")
Back to Journals » OncoTargets and Therapy » Volume 12
ARHGAP10 Inhibits the Proliferation and Metastasis of CRC Cells via Blocking the Activity of RhoA/AKT Signaling Pathway
Authors Liu L, Xie D, Xie H, Huang W, Zhang J, Jin W, Jiang W, Xie D
Received 9 July 2019
Accepted for publication 9 December 2019
Published 27 December 2019 Volume 2019:12 Pages 11507—11516
DOI https://doi.org/10.2147/OTT.S222564
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Lin Liu,1,* Dongyu Xie,2,3,* Haina Xie,4,* Wei Huang,1 Jingxian Zhang,5 Wenye Jin,5 Wenjing Jiang,1 Donghao Xie5,6
1Department of Pharmacy, Dahua Hospital, Shanghai, People’s Republic of China; 2Department of Spleen-Stomach, Zhenjiang Affiliated Hospital of Nanjing University of Chinese Medicine, Zhenjiang, People’s Republic of China; 3Department of Spleen-Stomach, Zhenjiang Hospital of Traditional Chinese Medicine, Zhenjiang, People’s Republic of China; 4School of Basic Medical Sciences, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China; 5Department of Pharmacy, Guanghua Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai, People’s Republic of China; 6School of Pharmacy, Jiangsu University, Zhenjiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Donghao Xie
Department of Pharmacy, Guanghua Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai, People’s Republic of China
Tel +86- 18221220090
Email [email protected]
Introduction: ARHGAP10 belongs to the ARHGAP family, which is downregulated in certain human tumors. However, the detailed function of ARHGAP10 remains unclear in human colon carcinoma (CRC). In the current study, we aimed to explore the role of ARHGAP10 in the growth and metastasis of CRC cells.
Methods: ARHGAP10 was induced silencing and overexpression using RNA interference (RNAi) and lentiviral-vector in CRC cells. Quantitative real-time PCR (qRT-PCR) and Western blot were used to quantify the mRNA and protein contents of ARHGAP10. Cell proliferation was determined by using Cell counting kit-8 (CCK-8). Transwell assay was utilized to examine the role of ARHGAP10 in the migration and invasion of CRC cells.
Results: Our results indicated that ARHGAP10 was downregulated in human CRC tissues and low expression of ARHGAP10 was associated with poor prognosis of patients with CRC. Moreover, ARHGAP10 overexpression significantly inhibited the proliferation and metastasis of CRC cells. Moreover, a PI3K/AKT inhibitor LY294002 was utilized to examine the connection between ARHGAP10 and AKT. Our findings demonstrated that the AKT inhibitor LY294002 could rescue the function of ARHGAP10 in CRC cells.
Discussion: It was the first time to elucidate that AKT involved in the ARHGAP10 signaling pathway and ARHGAP10 negatively mediated the phosphorylation of AKT (p-AKT) and RhoA activity in CRC cells. Interestingly, the Rho/MRTF/SRF inhibitor CCG-1423 significantly inhibited the phosphorylation of AKT in ARHGAP10 siRNA transfected CRC cells. Much importantly, overexpression of ARHGAP10 deeply suppressed the metastasis of CRC cells in the lung in vivo. Taken together, our findings not only enhanced the understanding of the anti-cancer effect of ARHGAP10 in CRC cells but also indicated its underlying pathway in CRC.
Keywords: ARHGAP10, AKT, RhoA activity, colon carcinoma
Introduction
Human colon carcinoma (CRC) is one of the major malignant cancers all over the world, which is a serious threat to human life.1 Due to the high mortality and incidence of CRC, the outcomes of chemotherapy and surgical resection are far from satisfactory. The 5-year overall survival rate of CRC patients is less than 11%.2 However, the underlying mechanism of CRC is rather less well understood.
ARGHGAP10 (RhoGTPase Activating Protein 10), also known as ARHGAP21, contains an N-terminal PDZ domain, a central pleckstrin homology (PH) domain and a C-terminally located Rho-GAP domain.3,4 It has been confirmed that Rho-GAP domains catalyze the transformation of active GTP-bound forms of small Rho-family GTPases to their inactive GDP-bound forms.5 Previous report has indicated that ARHGAP10 processes the GAP activity for RhoA and RhoC in the progression of prostate cancer progression.6 Moreover, ARHGAP10 has suppressed the migration of glioblastoma cells via mediating the activity of FAK.7 Further, overexpression of ARHGAP10 deeply inhibits the metastasis and proliferation of lung cancer cells and reduces tumorigenicity of ovarian cancer cells.8,9 However, the detailed function of ARHGAP10 is less investigated in CRC.
RhoA (Ras homologue family member A) is a member of small GTPases of the Rho family, which is a molecular switch that cycle between a GTP-bound active form and a GAP inactive form.10 The activation of RhoA plays a crucial role in actin cytoskeletal re-arrangement and contributes to the progression and invasion of CRC cells.11,12 Recently, evidence have identified that inactive RhoA promotes the progression and metastasis of CRC cells through enhancing the activity of Wnt/MYC signaling and presents tumor suppressor activity.13,14 Therefore, the more precise molecule mechanism of RhoA in CRC metastasis needs to be further investigated.
PI3K/AKT-signaling pathway is often hyper-activated in human CRC, which promotes the proliferation and metastasis of CRC cells.15,16 Thus, targeting AKT is reported as a promising anti-cancer strategy.17 It has been demonstrated that AKT phosphorylation (p-AKT) mediated the activation of RhoA in mouse embryos.18 Further, PI3K/AKT has been found downstream to RhoA in myoblast.19 However, the detailed connection between RhoA and AKT in CRC cells has not been confirmed yet.
In the present research, we induced silencing and overexpression of ARHGAP10 in CRC cells. Moreover, a PI3K/AKT inhibitor LY294002 was utilized to examine the connection between ARHGAP10 and AKT. Our analyses not only enhanced the understanding of ARHGAP10 but also indicated its potential signaling pathway in CRC cells.
Materials and Methods
Human CRC Tissues
The human CRC and adjacent-matched noncancerous tissues (n=80) were provided by Guanghua Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai, China. Moreover, all CRC patients involved in the retrospective study were diagnosed and treated at the Guanghua Hospital of Integrated Traditional Chinese and Western Medicine between May 2007 and April 2008. There were 43 men and 37 women enrolled in the present study, ranging in age from 33 to 73 years. The patients were followed up every 3 months after curative resection by an experienced nurse on clinic or by telephone interview. All patients were written informed consent. Our research was approved by the independent ethics committee of Guanghua Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai, China and was in accordance with the Declaration of Helsinki.
Cell Culture
Five human CRC cell lines (CACO2, RKO, HCT116, HT29 and LOVO) and one nonmalignant colon cell line FHC were purchased from the cell bank of the Shanghai Biology Institute (Shanghai, China). Among them, RKO, HCT116 and HT-29 cells have constitutive active PI3K, Cells were seeded in DMEM (SH30243.01, Hyclone, USA) and cultured in a 5% CO2 atmosphere at 37 °C. The AKT inhibitor LY294002 (25μmol/L; S1105, Selleck, USA) and the RhoA inhibitor CCG-1423 (1μmol/L; S7719, Selleck, USA) were dissolved in DMSO (D2650, Sigma, USA) and used to culture cells.
Immunohistochemistry Staining
The tissue sections were fixed in methanol (4%) for 30 min. Then, eliminated endogenous peroxidase activity was eliminated through incubating with H2O2 (3%) for 10 min. After that, the primary antibodies of ARHGAP10 (55139-1-AP, Proteintech, USA) were used to incubate the tissue sections at room temperature for 1h, followed by incubation with the HRP-labelled secondary antibody for 30 mins. Then, the sections were stained by DAB and re-stained by hematoxylin for 3 mins. An upright microscope (ECLIPSE Ni, NIKON, Japan) was used to take an image and analyzed by using the Microscopic image analysis system (DS-Ri2, NIKON, Japan), magnification, 200×. The high expression of ARHGAP10 was defined by the positive stain of tumor cells above 25% and the low expression was defined by the positive stain of tumor cells less than 25%.
Real-Time PCR
Total RNA were isolated from cell or tissue samples using the TRIzol Reagent kit (1596–026, Invitrogen, USA) and converted to the complementary DNA (cDNA) by using a cDNA synthesis kit (#K1622, Fermentas, Canada). Analyses were established on a Real-time Detection (ABI-7300, ABI, USA) using SYBR Green master mix (#K0223, Thermo, USA). The conditions of thermo-cycling were set as follows: 95°C for 10 mins followed by 40 cycles of 95°C for 15 s, 60°C for 45 s. 2−ΔΔCt method was used to quantify the relative gene expression. GAPDH was used as a control for mRNAs, whereas the RNU6B (U6) gene as a control for miRNAs. Three replications were needed for all reactions. Primer sequences were presented in Supplementary File 1.
Western Blot
Total protein was extracted using RIPA lysis buffer (JRDUN, Shanghai, China). BCA protein assay kit (PICPI23223, Thermo Fisher, USA) was utilized to measure total protein. Proteins that were adjusted to equal content (25μg) were fractionated in 10% SDS-PAGE and subsequently transferred onto PVDF nitrocellulose membrane (HATF00010, Millipore, USA) for 12h. Then, the membranes were probed with the primary antibodies at 4°C overnight followed by the appropriate HRP-conjugated goat anti-rabbit IgG (A0208, Beyotime, China) at 37°C for 60 min. Protein signals were detected using a chemiluminescence system (5200, Tanon, China). GAPDH served as an endogenous reference. Each analysis was established in triplicate. The product information of primary antibodies is listed in Supplementary Table 2.
Silencing and Overexpression of ARHGAP10
Three short interfering RNAs (siRNA) targeted to different regions of human ARHGAP10 (NM_024605.3) and a scramble siRNA (siNC) were linked with Lentiviral vector (pLKO.1). Then, the recombined vectors transiently transfected in degenerative NP cells, respectively, by Lipofectamine 2000 (Invitrogen, USA). The sequence information of siUSP15s is listed in Supplementary Table 1.
Meanwhile, pLVX-puro vector contained the full length of ARHGAP10 (NM_024605.3) cDNA sequence (oeARHGAP10) was transiently transfected in degenerative NP cells. The mock plasmid (oeNC) was functioned as a negative control (oeNC). Analyses were established at 48h after being transfected.
Cell Proliferation
Cell proliferation profile was examined by using Cell counting kit-8 (CCK-8) (CP002, SAB, USA) following the instruction given by the manufacturer. The OD450 value was quantified by a microplate reader (DNM-9602, Pulangxin, China). Three replications were needed for each time point.
Transwell
Cells were serum-starved in free-serum DMEM for 24h. Then, cells were filled in the upper transwell chamber (3422, COSTAR, USA) with (invasion assay) or without (migration assay) Matrigel-coated (BD Biosciences, MA). Then, 700 µL of medium supplemented with 10% fetal bovine serum (16000–044, GIBCO, USA) was injected into the lower chambers. After incubating at 37°C for 24h (migration assay) and 48h (invasion assay), cells were fixed in 4% formaldehyde for 10mins and followed by staining with 0.01% crystal violet (C8470, Solarbio, China) for 30mins. Then, the cells were photographed and counted under the microscope, magnification, 200× (XDS-500C, Caikon, Shanghai, China).
Rho GTPase Activation Assays
Cells were washed with ice-cold PBS and lysed and incubated with 30μg glutathione S-transferase–Rho binding domain of rhotekin (GST-RBD) beads (Cytoskeleton, Denver, CO, USA) for 1 h at 4 °C. Then, bound Rho proteins were analyzed by immunoblotting using an anti-RhoA antibody (Ab54835, Abcam, UK) after the beads being washed twice with washing buffer.
Animal Experiments
An equal number (1 ×106) of HT29 cells that transfected with oeNC and oeARHGAP10 were harvested and re-suspended in PBS, and subsequently intravenously injected through the tail vein into 5–6-week-old female BALB/c nude mice (weight: 16–18g, n=21 per group). Hematoxylin and eosin (H&E) was used to stain lung tissues removed from the xenograft mice (n = 6) on day 21. Mouse survival was examined daily for a total of 80 days and each injected group contained 15 nude mice. Animal experiments were approved by the institutional ethical committee of Guanghua Hospital of Integrated Traditional Chinese and Western Medicine and established according to the legal requirements. All procedures were followed by the laboratory animal’s welfare guidelines of Guanghua Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai, China.
Statistical Analysis
Statistical analyses were performed by using GraphPad Prism software Version 7.0 (CA, USA). All data represented as means ± SEM from three independent experiments. Statistical significance was assessed by using Student’s t-test and one-way analysis of variance (ANOVA). Statistically significant was accepted as the p-value <0.05.
Results
Low Expression of ARHGAP10 Is Associated with Poor Prognosis of Human CRC Patients
The relative mRNA level of ARHGAP10 was examined in 80 pairs of human CRC and adjacent-matched tissues. Our results suggested that the level of ARHGAP10 was significantly downregulated in CRC tissues than that in normal tissues (Figure 1A). Moreover, Immunohistochemistry assay was established to quantify the protein content of ARHGAP10 in human CRC or matched-normal tissues. Thirty-four tumor tissues presented high expression of ARHGAP10, whereas 46 cases showed low expression of ARHGAP10 (Figure 1B). Further, Kaplan-Meier overall survival (OS) curves for patients who had undergone curative surgery (n=80). As presented in Figure 1C, the OS of the low-ARHGAP10 group was much worse than the high-ARHGAP10 group. Therefore, the low expression of ARHGAP10 indicated the poor prognosis of CRC.
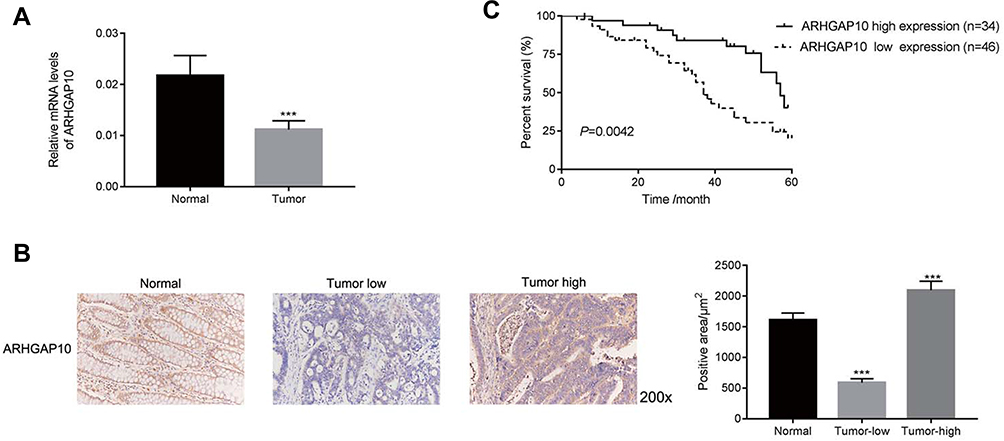 |
Figure 1 ARHGAP10 was downregulated in human CRC tissues. (A) The relative mRNA level of ARHGAP10 examined in human CRC and normal (n=20) tissues. ***p < 0.001 vs Normal. (B) Immunohistochemistry (IHC) assay was performed to examine the protein content of ARHGAP10 in human CRC or normal tissues, magnification, 200×. (C) Kaplan-Meier overall survival (OS) curves for patients who had undergone curative surgery (n=80). |
Silencing and Overexpression of ARHGAP10 in CRC Cells
To further assess the function of ARHGAP10, we quantified the level of ARHGAP10 in six CRC cells, including CACO2, HCT116, HT29, LOVO, RKO and FHC. Obviously, both the relative mRNA and protein levels of ARHGAP10 were much lower in HT29 and RKO cells. Meanwhile, the level of ARHGAP10 was significantly increased in HCT116 and FHC cells (Figure 2A and B).
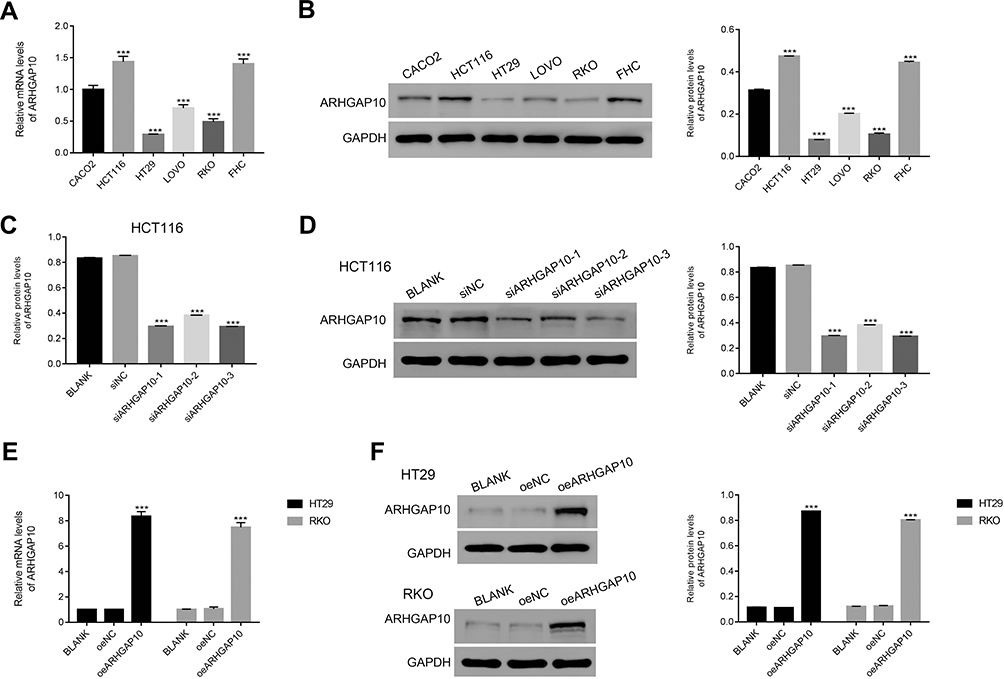 |
Figure 2 Silencing and overexpression of ARHGAP10 in CRC cells. (A, B). The relative mRNA and protein levels of ARHGAP10 examined in CACO2, HCT116, HT29, LOVO, RKO and FHC cells. ***p < 0.001 vs CACO2. (C, D). Both the relative mRNA and protein levels of ARHGAP10 were deeply suppressed by ARHGAP10 siRNAs. ***p < 0.001 vs siNC. (E, F). Both the relative mRNA and protein levels of ARHGAP10 were significantly upregulated in HT29 and RKO cells that transfected with oeARHGAP10. ***p < 0.001 vs oeNC. |
Next, three short interference RNAs (siRNA) that target different regions of human gene ARHGAP10 (NM_024605.3) were transfected into HT116 cells, respectively (siARHGAP10-1, siARHGAP10-2 and siARHGAP10-3). A scramble siRNA served as a negative control (siNC). The relative mRNA and protein levels of ARHGAP10 were deeply reduced in siARHGAP10s-transfected cells (Figure 2C and D). Therefore, all the ARHGAP10-1 siRNAs were well functioned to reduce the expression of endogenous ARHGAP10 in CRC cells.
Moreover, the full length of ARHGAP10 cDNA was inserted into the lentiviral vector (pLVX-Puro) and then the recombined vector (oeARHGAP10) was transfected into HT29 and RKO cells, respectively. A mock plasmid was transfected as a negative control (oeNC). As presented in Figure 2E and F, the level of ARHGAP10 was significantly upregulated in HT29 and RKO cells that transfected oeARHGAP10. Therefore, the oeARHGAP10 transfected cells were used in the following analyses.
Overexpression of ARHGAP10 Inhibited the Proliferation and Metastasis of CRC Cells
Then, CCK-8 assay was performed to determine the proliferation of HT29 and RKO cells that transfected with oeARHGAP10, respectively. The proliferation rate of oeARHGAP10 transfected cells showed no significant difference than that in oeNC transfected cells within 12h in two cell lines. After incubating for 48h, the proliferation rate of HT29 or RKO cells was significantly decreased by oeARHGAP10 (Figure 3A and B).
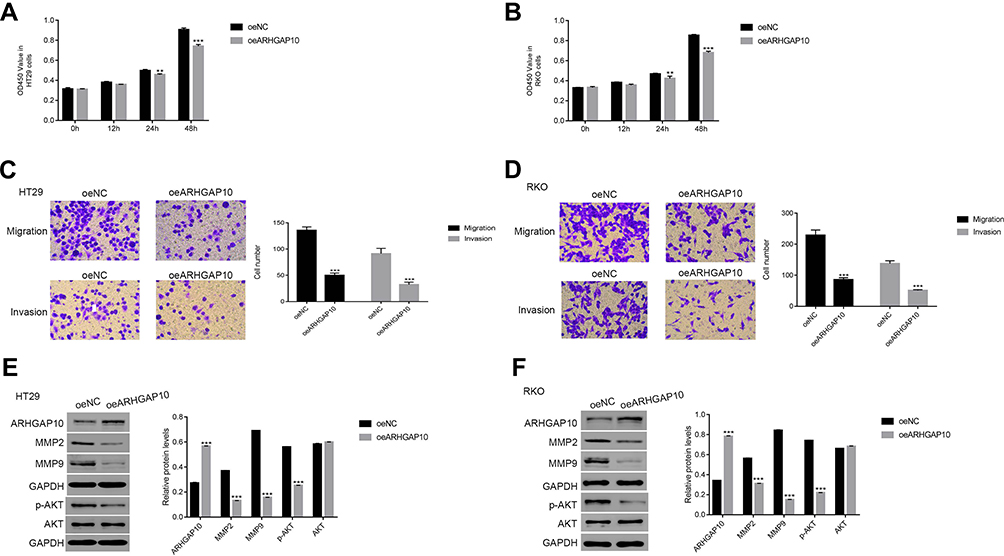 |
Figure 3 Overexpression of ARHGAP10 inhibited the proliferation and metastasis of CRC cells. (A, B). The proliferation rate was reduced in HT29 and RKO cells that transfected with oeARHGAP10. **p < 0.01 vs oeNC, ***p < 0.001 vs oeNC. (C, D). Overexpression of ARHGAP10 inhibited the migration and invasion of HT29 and RKO cells. ***p < 0.001 vs oeNC. (E, F). Western blot was used to examine the protein content of ARHGAP10, MMP2, MMP9, p-AKT and AKT in HT29 and RKO cells transfected with oeNC and oeARHGAP10, respectively, ***p < 0.001 vs oeNC. |
Next, we examined the migration and invasion of oeARHGAP10 transfected cells by using Transwell assay. Importantly, our results suggested that overexpression of ARHGAP10 significantly suppressed the migration and invasion of HT29 and RKO cells that transfected with oeARHGAP10 (Figure 3C and D). Thus, ARHGAP10 possessed the anti-metastasis property in human CRC cells.
Matrix metalloproteinases 2 (MMP2) and MMP9 belong to proteolytic enzyme family, which promote the metastasis of human CRC cells.20–22 In the current analyses, we also quantified the protein contents of MMP2 and MMP9 in oeARHGAP10 transfected cells. Both MMP2 and MMP9 were decreased in oeARHGAP10 transfected cells. Therefore, ARHGAP10 might target MMP2 and MMP9 in regulating the metastasis of CRC cells. More importantly, overexpression of ARHGAP10 deeply inhibited the phosphorylation of AKT (p-AKT) in HT29 and RKO cells (Figure 3E and F).
The PI3K/AKT Inhibitor LY294002 Rescued the Function of ARHGAP10
To further analyze the connection between ARHGAP10 and AKT in CRC cells, the PI3K/AKT inhibitor LY294002 (20μmol/L) was used to culture siARHGAP10 transfected CRC cells. Clearly, knockdown of ARHGAP10 significantly promoted the proliferation of HCT116 cells. However, this effect was deeply abolished by the PI3K/AKT inhibitor LY294002 (Figure 4A). Moreover, ARHGAP10 siRNA significantly promoted the migration and invasion number of HCT116 cells, whereas the PI3K/AKT inhibitor LY294002 strongly inhibited the migration and invasion of HCT116 cells (Figure 4B). Further, the protein contents of MMP2 and MMP9 were upregulated in siARHGAP10 transfected cells. However, the PI3K/AKT LY294002 deeply suppressed the expression of MMP2 and MMP9 in siARHGAP10 transfected cells. Overall, these results revealed that the PI3K/AKT inhibitor LY294002 could rescue the function of ARHGAP10 in siARHGAP10 transfected cells. Further, ARHAGP10 negatively mediated the phosphorylation of AKT in CRC cells (Figure 4C).
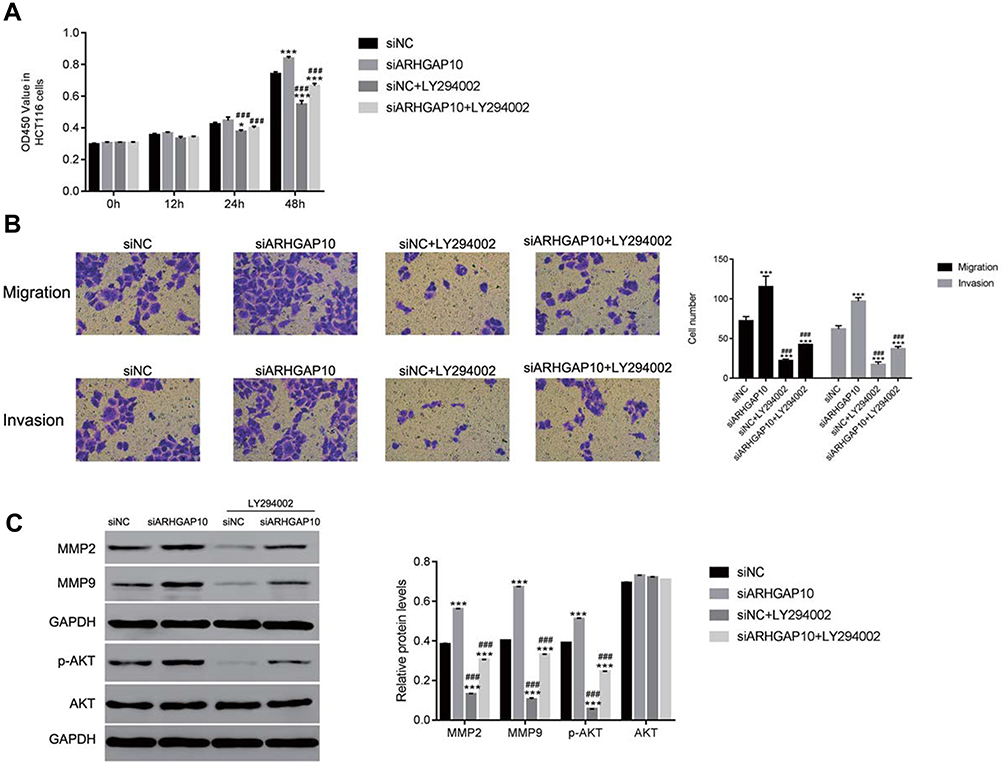 |
Figure 4 The AKT inhibitor LY294002 rescued the function of ARHGAP10 in CRC cells. (A). The proliferation rate of siARHGAP10 transfected cells was reduced in the presence of the AKT inhibitor LY294002. *p < 0.05 vs siNC, ***p < 0.001 vs siNC; ###p < 0.001 vs siARHGAP10. (B). The AKT inhibitor LY294002 inhibited the migration and invasion of siARHGAP10 transfected cells. ***p < 0.001 vs siNC; ###p < 0.001 vs siARHGAP10. (C). The protein content of MMP2, MMP9, p-AKT and AKT were reduced by the AKT inhibitor LY294002. ***p < 0.001 vs siNC; ###p < 0.001 vs siARHGAP10. |
ARHGAP10 Negatively Mediated RhoA Activity in CRC Cells
RhoA is a member of Rho GTPases, which improves the metastasis of CRC cells.23 Then, Rho GTPase activation assays were performed to examine the connection between RhoA and ARHGAP10 in CRC cells. As presented in Figure 5A–C, the protein content of RhoA-GTP was significantly suppressed in oeARHGAP10 transfected cells, whereas was significantly increased in siARHGAP10 transfected cells. These findings indicated that ARHGAP10 negatively modulated the activity of RhoA in human CRC cells.
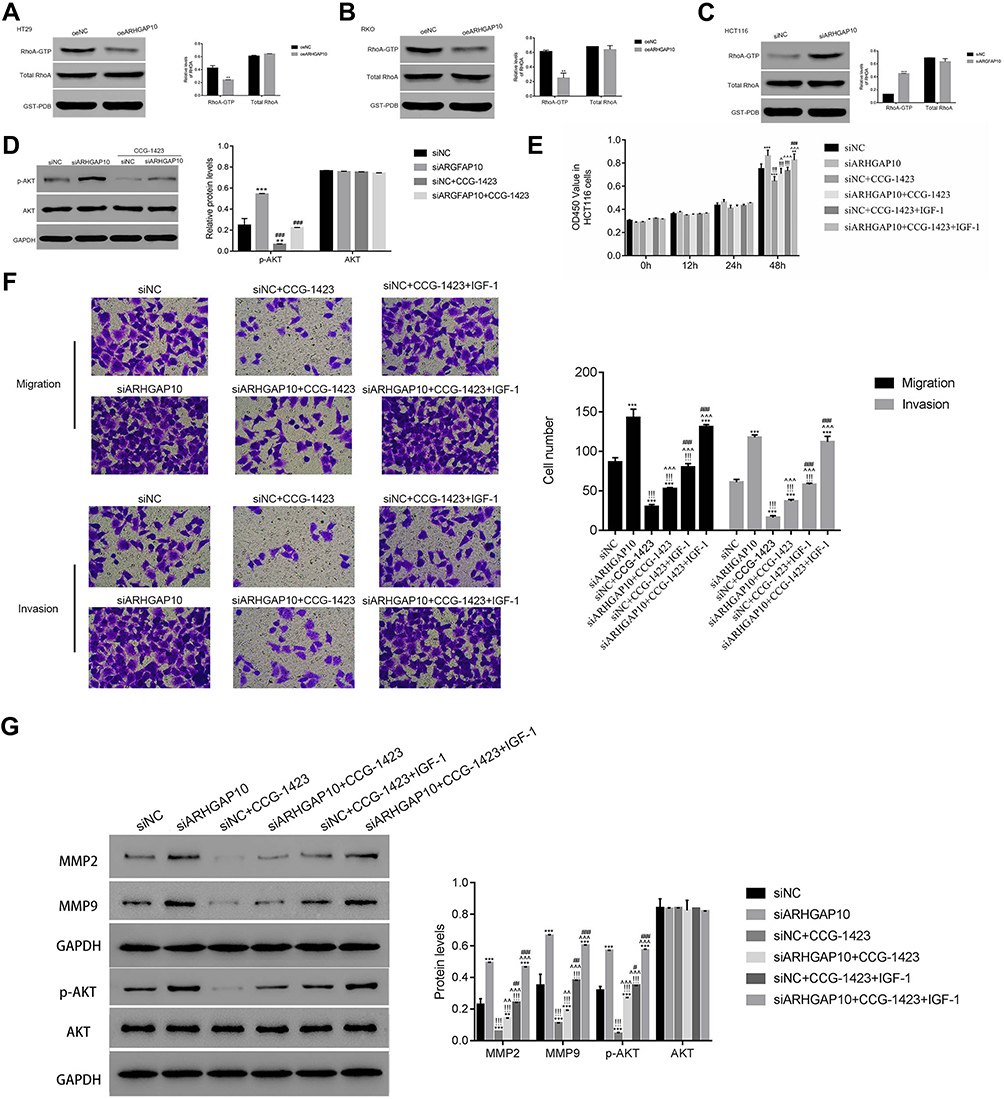 |
Figure 5 ARHGAP10 negatively correlated with the active RhoA in CRC cells. (A, B). The level of active RHOA was downregulated in HT29 and RKO transfected with oeARHGAP10. **p < 0.01 vs oeNC. (C). The protein content of active RhoA was upregulated in siARHGAP10 transfected cells. ***p < 0.001 vs siNC. (D). The protein content of p-AKT was suppressed by the RhoA inhibitor CCG-1423 in siARHGAP10 transfected cells. **p < 0.01 vs siNC, ***p < 0.001 vs siNC; ### p < 0.001 vs siARHGAP10. (E). The AKT agonist IGF-1 promoted the proliferation of siARHGAP10 transfected cells in the presence of RhoA inhibitor CCG-1423. **p < 0.01 vs siNC, ***p < 0.001 vs siNC; !!!p < 0.001 vs siARHGAP10; ^p < 0.05 vs siNC+ CCG-1423, ^^^p < 0.001 vs siNC+ CCG-1423; ###p < 0.001 vs siARHGAP10 + CCG-1423. (F). The AKT agonist IGF-1 released the suppression of the RhoA inhibitor CCG-1423 in the metastasis of siARHGAP10 transfected cells. ***p < 0.001 vs siNC; !!!p < 0.001 vs siARHGAP10; ^^^p < 0.001 vs siNC+ CCG-1423; ###p < 0.001 vs siARHGAP10 + CCG-1423. (G). The protein contents of MMP2, MMP9 and p-AKT were promoted by the AKT agonist IGF-1 in siARHGAP10 transfected cells that cultured in the presence of RhoA inhibitor CCG-1423. |
We also examined the protein content of p-AKT in siNC or siARHGAP10 transfected cells in the presence of Rho/MRTF/SRF inhibitor CCG-1423 (1 μmol/L). As presented in Figure 5D, Rho/MRTF/SRF inhibitor CCG-1423 deeply suppressed the phosphorylation of AKT in siARHGAP10 transfected cells.
The AKT Agonist IGF-1 Abolished the Effect of Rho/MRTF/SRF Inhibitor CCG-1423 in siARHGAP10 Transfected Cells
To further assess the connection between RhoA and AKT in siARHGAP10 transfected cells, the AKT agonist IGF-1 (100ng/mL) was used to culture cells as indicated. After being cultured for 48h, the proliferation of siARHGAP10 transfected cells was deeply suppressed by the Rho/MRTF/SRF inhibitor CCG-1423, but significantly recovered in the presence of the AKT agonist IGF-1 (Figure 5E). These results indicated that the AKT agonist attenuated the effect of the inhibitor CCG-1423 in siARHGAP10 transfected cells.
Moreover, the metastasis of siARHGAP10 transfected cells was also significantly reduced by the Rho/MRTF/SRF inhibitor CCG-1423. Importantly, the AKT agonist IGF-1 enhanced the migration and invasion of siARHGAP10 transfected cells in the presence of the Rho/MRTF/SRF inhibitor CCG-1423 (Figure 5F).
Further, we also examined the protein contents of MMP2, MMP9, p-AKT and AKT in different cells as indicated (Figure 5G). The protein contents of MMP2 and MMP9 were deeply suppressed by the inhibitor CCG-1432 in siARHGAP10 transfected cells, but the AKT agonist IGF-1 significantly relieved this suppression. The similar results were also obtained in quantifying the phosphorylation of AKT. Taken together, our findings illustrated that RhoA positively correlated with the phosphorylation of AKT in siARHGAP10 transfected cells. ARHGAP10 might attenuate the proliferation and metastasis of CRC cells through suppressing the activity of RhoA/AKT signaling pathway.
Overexpression of ARHGAP10 Reduced the Metastasis of CRC Cells in Lung in vivo
To address the function of ARHGAP10 in the metastasis of CRC cells in vivo, an equal number of oeNC and oeARHGAP10 transfected cells were injected into the caudal vein of nude mice. Results collected from H & E staining revealed that overexpression of ARHGAP10 significantly suppressed the nodule formation in vivo (Figure 6A). Moreover, the oeARHGAP10 transfected cells could prolong the survival time of mice compared with that of oeNC group (Figure 6B).
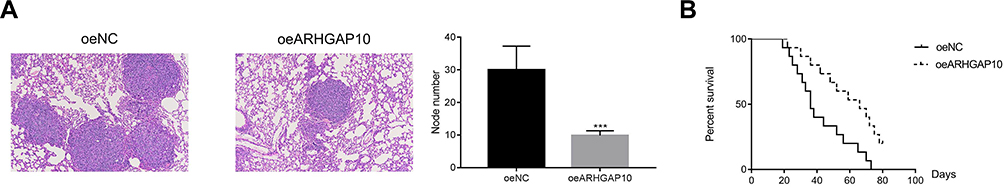 |
Figure 6 Overexpression of ARHGAP10 reduced the metastasis of CRC cells to pulmonary in vivo. (A). Overexpression of ARHGAP10 significantly suppressed the formation of nodules in vivo, ***p < 0.001 vs oeNC. (B). The survival time of mice that injected with oeARHGAP10 transfected cells was longer than that in the oeNC group. |
Discussion
Metastasis is the crucial reason for cancer-related deaths, which is a highly selective process.24 It has been indicated that the high rate of mortality of CRC is attributed to metastasis of neoplastic cells from the primary tumor to secondary organ sites.25 However, the underlying mechanism remains unclear. In the present study, ARHGAP10 was downregulated in CRC tissues compared with that in adjacent normal tissues. Moreover, low expression of ARHGAP10 was associated with poor prognosis of human CRC patients. Further, ARHGAP10 overexpression deeply suppressed the invasion and migration of CRC cells. Meanwhile, knockdown of ARHGAP10 significantly improved the migration and invasion of CRC cells. Therefore, ARHGAP10 was likely an anti-metastasis factor in CRC cells. Much more importantly, ARHGAP10 presented the clinical value as a novel prognosis marker in CRC.
MMP-2 and MMP-9 are closely associated with the progression of CRC.26 In this study, our results illustrated that ARHGAP10 was negatively correlated with MMP2 and MMP9 in CRC cells. Therefore, ARHGAP10 was a suppressor for MMP2 and MMP9. ARHGAP10 might inhibit the metastasis of CRC by blocking MMP2 and MMP9 in CRC cells.
Previous report has demonstrated that the activation of PI3K/AKT acts as an upstream signal in SOX2-induced MMP-2 expression in Hep-2 cells.27 Moreover, inhibiting the activity of PI3K/AKT signaling pathway leads to the inhibition of MMP-9 in CRC cells.28 In our analyses, we found the PI3K/AKT inhibitor LY294004 could rescue the function of ARHGAP10 in proliferation and metastasis of CRC cells. It was the first time to elucidate that ARHGAP10 involved in PI3K/AKT signaling pathway and ARHGAP10 negatively mediated the phosphorylation of AKT in CRC cells. Additionally, these findings suggested that ARHGAP10 might inhibit the migration and invasion of CRC cells by suppression of MMP-2 and MMP-9 via the PI3K/AKT signaling pathway.
RhoA is a member of Rho subfamily, which plays a critical role in the migration, invasion and proliferation of tumor cells.29 Moreover, it has been demonstrated that the expression of RhoA is modulated by the PI3K/AKT signaling pathway.18 Further, increasing the transduction of RhoA/ROCK pathway promotes the metastasis of breast cancer cells.30 In the present research, we found that ARHGAP10 inhibited RhoA activity in CRC cells. Moreover, the RhoA inhibitor CCG-1423 deeply suppressed the phosphorylation of AKT in siARHGAP10 transfected cells. Therefore, ARHGAP10 might be a novel component in RhoA/AKT signaling pathway. Overall, present research deepened insights into the anti-cancer effect of ARHGAP10 in CRC cells.
Conclusion
In this study, ARHGAP10 was induced by silencing and overexpression in CRC cells. Our analyses not only enhanced the understanding of ARHGAP10 but also indicated its potential signaling pathway in CRC cells.
Acknowledgments
We acknowledged the assistance supported by the Guanghua Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai, China and School of Pharmacy Jiangsu University, Zhenjiang, China. This research was supported by National Natural Science Foundation of China (NO.81774252).
Disclosure
All authors report no conflicts of interest in this work.
References
1. Sun P, Sun D, Wang X. Effects of Scutellaria barbata polysaccharide on the proliferation, apoptosis and EMT of human colon cancer HT29 cells. Carbohydr Polym. 2017;167:90–96. doi:10.1016/j.carbpol.2017.03.022
2. Li X, Peng S. Identification of metastasis-associated genes in colorectal cancer through an integrated genomic and transcriptomic analysis. Chin J Cancer Res. 2013;25(6):623–636. doi:10.3978/j.issn.1000-9604.2013.11.01
3. Basse`res DS, Tizzei EV, Duarte AA, Costa FF, Saad ST. ARHGAP10 a novel human gene coding for a potentially cytoskeletal RhoGTPase activating protein. Biochem Biophys Res Commun. 2002;294(3):0–585.
4. Rosa LRO, Soares GM, Silveira LR, Boschero AC, Barbosa-Sampaio HCL. ARHGAP21 as a master regulator of multiple cellular processes. J Cell Physiol. 2018;233(11):8477–8481. doi:10.1002/jcp.26829
5. Sousa S, Cabanes D, Archambaud C, et al. ARHGAP10 is necessary for alpha-catenin recruitment at adherens junctions and for Listeria invasion. Nat Cell Biol. 2005;7(10):954–960. doi:10.1038/ncb1308
6. Lazarini M, Traina F, Machado-Neto JA, et al. ARHGAP21 is a RhoGAP for RhoA and RhoC with a role in proliferation and migration of prostate adenocarcinoma cells. Biochim Biophys Acta. 2013;1832(2):365–374. doi:10.1016/j.bbadis.2012.11.010
7. Bigarella CL, Borges L, Costa FF, Saad ST. ARHGAP21 modulates FAK activity and impairs glioblastoma cell migration. Biochim Biophys Acta. 2009;1793(5):806–816. doi:10.1016/j.bbamcr.2009.02.010
8. Teng J-P, Yang Z-Y, Zhu Y-M, Ni D, Zhu Z-J, Li X-Q. The roles of ARHGAP10 in the proliferation, migration and invasion of lung cancer cells. Oncol Lett. 2017;14(4):4613–4618. doi:10.3892/ol.2017.6729
9. Luo N, Guo J, Chen L, Yang W, Qu X, Cheng Z. ARHGAP10, downregulated in ovarian cancer, suppresses tumorigenicity of ovarian cancer cells. Cell Death Dis. 2016;7(3):. doi:10.1038/cddis.2015.401.
10. Haga RB, Ridley AJ. Rho GTPases: regulation and roles in cancer cell biology. Small GTPases. 2016;7(4):207–221. doi:10.1080/21541248.2016.1232583
11. Li H, Peyrollier K, Kilic G, Brakebusch C. Rho GTPases and cancer. BioFactors. 2014;40(2):226–235. doi:10.1002/biof.1155
12. Yu X, Wang D, Wang X, et al. CXCL12/CXCR4 promotes inflammation-driven colorectal cancer progression through activation of RhoA signaling by sponging miR-133a-3p. J Exp Clin Cancer Res. 2019;38(1):32. doi:10.1186/s13046-018-1014-x
13. Rodrigues P, Macaya I, Bazzocco S, et al. RHOA inactivation enhances Wnt signaling and promotes colorectal cancer. Nat Commun. 2014;5(5):5458. doi:10.1038/ncomms6458
14. Dopeso H, Rodrigues P, Bilic J, et al. Mechanisms of inactivation of the tumour suppressor gene RHOA in colorectal cancer. Br J Cancer. 2018;118(1):106–116. doi:10.1038/bjc.2017.420
15. Xu C-L, Wang J-Z, Xia X-P, et al. Rab11-FIP2 promotes colorectal cancer migration and invasion by regulating PI3K/AKT/MMP7 signaling pathway. Biochem Biophys Res Commun. 2016;470(2):397–404. doi:10.1016/j.bbrc.2016.01.031
16. Tan X, Chen S, Wu J, et al. PI3K/AKT-mediated upregulation of WDR5 promotes colorectal cancer metastasis by directly targeting ZNF407. Cell Death Dis. 2017;8(3):. doi:10.1038/cddis.2017.111.
17. Brown JS, Banerji U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol Ther. 2016;172:101–115. doi:10.1016/j.pharmthera.2016.12.001
18. Liu L, Wang Y, Yu Q. The PI3K/Akt signaling pathway exerts effects on the implantation of mouse embryos by regulating the expression of RhoA. Int J Mol Med. 2014;33(5):1089–1096. doi:10.3892/ijmm.2014.1701
19. Reuveny M, Heller H, Bengal E. RhoA controls myoblast survival by inducing the phosphatidylinositol 3-kinase-Akt signaling pathway. FEBS Lett. 2004;569(1–3):129–134. doi:10.1016/j.febslet.2004.05.035
20. Banday MZ, Sameer AS, Mir AH, Mokhdomi TA, Chowdri NA, Haq E. Matrix metalloproteinase (MMP) −2, −7 and −9 promoter polymorphisms in colorectal cancer in ethnic Kashmiri population - A case-control study and a mini review. Gene. 2016;589(1):81–89. doi:10.1016/j.gene.2016.05.028
21. Dong W, Li H, Zhang Y, et al. Matrix metalloproteinase 2 promotes cell growth and invasion in colorectal cancer. Acta Biochim Biophys Sin. 2011;43(11):840–848. doi:10.1093/abbs/gmr085
22. Yang X, Cui S, Zeng L, et al. Overexpression of Rab1B and MMP9 predicts poor survival and good response to chemotherapy in patients with colorectal cancer. Aging. 2017;9(3):914–931. doi:10.18632/aging.v9i3
23. Zhang G-Y, Yang W-H, Chen Z. Upregulated STAT3 and RhoA signaling in colorectal cancer (CRC) regulate the invasion and migration of CRC cells. Eur Rev Med Pharmacol Sci. 2016;20(10):2028–2037.
24. Fidler IJ. Origin and biology of cancer metastasis. Cytometry Part A. 2010;10(6):673–680. doi:10.1002/cyto.990100602
25. Schwartz B, Bresalier R, Kim YS. The role of mucin in colon-cancer metastasis. Int J Cancer. 1992;52(1):60–65. doi:10.1002/ijc.2910520113
26. Hritz I, Varga Z, Juhász M, Miheller P, Tulassay Z, Herszényi L. Increased serum MMP-2, MMP-7, MMP-9, TIMP-1 and TIMP-2 levels in colorectal cancer development. Gastroenterology. 2011;140(5):S–343. doi:10.1016/S0016-5085(11)61393-0
27. Yang N, Hui L, Wang YAN, Yang H, Jiang X. SOX2 promotes the migration and invasion of laryngeal cancer cells by induction of MMP-2 via the PI3K/Akt/mTOR pathway. Oncol Rep. 2014;31(6):2651–2659. doi:10.3892/or.2014.3120
28. Chen S, Chen WEI, Zhang X, Lin S, Chen Z. Overexpression of KiSS-1 reduces colorectal cancer cell invasion by downregulating MMP-9 via blocking PI3K/Akt/NF-κB signal pathway. Int J Oncol. 2016;48(4):1391–1398. doi:10.3892/ijo.2016.3368
29. Timpson P, McGhee EJ, Morton JP, et al. Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53. Cancer Res. 2011;71(3):747–757. doi:10.1158/0008-5472.CAN-10-2267
30. Zhang C, Wang H-J, Bao Q-C, Wang L, Guo T-K. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget. 2016;7(45):73593–73606. doi:10.18632/oncotarget.12435
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.