Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Antimicrobial Peptides SLPI and Beta Defensin-1 in Sputum are Negatively Correlated with FEV1
Authors Cane J ![]() , Tregidgo L, Thulborn S, Finch D, Bafadhel M
, Tregidgo L, Thulborn S, Finch D, Bafadhel M ![]()
Received 13 January 2021
Accepted for publication 19 April 2021
Published 28 May 2021 Volume 2021:16 Pages 1437—1447
DOI https://doi.org/10.2147/COPD.S301622
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Jennifer Cane,1,2 Laura Tregidgo,1 Samantha Thulborn,1,2 Donna Finch,3 Mona Bafadhel1,2
1Respiratory Medicine Unit, Nuffield Department of Medicine, University of Oxford, Oxford, UK; 2Oxford NIHR Biomedical Research Centre, University of Oxford, Oxford, UK; 3Alchemab Therapeutics, Cambridge, UK
Correspondence: Jennifer Cane Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) and asthma have heterogeneous inflammation with inhaled corticosteroids (ICS) as a mainstay of treatment. There is increased prevalence of non-typeable Haemophilus influenzae (NTHi) persistence in airways of patients with neutrophilic airway inflammation, potentially due to suppressed host defence after corticosteroid treatment. Antimicrobial peptides (AMPs) have antimicrobial activity against pathogens and immunomodulatory effects. We investigated whether AMPs associate with NTHi presence in COPD and asthma, and whether ICS alter this.
Methods: Secretory leukocyte protease inhibitor (SLPI), osteopontin, elafin and beta defensin-1 were measured in sputum supernatants from healthy donors (n=9), asthmatics (n=21) and patients with COPD (n=14). Elafin and beta defensin-1 were measured in a primary human bronchial epithelial cells (HBECs) from healthy and COPD donors infected with NTHi and pre-treated with fluticasone propionate (FP) and budesonide (BUD). Internalised NTHi was quantified by qPCR.
Results: Sputum SLPI was negatively correlated with FEV1 (p< 0.001, r=− 0.610), FEV1% predicted (p< 0.001, r=− 0.583) and FEV1/FVC (p=0.001, r=− 0.528). Sputum beta defensin-1 was negatively associated with FEV1 (p< 0.001***r=− 0.594). SLPI and beta defensin-1 levels in sputum were higher in the healthy controls and COPD group compared to the asthma group (p=0.001 and p=0.014) and (p< 0.001 and p=0.007, respectively). ICS use was associated with higher sputum osteopontin compared to those with no ICS use. NTHi infection of COPD HBECs produced higher levels of beta defensin-1 compared to healthy donors (mean (SD) release: 45.1pg/mL (7.3) vs 21.2pg/mL (7.3) respectively, p=0.014). Elafin release from HBECs from COPD donors did not change following NTHi infection; however, elafin from healthy donors was significantly reduced (%mean reduction: 23.7%, 95% confidence intervals (CI) of reduction: 5.3– 38.4%, p< 0.01).
Conclusion: Sputum SLPI and beta defensin-1 may be markers to identify those patients with declining lung function. ICS use was associated with higher sputum osteopontin compared to those with no ICS use.
Keywords: antimicrobial peptide, non-typeable haemophilus influenzae, chronic obstructive pulmonary disease
Background
The airway epithelium is the first line of defence against inhaled particles. Disease characteristic epithelial traits are maintained in in vitro culture, as seen by abnormal junction formation1,2 and delayed wound repair3,4 in both asthma and chronic obstructive pulmonary disease (COPD). Part of the host response includes the release of antimicrobial peptides (AMPs) from the airway epithelium. AMPs (including secretory leukocyte protease inhibitor (SLPI), elafin and beta defensin-1) are one line of defence to limit microbial infection of the human mucosae. Through electrostatic forces, AMPs are able to destroy bacteria by disrupting the bacterial membranes.5 Reduced AMP level and function can weaken this defence and may allow colonisation of the host. Osteopontin is a protein known to co-localise with AMPs and has been shown to disrupt AMP antimicrobial action by reducing lysozyme-induced bacterial killing.6 Beta defensin-1 production has previously been shown to be upregulated in epithelial cells in COPD.7
It is known that inflammation in COPD and asthma is heterogeneous.8,9 Airway inflammatory phenotypes of airway disease (neutrophilic, eosinophilic, mixed granulocytic and pauci-inflammatory)10,11 can lend to differential treatment options.12–14 Non-typeable Haemophilus influenzae (NTHi) is the most commonly found bacteria in the airways of COPD and asthmatic patients at stable state15–17 and is associated with a pro-inflammatory neutrophilic response.8 Levels of SLPI and elafin have previously shown inverse correlation with bacterial load.18,19 Inhaled corticosteroids (ICS) are often used in the treatment of asthma and COPD,2 but have been shown to correlate with airway bacterial load,15 particularly after rhinovirus infection in murine models20 and act to impair the cathelicidin response in COPD.21
The effect of acquisition of a new pathogenic bacteria strain on levels of AMPs lysozyme, lactoferrin, cathelicidin and SLPI in the airways of patients with COPD have been investigated. Lysozyme levels are lower during colonisation with NTHi and M. catarrhalis, whilst SLPI levels are lower during exacerbation22 due to NTHi.23 In murine models, ICS pre-treatment prior to infection with rhinovirus20 can affect AMP production, as shown for SLPI and pentraxin-3.
Despite knowledge of the effect of chronic bacterial infection in the airway, the effect of AMPs with neutrophilic airway inflammation in patients with airways disease, especially COPD, has not been studied. Furthermore, whether the increased risk of microbial infection in specific inflammatory airways disease phenotypes and ICS use renders a defect in the host-response to microbial infection has yet to be confirmed.
In this study, we investigate in-vitro and ex-vivo host-response in neutrophil-high compared to neutrophil-low phenotypes of COPD, compared to asthma and healthy controls. We hypothesise the neutrophilic airway disease phenotype has a deficiency of AMPs, and that NTHi alters the host AMP response to facilitate pathogen survival in COPD patients.
Methods
Participants
Participants with clinical diagnoses of COPD or asthma according to British Thoracic Society/European Respiratory Society guidelines24 and healthy controls were recruited from the Churchill Hospital, part of the Oxford University Hospitals NHS Foundation Trust. Participants provided induced sputum to standard procedures.25 Participants performed spirometry (peak flow and/or forced expiratory volume in one second (FEV1)) according to American Thoracic Society/European Respiratory Society’s guidelines.26 All participants provided informed written consent and the study had national ethical approval (08/H0406/189). This study was conducted in accordance with the Declaration of Helsinki.
Bronchial Epithelial Cell Culture
Human bronchial epithelial cells (HBECs) from three non-smoking healthy donors and three COPD donors (Lonza, Basel, Switzerland) were cultured in Airway Epithelial Cell Medium (Promocell, Heidelberg, Germany) as previously described.27 Further detail is included in the supplement. Characteristics of HBEC donors are detailed in Table E1.
Treatment of Bronchial Epithelial Cells
HBECs were treated with differing concentrations of budesonide (BUD) and fluticasone propionate (FP) at high, medium or low concentrations (16nM, 1.6nM and 0.16nM, and 10nM, 1nM and 0.1nM, respectively), or with dimethyl sulfoxide (DMSO) as vehicle control. Different ICS doses reflect potency equivalence. Cells were incubated for two hours before being infected with 1×106 colony forming units NTHi (strain 398, from patient with COPD) and incubated for a further two hours. Supernatants were collected from cells and stored at −80°C prior to measurement of AMP release. Cells were assessed visually by light microscopy to ensure no morphological changes had occurred. Cells were then treated with 200µg/mL gentamycin (Sigma-Aldrich, St Louis, USA) in media for one hour to remove external bacteria. Cells were lysed with 0.025% saponin (Sigma-Aldrich) for ten minutes, and bacterial DNA was extracted from the solution for qPCR analysis. Experimental conditions were run in duplicate for each test. Experiments were conducted with three repeats per cell donor.
Microbial Quantification
Ten-fold dilutions of sputum cell-suspensions were plated onto blood agar and chocolate agar and incubated at 37°c for twenty-four hours. Colonies were counted and the colony forming units (CFU) calculated and added to give the total CFU. Bacterial DNA was isolated from sputum plugs and NTHi- treated HBECs with DNeasy Blood and Tissue kit (Qiagen, Venlo, Netherlands). Primers and probe for detection of the NTHi Omp P6 gene are: Forward primer: CCAGCTGCTAAAGTATTAGTAGAAG, Reverse primer: TTCACCGTAAGATACTGTGCC, Probe: aca[+A]cg[+T]cg[+T]gc[+A]gatgc. Levels of NTHi were quantified against standard plasmid concentrations.
Peptide Measures
Levels were measured by Enzyme Linked Immunosorbent Assay (ELISA): beta-defensin-1 (Peprotech, Cranbury, New Jersey, USA), elafin (DuoSet R&D Systems, Minneapolis, Minnesota, USA), SLPI and osteopontin (Quantikine R&D Systems) (Lower limit of detection: 0.0004, 0.0313, 0.0625 and 0.011ng/mL, respectively). ELISAs were carried out as per the manufacturer’s instructions. All measures were conducted in PBS sputum supernatants, while beta defensin-1 and elafin were measured in HBEC supernatant.
Statistical Analyses
Statistical analysis was completed with Prism (Version 7.03, GraphPad Software Inc., La Jolla, CA). Data is reported as mean (SD) for normally distributed data tested by students t test, and geometric mean (95% confidence interval (CI)) for log normalised data. Comparisons between basal and treated release are calculated as a mean percentage of basal release (95% CI). One-way analysis of variance was used to test differences between sputum measures in the three disease groups. T-tests were used to test difference between basal and stimulated cell AMP release. Associations were assessed using Spearman’s rank correlation analysis on non-log normalised data. Independence of categorical variables was tested by chi squared test. Differences were considered statistically different at p<0.05. Percentage NTHi internalisation was calculated from the amount of NTHi quantified in DNA extracted from HBECs and the total amount of NTHi added.
Results
Fourteen patients with COPD, twenty-one asthmatics and nine healthy individuals were sampled with sputum induction. Participant characteristics are shown in Table 1. COPD participants were older, had worse lung function and more likely to be current or ex-smokers. No correlation of sputum AMP measures were seen with gender, pack-year history, or smoking status (Table 2). SLPI was negatively correlated with FEV1 (p<0.001, r=−0.610), FEV1% predicted (p<0.001, r=−0.583) and FEV1/FVC (p=0.001, r=−0.528). Beta defensin-1 level correlated positively with age (p=0.024, r=0.341), and negatively with FEV1 (p<0.001, r=−0.594).
 |
Table 1 Patient Characteristics of Participants with Available Sputum |
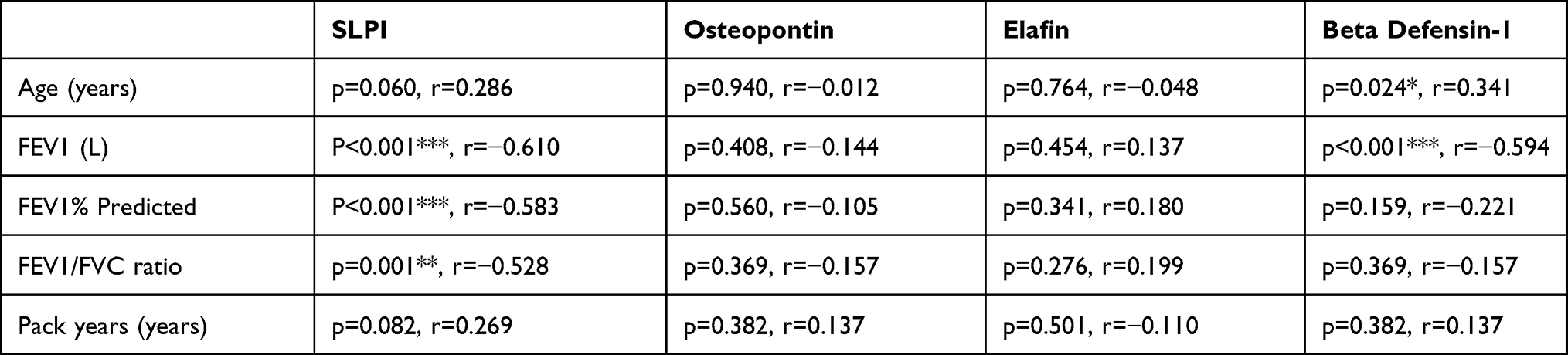 |
Table 2 Correlation Analysis of Participant Characteristics and Sputum AMP Concentrations in All Participants. Significant Correlation Where *p<0.5, **p<0.01, ***p<0.001 |
Sputum AMP Levels from Health and Disease
Sputum SLPI was negatively correlated with FEV1 (p<0.001, r=−0.610), FEV1% predicted (p<0.001, r=−0.583) and FEV1/FVC (p=0.001, r=−0.528). Sputum beta defensin-1 was negatively associated with FEV1 (p<0.001***r=−0.594). SLPI and beta defensin-1 levels in sputum were higher in the healthy controls and COPD group compared to the asthma group (p=0.001 and p=0.014) and (p<0.001 and p=0.007, respectively) (Figure 1). Osteopontin and elafin levels were not different between COPD, asthma and healthy controls. ICS use compared to no ICS in COPD and asthma patients was associated with increased osteopontin release (Difference of geometric means: 233.3pg/mL, p=0.024, Figure E1). Never smokers had lower levels of sputum SLPI compared to current smokers (p=0.022) and ex-smokers (p=0.038) (Figure E2).
 |
Figure 1 Comparison of peptide levels in sputum from healthy donors (n=9), asthmatics (n=21) and patients with COPD (n=14) tested in duplicate. (A) SLPI, (B) osteopontin, (C) elafin and (D) beta defensin-1. Data shown as geometric mean and 95% confidence intervals. Significant differences represented as *p<0.05, **p<0.01, ****p<0.0001. |
Sputum AMP Levels and NTHi Presence
There was a non-significant negative correlation of SLPI and osteopontin and to NTHi (p=0.222, r=−0.349 and p=0.249, r=−0.380, respectively); and a non-significant positive correlation between NTHi and beta defensin-1 (p=0.265, r=0.320) and elafin (p=0.520, r=0.332) (Figure 3). Sputum SLPI showed positive correlation with neutrophil count (p=0.046, r=0.345). No differences were seen in any AMP measured in sputum when stratified by neutrophilic inflammation, with a cut off of 70% sputum neutrophils (Figure 2, Table E2).
 |
Figure 2 Peptide levels in sputum from asthmatics and COPD patients grouped into sputum neutrophil high and low groups are similar. (A) SLPI, (B) osteopontin, (C) elafin and (D) beta defensin-1. Graph showing asthmatic patients (n=21) (o) and COPD patients (n=14) (●), tested in duplicate. Data shown as geometric mean and 95% confidence intervals. |
 |
Figure 3 Correlation of NTHi presence in the airway and peptide levels in healthy donors (n=7, white dots), patients with COPD (n=9, grey dots) and asthma (n=2, black dots), tested in duplicate. (A) SLPI, (B) osteopontin, (C) elafin and (D) beta defensin-1. |
Effect of NTHi Infection on AMP Release from Bronchial Epithelial Cells
Elafin release from healthy donor epithelial cells was reduced by NTHi infection (mean reduction: 23.7%, 95% CI of reduction: 5.3–38.4%, p<0.01) (Figure 4). No significant difference in elafin release was seen upon infection of epithelial cells from patients with COPD. NTHi induction of protein changes in the conditions used was confirmed by measures of IL-8 in healthy HBEC donors, by increasing IL-8 release from untreated (174.9 to 509.2pg/mL, p=0.037) (Figure E5).
 |
Figure 4 (A) Beta defensin-1 release from HBECs when untreated and infected with NTHi. (B) Elafin release from HBECs when untreated and infected with NTHi. Results are shown as a mean percentage change from untreated cell release. Tests were run in duplicate per experiment, from three separate experiments for each donor. Donors: health (n=3, white bars) and COPD (n=3, grey bars). Significant changes form basal release shown as * p<0.05 and ** p<0.01. |
Comparison of AMP Release from Epithelial Cells from Health and COPD
Neither beta defensin-1 or elafin basal release differed between health and COPD (Figure 4). Upon infection with NTHi, epithelial cells from COPD patients released more beta defensin-1 than NTHi infected healthy donors (healthy donor release mean (SD): 21.2pg/mL (7.26), COPD donor release: 45.1pg/mL (12.9), p=0.014).
Effect of Steroids on AMP Release from Bronchial Epithelial Cells
Upon treatment with BUD only, beta defensin-1 release from HBECs from healthy donors showed a non-significant trend to increase from basal level by 69.7% (95% CI: 2.1–137.3%) (Figure 5A). BUD treatment caused no changes in COPD donor epithelial cell release of beta defensin-1. No change in elafin release was observed (Figure 5B). Combined steroid and NTHi treatment of HBECs is discussed in the supplementary material (Figure E3). NTHi internalisation into HBECs was not affected by steroid treatment (Figure E4).
 |
Figure 5 (A) Beta defensin-1 release from HBECs treated with high steroid concentrations. (B) Elafin release from HBECs treated with high steroid concentrations. Results are shown as a mean percentage change from untreated cell release. Tests were run in duplicate per experiment, from three separate experiments for each donor. High Budesonide (Bud): 16nM, High Fluticasone propionate (FP): 10nM. Donors: health (n=3, white bars), COPD (n=3, grey bars). |
Discussion
In this study, we have examined AMPs in the sputum of patients with COPD, asthma and compared these to healthy individuals ex-vivo. We have also looked at the effect of ICS and NTHi on epithelial cells in-vitro.
We found a negative correlation between sputum SLPI and beta defensin-1 and FEV1. Recently, cathelicidin has also been found to negatively correlate with FEV1 in stable COPD patients,28 highlighting the potential for these AMPs to identify patients at greater risk of lung function decline.
Our results show that SLPI and beta defensin-1 levels are low in sputum from asthmatics compared to health and COPD, while no difference in osteopontin and elafin were seen between disease groups. The AMPs selected in this study have been investigated previously. Contradicting our results, studies have demonstrated that beta defensin-1, elafin and osteopontin are elevated in COPD patient sputum compared to healthy controls.6,7,19 These differences in findings may be explained by our low number of healthy controls included in the study, and groups not being age matched.
We found that elafin and beta defensin-1 showed a non-significant trend to positive correlation with NTHi presence in the airways as measured by sputum plug NTHi, while SLPI and osteopontin showed a non-significant negative correlation with NTHi presence. This indicates that patients with NTHi infection may not be lacking in release of the AMPs tested in our study. Levels of SLPI and elafin have previously shown inverse correlation with bacterial load.18,19 While this reflects our observation of SLPI, the different finding with elafin may be accounted for by only measuring NTHi levels in this study. It is conceivable that different bacteria affect AMP levels in the airways. Interestingly, SLPI has been shown to be present in lower levels in COPD patient sputum supernatants during exacerbation associated with NTHi compared to baseline.23 As 40–50% of COPD exacerbations are associated with bacteria,29 this again identifies SLPI as an AMP modifiable by bacterial presence.
As patients with high neutrophils and low eosinophils more frequently experience persistent airway infection of NTHi than other inflammatory groups, we hypothesised that this may be due to an impairment in AMP defence. Our results however suggest that there is no AMP deficiency in patients with low neutrophils compared to those with higher sputum neutrophils. No difference in beta defensin-1 presence in the airways between inflammatory groups has been observed previously.7
In vitro Model of Bronchial Epithelial NTHi Infection
Firstly, we found that HBECs from patients with COPD produce higher levels of beta defensin-1 than epithelial cells from healthy donors when infected in vitro with NTHi. This suggests that bronchial epithelium in COPD has a heightened reaction to the bacteria compared to the epithelium of healthy. This also indicates that this host response is not impaired in COPD. Beta defensin-1 production has previously been shown to be upregulated in epithelial cells in COPD.7 The differences in response of epithelial cells from healthy individuals and patients with COPD have also been noted, with cigarette smoke producing a decrease in beta defensin-1 release from healthy donor cells, while triggering increased production from COPD patient cells.7
Secondly, we found that NTHi infection of HBECs downregulates their production of elafin in health but not disease. Thirdly, we show that pre-treatment of epithelial cells with the steroids budesonide and fluticasone propionate caused no difference in beta defensin-1 or elafin release, other than high concentrations of steroids reducing the amount of elafin released compared to basal release from healthy donors. Corticosteroids act to reduce airway inflammation by binding to glucocorticoid receptors and regulating gene expression.30 Steroids have previously been shown to not affect epithelial cell release of elafin,31 and here we specifically show that this is true for the inhaled corticosteroids tested in this study.
Our measures of internalised NTHi within epithelial cells have shown no significant increase in internalisation after corticosteroid treatment. NTHi internalisation is thought to occur through attachment and penetration.32 Corticosteroid treatment has previously been seen to improve epithelial barrier function in vitro to a greater extent in health compared to COPD.33 Therefore, our result was expected as barrier integrity is not weakened by corticosteroid treatment.
There is a disparity between our ex vivo sputum analysis which indicates that NTHi levels are not associated with decreased levels of either AMP, compared with our in vitro work. This can be attributed to the contributions by immune cells to the in vivo environment, as immune cells such as neutrophils also release elafin,34,35 while monocytes and dendritic cells36 produce beta defensin-1 peptide.
Our study has some limitations. Firstly, we did not have information regarding the inflammatory phenotypes of the HBEC donors to determine whether differences in AMP release are seen between donors with different inflammatory states. Secondly, there are many strains of NTHi, differing in their outer membrane protein expression.37 Our study was carried out with just one strain of NTHi, and we cannot be certain that other strains would provide the same results. Furthermore, acquisition of different bacterial strains is associated with exacerbation and immune response generation,38 and we do not have information about the epithelial cell donors to confirm whether they had previously encountered the NTHi strain used. We have investigated one bacterial species; however, other bacterial species are present and may be causing an effect on AMPs. We also acknowledge the small sample size included in the study and variability of HBEC responses between donors may affect the validity of our results. Further, our in vitro model would be more physiologically relevant if cells were cultures to air-liquid interface. Measures of an AMP known to be induced by NTHi would have provided a further control to ensure previous findings were replicable in our study alongside our new findings. Measures of all AMPs investigated in the study for HBEC response experiments would have provided a more complete representation of the epithelial response to NTHi and ICS treatment.
Conclusions
We hypothesised acquisition of NTHi may cause down-regulation of AMP release, resulting in the bacteria facilitating its ability to infect the host. We found sputum SLPI and beta defensin-1 to negatively correlate with FEV1, indicating their potential as markers of lung health. Our work suggests that NTHi does impact the bronchial epithelium’s contribution to AMP presence in the airways; however, this was not reflected in sputum measures. Future studies should aim to further understand interactions of AMPs with each other and surrounding molecules to get a better picture of their action in the airways.
Abbreviations
AMP, Antimicrobial peptide; BD-1, Beta defensin-1; BUD, Budesonide; CFU, Colony forming units; CI, Confidence interval; DMSO, Dimethyl sulfoxide; DNA, Deoxyribonucleic acid; FEV1, Forced expiratory volume in one second; FP, Fluticasone propionate; HBEC, human bronchial epithelial cell; ICS, Inhaled corticosteroid; n, number; ng/mL, nanograms per millilitre; NHS, National Health Service; nM, nano molar; NTHi, Non-typeable Haemophilus influenzae; pg/mL, Picograms per millilitre; qPCR, quantitative polymerase chain reaction; SD, Standard deviation; SLPI, Secretory leukocyte protease inhibitor.
Data Sharing Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
All participants provided informed written consent and the study had ethical approval (08/H0406/189), approved by National Research Ethics Service, Leicestershire, Northamptonshire & Rutland C.
Consent for Publication
Not applicable
Acknowledgments
We thank all volunteers for taking part, and we thank Clare Connelly and Catherine Borg for recruiting and assessing participants.
Funding
MRC Proximity to Discovery Grant, National Institute of Health Research, Oxford Health Service Research Committee, Pfizer.The research was supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. This report was independent research supported by the National Institute for Health Research (Dr Mona Bafadhel, PDF-2013-06-052). The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health.
Disclosure
The authors declare that they have no competing interests.
References
1. Heijink IH, Noordhoek JA, Timens W, van Oosterhout AJ, Postma DS. Abnormalities in airway epithelial junction formation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;189(11):1439–1442. doi:10.1164/rccm.201311-1982LE
2. Xiao C, Puddicombe SM, Field S, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. 2011;128(3):
3. Perotin JM, Adam D, Vella-Boucaud J, et al. Delay of airway epithelial wound repair in COPD is associated with airflow obstruction severity. Respir Res. 2014;15:151. doi:10.1186/s12931-014-0151-9
4. Stevens PT, Kicic A, Sutanto EN, Knight DA, Stick SM. Dysregulated repair in asthmatic paediatric airway epithelial cells: the role of plasminogen activator inhibitor-1. Clin Exp Allergy. 2008;38(12):1901–1910. doi:10.1111/j.1365-2222.2008.03093.x
5. Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415(6870):389–395. doi:10.1038/415389a
6. Gela A, Kasetty G, Morgelin M, et al. Osteopontin binds and modulates functions of eosinophil-recruiting chemokines. Allergy. 2016;71(1):58–67. doi:10.1111/all.12771
7. Baines KJ, Wright TK, Simpson JL, et al. Airway beta-defensin-1 protein is elevated in COPD and severe asthma. Mediators Inflamm. 2015;2015:8. doi:10.1155/2015/407271
8. Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184(6):184.
9. Bhowmik A, Seemungal TAR, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax. 2000;55(2):114–120. doi:10.1136/thorax.55.2.114
10. Bafadhel M, McKenna S, Terry S, et al. Blood eosinophils to direct corticosteroid treatment of exacerbations of chronic obstructive pulmonary disease: a randomized placebo-controlled trial. Am J Respir Crit Care Med. 2012;186(1):186. doi:10.1164/rccm.201108-1553OC
11. Simpson JL, Scott R, Boyle MJ, Gibson PG. Inflammatory subtypes in asthma: assessment and identification using induced sputum. Respirology. 2006;11(1):54–61. doi:10.1111/j.1440-1843.2006.00784.x
12. Agusti A, Bel E, Thomas M, et al. Treatable traits: toward precision medicine of chronic airway diseases. Eur Respir J. 2016;47(2):410–419. doi:10.1183/13993003.01359-2015
13. Gibson PG, Yang IA, Upham JW, et al. Effect of azithromycin on asthma exacerbations and quality of life in adults with persistent uncontrolled asthma (AMAZES): a randomised, double-blind, placebo-controlled trial. The Lancet. 2017;390(10095):659–668. doi:10.1016/S0140-6736(17)31281-3
14. Brusselle GG, Vanderstichele C, Jordens P, et al. Azithromycin for prevention of exacerbations in severe asthma (AZISAST): a multicentre randomised double-blind placebo-controlled trial. Thorax. 2013;68(4):322–329. doi:10.1136/thoraxjnl-2012-202698
15. Garcha DS, Thurston SJ, Patel AR, et al. Changes in prevalence and load of airway bacteria using quantitative PCR in stable and exacerbated COPD. Thorax. 2012;67(12):67. doi:10.1136/thoraxjnl-2012-201924
16. Bafadhel M, Haldar K, Barker B, et al. Airway bacteria measured by quantitative polymerase chain reaction and culture in patients with stable COPD: relationship with neutrophilic airway inflammation, exacerbation frequency, and lung function. Int J Chron Obstruct Pulmon Dis. 2015;10:10. doi:10.2147/COPD.S80091
17. Hilty M, Burke C, Pedro H, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578. doi:10.1371/journal.pone.0008578
18. Mallia P, Footitt J, Sotero R, et al. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(11):1117–1124. doi:10.1164/rccm.201205-0806OC
19. Tsoumakidou M, Bouloukaki I, Thimaki K, Tzanakis N, Siafakas NM. Innate immunity proteins in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Exp Lung Res. 2010;36(6):373–380. doi:10.3109/01902141003690389
20. Singanayagam A, Glanville N, Girkin JL, et al. Corticosteroid suppression of antiviral immunity increases bacterial loads and mucus production in COPD exacerbations. Nat Commun. 2018;9(1):2229. doi:10.1038/s41467-018-04574-1
21. Singanayagam A, Glanville N, Cuthbertson L, et al. Inhaled corticosteroid suppression of cathelicidin drives dysbiosis and bacterial infection in chronic obstructive pulmonary disease. Sci Transl Med. 2019;11(507):eaav3879. doi:10.1126/scitranslmed.aav3879
22. Tangedal S, Nielsen R, Aanerud M, et al. Sputum microbiota and inflammation at stable state and during exacerbations in a cohort of chronic obstructive pulmonary disease (COPD) patients. PLoS One. 2019;14(9):e0222449. doi:10.1371/journal.pone.0222449
23. Parameswaran GI, Sethi S, Murphy TF. Effects of bacterial infection on airway antimicrobial peptides and proteins in COPD. Chest. 2011;140(3):611–617. doi:10.1378/chest.10-2760
24. Celli BR, MacNee W, Agusti A, et al. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932–946. doi:10.1183/09031936.04.00014304
25. Weiszhar Z, Horvath I. Induced sputum analysis: step by step. Breathe. 2013;9(4):300–306. doi:10.1183/20734735.042912
26. Brusasco V, Crapo R, Viegi G. Coming together: the ATS/ERS consensus on clinical pulmonary function testing. Eur Respir J. 2005;26(1):1–2. doi:10.1183/09031936.05.00034205
27. Slater M, Torr E, Harrison T, et al. The differential effects of azithromycin on the airway epithelium in vitro and in vivo. Physiol Rep. 2016;4(18):e12960. doi:10.14814/phy2.12960
28. Burkes RM, Ceppe AS, Couper DJ, et al. Plasma cathelicidin is independently associated with reduced lung function in COPD: analysis of the subpopulations and intermediate outcome measures in COPD study cohort. Chronic Obstr Pulm Dis. 2020;7(4):370–381. doi:10.15326/jcopdf.7.4.2020.0142
29. Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184(6):662–671.
30. Suissa S, McGhan R, Niewoehner D, Make B. Inhaled corticosteroids in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2007;4(7):535–542. doi:10.1513/pats.200701-024FM
31. van Wetering S, van der Linden AC, van Sterkenburg MA, et al. Regulation of SLPI and elafin release from bronchial epithelial cells by neutrophil defensins. Am J Physiol Lung Cell Mol Physiol. 2000;278(1):L51–58. doi:10.1152/ajplung.2000.278.1.L51
32. Ikeda M, Enomoto N, Hashimoto D, et al. Nontypeable Haemophilus influenzae exploits the interaction between protein-E and vitronectin for the adherence and invasion to bronchial epithelial cells. BMC Microbiol. 2015;15:263. doi:10.1186/s12866-015-0600-8
33. Heijink I, van Oosterhout A, Kliphuis N, et al. Oxidant-induced corticosteroid unresponsiveness in human bronchial epithelial cells. Thorax. 2014;69(1):5–13. doi:10.1136/thoraxjnl-2013-203520
34. van Wetering S, van der Linden AC, van Sterkenburg MA, Rabe KF, Schalkwijk J, Hiemstra PS. Regulation of secretory leukocyte proteinase inhibitor (SLPI) production by human bronchial epithelial cells: increase of cell-associated SLPI by neutrophil elastase. J Invest Med. 2000;48(5):359–366.
35. Sallenave JM, Si Tahar M, Cox G, Chignard M, Gauldie J. Secretory leukocyte proteinase inhibitor is a major leukocyte elastase inhibitor in human neutrophils. J Leukoc Biol. 1997;61(6):695–702. doi:10.1002/jlb.61.6.695
36. Ryan LK, Dai J, Yin Z, et al. Modulation of human beta-defensin-1 (hBD-1) in plasmacytoid dendritic cells (PDC), monocytes, and epithelial cells by influenza virus, Herpes simplex virus, and Sendai virus and its possible role in innate immunity. J Leukoc Biol. 2011;90(2):343–356. doi:10.1189/jlb.0209079
37. St Geme JW
38. Sethi S. Bacteria in exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1(2):109–114. doi:10.1513/pats.2306029
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.