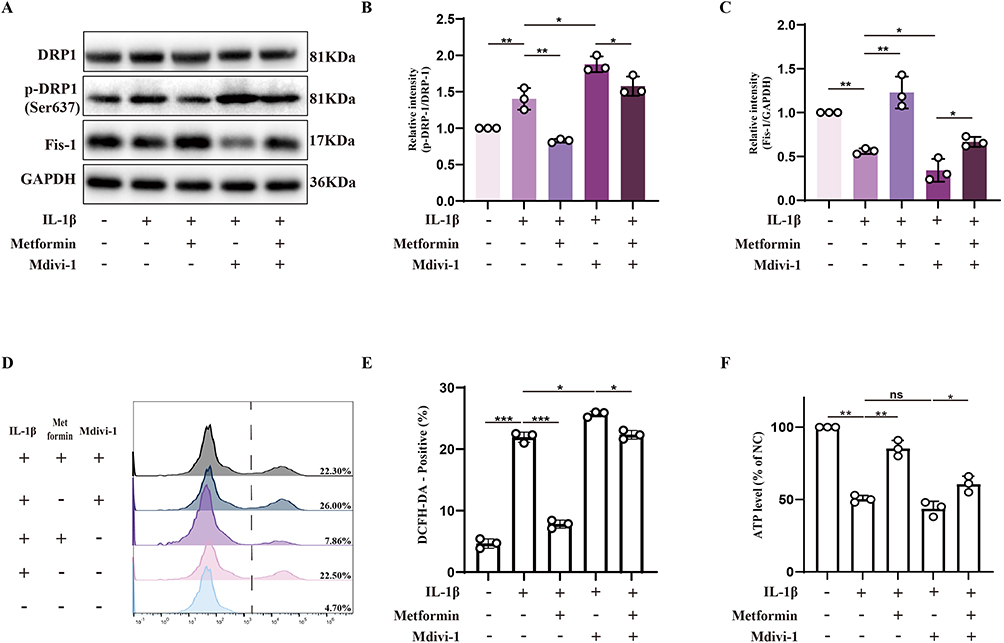 |
Figure 3 Metformin promotes mitochondrial fission to preserve mitochondrial function. Chondrocytes were pretreated with or without IL-1β (10 ng/mL) for 24 h, followed by metformin (4 mM) treatment for 6 h or Mdivi-1 (20 μM) treatment for 2 h. (A) Western blot analysis of Drp1, phosphorylated Drp1 at Ser637 (p-Drp1 Ser637), and Fis-1 protein expression levels. (B and C) Densitometric quantification of the p-Drp1 (Ser637)/Drp1 ratio and Fis-1 expression levels normalized to GAPDH. (D and E) Intracellular ROS levels were measured using the DCFH-DA probe and analyzed by flow cytometry. The proportion of DCFH-DA–positive cells was quantified. (F) Intracellular ATP levels were measured across treatment groups. All data were normalized to untreated chondrocytes (NC group). Statistical analysis was performed using one-way ANOVA followed by Tukey’s post-hoc multiple comparisons test: ns, P > 0.05; * P < 0.05; ** P < 0.01; *** P < 0.001. The data represent the mean ± SD from at three independent experiments. |
To further explore the relationship between mitochondrial fission and cellular function, intracellular ROS levels were quantified. Results showed that Mdivi-1 increased ROS levels, while metformin significantly reduced this ROS accumulation (Figure 3D and E). ATP levels measured by colorimetric assay showed that metformin increased ATP content in IL-1β-treated chondrocytes and reversed ATP synthesis impairment caused by Mdivi-1 (Figure 3F).
In conclusion, metformin promotes mitochondrial fission by inhibiting p-Drp1 (Ser637), thereby reducing ROS accumulation and enhancing ATP production, alleviating oxidative stress in chondrocytes. This effect may be mediated by metformin’s regulation of mitochondrial dynamics and fission-dependent mitophagy.
Metformin Enhances Mitophagy in Chondrocytes by Promoting Drp1-Mediated Mitochondrial Fission
To first evaluate the regulatory effect of metformin on cartilage mitophagy in vivo, we examined mitophagy-related proteins in primary chondrocytes isolated from Sham, ACLT, and ACLT + Metformin mice. ACLT markedly reduced the expression of PINK1 and Parkin, indicating impaired mitophagy in cartilage tissue, whereas metformin treatment partially restored the levels of these key proteins(Supplementary Figure S1).
Building on these in vivo observations, we further investigated whether metformin promotes mitophagy through Drp1-mediated mitochondrial fission in vitro. Western blot analysis revealed that IL-1β stimulation led to increased P62 accumulation, impaired LC3-I to LC3-II conversion, and reduced recruitment of Pink-1 and Parkin to the outer mitochondrial membrane, indicating attenuated mitophagy under stress conditions. Upon treatment with the Drp1 inhibitor Mdivi-1, these changes were more pronounced, suggesting that Drp1 inhibition further suppresses mitophagy. Notably, metformin treatment significantly enhanced mitophagy in IL-1β-treated cells, as indicated by decreased P62, increased LC3-II, and restored Pink1 and Parkin mitochondrial accumulation. Furthermore, metformin partially rescued the mitophagy defects induced by Mdivi-1–mediated Drp1 inhibition (Figure 4A–F).
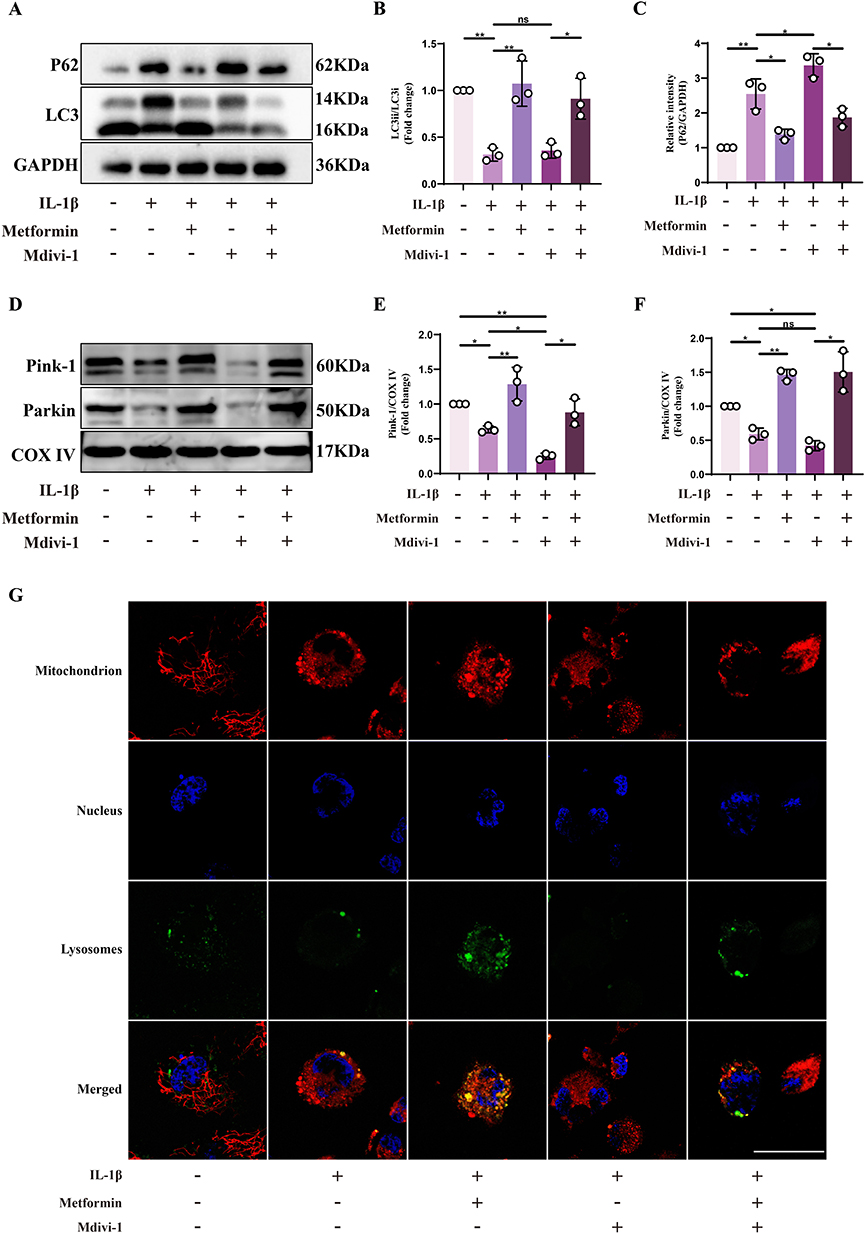 |
Figure 4 Metformin enhances mitophagy in chondrocytes by promoting Drp1-mediated mitochondrial fission. Chondrocytes were pretreated with or without IL-1β (10 ng/mL) for 24 h, followed by metformin (4 mM) treatment for 6 h or Mdivi-1 (20 μM) treatment for 2 h. (A) Western blot analysis of LC3 and P62 protein expression in chondrocytes under different treatment conditions. (B and C) Quantification of LC3-II/LC3-I ratio and P62 expression levels, normalized to GAPDH. (D) Mitochondrial proteins were isolated from chondrocytes of each treatment group, followed by Western blot analysis of Pink1 and Parkin expression. (E and F) Densitometric quantification of Pink1 and Parkin protein levels, normalized to COX IV as the mitochondrial loading control. (G) Confocal microscopy images showing mitochondria (red), lysosomes (green), and nuclei (blue) in chondrocytes under each treatment condition. Yellow signals represent mitochondrial–lysosomal colocalization, indicative of mitophagy. Scale bar: 20 μm. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post-hoc multiple comparisons test: ns, P > 0.05; * P < 0.05; ** P < 0.01. The data represent the mean ± SD from at three independent experiments. |
Confocal microscopy was used to examine mitochondrial–lysosomal colocalization, and the results were consistent with Western blot data, showing that metformin significantly enhanced colocalization, indicating increased mitophagy (Figure 4G).
In summary, metformin regulates Drp1-mediated mitochondrial fission to restore impaired mitophagy in chondrocytes, thereby exerting anti-aging and protective effects.
Metformin Mitigates Chondrocyte Damage via the AMPK/Drp1 Signaling Axis
To determine whether metformin regulates Drp1-mediated mitochondrial fission via AMPK signaling, AMPK expression was silenced in chondrocytes using siRNA. Western blot showed that AMPK expression was significantly reduced after siRNA treatment, indicating effective silenced (Figure 5A and B).
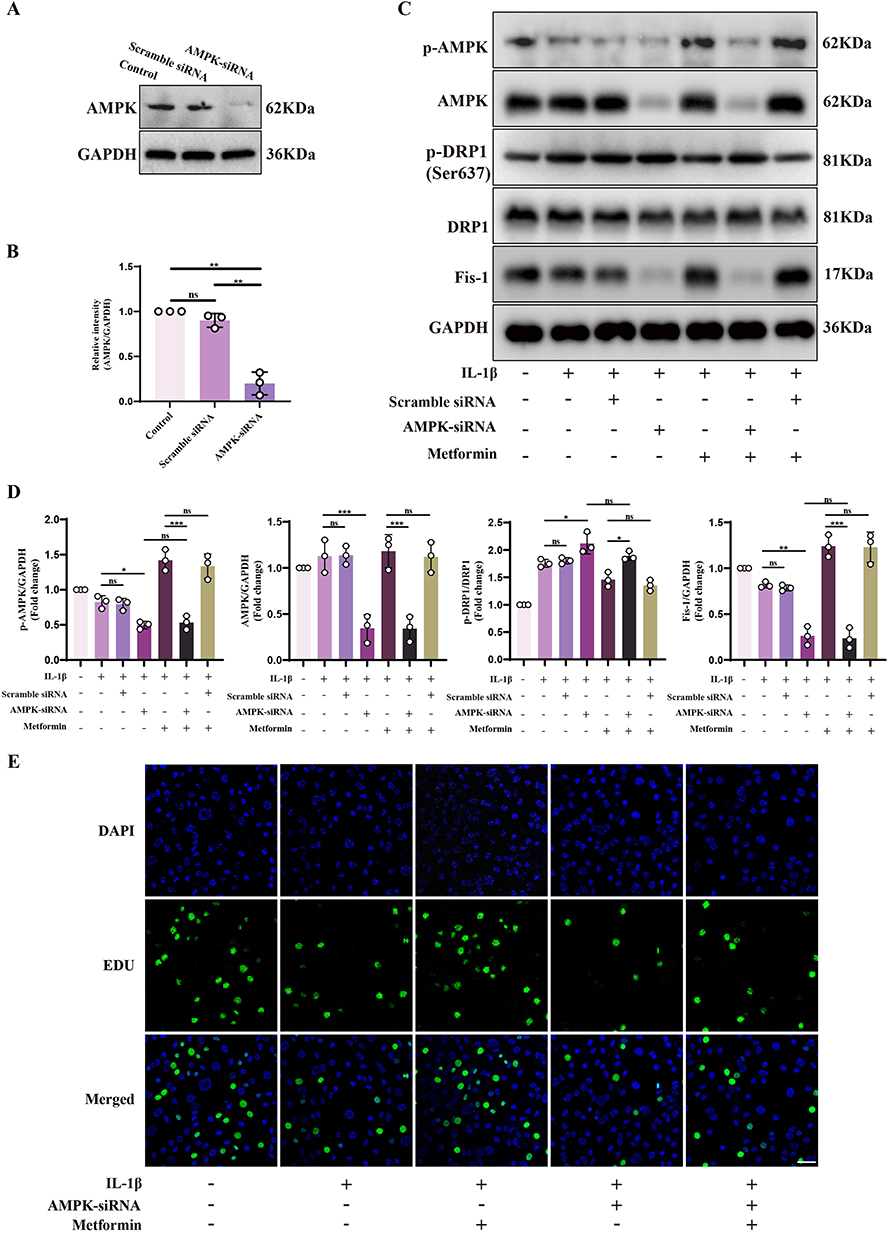 |
Figure 5 Metformin mitigates chondrocyte damage via the AMPK/Drp1 signaling axis. (A and B) AMPK expression was silenced in chondrocytes using AMPK siRNA, and AMPK protein levels were evaluated by Western blot. Quantification was performed relative to GAPDH. (C) Chondrocytes were transfected with AMPK siRNA or Scramble siRNA, followed by treatment with or without IL-1β (10 ng/mL) for 24 h, and then metformin (4 mM) for 6 h. Western blot analysis was conducted to assess the expression of AMPK, p-AMPK, Drp1, p-Drp1 (Ser637), and Fis-1. (D) Densitometric analysis of AMPK, p-AMPK, and Fis-1 protein levels normalized to GAPDH. The p-Drp1 (Ser637)/Drp1 ratio was also quantified. (E) EdU staining was performed to assess DNA synthesis as an indicator of chondrocyte proliferative capacity under each treatment condition. DAPI was used to stain nuclei. Scale bar: 20 μm. Scramble siRNA served as the non-specific siRNA control for comparison with AMPK-targeting siRNA. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post-hoc multiple comparisons test: ns, P > 0.05; * P < 0.05; ** P < 0.01; *** P < 0.001. The data represent the mean ± SD from at three independent experiments. |
Compared to control and control siRNA groups, AMPK silenced resulted in further increased p-Drp1 (Ser637) and decreased Fis-1 expression under IL-1β stimulation. The inhibitory effect of metformin on p-Drp1 (Ser637) was significantly weakened, with p-Drp1 remaining elevated, suggesting that metformin cannot suppress Ser637 phosphorylation in the absence of AMPK. Moreover, changes in Fis-1 expression supported this finding: after AMPK silenced, metformin’s effect on increasing Fis-1 was significantly reduced, with expression levels comparable to the IL-1β + AMPK-siRNA group, indicating impaired recovery of mitochondrial fission (Figure 5C and D). These results indicate that metformin suppresses Drp1 (Ser637) phosphorylation via AMPK signaling, thereby promoting mitochondrial fission and mitophagy, reducing ROS, and alleviating oxidative stress.
Additionally, chondrocyte proliferation under various conditions was evaluated using EdU-488 incorporation assays. IL-1β significantly inhibited proliferation, while metformin effectively restored it. However, after AMPK silenced, the pro-proliferative effect of metformin was significantly reduced (Figure 5E and Supplementary Figure S2).
In conclusion, these results confirm that metformin activates AMPK to inhibit Drp1 (Ser637) phosphorylation, promote mitochondrial fission and autophagy, maintain mitochondrial homeostasis and energy production, enhance proliferation, reduce apoptosis, and delay OA progression.
Discussion
OA is a chronic degenerative joint disorder primarily characterized by progressive articular cartilage deterioration and oxidative stress within chondrocytes.27 Currently, more than 500 million people worldwide are affected by OA, making it one of the most prevalent chronic diseases among the elderly and a major contributor to age-related disability.28 However, no curative agents or disease-modifying drugs are currently available to effectively halt or reverse OA progression. Accumulating evidence suggests that oxidative stress-induced inflammatory responses and concurrent mitochondrial dysfunction in chondrocytes are hallmark drivers of OA pathogenesis and progression.29,30 Therefore, targeting oxidative inflammation and restoring mitochondrial homeostasis in chondrocytes represent promising therapeutic strategies for OA management. In this study, we demonstrated that metformin robustly activated AMPK signaling in chondrocytes and inhibited the phosphorylation of Drp1 at Ser637, a modification known to suppress mitochondrial fission. The downregulation of p-Drp1 (Ser637) restored mitochondrial fission, thereby facilitating the selective removal of dysfunctional mitochondria through mitophagy. Furthermore, metformin-induced mitophagy significantly reduced intracellular ROS levels and protected mitochondrial structure and cellular function against oxidative stress. The suppression of ROS accumulation contributed to the maintenance of chondrocyte proliferative activity and functional stability. Consistently, histological evaluation revealed attenuated cartilage matrix degradation and reduced chondrocyte loss.
Mitochondria serve as central regulators of cellular metabolism, not only supplying energy through ATP synthesis but also modulating ROS and metabolic intermediates involved in proliferation, senescence, and apoptosis.31 Therefore, preserving mitochondrial homeostasis and functional integrity is critical for sustaining cellular viability and tissue stability. Such homeostasis depends on tightly regulated mitochondrial dynamics, particularly the delicate balance between fission and fusion events.32 Mitochondrial fission plays a particularly critical role in maintaining organelle quality, as it not only facilitates the separate of damaged mitochondrial fragments but also primes the initiation of mitophagy.8 Mitochondrial fission is initiated by contact between the endoplasmic reticulum and mitochondria, which promotes localized actin polymerization at constriction sites and facilitates initial outer membrane narrowing. Subsequently, the dynamin-related GTPase Drp1 is recruited to the mitochondrial surface through adaptor proteins including Fis-1 and mitochondrial fission factor (Mff). Activated Drp1 uses GTPase activity to self-assemble into spirals, promoting further membrane constriction via GTP hydrolysis, in coordination with OPA1-regulated inner membrane division. The resulting mitochondrial fragments are selectively sequestered by autophagosomes and degraded via mitophagy, thereby ensuring mitochondrial quality control.33,34 Previous studies have shown that inhibiting Drp1 markedly blocks mitophagy initiation. Inhibitors such as Mdivi-1 not only block mitochondrial fission but also reduce membrane potential, disrupt energy metabolism, and induce apoptosis, highlighting the essential role of Drp1 in mitochondrial and cellular homeostasis.35–37 This phenomenon was further confirmed in our experimental model. Our results showed that oxidative stress suppressed Drp1 activity in chondrocytes, leading to impaired mitochondrial fission and mitophagy, accumulation of dysfunctional mitochondria and ROS, and subsequent overactivation of the NLRP3 inflammasome. This process exacerbated inflammation and functional decline in chondrocytes, suggesting that impaired Drp1-mediated fission is a key event in OA progression.
Drp1 is intricately regulated by multiple protein kinases. Cyclin-dependent kinase 1 (Cdk1) enhances mitochondrial fission by phosphorylating Drp1 at Ser616.38 Similarly, in response to oxidative stress or growth factor stimulation, extracellular signal-regulated kinases 1/2 (ERK1/2) phosphorylate Drp1 at Ser616, thereby enhancing its activity and promoting mitochondrial fission.39 In contrast, Drp1 is also subject to negative regulatory mechanisms. Protein kinase A (PKA) phosphorylates Drp1 at Ser637, which impairs its interaction with outer mitochondrial membrane adaptors such as Fis-1 and Mff, thereby inhibiting its mitochondrial recruitment and suppressing fission.40 Moreover, AMPK, a key metabolic stress sensor, facilitates mitochondrial fission by phosphorylating Mff and enhancing its affinity for Drp1.41 Notably, in aging-related pathological conditions, senescence-associated factors also regulate Drp1 activity. Studies have shown that aging cells release factors such as p53, which upregulate Drp1 phosphorylation at Ser637 and inhibit its fission function, leading to imbalance in mitochondrial dynamics.42 Consistent with these findings, we observed a marked increase in Drp1 phosphorylation at Ser637 in IL-1β–treated chondrocytes, an in vitro model of OA, suggesting suppressed Drp1 activity and impaired mitochondrial fission capacity. This finding further confirms the impaired mitochondrial fission in OA pathogenesis and provides a theoretical basis for targeting upstream regulators to restore mitochondrial homeostasis.
As a key sensor of cellular energy homeostasis, AMPK plays a central role in regulating metabolic balance, and suppressing oxidative stress and inflammation.43 In addition to regulating mitochondrial function, AMPK can activate ULK1 and inhibit the mTOR pathway to induce and maintain autophagy.15,16 AMPK dysregulation is associated with various diseases, including diabetes, cardiovascular disease, cancer, chronic inflammation, and osteoarthritis, making it an important therapeutic target. Previous studies have demonstrated that ligustilide attenuates inflammatory injury following ischemic stroke via AMPK activation, while osthole suppresses OA progression through the AMPK signaling pathway.44,45 Additionally, other study has confirmed that metformin exerts protective effects against OA through AMPK activation.20 Our study further verified this conclusion and revealed more specific molecular mechanisms. We demonstrated that metformin significantly enhances AMPK phosphorylation at Thr172, which in turn attenuates cartilage degeneration in OA. Importantly, AMPK activation was shown—through siRNA-mediated interference—to markedly suppress Drp1 phosphorylation at Ser637, thereby promoting mitochondrial fission and subsequent mitophagy, ultimately alleviating intracellular ROS buildup and inflammatory signaling. However, the regulatory effects of AMPK on Drp1 appear to be highly context-dependent, with potential for bidirectional modulation depending on the physiological or pathological milieu. For instance, in a model of lead-induced mitochondrial hyperfission, AMPK activation suppresses Ser616 phosphorylation of Drp1, thereby inhibiting pathological mitochondrial division; conversely, in carbon tetrachloride–induced liver fibrosis, AMPK enhances Ser616 phosphorylation to promote mitochondrial biogenesis.13,46 These findings underscore the context-specific nature of AMPK-mediated regulation of Drp1. Our study extends the understanding of AMPK–Drp1 signaling in chondrocytes by demonstrating that, under OA-associated oxidative stress, AMPK suppresses inhibitory phosphorylation of Drp1 at Ser637, thereby restoring mitochondrial dynamics, enhancing mitophagy, and mitigating inflammatory responses. This finding provides a new theoretical basis for targeting the AMPK–Drp1 axis to treat cartilage degeneration and OA progression.
Although this study elucidated the critical role of the AMPK/Drp1/mitochondrial fission pathway in the effect of metformin on delaying OA progression, there are still some issues worthies of further investigation. First, the balance between mitochondrial fission and autophagy is critical for preserving mitochondrial integrity and cellular homeostasis. Previous studies have indicated that excessive activation of mitochondrial fission can disrupt mitochondrial quality control, leading to metabolic dysregulation and cellular injury.47 Metformin has been reported to inhibit excessive mitochondrial fission under certain pathological conditions.13,48,49 Therefore, whether metformin can dynamically regulate the AMPK/Drp1 pathway during OA treatment to achieve moderate regulation of mitochondrial fission and sustained cellular protection requires further study. Second, although our investigation primarily focused on the protective effects of metformin in chondrocytes, OA is a multifactorial disease involving the interplay of various joint components, including cartilage, synovium, and subchondral bone. Whether metformin can also exert synergistic protective effects by targeting other joint-resident cells, such as synoviocytes, osteoblasts, or immune cells, remains to be elucidated. Moreover, while we demonstrated that AMPK activation suppresses Drp1 phosphorylation at Ser637 to restore mitochondrial fission, it remains unclear whether AMPK directly acts on Drp1 or regulates its phosphorylation indirectly through additional co-factors or intermediary signaling molecules. This study utilized only male mice in the ACLT‐induced osteoarthritis model, which may limit the generalizability of the findings. Previous studies have indicated that the incidence, inflammatory response, and progression of OA may differ between sexes; therefore, the present work cannot fully assess the potential influence of sex on the therapeutic effects of metformin or on OA progression.50 Future studies will incorporate both male and female animals to more comprehensively evaluate sex-dependent differences in metformin’s efficacy and underlying mechanisms. In addition, the metformin dosage used in this study was determined based on human equivalent dose conversion using body surface area. However, doses applied in animal experiments cannot be directly translated to clinical practice. Future investigations should include systematic dose–response studies to determine the safe and effective therapeutic range of metformin for osteoarthritis.
Conclusion
In summary, our study demonstrates that metformin delays OA progression by regulating mitochondrial fission and mitophagy via the AMPK/Drp1 signaling axis, thereby mitigating chondrocyte senescence and oxidative stress. These findings not only deepen our understanding of OA pathogenesis, but also offer novel pharmacological insights into the therapeutic potential of metformin, thereby providing a theoretical rationale for its future clinical application in OA management.
Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
This study used ChatGPT4.0 to assist the author in improving the language of the first draft after its completion, to enhance the fluency and readability of the text. However, the accuracy of the content, viewpoints, and responsibility for the final article are assumed by the author.
Data Sharing Statement
Data will be made available on request.
Ethics Approval Statement
All animal experiments were approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (Approval No. 2021-526). All surgical interventions were performed under sodium pentobarbital anesthesia, and every effort was made to minimize animal suffering.
Acknowledgement
The graphical abstract was created using BioRender.com.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Science and Technology Research Program of Chongqing Municipal Education Commission (Grant No. KJQN202415131); Chongqing Science and Health Joint Development Program of Chinese Medicine Technology Innovation and Application (Grant No.2021ZY014325); Chongqing’s Special Funding for Postdoctoral Research Projects (Grant No. 2023CQBSHTB3124).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Goldring SR, Goldring MB. Changes in the osteochondral unit during osteoarthritis: structure, function and cartilage–bone crosstalk. Nat Rev Rheumatol. 2016;12(11):632–17. doi:10.1038/nrrheum.2016.148
2. Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12(7):412–420. doi:10.1038/nrrheum.2016.65
3. Lepetsos P, Papavassiliou AG. ROS/oxidative stress signaling in osteoarthritis. Biochimica et Biophysica Acta (BBA). 2016;1862(4):576–591. doi:10.1016/j.bbadis.2016.01.003
4. Harper JW, Ordureau A, Heo JM. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol. 2018;19(2):93–108. doi:10.1038/nrm.2017.129
5. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20(9):1013–1022. doi:10.1038/s41556-018-0176-2
6. Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13(2):148–159. doi:10.1038/cmi.2015.95
7. He R, Wei Y, Peng Z, et al. α-Ketoglutarate alleviates osteoarthritis by inhibiting ferroptosis via the ETV4/SLC7A11/GPX4 signaling pathway. Cell Mol Biol Lett. 2024;29(1):88. doi:10.1186/s11658-024-00605-6
8. Li GB, Zhang HW, Fu RQ, et al. Mitochondrial fission and mitophagy depend on cofilin-mediated actin depolymerization activity at the mitochondrial fission site. Oncogene. 2018;37(11):1485–1502. doi:10.1038/s41388-017-0064-4
9. Shahni R, Cale CM, Anderson G, et al. Signal transducer and activator of transcription 2 deficiency is a novel disorder of mitochondrial fission. Brain. 2015;138(10):2834–2846. doi:10.1093/brain/awv182
10. Murakawa T, Yamaguchi O, Hashimoto A, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015;6(1):7527. doi:10.1038/ncomms8527
11. Liu S, Cheng S, Chen B, et al. Microvesicles-hydrogel breaks the cycle of cellular senescence by improving mitochondrial function to treat osteoarthritis. J Nanobiotechnology. 2023;21(1):429. doi:10.1186/s12951-023-02211-8
12. Xie N, Yuan K, Zhou L, et al. PRKAA/AMPK restricts HBV replication through promotion of autophagic degradation. Autophagy. 2016;12(9):1507–1520. doi:10.1080/15548627.2016.1191857
13. Yang L, Li X, Jiang A, et al. Metformin alleviates lead-induced mitochondrial fragmentation via AMPK/Nrf2 activation in SH-SY5Y cells. Redox Biol. 2020;36:101626. doi:10.1016/j.redox.2020.101626
14. Liu X, Xiao ZD, Han L, et al. lncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat Cell Biol. 2016;18(4):431–442. doi:10.1038/ncb3328
15. Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461. doi:10.1126/science.1196371
16. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi:10.1038/ncb2152
17. Song SB, Park JS, Jang SY, Hwang ES. Nicotinamide treatment facilitates mitochondrial fission through Drp1 activation mediated by SIRT1-induced changes in cellular levels of cAMP and Ca2+. Cells. 2021;10(3):612. doi:10.3390/cells10030612
18. He M, Lu B, Opoku M, et al. Metformin prevents or delays the development and progression of osteoarthritis: new insight and mechanism of action. Cells. 2022;11(19):3012. doi:10.3390/cells11193012
19. Feng X, Pan J, Li J, et al. Metformin attenuates cartilage degeneration in an experimental osteoarthritis model by regulating AMPK/mTOR. Aging. 2020;12(2):1087–1103. doi:10.18632/aging.102635
20. Li J, Zhang B, Liu WX, et al. Metformin limits osteoarthritis development and progression through activation of AMPK signalling. Ann Rheumatic Dis. 2020;79(5):635–645. doi:10.1136/annrheumdis-2019-216713
21. Zhu Z, Huang Y, Li J, et al. AMPK activator decelerates osteoarthritis development by inhibition of β-catenin signaling in chondrocytes. J Orthop Translat. 2023;38:158–166. doi:10.1016/j.jot.2022.10.005
22. Qin L, Yang J, Su X, et al. The miR-21-5p enriched in the apoptotic bodies of M2 macrophage-derived extracellular vesicles alleviates osteoarthritis by changing macrophage phenotype. Genes Dis. 2023;10(3):1114–1129. doi:10.1016/j.gendis.2022.09.010
23. Lei Y, Wang X, Liao J, et al. Shear-responsive boundary-lubricated hydrogels attenuate osteoarthritis. Bioact Mater. 2022;16:472–484. doi:10.1016/j.bioactmat.2022.02.016
24. Zhu Z, Gao S, Chen C, et al. The natural product salicin alleviates osteoarthritis progression by binding to IRE1α and inhibiting endoplasmic reticulum stress through the IRE1α-IκBα-p65 signaling pathway. Exp Mol Med. 2022;54(11):1927–1939. doi:10.1038/s12276-022-00879-w
25. Zeng C, Duan F, Hu J, et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 2020;34:101523. doi:10.1016/j.redox.2020.101523
26. Han J, Park H, Maharana C, et al. Alzheimer’s disease-causing presenilin-1 mutations have deleterious effects on mitochondrial function. Theranostics. 2021;11(18):8855–8873. doi:10.7150/thno.59776
27. Deng C, Zhou Q, Zhang M, et al. Bioceramic scaffolds with antioxidative functions for ROS scavenging and osteochondral regeneration. Adv Sci. 2022;9(12):2105727. doi:10.1002/advs.202105727
28. Kloppenburg M, Namane M, Cicuttini F. Osteoarthritis. Lancet. 2025;405(10472):71–85. doi:10.1016/S0140-6736(24)02322-5
29. Kang D, Lee J, Jung J, et al. Selenophosphate synthetase 1 deficiency exacerbates osteoarthritis by dysregulating redox homeostasis. Nat Commun. 2022;13(1):779. doi:10.1038/s41467-022-28385-7
30. Chen P, Zheng L, Wang Y, et al. Desktop-stereolithography 3D printing of a radially oriented extracellular matrix/mesenchymal stem cell exosome bioink for osteochondral defect regeneration. Theranostics. 2019;9(9):2439–2459. doi:10.7150/thno.31017
31. Chen M, Chen Z, Wang Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12(4):689–702. doi:10.1080/15548627.2016.1151580
32. Kasahara A, Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014;24(12):761–770. doi:10.1016/j.tcb.2014.08.005
33. Chen W, Zhao H, Li Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Sig Transduct Target Ther. 2023;8(1):333. doi:10.1038/s41392-023-01547-9
34. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. doi:10.1038/nrm3028
35. Givvimani S, Munjal C, Tyagi N, Sen U, Metreveli N, Tyagi SC. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PLoS One. 2012;7(3):e32388. doi:10.1371/journal.pone.0032388
36. König J, Ott C, Hugo M, et al. Mitochondrial contribution to lipofuscin formation. Redox Biol. 2017;11:673. doi:10.1016/j.redox.2017.01.017
37. Sumida M, Doi K, Ogasawara E, et al. Regulation of mitochondrial dynamics by dynamin-related protein-1 in acute cardiorenal syndrome. J Am Soc Nephrol. 2015;26(10):2378–2387. doi:10.1681/ASN.2014080750
38. Zaja I, Bai X, Liu Y, et al. Cdk1, PKCδ and calcineurin-mediated Drp1 pathway contributes to mitochondrial fission-induced cardiomyocyte death. Biochem Biophys Res Commun. 2014;453(4):710–721. doi:10.1016/j.bbrc.2014.09.144
39. She H, Zhu Y, Deng H, et al. Protective effects of dexmedetomidine on the vascular endothelial barrier function by inhibiting mitochondrial fission via ER/Mitochondria contact. Front Cell Dev Biol. 2021;9:636327. doi:10.3389/fcell.2021.636327
40. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8(10):939–944. doi:10.1038/sj.embor.7401062
41. Hu Y, Chen H, Zhang L, et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy. 2021;17(5):1142–1156. doi:10.1080/15548627.2020.1749490
42. Kim YY, Um JH, Yoon JH, et al. p53 regulates mitochondrial dynamics by inhibiting Drp1 translocation into mitochondria during cellular senescence. FASEB J. 2020;34(2):2451–2464. doi:10.1096/fj.201901747RR
43. Luan M, Shi SS, Shi DB, et al. TIPRL, a novel tumor suppressor, suppresses cell migration, and invasion through regulating AMPK/mTOR signaling pathway in gastric cancer. Front Oncol. 2020;10:1062. doi:10.3389/fonc.2020.01062
44. Wu Q, Liu J, Mao Z, et al. Ligustilide attenuates ischemic stroke injury by promoting Drp1-mediated mitochondrial fission via activation of AMPK. Phytomedicine. 2022;95:153884. doi:10.1016/j.phymed.2021.153884
45. Ma T, Wang X, Qu W, et al. Osthole suppresses knee osteoarthritis development by enhancing autophagy activated via the AMPK/ULK1 pathway. Molecules. 2022;27(23):8624. doi:10.3390/molecules27238624
46. Kang JW, Hong JM, Lee SM. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J Pineal Res. 2016;60(4):383–393. doi:10.1111/jpi.12319
47. Guo L, Cui C, Wang J, et al. PINCH-1 regulates mitochondrial dynamics to promote proline synthesis and tumor growth. Nat Commun. 2020;11(1):4913. doi:10.1038/s41467-020-18753-6
48. Wang Q, Zhang M, Torres G, et al. Metformin suppresses diabetes-accelerated atherosclerosis via the Inhibition of Drp1-mediated mitochondrial fission. Diabetes. 2017;66(1):193–205. doi:10.2337/db16-0915
49. Hu Y, Zhou Y, Yang Y, et al. Metformin protects against diabetes-induced cognitive dysfunction by inhibiting mitochondrial fission protein DRP1. Front Pharmacol. 2022;13:832707. doi:10.3389/fphar.2022.832707
50. Cui A, Li H, Wang D, Zhong J, Chen Y, Lu H. Global, regional prevalence, incidence and risk factors of knee osteoarthritis in population-based studies. EClinicalMedicine. 2020;29–30:100587. doi:10.1016/j.eclinm.2020.100587
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Electroacupuncture Exerts Chondroprotective Effect in Knee Osteoarthritis of Rabbits Through the Mitophagy Pathway
Xing L, Chen X, Guo C, Zhu W, Hu T, Ma W, Du M, Xu Y, Guo C
Journal of Pain Research 2023, 16:2871-2882
Published Date: 21 August 2023
Network Analysis of Osteoarthritis Progression Using a Steiner Minimal Tree Algorithm
Xie Y, Shao F, Ji Y, Feng D, Wang L, Huang Z, Wu S, Sun F, Jiang H, Miyamoto A, Wang H, Zhang C
Journal of Inflammation Research 2024, 17:3201-3209
Published Date: 18 May 2024