")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 9
ARFIP2 Regulates EMT and Autophagy in Hepatocellular Carcinoma in Part Through the PI3K/Akt Signalling Pathway
Authors Huang K, Lin Y, Wang K, Shen J, Wei D
Received 3 October 2022
Accepted for publication 9 December 2022
Published 20 December 2022 Volume 2022:9 Pages 1323—1339
DOI https://doi.org/10.2147/JHC.S392056
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Jörg Trojan
Kaida Huang,1 Yubiao Lin,1 Keyin Wang,2 Jianfen Shen,3 Dahai Wei2– 4
1Department of Oncology, Xiamen Haicang Hospital, Xiamen, People’s Republic of China; 2Department of Infectious Diseases, Affiliated Hospital of Jiaxing University, Jiaxing, People’s Republic of China; 3Department of Central Laboratory, Affiliated Hospital of Jiaxing University, Jiaxing, People’s Republic of China; 4Institute of Hepatology, Affiliated Hospital of Jiaxing University, Jiaxing, People’s Republic of China
Correspondence: Dahai Wei, Institute of Hepatology, Affiliated Hospital of Jiaxing University, Jiaxing, People’s Republic of China, Tel/Fax +86-573-89975669, Email [email protected]
Purpose: ARFIP2, a canonical BAR domain-containing protein, is closely associated with regulating cargo exit from the Golgi. However, the potential biological functions of ARFIP2 in hepatocellular carcinoma (HCC) have not been well investigated. This study aimed to explore the critical role of ARFIP2 in HCC cells.
Methods: The expression of proteins related to epithelial to mesenchymal transition (EMT) and cell autophagy in HCC cells and tissues was assayed by quantitative real-time PCR, Western blotting, immunohistochemistry and immunofluorescence staining. The ability of cells to proliferate, migrate and invade was detected by Cell Counting Kit-8, Transwell migration and invasion assays. In addition, the function of ARFIP2 in vivo was assessed using a tumour xenograft model.
Results: ARFIP2 expression is significantly upregulated in early recurrent and metastatic HCC patients and was positively correlated with a poor prognosis. ARFIP2 overexpression promoted cell proliferation, migration, and invasion by inducing EMT and inhibiting autophagy in vitro. Furthermore, the regulatory effects of ARFIP2 on autophagy and EMT were partially attributed to its regulation of the PI3K/AKT signalling pathway. The in vivo results also showed that ARFIP2 modulates HCC progression.
Conclusion: Our results substantiate a novel mechanism by which ARFIP2 can regulate the activity/phosphorylation of Akt to promote EMT and inhibit autophagy in part via the PI3K/Akt signalling pathway. The ARFIP2/PI3K/Akt axis may be a potential diagnostic biomarker and therapeutic target for HCC.
Keywords: hepatocellular carcinoma, ARFIP2, PI3K/AKT, EMT, autophagy
Graphical Abstract:
Introduction
Hepatocellular carcinoma (HCC) is the most frequently occurring human digestive system neoplasm, which are characterized by a high degree of malignancy, a high rate of recurrence, difficult early diagnosis, and a poor prognosis.1 Currently, despite the significant progress in diagnosis and treatment in recent years, this cancer still remains an important barrier to increasing life expectancy across the world.1,2 According to GLOBOCAN 2020, HCC represents the 5th most common disease and ranks as the third-leading cause of cancer related deaths following lung and colorectal cancers, accounting for an estimated 80,000 and 70,000 related deaths annually worldwide.2,3 Abundant evidence clearly indicates that therapy is likely to be less extensive and more successful when HCC is detected at an early stage.4–6 Therefore, efforts should be made to explore more effective therapeutic targets and biomarkers for early and accurate diagnosis to prevent and control HCC.7 In our previous study, we applied a comparative proteomic approach to identify the proteome differences between HCC induced by hepatitis B virus (HBV) genotype B-versus genotype C. These efforts led to the identification of a BAR (Bin/Amphiphysin/Rvs) domain membrane-shaping protein, ADP ribosylation factor interacting protein 2 (ARFIP2, also known as POR1) as a potential diagnostic biomarker to distinguish HBV-related HCC among patients infected with different genotypes and a regulator of the PI3K/Akt signalling pathway.8 ARFIP2, which was originally detected as a binding partner for Rac1-interacting protein and GTP-bound ADP-ribosylation factors, plays a key role in intracellular transport and especially in endocytosis through the trans-Golgi network (TGN) by a PI(4)P-dependent reaction.9,10 It has been reported that ARFIP2 is regulated by Arl1 on Golgi membranes and that its overexpression enhances the formation of tubular structures emanating from the TGN.10,11 In addition, another study showed that ARFIP2 is a substrate of Akt kinase and is associated with neuroprotection by regulating aggregate formation of mutant huntingtin in Huntington disease.12 Recently, ARFIP2 was found to be a component of ATG9A-positive membranes and can serve as a molecular scaffold to regulate ATG9A vesicle formation, distribution and activation of binding partners of ATG9A, in particular PI4KIIIβ.13 Remarkably, although previous studies have shown that ARFIP2 is required for mediating multiple functions, such as the formation of tubular structures, the activity of Akt kinase, and the initiation of autophagy, whether these functions are also related to the regulation of the occurrence and progression of HCC remains unclear.11–13
The PI3K/Akt signalling pathway is one of the classic signalling pathways that mediates the cell cycle, and exerts a major effect on transcription, translation, proliferation, growth, and survival in response to extracellular signals through its numerous downstream targets.14 The abnormal activation of Akt, which acts as a downstream molecule of PI3K and belongs to the AGC family of protein kinases that phosphorylates several substrates and downstream effectors, is very frequently observed in the pathogenesis of many human cancers.12,14 A large number of studies from different groups have indicated that the PI3K/Akt signalling pathway can regulate multiple biological processes, such as the epithelial to mesenchymal transition (EMT) and autophagy, which play a critical function in tumour progression in several malignancies including HCC.15–19 EMT, a multistep biological process of cell transdifferentiation in which epithelial cancer cells are transcriptionally reprogrammed and acquire mesenchymal traits, has also been reported to modulate tumour progression in multiple types of cancers.20–22 For HCC progression, EMT has been considered to be a key program closely associated with cellular adhesion, cellular motility, and invasive properties.18,21
Autophagy, which is an evolutionarily conserved catabolic degradation process as well as a response to several types of stresses, involves aggregated proteins, damaged organelles, cytoplasmic macromolecules and invading pathogens through the lysosomal degradation pathway for differentiation, development, homeostasis, and survival.16,19,23 According to reports in the literature, malfunction of autophagy contributes to the pathophysiological onset and progression of many harmful diseases ranging from neurodegeneration, autoimmunity, ageing, heart disease, and infection to cancer, including HCC.23,24 During the development of HCC, autophagy plays dual roles as a tumour inhibitor in the dysplastic stage of initiation or as a tumour promotor in the subsequent progression stages.25–27 However, a large number of studies show that suppression of the PI3K/Akt signalling pathway plays a crucial role in the induction of autophagic progression, and then inhibits the migration and proliferation of HCC cells.19,26–28
To our knowledge, the role of ARFIP2 in HCC has not been reported previously. Therefore, the purpose of this study was to examine the potential function of ARFIP2 in HCC occurrence and progression. In addition, we further explored whether ARFIP2 affects EMT and autophagy in HCC through the PI3K/Akt signalling pathway. The results of these experiments may provide a potential diagnostic biomarker and treatment target for patients with HCC, and also provide novel insight into the role and molecular mechanism of ARFIP2 in HCC progression.
Materials and Methods
Clinical Samples
In our study, thirty-six fresh tumour tissues collected at the time of surgery from primary HCC were used to detect the mRNA and protein expression of ARFIP2 and other proteins. In addition, a total 86 formalin-fixed and paraffin-embedded HCC tissues from two cohorts of consecutive patients who underwent surgical resection were retrieved for immunohistochemical (IHC) staining. The retrospective clinical data and follow-up information were collected by reviewing patient medical records. According to a previous study, the time to recurrence/metastasis was defined as the interval from the date of primary surgery to the date when tumour recurrence was diagnosed.29 The project design and study protocols complied with the declaration of Helsinki were approved by the Medical Ethics Committee Shanghai Jiao Tong University School of Medicine and Affiliated Hospital of Jiaxing University (LS2020-016). Written informed consent was received from each participant in this study after being informed about the research purposes. The experiments were carried out in accordance with the relevant guidelines and regulations.
Cell Culture and Transfection
Human HCC cell lines (HepG2, Huh-7, SMMC-7721, and HCCLM3) were purchased from the Shanghai Institutes for Biological Sciences at the Chinese Academy of Science (Shanghai, China). Among them, HepG2 and HCC cell lines were used for in vitro experiments, while HCCLM3 cell line was used for the in vivo experiments. All of the cell lines were maintained in high-glucose DMEM supplemented with 10% heat-inactivated foetal bovine serum (FBS, Gibco, USA) under standard conditions at 37°C containing 5% CO2. For transient knockdown of human ARFIP2, a synthetic ARFIP2 siRNA (Supplementary Table S1) was purchased from GenePharma Co., Ltd. (Shanghai, China). To create the ARFIP2 expression construct, complementary DNA of the protein coding sequence region of full-length human ARFIP2 (GenBank Accession Number, BC000392) was generated by conventional PCR cloning techniques, and inserted into EcoRI/XbaI restriction sites of the lentiviral expression vector pLVX-puro. The empty vector was used as a negative control. All transfection procedures were performed using Lipofectamine 3000 (Invitrogen, CA, USA) according to the manufacturer’s instructions.
Quantitative Real-Time PCR (qPCR)
The expression of ARFIP2 RNA was analyzed by quantitative reverse transcription-PCR conducted using total RNA samples and gene-specific primers and SYBRGreen PCR Master Mix (Life Tech) on a Step One Plus Real-Time PCR system (Applied Biosystems). β-actin was used as an internal control. The sequences of the gene-specific primers used to detect the expression of these genes are described in Supplementary Table S2. After RT‒PCR, the relative abundance of each cellular target was determined by comparing the threshold cycle (Ct) values between each sample and the internal control, and the values were normalized to the geometric mean of the reference gene.
Western Blotting (WB) Analysis
Proteins expression was subjected to Western blot analysis as previously described.8,30 Briefly, the same amount of total denatured protein (30 µg) was fractionated on SDS‒PAGE via conventional procedures and transferred to NC membranes (Millipore, Bradford, MA) using BIO-RAD system. Afterwards, the membranes were blocked for 2 h in PBST buffer containing 5% BSA for nonspecific binding sites, and then probed with the corresponding primary antibodies overnight at 4°C. The primary antibodies used are listed in Supplementary Table S3. After three rinses with PBST buffer for 10 min each, the membranes were incubated with the appropriate HRP-labelled goat anti-rabbit or anti-mouse IgG secondary antibodies (1:5000 dilution, TransGen Biotech) for 1 h at room temperature. After washing the membranes four times again in TBST buffer, the protein expression levels were detected using enhanced chemiluminescence and visualized with the highly sensitive GeneGnome XRQ Chemical Imaging System (Gene Company, Hong Kong) by autoradiography.
Immunohistochemistry (IHC)
IHC was conducted according to the protocol. Briefly, the HCC tissue samples were fixed with formalin. After routine deparaffinization and dehydration, the sections were covered with peroxyacetic acid, sent to antigen retrieval, and then incubated with the primary anti-ARFIP2 polyclonal antibody (1:100 dilution, #14548-1-AP, Proteintech) at 4°C overnight, followed by submergence in the corresponding biotinylated secondary antibody at room temperature for another 1 h and visualization using DAB. The IHC intensity score was graded as follows: 0 (negative staining); 1 (minimal staining); 2 (moderate staining) and 3 (strong staining).
Cell Proliferation Assay
Cell proliferation viability assays were performed using Cell Counting Kit-8 (CCK-8). Briefly, cells (2 × 103/well) were seeded into 96-well plates. Then, 10 µL of CCK-8 solution was added and incubated for an additional 2 hours at 37°C. After that, the absorbance of each sample was detected at 450 nm.
To assess the colony formation potential, tumour cells (1 × 103/well) were inoculated into 6-well plates and incubated for 2 weeks. Colonies were methanol-fixed with 4% formaldehyde for 30 min and later stained using crystal violet (0.1%) for 5 min, and colonies with > 50 cells were counted.
Transwell Assay
Tumour cell invasion and migration analyses were performed using a transwell system (8-μm pore, BD Bioscience) precoated with or without Matrigel. HCC cells were resuspended in serum-free medium (200 μL) at a density of 1×105 per well and added to the top chamber precoated with 50 μL Matrigel. Medium (700 μL) with 10% FBS was added to the lower chamber. After further incubation, the cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet, and the number of cells in five different fields was photographed and counted.
Cell Apoptosis Analysis
Cell apoptosis was assessed by flow cytometry with an Annexin V and propidium iodide (PI) double-staining method, using a FITC Annexin V Apoptosis Detection Kit (BD Biosciences), according to the manufacturer’s protocol. Briefly, HCC cells were seeded in a six-well plate at a density of 3 × 105 cells/well, grown for 48 h, and washed, digested, centrifuged, and resuspended in 100 μL binding buffer containing 5 μL Annexin V-FITC and 5 μL PI. After staining for 15 min in the dark at room temperature, apoptosis was detected by flow cytometry and the apoptotic rate was assessed with BD FACS software.
Immunofluorescence (IF) Staining
Cells were cultured on appropriate coverslips in 24-well plates for 24 h, fixed with 4% formaldehyde for 15 min, blocked with 5% bovine serum albumin, and permeabilized with 0.5% Triton X-100 at room temperature for 10 min. After that, the corresponding diluted primary antibodies (Table S3) were added to the coverslips at 4 °C overnight. Incubation with the appropriate secondary antibody was carried out in a wet box at 37 °C for 1 h under dark conditions. The nuclei were stained with DAPI in the dark for 10 min, and slides were observed using a fluorescence microscope to capture images within 4 h.
Monodansylcadaverine (MDC) Staining
Autophagy Staining Assay Kit with MDC (Beyotime, Shanghai, China) was used to detect autophagic vacuoles. HCC cells were seeded in 24-well plates and probed with a 1:1000 dilution of MDC for 30 min at 37°C in the dark. Then, the cells were washed three times with assay buffer and immediately detected using fluorescence microscopy to evaluate the level of autophagy.
Vivo Tumorigenesis
All animal experiments were conducted with the approval of the Committee on the Ethics of Animal Experiments of Jiaxing University with the use of experimental animals in strict accordance with the Guide for Care and Use of Laboratory Animals (US National Institutes of Health). In our study, four week-old male BALB/c nude mice were provided by the Laboratory Animal Center, Jiaxing University (Jiaxing, China). For in vivo tumorigenesis observations, 12 BALB/c nude mice were randomized into two groups with six mice per group. Mice were injected with 2 × 106 stably-transfected SMMC-7721 pLVX-vector cells or 2×106 SMMC-7721 pLVX-ARFIP2 cells suspended in 200 μL PBS via the right flank of the nude mice to induce the formation of xenograft tumours. The tumour size was measured every 4 days using a Vernier calliper. After 26 days, the mice were killed, and the tumours were then excised for further analysis.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 9 (GraphPad Software, San Diego, California, USA), and the values are presented as the mean ± SD. Student’s t-test was used for comparisons between two groups. p < 0.05 was assumed to indicate a statistically significant difference.
Results
High ARFIP2 Expression in HCC Tissues is Correlated with Early Recurrence/Metastasis
In our previous study, we found that ARFIP2 was downregulated in the surrounding noncancerous tissues of HBV genotype B induced HCC, and ARFIP2 could be a potential biomarker and therapeutic target for HBV genotypes B and C-induced HCC (Supplementary Figure S1A and B). To explore whether ARFIP2 is an important factor in determining the clinical outcomes of HCC patients, we assessed its expression in three HCC patient groups with different recurrence/metastasis times by qPCR. As shown Figure 1A, qPCR analysis revealed that ARFIP2 was upregulated 3.83-fold in the early recurrence/metastasis group (R/M≤12 months, n = 12, P = 0.003) and 2.18-fold in the late recurrence and metastasis group (R/M12–24 months, n = 12, P = 0.034) compared with the NR/M group (n = 12). To validate this at the protein level, additional Western blot analysis was carried out. As shown in Figure 1B, ARFIP2 levels were significantly lower in the NR/M group than in both the R/M≤12 months group and the R/M12–24 months group. To further investigate whether ARFIP2 plays an important role in predicting the clinical progression of HCC, the expression of ARFIP2 in 86 paraffin-embedded HCC specimens was detected through IHC analysis and assessed for its correlation with the clinicopathological features of HCC patients (Table 1). Among these 102 HCC samples, 52 had high ARFIP2 expression, and 34 had low ARFIP2 expression (representative images shown in Figure 1C). Higher expression of ARFIP2 was positively associated with tumour aggressive phenotypes, including multiple tumour number (P = 0.031), microvascular invasion (P = 0.008), poor tumour differentiation (P = 0.036), and advanced TNM stage (P = 0.027), but was not significantly associated with age, AFP, or tumour size. Overall, these results clearly indicated that ARFIP2 expression is significantly increased at both the mRNA and protein levels in recurrent/metastatic HCC tumours, especially in HCC tumours with early recurrence/metastasis.
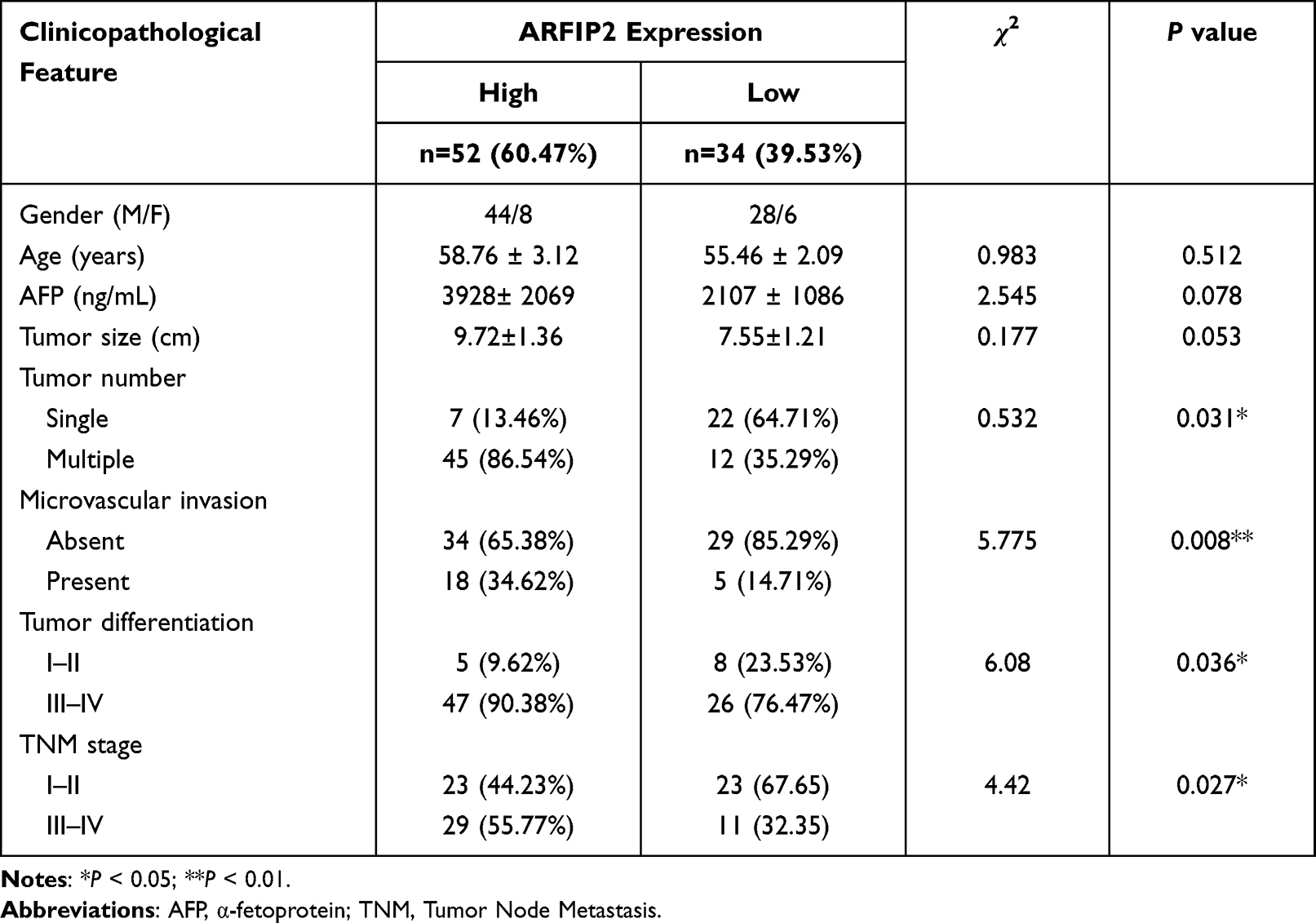 |
Table 1 Correlations Between ARFIP2 Expression and Clinicopathological Characteristics in 87 HCC Patients |
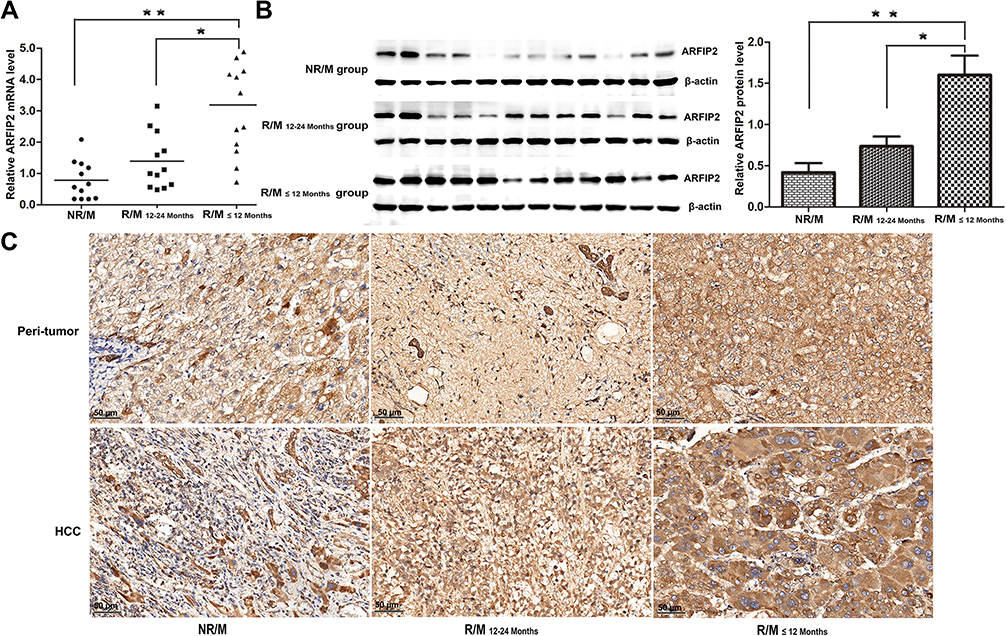 |
Figure 1 High ARFIP2 expression was significantly associated with early recurrence/metastasis in HCC. (A) qPCR analysis of ARFIP2 expression in three HCC patient groups with different recurrence/metastasis times. (B) WB analysis of ARFIP2 expression in three HCC patient groups with different recurrence/metastasis times. (C) Representative image of ARFIP2 immunohistochemical staining in HCC tumour tissues and peritumor tissues. *P < 0.05, **P < 0.01; NR/M, patients with no r recurrence/metastasis within 24 months; R/M12–24 months, patients with recurrence/metastasis within 12–24 months; R /M≤12 months, patients with recurrence/metastasis within 12 months. |
Low ARFIP2 Expression Can Inhibit Cell Proliferation, Invasion and Migration in vitro
As cancer cell proliferation, invasion and migration are tightly associated with the recurrence, metastasis, differentiation, and microvascular invasion of tumours, we further assessed whether the expression of ARFIP2 could affect HCC cell proliferation, invasion and migration in vitro. Using qPCR and WB, we examined the expression of ARFIP2 in four HCC cell lines (HepG2, Huh7, HCCLM3 and SMMC-7721) for this purpose. The results indicated that the protein and mRNA levels of ARFIP2 were higher in HepG2 cells than in the three other HCC cell lines (Figure 2A); therefore, this cell line was used to assess the effects of ARFIP2 on the proliferation, invasion and migration of HCC cells in this study. Since HepG2 cells were selected for in vitro studies, we transfected HepG2 cells with ARFIP2 siRNA by siRNA-mediated loss-of-function analysis, which resulted in over 70% inhibition of ARFIP2 by HCC cells, suggesting effective knockdown by ARFIP2 siRNA (Figure 2B). Cell colony formation assays and CCK8 assays were performed to examine the proliferation of HepG2 cells (Figure 2C). Both experiments demonstrated that low expression of ARFIP2 led to an increase in the proliferation of HepG2 cells. Next, we carried out Transwell assays to analyse the invasion and migration of HCC cells after ARFIP2 was knocked down and to further examine the role of ARFIP2 in tumour invasion and migration. In this study, we proved that low expression of ARFIP2 could significantly inhibit HCC cell invasion (P < 0.01, Figure 2D) and migration (P < 0.01, Figure 2E) through Matrigel. These results are consistent with the clinical results.
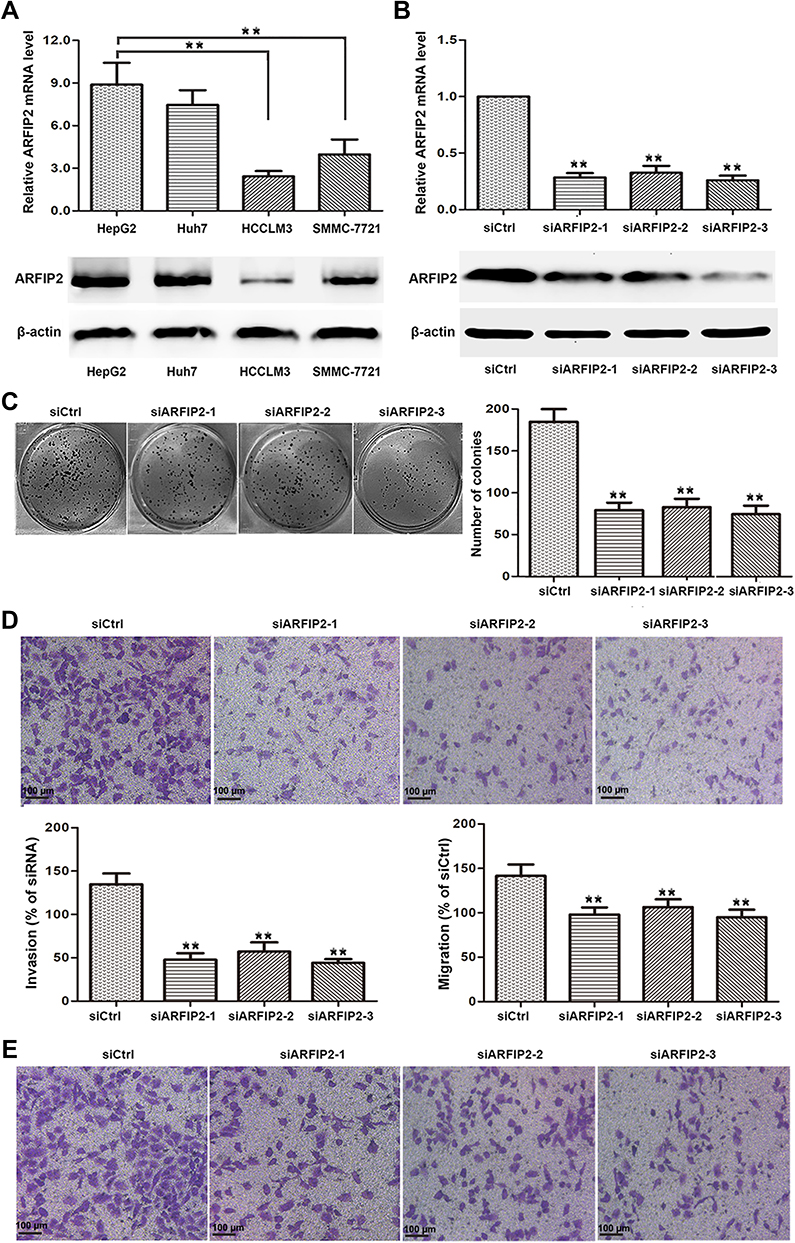 |
Figure 2 ARFIP2 deficiency inhibits the proliferation, invasion and migration of HCC cells. (A) qPCR and WB analyses were used to detect the expression of ARFIP2 in four HCC cell lines. (B) qPCR and WB analysis of ARFIP2 expression in HepG2 cells transfected with siARFIP2 #1, #2, or #3 and siCtrl. (C) A colony formation assay was performed to assess the proliferation ability of HepG2 cells following transfection with control siRNA or ARFIP2 siRNA. (D) Transwell assays were used to detect the potential effect of ARFIP2 on invasion. (E) Transwell assays were used to detect the potential effect of ARFIP2 on migration. **P < 0.01. |
ARFIP2 Regulates of the Akt Signalling Pathway
Previously, it has been reported that ARFIP2 is involved in the activity of Akt kinase and may regulate the PI3K/Akt signalling pathway.8,12 In line with the results of our previous experiment, ARFIP2 is one of the differentially expressed proteins related to the tumorigenesis and development of HBV genotypes B- and C-induced HCC, suggesting that ARFIP2 can regulate the activity of PI3K/Akt to modulate HCC occurrence and progression. To corroborate this finding, we established stable overexpression cell lines via lentiviral infection of HepG2 and Huh7 cells with control and pLVX-ARFIP2 vector, as these cells exhibited relatively low endogenous ARFIP2 levels, and determined whether the observed effect of ARFIP2 on HCC progression is Akt-dependent. As demonstrated in Figure 3A, the expression levels of P62 and phosphorylated Akt (p-Akt) increased markedly after ARFIP2 was overexpressed, while no significant change in Akt protein expression was observed. Since ARFIP2 can regulate the activity/phosphorylation of Akt and then affect the PI3K/Akt signalling pathway, which plays a crucial role in affecting autophagy and EMT, we further explored the relationship between ARFIP2 and autophagy and EMT-related protein expression in HCC cells. Western blot assays showed that the expression levels of Beclin1 and LC3B decreased most markedly after ARFIP2 was overexpressed, while N-cadherin protein expression was significantly increased (Figure 3A). To determine where ARFIP2 regulates p-Akt activity, we ectopically expressed individual components of the PI3K/Akt signalling pathway and assessed how the induction of p-Akt activity was altered by overexpression of ARFIP2. Compared with control HepG2 or Huh7 cells, cells with stable ARFIP2 overexpression exhibited substantially paired p-Akt expression following treatment with the PI3K inhibitor LY294002 or the PI3K activator 740 Y-P (Figure 3B). In contrast, p-PI3K expression driven by LY294002 or 740 Y-P was not affected, suggesting that ARFIP2 acts downstream of the Akt kinase and proximal to Akt.
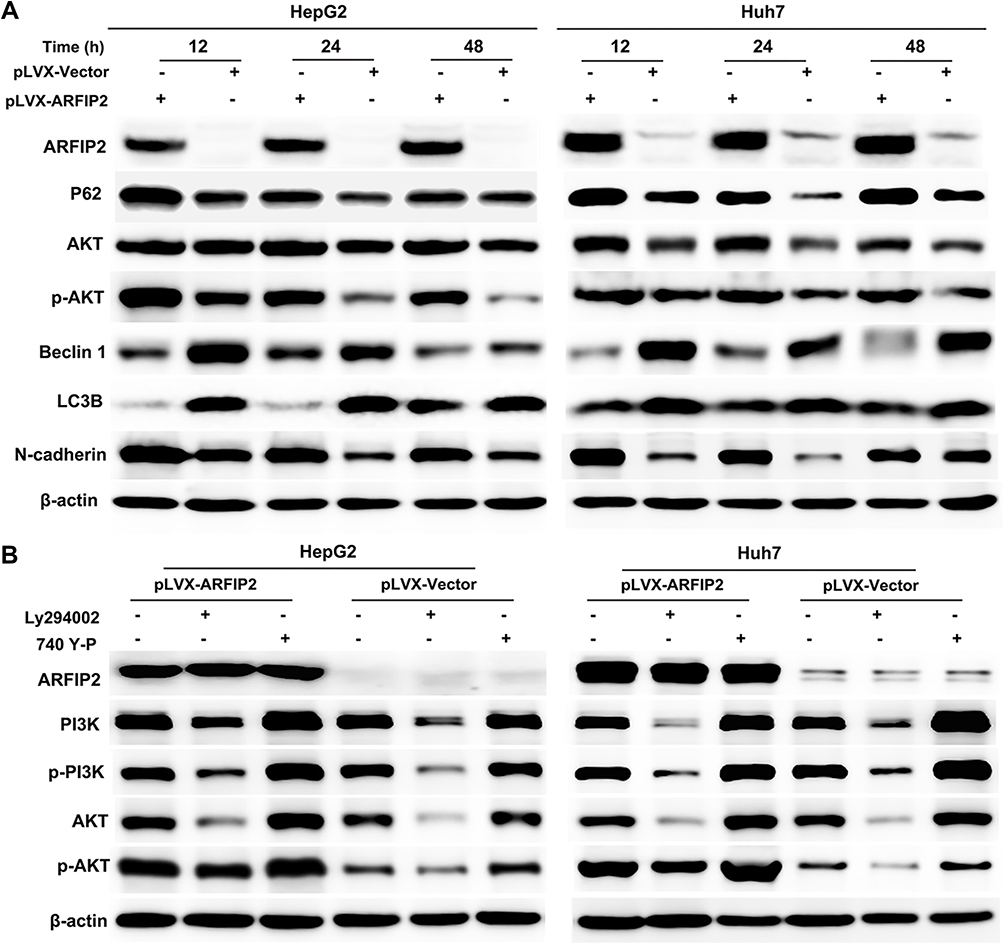 |
Figure 3 The effects of ARFIP2 on proliferation, invasion and migration are partially dependent upon the PI3K/Akt signalling pathway in HCC cells. (A) Levels of P62, AKT, p-AKT, Beclin 1, LC3B, and N-cadherin proteins were assessed in HepG2 and Huh7 cells stably-transfected with ARFIP2. (B) Effects of ARFIP2 on the PI3K/AKT signalling pathway after treatment with 20 µM LY294002 and 20 µM 740 Y-P. |
ARFIP2 Inhibits Apoptosis in HCC Cells
It is well known that the Akt signalling pathway is constitutively active in HCC and plays an important role in mediating HCC cell apoptosis, EMT and autophagy.14,15 We therefore hypothesized that ARFIP2 might contribute to mediating HCC cell apoptosis through the Akt signalling pathway. To determine whether ARFIP2-induced promotion of the Akt signalling pathway is related to apoptosis, we used flow cytometry with annexin-V and PI staining to determine the number of cells undergoing apoptosis. Compared to the control vector, pLVX-ARFIP2 strongly suppressed apoptosis in HepG2 and Huh7 cells (P < 0.01, Figure 4A). Correspondingly, we also investigated the induction of apoptotic marker expression with ARFIP2 overexpression by WB and qPCR analysis. As shown in Figure 4B and C, ARFIP2 transfection induced the activation of apoptosis markers, such as cleaved caspase-3, Bax and cleaved PARP and it also reduced Bcl-2 levels in HepG2 and Huh7 cells. These results clearly proved that ARFIP2 transfection significantly suppressed apoptosis in HCC cells compared with the control cells.
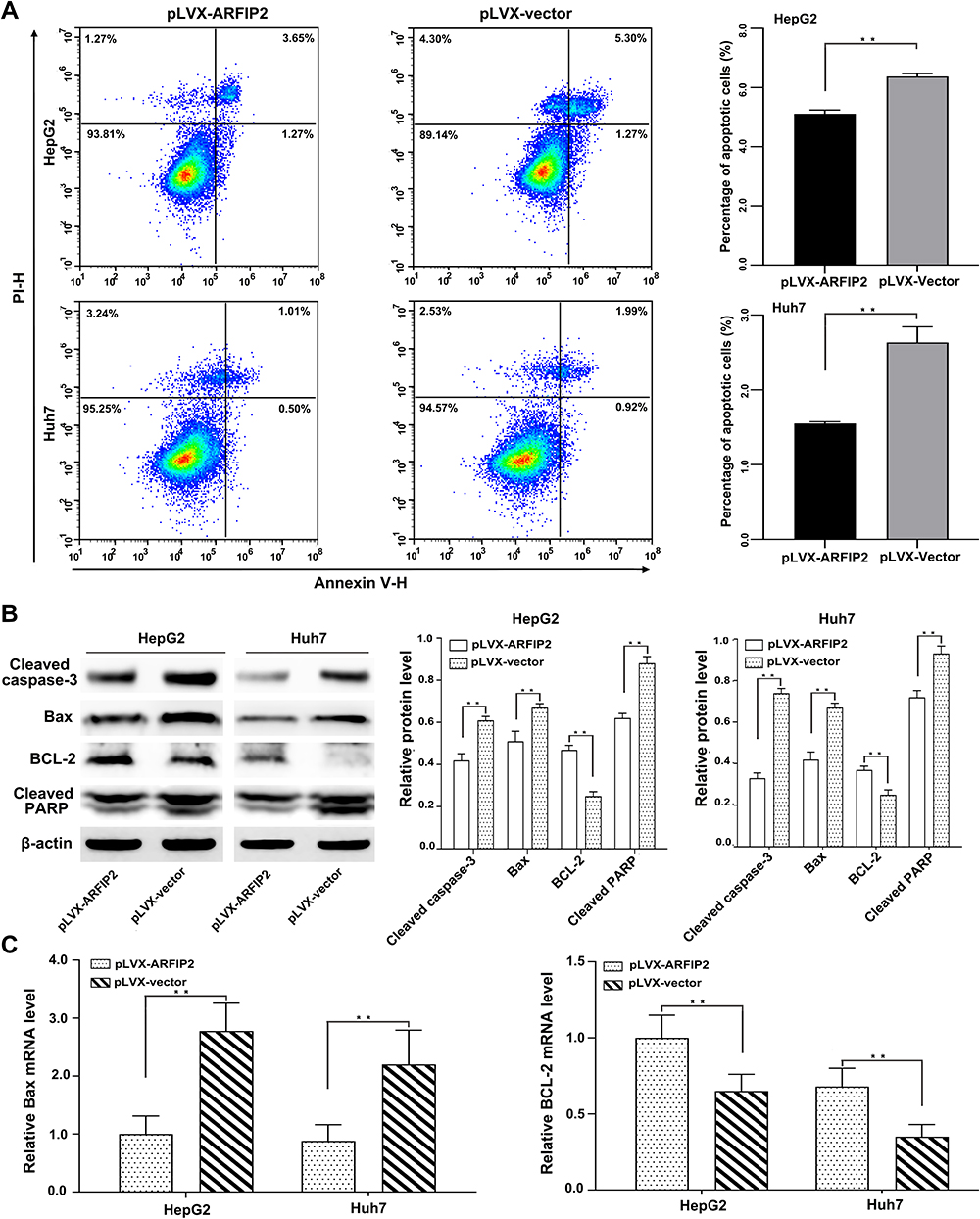 |
Figure 4 ARFIP2 supresses apoptosis in HCC cells. (A) Flow cytometry analysis of apoptosis induced by ARFIP2. (B) Effects of ARFIP2 on the protein expression of apoptosis-related proteins in HepG2 and Huh7 cells. (C) Effects of ARFIP2 on the mRNA expression of apoptosis-related proteins in HepG2 and Huh7 cells. **P < 0.01. |
ARFIP2 Repressed EMT in HCC Cells
To elucidate the potential molecular mechanisms underlying ARFIP2 in repressing HCC recurrence/metastasis, we assessed whether EMT markers were regulated. E-cadherin, vimentin and N-cadherin are classical EMT markers, and the IF results demonstrated that overexpression of ARFIP2 decreased E-cadherin expression levels and increased vimentin and N-cadherin expression levels in HepG2 and Huh7 cells compared with the levels in the corresponding control cells (Figure 5A). We then performed WB to confirm that ARFIP2 could downregulate the protein expression of E-cadherin and promote that of vimentin and N-cadherin, as well as that of β-catenin, a component of cell–cell adhesion machinery (Figure 5B). Therefore, these data suggested that ARFIP2 might be an inducer of EMT in HCC cells.
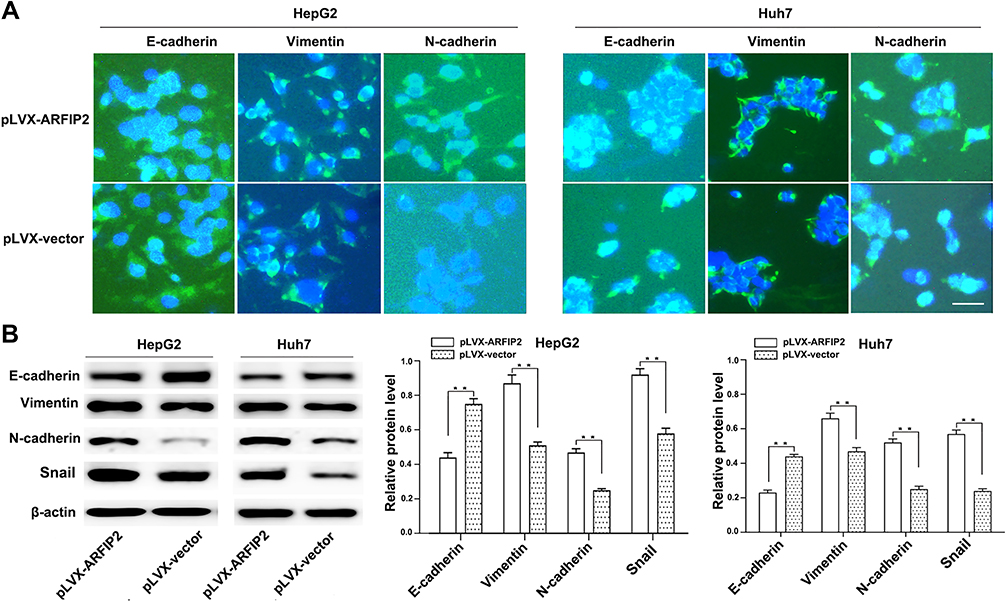 |
Figure 5 ARFIP2 can regulate EMT in HCC cells. (A) Representative immunofluorescence images showing the expression of EMT marker proteins in ARFIP2-overexpressing HepG2 and Huh7 cells. Blue, DAPI. Scale bar, 20 µm. (B) WB analysis for detection of the indicated proteins in ARFIP2-overexpressing HepG2 and Huh7 cells. **P < 0.01. |
ARFIP2 Induces Autophagy in HCC Cells
Based on the above-described results, we also hypothesized that ARFIP2 played an important role in autophagy by affecting the tumour characteristics of HCC cells and that this function partially relies on the Akt signalling pathway. Therefore, we detected the changes in the activity of autophagy by visualizing the fluorescence of MDC, which has been reported to be a specific in vivo marker for autophagic vacuoles. To explore the relationship between ARFIP2 and autophagy in HCC cells that were stably-transfected with pLVX-ARFIP2, autophagosomes in cells with overexpressing ARFIP2 and in control cells were observed by MDC staining. As shown in Figure 6A, MDC accumulated in autophagic vacuoles in the corresponding control cells, which exhibited distinct dot-like structures distributed within the cytoplasm or localized in the perinuclear regions, compared with the levels in the ARFIP2-overexpressing cells. This observation was further supported by decreased levels of autophagy-related protein expression, compared with the control group. As expected, the expression levels of ATG7, Beclin1 and LC3B decreased markedly after ARFIP2 was overexpressed, while P62 protein expression was significantly increased (Figure 6B and C). The above results suggest that the overexpression of ARFIP2 can significantly decrease autophagy in HCC cells.
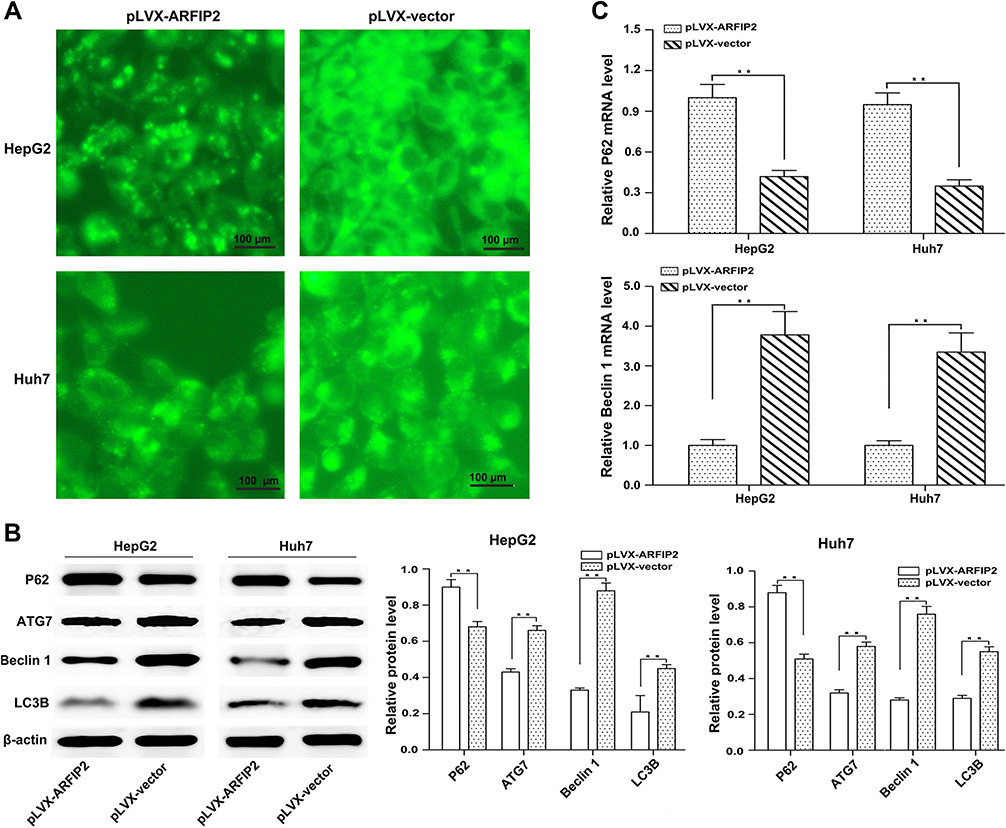 |
Figure 6 ARFIP2 can inhibit autophagy in HCC cells. (A) Autophagic vacuoles in cells overexpressing ARFIP2 were observed by MDC staining. (B) WB analysis was used to analyze the protein expression of autophagy-related markers. (C) qPCR analyse was used to analyze the mRNA expression of autophagy-related markers. **P < 0.01. |
ARFIP2 Modulates HCC Tumorigenesis in Mice
In the above in vitro experiments, our results showed that ARFIP2 induced HCC cell proliferation, inhibited apoptosis, promoted EMT, and repressed autophagy partly through modulation of the AKT signalling pathway. Thus, we constructed stably transfected SMMC-7721 cells with pLVX-ARFIP2 and subcutaneously injected them into the left flanks of nude mice to validate the function of endogenous ARFIP2 in vivo. Specifically, tumour xenograft mouse models were successfully obtained with ARFIP2-overexpressing SMMC-7721 cells. As shown in Figure 7A and B, overexpression of ARFIP2 promoted the proliferation of SMMC-7721 cells in this mouse tumour model and induced the expression of Ki67 protein in vivo compared to that in the control groups. WB analysis showed that the xenografts derived from ARFIP2-overexpressing cells exhibited decreased levels of Beclin 1 and LC3B and increased levels of P62, p-AKT and N-cadherin, but did not show significant in AKT levels (Figure 7C). In conclusion, these results are consistent with the finding that overexpression of ARFIP2 suppresses cell proliferation in vitro, suggesting that ARFIP2 promotes HCC progression in vivo and in vitro.
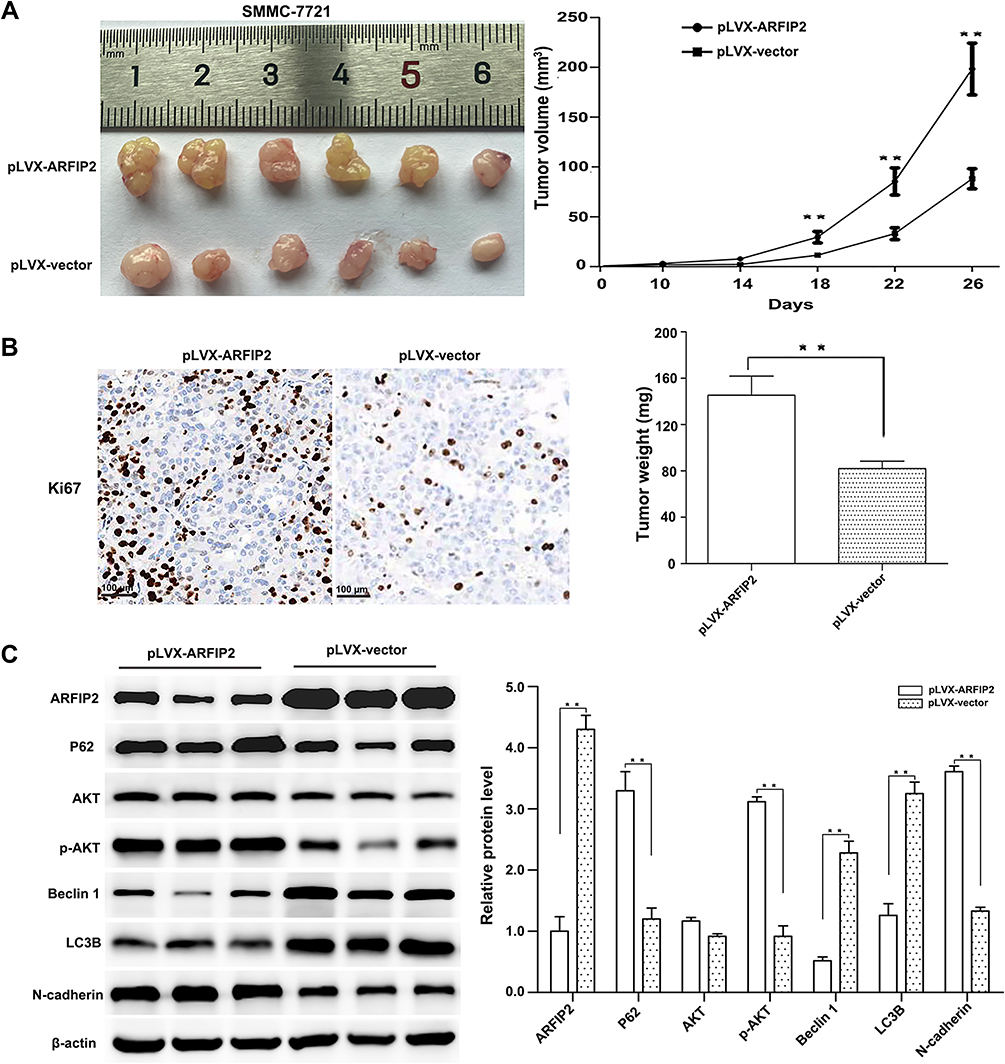 |
Figure 7 The effect of ARFIP2 overexpression on tumorigenesis in vivo. (A) Photographs of tumours excised from mice injected with control cells or cells overexpressing ARFIP2 (n = 6 per group). After 26 days, the mice were sacrificed, and xenograft size and weight were measured. (B) Levels of Ki167 protein in tumours from nude mice. (C) WB analysis of the expression of the relative proteins from nude mice. **P < 0.01. |
Discussion
Despite significant advances in therapy that have been applied to prolong the survival rate, HCC continues to be a major aggressive malignancy characterized by rapid intrahepatic recurrence and extrahepatic metastasis.1,3,31 As such, there is an urgent need to screen the key indicators and understand the molecular mechanisms underlying the recurrence and metastasis of HCC. Using our own patient cohort, we first showed that the promotion of ARFIP2 in HCC patients was closely associated with aggressive tumour features, such as the presence of multiple tumour numbers, microvascular invasion, tumour differentiation and advanced TNM stage (Table 1). Consistently, the ARFIP2 expression level is an independent predictor of the recurrence and metastasis of HCC; HCC patients with higher ARFIP2 expression exhibited earlier recurrence and metastasis than those with lower ARFIP2 expression (Figure 1). On the other hand, knockdown of ARFIP2 remarkably repressed the proliferation, invasion, and migration of HCC cells while promoting apoptosis in HCC cells (Figures 2 and 4). These observations suggest that ARFIP2 plays a crucial role in blocking HCC progression, particularly by preventing HCC recurrence and metastasis. However, the definite molecular mechanisms of ARFIP2 in HCC progression need to be elucidated. This study, for the first time, provides reliable evidence to verify that ARFIP2 is linked to HCC progression and can be used as an independent prognostic indicator of HCC recurrence and metastasis.
There is strong evidence that the PI3K/AKT signalling pathway plays an essential role in the pathogenesis of many human cancers, and as such, it is being vigorously pursued as a potential therapeutic target.32 AKT, a downstream PI3K effector, has been implicated in the regulation of the cell cycle, cell proliferation, apoptosis, metabolism, and angiogenesis by communicating with its related downstream molecules and is dysregulated in many cancer types, including HCC.14 Moreover, AKT has been reported to interact with ARFIP2 through bioinformatics and protein‒protein interaction analyses.8,12 However, studies on the cause-and-effect nature of the association between ARFIP2 and AKT in HCC pathogenesis are currently lacking. To further explore the relationship between ARFIP2 and Akt in HCC cell lines, we transfected HepG2 cells with pLVX-ARFIP2 and found that the levels of PI3K and total Akt were not significantly changed, but the level of p-Akt was induced, and was positively correlated with ARFIP2 level (Figure 3A). In addition, to obtain a better understanding of this molecular mechanism and to determine whether PI3K/AKT participates in ARFIP2-regulated HCC tumorigenesis and progression, we used the specific PI3K inhibitor LY294002 or the specific PI3K activator 740Y-P to treat ARFIP2 cells and then detected PI3K, p- PI3K, AKT and p-AKT expression by WB (Figure 3B). Interestingly, the effects of activation of p-Akt induced by ARFIP2 overexpression in HCC cells were not counteracted by LY294002 or 740Y-P. These findings strongly suggest that the promotory effect of ARFIP2 on HCC progression is partly mediated by the regulation of the Akt signalling pathway. However, it remains to be determined how ARFIP2 inhibits the activity of p-Akt.
The Akt signalling pathway was revealed to modulate the process of EMT and autophagy, and has attracted widespread attention as a potential target for the prevention and treatment of different cancer types.33,34 EMT is also widely considered a reversible physiological process for oncogenesis that mediates the aggressive features in the invasion and migration of neoplastic malignancies.35 This process can be assessed based on the expression levels of several additional EMT-associated molecular markers, which are independent of cancer type and site.34,36 Here, we demonstrated that low levels of ARFIP2 inhibit the invasion and migration of HepG2 cells, which suggests that ARFIP2 may promote EMT (Figure 2D and E). In contrast, ARFIP2 expression in HepG2 and Huh7 cells induced the EMT process, which was characterized by upregulation of the most important epithelial marker E-cadherin, and downregulation of mesenchymal markers, such as vimentin and N-cadherin (Figure 5). Accordingly, ARFIP2 overexpression modulated the expression of the associated EMT molecular biomarkers and promoted EMT, suggesting a potential role of ARFIP2 in HCC cells.
As mentioned above, we speculated that ARFIP2 suppresses the autophagy process through the Akt signalling pathway to suppress HCC cell proliferation and growth based on previous reports. To verify this hypothesis, we first assessed the protein expression of the autophagy-related markers p62, ATG7, Beclin1 and LC3B and found that ATG7, Beclin1 and LC3B levels were decreased while p62 levels were increased in both ARFIP2-overexpressing stable cell lines, which were established by transducing lentivirus into HepG2 and Huh7 cells (Figure 6). In this study, we confirmed that ARFIP2 overexpression resulted in significant increase in Akt phosphorylation levels and a decrease in Beclin1 and LC3B protein expression levels, suggesting that ARFIP2 is involved in Beclin1- and LC3B-mediated autophagy in HCC cells through the Akt signalling pathway. In addition, autophagy, as a type II programmed cell death, plays important roles with autophagy-related (ATG) proteins in tumorigenesis. The autophagic response is mediated by a complex network of proteins and lipids, most of from which are members of the ATG protein family. Among those, ATG9A is particularly interesting since it is the only transmembrane ATG protein required for autophagosome formation.37,38 A recent study revealed that ARFIP2 coupled with ATG9A promoted autophagy by positively regulating ATG9A exit from the Golgi complex, thereby incorporating and delivering PI4KIIIβ and PI4P to the autophagosome initiation site in HEK293A cells.13 Inconsistent with these finding, we found that the overexpression of ARFIP2 expression in HCC cells could not increase the levels of LC3B and decrease P62 accumulation, as demonstrated by our IHC and WB results, indicating that ARFIP2 may not be the direct factor which promotes autophagy in HCC by upregulating ATG9A. However, it remains to be determined how ARFIP2 mediates autophagy.
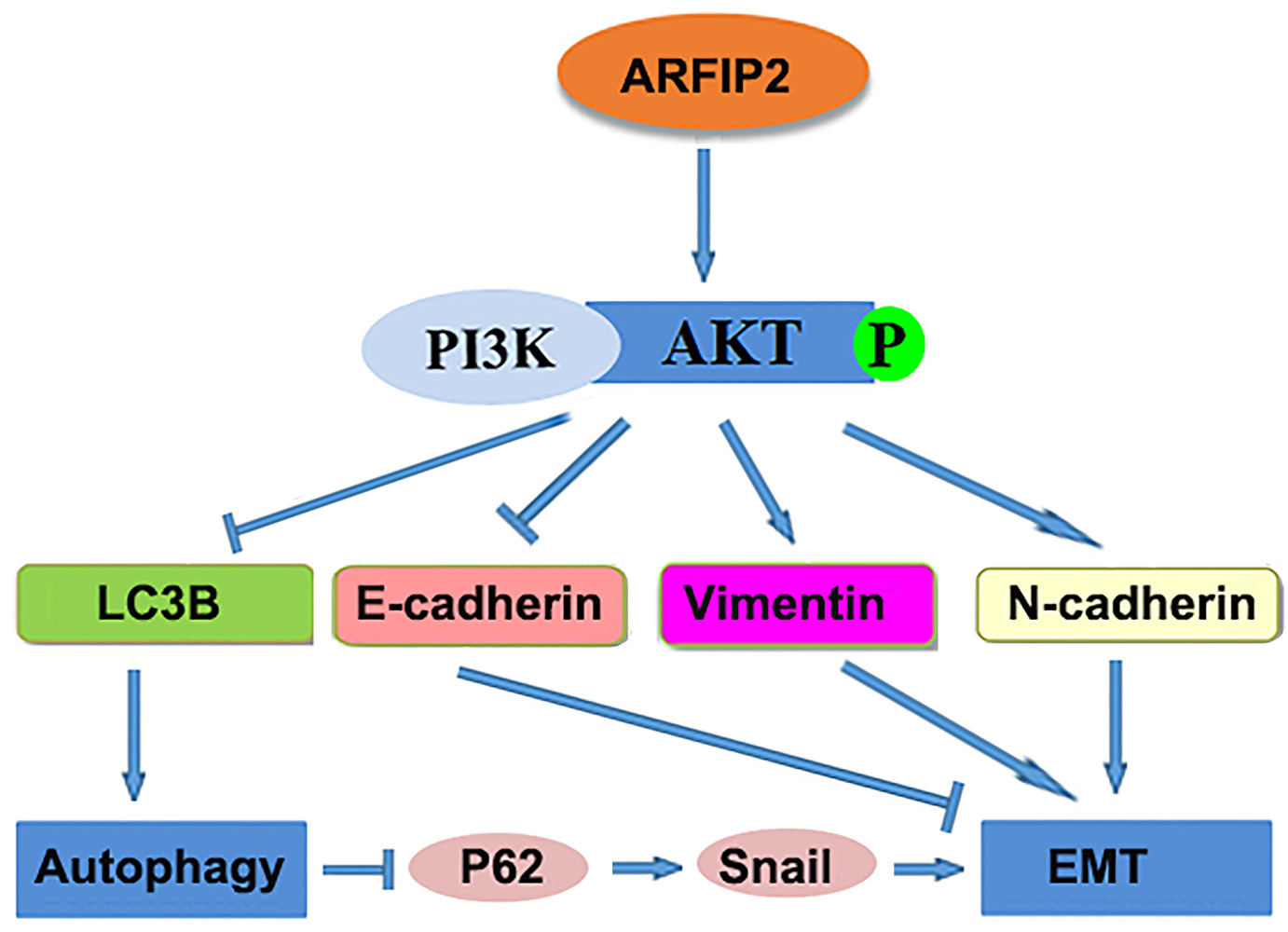
As stated above, autophagy and EMT are the two principal biological processes in the occurrence and progression of cancer. According to previous studies, there is a complex relationship between these two important processes.39,40 On the one hand, EMT is critical for cancer cell invasion and migration by inhibiting the autophagy process. On the other hand, there is increasing evidence indicating that EMT reverses autophagy in cancer cells.41 In our studies, we observed that suppression of autophagy results in P62 induction; P62 stabilizes Snail, so this increase in P62 expression leads to decreased and increased degradation of Snail, which leads to a increased EMT and is consistent with previous reports.18,42 Based on our findings, we propose a schematic model showing how ARFIP2-inhibited autophagy promote EMT in HCC cells (Figure 8). In this model, ARFIP2 promotes the Akt signalling pathway, leading to autophagy inhibition and EMT activation, and then induces HCC cell invasion, migration and metastasis.
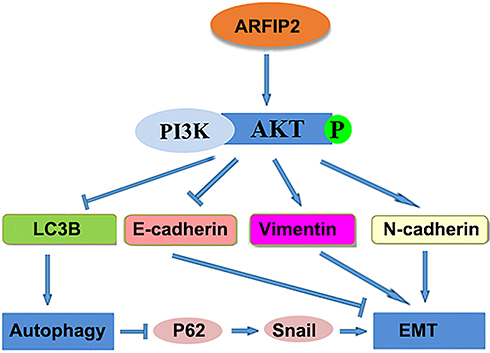 |
Figure 8 A proposed model of the mechanism by which ARFIP2 modulates the PI3K/Akt signalling pathway to regulate EMT and autophagy in HCC cells. |
The present study is a preliminary investigation of the role of ARFIP2 and there were also certain limitations. We have preliminarily studied and proved the effects of ARFIP2 through PI3K/ Akt signalling pathway in HCC progression, but we have not explored it very deeply, especially the relationship between ARFIP2 and the activity of p-Akt. Most important, our study did not clarify the underlying mechanism in detail how ARFIP2 inhibited the activity of p-Akt in HCC cells. Besides, only the expression levels of PI3K/Akt signalling pathway were detected, which was not conclusive. So, it is necessary to determine whether ARFIP2 modulated EMT and autophagy via other signaling pathways.
Conclusions
In summary, this study clearly shows that ARFIP2 is a tumour inducer in HCC and that high ARFIP2 expression is associated with more aggressive tumour characteristics and poorer patient prognosis. Moreover, ARFIP2 silencing inhibits HCC cell proliferation, invasion and migration, whereas the overexpression of ARFIP2 induces EMT and suppresses apoptosis and autophagy partly via the Akt signalling pathway. Furthermore, the stimulatory effects of ARFIP2 on EMT occur through the downregulation of E-cadherin and the upregulation of vimentin, Snail and N-cadherin and through the effects of ARFIP2 on autophagy, as evidenced by the detected changes in the expression levels of LC3B and P62. Therefore, we conclude that ARFP2 regulates the activity/phosphorylation of AKT to promote EMT and inhibit autophagy, and these effects are partly achieved through the Akt signalling pathway. Thus, as expected, our work identified a new role for ARFIP2 in HCC progression and revealed the underlying molecular mechanisms; importantly, this study provides a basis for the future development of ARFIP2-targeting therapeutic strategies in HCC.
Abbreviations
HCC, hepatocellular carcinoma; HBV, hepatitis B virus; BAR, Bin/Amphiphysin/Rvs; ARFIP2, ADP ribosylation factor interacting protein 2; TGN, trans-Golgi network; EMT, epithelial to mesenchymal transition; IHC, immunohistochemical; qPCR, quantitative real-time PCR; WB, Western blotting; IF, Immunofluorescence; MDC, Monodansylcadaverine; ATG, autophagy-related.
Ethics Approval and Consent to Participate
The project was approved by the Medical Ethics Committee Shanghai Jiao Tong University School of Medicine and Affiliated Hospital of Jiaxing University and informed consents were received from all participants in this study.
Consent for Publication
All authors have seen and approved the manuscripts and consent for publication.
Funding
This work was supported by Zhejiang Provincial Natural Science Foundation of China under Grant No. LY20C010004, the Science and Technology Program of Haicang District of Xiamen city under Grant No.350205Z20184003, and the Construction Project of Key Laboratory of Infectious Disease Involved in Viruses in Jiaxing City.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3
2. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
3. Jindal A, Thadi A, Shailubhai K. Hepatocellular Carcinoma: etiology and current and future drugs. J Clin Exp Hepatol. 2019;9(2):221–232. doi:10.1016/j.jceh.2019.01.004
4. Kanwal F, Singal AG. Surveillance for hepatocellular carcinoma: current best practice and future direction. Gastroenterology. 2019;157(1):54–64. doi:10.1053/j.gastro.2019.02.049
5. Tapper EB, Parikh ND. Mortality due to cirrhosis and liver cancer in the United States, 1999–2016: observational study. BMJ. 2018;362:k2817. doi:10.1136/bmj.k2817
6. Tsuchiya N, Sawada Y, Endo I, et al. Biomarkers for the early diagnosis of hepatocellular carcinoma. World J Gastroenterol. 2015;21(37):10573–10583. doi:10.3748/wjg.v21.i37.10573
7. Nault JC, Villanueva A. Biomarkers for hepatobiliary cancers. Hepatology. 2021;73(Suppl 1):115–127. doi:10.1002/hep.31175
8. Wei D, Zeng Y, Xing X, et al. Proteome differences between hepatitis B virus genotype-B- and genotype-C-induced hepatocellular carcinoma revealed by iTRAQ-Based quantitative proteomics. J Proteome Res. 2016;15(2):487–498. doi:10.1021/acs.jproteome.5b00838
9. Cruz-Garcia D, Ortega-Bellido M, Scarpa M, et al. Recruitment of arfaptins to the trans-Golgi network by PI(4)P and their involvement in cargo export. EMBO J. 2013;32(12):1717–1729. doi:10.1038/emboj.2013.116
10. Ambroggio EE, Sillibourne J, Antonny B, et al. Arf1 and membrane curvature cooperate to recruit Arfaptin2 to liposomes. PLoS One. 2013;8(4):e62963. doi:10.1371/journal.pone.0062963
11. Nakamura K, Man Z, Xie Y, et al. Structural basis for membrane binding specificity of the Bin/Amphiphysin/Rvs (BAR) domain of Arfaptin-2 determined by Arl1 GTPase. J Biol Chem. 2012;287(30):25478–25489. doi:10.1074/jbc.M112.365783
12. Rangone H, Pardo R, Colin E, et al. Phosphorylation of arfaptin 2 at Ser260 by Akt inhibits PolyQ-huntingtin-induced toxicity by rescuing proteasome impairment. J Biol Chem. 2005;280(23):22021–22028. doi:10.1074/jbc.M407528200
13. Judith D, Jefferies HBJ, Boeing S, et al. ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIβ. J Cell Biol. 2019;218(5):1634–1652. doi:10.1083/jcb.201901115
14. Revathidevi S, Munirajan AK. Akt in cancer: mediator and more. Semin Cancer Biol. 2019;59:80–91. doi:10.1016/j.semcancer.2019.06.002
15. Karimi Roshan M, Soltani A, Soleimani A, et al. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie. 2019;165:229–234. doi:10.1016/j.biochi.2019.08.003
16. Zhang L, Zhang Y, Shen D, et al. RNA binding motif protein 3 promotes cell metastasis and epithelial-mesenchymal transition through STAT3 signaling pathway in hepatocellular carcinoma. J Hepatocell Carcinoma. 2022;9:405–422. doi:10.2147/JHC.S351886
17. Ma Z, Lou S, Jiang Z. PHLDA2 regulates EMT and autophagy in colorectal cancer via the PI3K/AKT signaling pathway. Aging. 2020;12(9):7985–8000. doi:10.18632/aging.103117
18. Dian MJ, Li J, Zhang XL, et al. MST4 negatively regulates the EMT, invasion and metastasis of HCC cells by inactivating PI3K/AKT/Snail1 axis. J Cancer. 2021;12(15):4463–4477. doi:10.7150/jca.60008
19. Zhang M, Liu S, Chua MS, et al. SOCS5 inhibition induces autophagy to impair metastasis in hepatocellular carcinoma cells via the PI3K/Akt/mTOR pathway. Cell Death Dis. 2019;10(8):612. doi:10.1038/s41419-019-1856-y
20. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. doi:10.1038/nrm3758
21. Giannelli G, Koudelkova P, Dituri F, et al. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65(4):798–808. doi:10.1016/j.jhep.2016.05.007
22. Jia Y, Feng Q, Tang B, et al. Decorin suppresses invasion and EMT phenotype of glioma by inducing autophagy via c-Met/Akt/mTOR Axis. Front Oncol. 2021;11:659353. doi:10.3389/fonc.2021.659353
23. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–542. doi:10.1038/nrc.2017.53
24. Singh SS, Vats S, Chia AY, et al. Dual role of autophagy in hallmarks of cancer. Oncogene. 2018;37(9):1142–1158. doi:10.1038/s41388-017-0046-6
25. Ascenzi F, De Vitis C, Maugeri-Saccà M, et al. SCD1, autophagy and cancer: implications for therapy. J Exp Clin Cancer Res. 2021;40(1):265. doi:10.1186/s13046-021-02067-6
26. Li TT, Zhu D, Mou T, et al. IL-37 induces autophagy in hepatocellular carcinoma cells by inhibiting the PI3K/AKT/mTOR pathway. Mol Immunol. 2017;87:132–140. doi:10.1016/j.molimm.2017.04.010
27. Liu L, Liao JZ, He XX, et al. The role of autophagy in hepatocellular carcinoma: friend or foe. Oncotarget. 2017;8(34):57707–57722. doi:10.18632/oncotarget.17202
28. He J, Deng L, Liu H, et al. BCL2L10/BECN1 modulates hepatoma cells autophagy by regulating PI3K/AKT signaling pathway. Aging. 2019;11(2):350–370. doi:10.18632/aging.101737
29. Cai Z, Zeng Y, Xu B, et al. Galectin-4 serves as a prognostic biomarker for the early recurrence/metastasis of hepatocellular carcinoma. Cancer Sci. 2014;105(11):1510–1517. doi:10.1111/cas.12536
30. Wei D, Li NL, Zeng Y, et al. The molecular chaperone GRP78 contributes to toll-like receptor 3-mediated innate immune response to hepatitis C virus in hepatocytes. J Biol Chem. 2016;291(23):12294–12309. doi:10.1074/jbc.M115.711598
31. Chen Y, Wei D, Deng M. Comparative analysis of serum proteins between hepatitis B virus genotypes B and C infection by DIA-based quantitative proteomics. Infect Drug Resist. 2021;14:4701–4715. doi:10.2147/IDR.S335666
32. Slomovitz BM, Coleman RL. The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clin Cancer Res. 2012;18(21):5856–5864. doi:10.1158/1078-0432.CCR-12-0662
33. Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr. 2015;9(4):317–324. doi:10.1080/19336918.2015.1016686
34. Padmanaban V, Krol I, Suhail Y, et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature. 2019;573(7774):439–444. doi:10.1038/s41586-019-1526-3
35. Sengez B, Carr BI, Alotaibi H. EMT and inflammation: crossroads in HCC. J Gastrointest Cancer. 2022;1–9. doi:10.1007/s12029-021-00801-z
36. Canel M, Serrels A, Frame MC, et al. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J Cell Sci. 2013;126(Pt 2):393–401. doi:10.1242/jcs.100115
37. Mailler E, Guardia CM, Bai X, et al. The autophagy protein ATG9A enables lipid mobilization from lipid droplets. Nat Commun. 2021;12(1):6750. doi:10.1038/s41467-021-26999-x
38. Guardia CM, Tan XF, Lian T, et al. Structure of human ATG9A, the only transmembrane protein of the core autophagy machinery. Cell Rep. 2020;31(13):107837. doi:10.1016/j.celrep.2020.107837
39. Gugnoni M, Sancisi V, Manzotti G, et al. Autophagy and epithelial-mesenchymal transition: an intricate interplay in cancer. Cell Death Dis. 2016;7(12):e2520. doi:10.1038/cddis.2016.415
40. Chen HT, Liu H, Mao MJ, et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol Cancer. 2019;18(1):101. doi:10.1186/s12943-019-1030-2
41. Wang Y, Xiong H, Liu D, et al. Autophagy inhibition specifically promotes epithelial-mesenchymal transition and invasion in RAS-mutated cancer cells. Autophagy. 2019;15(5):886–899. doi:10.1080/15548627.2019.1569912
42. Grassi G, Di Caprio G, Santangelo L, et al. Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting Snail degradation. Cell Death Dis. 2015;6(9):e1880. doi:10.1038/cddis.2015.249
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.