Back to Archived Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 14
Antimicrobial Activity, in silico Molecular Docking, ADMET and DFT Analysis of Secondary Metabolites from Roots of Three Ethiopian Medicinal Plants
Authors Anza M, Endale M ![]() , Cardona L, Cortes D, Eswaramoorthy R
, Cardona L, Cortes D, Eswaramoorthy R ![]() , Zueco J, Rico H, Trelis M, Abarca B
, Zueco J, Rico H, Trelis M, Abarca B
Received 7 June 2021
Accepted for publication 10 August 2021
Published 20 August 2021 Volume 2021:14 Pages 117—132
DOI https://doi.org/10.2147/AABC.S323657
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Juan Fernandez-Recio
Mathewos Anza,1 Milkyas Endale,1 Luz Cardona,2 Diego Cortes,3 Rajalakshmanan Eswaramoorthy,1 Jesus Zueco,4 Hortensia Rico,4 Maria Trelis,5 Belen Abarca2
1Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University, Adama, Ethiopia; 2Department of Organic Chemistry, Faculty of Chemistry, University of Valencia, Burjassot, Spain; 3Department of Pharmacology, Faculty of Pharmacy, University of Valencia, Burjassot, Spain; 4Department of Microbiology and Ecology, Faculty of Pharmacy, University of Valencia, Burjassot, Spain; 5Parasites and Health Research Group, Department of Pharmacy, Pharmaceutical Technology and Parasitology, Faculty of Pharmacy, University of Valencia, Burjassot, Spain
Correspondence: Mathewos Anza; Milkyas Endale
Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University, P.O. Box 1888, Adama, Ethiopia
Email [email protected]; [email protected]
Background: Uvaria scheffleri (Annonaceae), Clematis burgensis (Ranunculaceae), and Euphorbia schimperiana (Euphorbiaceae) are medicinal plants traditionally used to treat cough, tuberculosis, asthma, sore throat and skin infections.
Methods: Silica gel column chromatographic separation was used to isolate compounds. Crude extract and isolated compounds were evaluated for antimicrobial activity against Staphylococcus aureus, Escherichia coli, and Candida albicans via the broth dilution method. Docking studies were performed with E. coli DNA-Gyrase B and human DNA topoisomerase IIα by using AutoDock Vina. ADMET were predicted by SwissADME, PreADMET, and OSIRIS Property predictions. The optimized structures and molecular electrostatic potential surface of the isolated compounds were predicted by DFT analysis using B3LYP/6-31G basis levels.
Results: Silica gel column chromatographic separation afforded five compounds 1– 5 of which N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol ( 1) is reported herein for the first time, along with known C-benzylated dihydrochalcone uvaretin ( 2), bis(2-ethylheptyl) phthalate ( 3), lupeol ( 4) and suberosin derivative ( 5). Dichloromethane roots extract of U. scheffleri showed potent antibacterial activity against S. aureus (MIC = 6.25 μg/mL) compared to gentamicin (MIC=5 μg/mL). In silico, molecular docking analysis of compounds ( 1and 3– 5) showed strong interaction with E. coli DNA gyrase B with a binding energy value ranging from − 6.9 to − 6.0 kcal/mol compared to ciprofloxacin − 7.2 kcal/mol, whereas analysis against human topoisomerase IIα showed binding energy value ranging from − 5.9 to − 5.3 kcal/mol compared to vosaroxin (− 6.2 kcal/mol).
Conclusion: The results obtained suggest that N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol ( 1) and coumarin ( 5) are potential topoisomerase II α inhibitors and might be used as anticancer agents. The ADMET studies showed the highest drug-likeness properties for studied compounds other than bis(2-ethylheptyl) phthalate ( 3). DFT calculations suggested that studied compounds showed the lowest gap energy and were chemically reactive, and isolated compounds may serve as potential drug candidates that corroborate with the traditional uses of studied plants.
Keywords: Uvaria scheffleri, Clematis burgensis, Euphorbia schimperiana, antimicrobial, molecular docking, ADMET, DFT analysis
Introduction
Medicinal plants have played a pivotal role in primary health care and offer a rich source of novel bioactive compounds in drug discovery and development.1 Traditional medicine appears to be the source of healthcare particularly in the rural majority communities of Africa due to its intrinsic qualities, unique and holistic approaches as well as its accessibility and affordability.2 Infectious diseases are public health problems and a significant cause of death worldwide.3 Infections due to pathogenic microorganisms cause a severe concern to human health. Increasing cases of drug resistance, unwanted side effects of existing antibiotics, and the reappearance of earlier known infections have demanded the need for new, safe and effective antimicrobial agents.4–6 In drug development, virtual screening like drug-likeness, ADMET, and DFT analysis are a computational method to find compounds that are likely to exhibit physiological activity in a short time and at a low cost using various in silico simulation methods.7
Uvaria scheffleri Diels (Annonaceae) (Boyyiniya, Wolaitigna, Ethiopia) is widely distributed in East Africa, and the root decoction is used to treat cough, tuberculosis, asthma, and sore throat.8 In Ethiopia, the fruit is edible and used for the treatment of skin infections.9 Uvarindole alkaloids and chalcones are among the most cited compounds reported from the genus with a broad spectrum of biological activities. Clematis burgensis Engl. (Ranunculaceae) (Fitti, Oromiffa, Ethiopia) is endemic to Ethiopia and grows in open montane forest and forest borders, along roads and streams. Leaves of C. burgensis are used for the treatment of otorrhoea and eczema.10 Euphorbia schimperiana (Euphorbiaceae) (Benjjile, Sidamigna, Ethiopia) is a medicinal plant where leaves and roots decoction treat parasitic infections venereal diseases, and latex as ear drops is used to treat otitis.11
Despite a broad spectrum of ethnomedicinal uses, there are limited scientific studies done on these plants. Thus, we report herein novel Uvarindole-C derivative named as N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol (1) along with known dihydrochalcone chalcone Uvartin (2) from roots extract of U. sheffleri (Figure 1, 1 and 2), bis (2-ethylheptyl) phthalate (3) and lupeol (4) from roots of C. burgensis (Figure 1, 3 and 4) and prenylated coumarin (5) from roots of E. schimperiana (Figure 1, 5) along with antibacterial activity of dichloromethane crude extract and isolated compounds, in silico molecular docking, ADMET and DFT analysis for the first time.
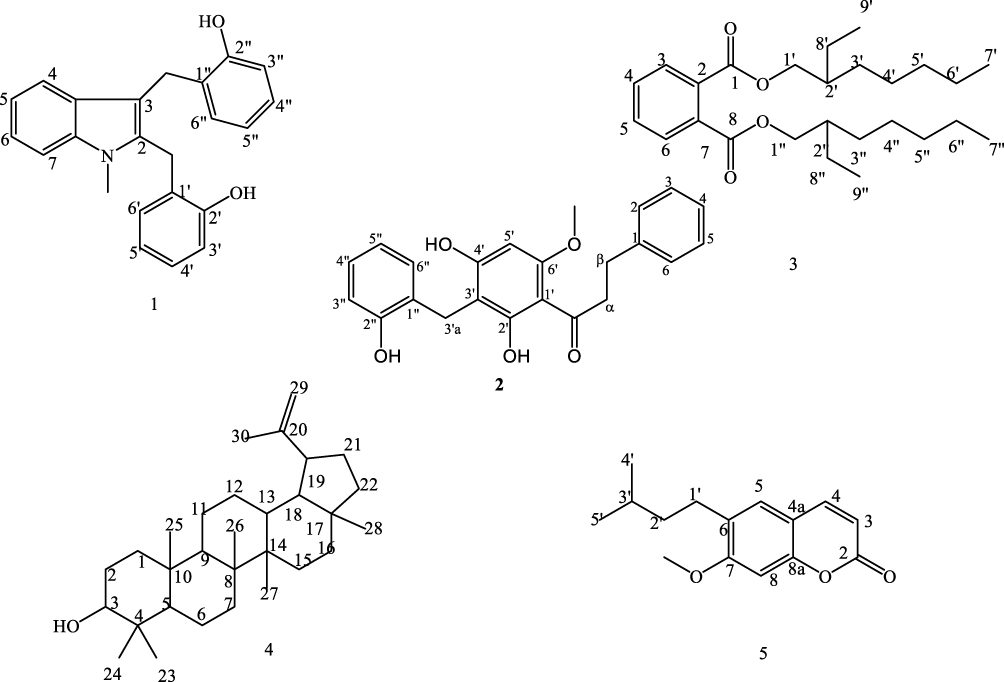 |
Figure 1 Structure of compounds from the roots extract of U. scheffleri 1–2,C.burgensis 3–4, and E. shemperiana 5. |
Materials and Methods
General
1H NMR, 13C NMR spectra were recorded on Bruker Avance 500 DPX and Bruker AV-300 spectrometers operating at frequencies of 500 and 300 MHz for 1H and 125 MHz and 75 MHz 13C, respectively. Chemical shifts are expressed in ppm, and J values are given in Hz. The HRMS was determined on a SCIEX, 5800 MALDI TOF (SCIEX, USA). Analytical TLC plate with silica gel 60 F254 TLC (Merck, Germany) was used to determine the TLC profile. The spots on TLC plates were visualized using a UV lamp (254 and 365 nm). Column chromatography was performed on silica gel (60–120 mesh). All chemicals, solvents, and reagents were used analytical grade level.
Collection and Identification
The roots of U. scheffleri, C. burgensis and E. schimperiana were collected from Wolaita zone SNNPR of Ethiopia in November 2018 and identified with the help of Botanist Shambel Alemu in comparison with the specimens at the National Herbarium, Addis Ababa University, Ethiopia (Voucher code MAUs-001/11, MACb-004/11 and MAEs-005/11 respectively). Freshly collected roots were transported to the Organic Chemistry Laboratory of Wolaita Sodo University, Ethiopia. The collected plant materials were air-dried and grounded into a fine powder at room temperature.
Extraction and Isolation
Grounded roots of U. scheffleri (500 g) and E. schimperiana (400 g) were extracted with dichloromethane for 24 h by maceration while shaking using an electronic shaker at a speed of 230 rpm at room temperature. The filtrate was concentrated using a vacuum rotary evaporator to yield 32 g (8%) and 22 g (5.5%) crude extracts, respectively. Similarly, grounded roots powder of C. burgensis (800 g) was extracted with dichloromethane/methanol (1:1, 2 L) for 24 h by maceration while shaking at room temperature. The filtrate was concentrated using a vacuum rotary evaporator to yield 32.30 g (4%) crude extract.
The dried root extract of U. scheffleri (30 g) was subjected to silica gel flash column chromatography and eluted with an increasing gradient of ethyl acetate in n-hexane. A total of 75 fractions were collected (each 50 mL). Fraction 20 (eluted with 45% ethyl acetate) afforded N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol (1, 12 mg) as pale gray crystals. Fraction 22–24 were combined and subjected to silica gel column chromatography and eluted with an increasing gradient of ethyl acetate in n-hexane. A total of 22 fractions (25 mL each) were collected. Subfraction 12 eluted with 40% ethyl acetate in n-hexane afforded Uvaretin (2, 9 mg). In the same way, the dried crude extract of C. burgensis (28 g) was subjected to silica gel column chromatography and eluted with an increasing gradient of ethyl acetate in petroleum ether followed by an increasing gradient of methanol in dichloromethane. A total of 104 fractions (50 mL each) were collected. Fraction 20 and 21 eluted with 85% ethyl acetate in petroleum ether afforded bis (2-ethyl heptyl) phthalate (3, 40 mg). Fraction 22–30 combined and further purified by silica gel column chromatography using an increasing gradient of ethyl acetate in petroleum ether to give lupeol (4, 45 mg). The root extract of E. schimperiana (20 g) was subjected to silica gel flash column chromatography and eluted with an increasing gradient of ethyl acetate in n-hexane. A total of 42 fractions were collected (each 25 mL). Fraction 9 (eluted with 15% ethyl acetate) afforded compound 5 (12 mg) as yellow amorphous powder.
Spectra Data of Isolated Compounds (1-5)
N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol (1)
Pale gray crystals. HRMS value [M]+ = 343.2953 (calculated for the molecular formula C23H21NO2= 343.2972). 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 8.2 Hz, 1H, H-4), 8.10 (m, 1H, H-5), 7.78 (t, 1H, H-6), 8. 09 (m, 1H, H-7), 2.66 (s, 3H, -NCH3), 4.61 (s, 2H, 2a-CH2), 7.31 (dd, J = 8.2,1.7 Hz, 1H, H-3ʹ), 7.38 (d, J = 8.1 Hz, 1H, H-4ʹ), 7.53 (m, 1H, H-5ʹ), 8.10 (m, 1H, H-6ʹ), 4.44 (s, 2H, 3a-CH2), 7.67 (d, J = 8.6 Hz, 1H, H-3”), 8.43 (dd, J = 7.9, 1.4 Hz, 1H, H-4”), 7.86 (d, J = 8.2 Hz, 1H, H-5”), 7.53 (m, 1H, H-6”), 3.42 (s, 1H, OH), 5.89 (s, 1H, OH). 13C NMR (125 MHz, CDCl3) δ 140.7 (C-2), 119.8 (C-3), 122.9 (C-3a), 118.6 (C-4), 121.0 (C-5), 129.2 (C-6), 111.8 (C-7), 143.0 (C-7a), 42.8 (2a-CH2), 132.9 (C-1ʹ) 158.9 (C-2ʹ), 122.0 (C-3ʹ), 130.1 (C-4ʹ), 119.7 (C-5ʹ), 137.3 (C-6ʹ), 40.8 (3a- CH2), 134.1 (C-1”), 151.2 (C-2ʹ), 113.7 (C-3”), 128.9 (C-4”), 122.8 (C-5”), 124.5 (C-6”), and 36.1 (-N-CH3).
Uvaretin (2)
Pale gray needles. ESI-MS m/z = 378.12. 1H NMR (300 MHz, CDCl3) δ 11.35 (s, 1H, OH), 7.77 (d, J = 8.2, 1H, H-2), 7.81 (dd, J = 8.1, 1.7 Hz, 1H, H-3), 7.56 (t, 1H, H-4), 7.30 (m, 1H, H-5), 7.49 (dd, J = 7.5, 1.7 Hz, 1H, H-6), 7.23 (s, 1H, H-5ʹ), 7.12 (d, J = 8.2 Hz, 1H, H-6”), 6.97 (m, 1H, H-3”), 6.91 (m, 1H, H-4”), 6.82 (dd, J = 7.9, 1.3 Hz, 1H, H-5”), 4.90 (s, 2H, H-3ʹa’), 4.37 (s, 3H, 6ʹ-OCH3), 3.15 (m, 2H, α-CH2) and 2.85 (m, 2H, β-CH2). 13C NMR (75 MHz, CDCl3) δ 140.6 (C-1), 128.6 (C-2), 128.5 (C-3), 126.6 (C-4), 128.4 (C-5), 136.8 (C-6), 30.6 (α-CH2), 41.0 (β-CH2), 203.4 (C=O), 118.2 (C-1ʹ), 162.9 (C-2ʹ), 121.2 (C-3ʹ), 155.4 (C-4ʹ), 112.1 (C-5ʹ), 155.0 (C-6ʹ), 28.2 (C-3ʹa), 122.0 (C-1”), 154.0 (C-2”), 116. 6 (C-3”), 119. 9 (C-4”), 119.5 (C-5”), 131.3 (C-6”).
Bis 2-Ethylheptyl) Phthalate (3)
Yellow amorphous crystal, HRMS [M+H]+ = 419.3363 (calculated for the molecular formula C26H42O4= 419.3183). 1H NMR (300 MHz, CDCl3) δ 7.64 (2H, dd, J = 8.8, 2.2 Hz, H-3, 6), 7.46 (2H, dd, J = 8.8, 2.2 Hz, H-4, 5), 4.21 (4H, t, J = 6.2 Hz, H-1ʹ, 1”), 1.65 (4H, m, H-2ʹ, 2”), 1.25 (4H, m, H-3ʹ, 3”), 1.29 (8H, m, H- 4ʹ, 4”, 5ʹ, 5”), 1.31 (4H, m, H-6ʹ, 6”), 0.89 (6H, m, H-7ʹ, 7”), 1.40 (4H, m, H-8ʹ, 8”), 0.90 (6H, m, H-9ʹ, 9”); 13C NMR (75 MHz, CDCl3) 167.7 (C-1, 8), 132.2 (C-2, 7), 130.9 (C-3, 6), 128.8 (C-4, 5), 68.1 (C-1ʹ,1”), 38. 7 (C-2ʹ, 2”), 30.3 (C-3ʹ, 3”), 28.9 (C-4ʹ, 4”), 29.7 (C-5ʹ, 5”), 23.7 (C-8ʹ, 8”), 22.9 (C-6ʹ, 6”), 14.0 (C-7ʹ, 7”), 10.9 (C-9ʹ, 9”).
Lupeol (4)
White powder, [M]+ m/z = 424. 1H NMR (300 MHz, CDCl3) δ 3.18 (dd, J = 10.9, 5.4 Hz, 1H, H-3), 4.68 (d, J = 2.5 Hz, 1H, H-29a), 4.56 (dd, J = 2.6, 1.4 Hz, 1H, H-29b), 2.37–1.39 (m, 25H), 1.66 (s, 3H, H-30), 1.05 (s, 3H, H-28), 0.96 (s, 3H, H-27), 0.94 (s, 3H, H-26), 0.84 (s, 3H, H-25), 0.85 (s, 3H, H-24) and 0.76 (s, 3H, H-23). 13C-NMR (75MHz, CDCl3) δ 38.9 (C-1), 27.6 (C-2), 79.2 (C-3), 39.8 (C-4), 55.5 (C-5), 18.5 (C-6), 34.5 (C-7), 41.0 (C-8), 50.6 (C-9), 37.4 (C-10), 21.1 (C-11), 25.3 (C-12), 38.2 (C-13), 43.0 (C-14), 27.6 (C-15), 35.8 (C-16), 43.2 (C-17), 48.5 (C-18), 48.2 (C-19),151.2 (C-20), 30.0 (C-21), 40.2 (C-22), 28.2 (C-23), 16.2 (C-24), 16.3 (C-25), 15.6 (C-26), 14.7 (C-27).18.5 (C-28), 109.5 (C-29), 19.5 (C-30).
Compound (5)
Yellow needles, [M]+m/z = 246.1, 1H NMR δ 0.88 (6H, d, J = 5.3 Hz, 2x Me), 2.02 (3H, m, H-2ʹ, H-3ʹ), 2 31 (2H, m, H-1ʹ), 3 95 (3H, s, 7-OMe), 6 26 (1H, d, J = 9 0.5 Hz, H-3), 6.84 (1H, s, H-8), 6.91 (1H, s, H-5), and 7.61 (1H, d, J = 9.5 Hz, H-4). 13C NMR (75 MHz, CDCl3) δ 161.5 (C-2), 150.2 (C-7), 149.7 (C-6), 143.3 (C-4), 114.0 (C-4a), 113.3 (C-3), 111.4 (C-5), 107.5 (C-8), 103.2 (C-8a), 56.4 (-OCH3), 31.9 (C-1ʹ), 31.6 (C-2ʹ), 23.4 (C-3ʹ), 22.7 (C-4ʹ), 14.8 (C-5ʹ).
Antimicrobial Study
Microorganism Strain
In vitro antibacterial activity of dichloromethane root extract of U. scheffleri and isolated compounds were evaluated against Staphylococcus aureus (CECT 59) and Escherichia coli (CECT 434), whereas antifungal activity was examined against Candida albicans (ATCC 26555) strains.
Antimicrobial Activity Assay
The assays of extract and isolated compounds were measured by the broth dilution technique as previous described by Valgas et al.12 A microdilution method in 96-well plates, using Mueller–Hinton broth media for bacteria and Yeast Extract Peptone Dextrose media (YEPD) for C. albicans, was performed in order to determine minimum inhibitory concentrations. Extracts and isolated compounds were dissolved in DMSO (Sigma, USA) to prepare stock solutions at a concentration of 100 µg/mL. Further half-fold serial dilutions were performed by adding culture broth to reach concentrations ranging from 100 to 1.56 µg/mL, which were distributed in 96-well plates, as well as a sterility/negative control and growth control (containing culture broth plus DMSO, without antimicrobial substance). Each test and growth control well was inoculated with 10 µL of a bacterial suspension giving a concentration of 105 CFU/mL in the case of bacteria and 104 CFU/mL in the case of C. albicans. The medium without strain was used as a negative control, and gentamicin and fluconazole for bacterial and fungal strain, respectively, were used as positive controls. All experiments were performed in triplicate, and the microdilution trays were incubated at 37 °C for bacterial strains, at 28 °C fungal strain for 24 h. The microbial growth was detected by optical density measurement at 595nm (ELISA reader, CLX800-BioRAD Instruments). MIC values were defined as the lowest concentration of each compounds, which completely inhibited microbial growth. The results were expressed in micrograms per milliliter.12
Computational Study
Molecular Docking
AutoDock Vina with standard protocol was used to dock the proteins (PDB ID: 6F86, and PDB ID: 3QX3) and isolated chemical constituents (1–5) into the active site of proteins.13,14 The chemical structures of compounds 1–5 were drawn using ChemOffice tool (Chem Draw 16.0) assigned with proper 2D orientation, and energy of each molecule was minimized using ChemBio3D. The energy minimized ligand molecules were then subjected to quantum mechanical treatment for geometry optimization and frequency calculation employing B3LYP/6-31G (d,p) level of theory. The DFT optimized structures were used as input for AutoDock Vina, in order to carry out the docking simulation.15–17 The crystal structure of receptor molecule E. coli gyrase B (PDB ID: 6F86), and human topoisomerase II alpha (PDB ID: 3QX3) were downloaded from protein data bank. The protein preparation was done using the reported standard protocol18 by removing the co-crystallized ligand, selected water molecules and cofactors, the target protein file was prepared by leaving the associated residue with protein by using auto preparation of target protein file Auto Dock 4.2 (MGL tools1.5.7). The graphical user interface program was used to set the grid box for docking simulations. The grid box was constructed using 40, 40, and 40, pointing in x, y, and z directions, respectively, with a grid point spacing of 0.375 Å. The center grid box are (13.52 Å, 15.68 Å and 19.12 Å), (14.24 Å, 17.49 Å and 12.27 Å), and (23.26 Å, and 18.59 Å) for 6f86, and 3qx3, respectively. The docking algorithm provided with Auto Dock Vina was used to search for the best docked conformation between ligand and protein. During the docking process, a maximum of nine conformers were considered for each ligand. The conformations with the most favorable (least) free binding energy were selected for analyzing the interactions between the target receptor and ligands by Discovery studio visualizer and PyMOL. The ligands are represented in different color, H-bonds and the interacting residues are represented in stick model representation.
In-silico Drug-Likeness and Toxicity Predictions
Drug-likeness of isolated compounds was predicted based on an already established concept by Lipinski et al.19 The structures of isolated compounds (1, 3–5) were converted to their canonical simplified molecular-input line-entry system (SMILE). They submitted to the SwissADME and PreADMET tool to estimate in silico pharmacokinetics, such as the number of hydrogen donors, hydrogen acceptors, rotatable bonds, and total polar surface area of a compound. The organ toxicities and toxicological endpoints of the isolated compounds were predicted using PreADMET and OSIRIS Property.20 The selection of compounds as drug candidates were determined by a parameter called drug score. The higher the drug score value, the higher the compounds chance of being considered a drug candidate.21
Quantum Computational Studies
Density functional theory (DFT) is emerging as an effective technique to study biomolecular systems to know the reaction coordinates and of the transition state of a reaction is fundamental for the development of mechanism-based inhibitors that usually mimic the transition state.22 In the present study, the DFT analysis of compounds (1, 3–5) was performed using Gaussian 09 and visualized through Gauss view 6.0.15–17 The structural coordinates were optimized using B3LYP/6-31 G (d,p) level basis set without any symmetrical constraints. The molecular electrostatic potential map and energies of the compounds were obtained from the optimized geometry. The Koopman’s approximation was used to estimate the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), energy gap, and related reactive parameters (electronegativity, chemical potential, hardness, softness, electrophilicity).23
Results and Discussion
The roots extract of U. scheffleri, C. burgensis, and E. shemperiana after silica gel column chromatography furnished a new Uvarindole-C derivative named N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol (1) along with known dihydrochalcone uvaretin (2), bis (2-ethylheptyl) phthalate (3), lupeol (4), and prenylated coumarin (5). The structures of compounds (1–5) were characterized by 1H NMR, 13C NMR, DEPT-135, 2D NMR (HSQC, COSY, and HMBC), MS techniques and comparison with literature data. The detailed characterizations of these compounds are presented below.
Compound (1) was obtained as pale gray crystals and positive reaction to Dragendorff's reagent inductive to alkaloid. Its HRMS displayed [M]+ m/z peak at 343.2953 (calculated 343.2972 for the chemical formula C23H21NO2). The 1H NMR spectrum showed two singlets at δ 4.44 and 4.61 (2 x C-CH2) attributed to methylene moiety. Methyl attached to nitrogen was observed at δ 2.66 (N-CH3), showing a 1J correlation with δ 36.5 peaks in the HSQC spectrum and a 3J HMBC correlation with C-2 (140.7) and C-7a (δ 143.0) (Figure 2, and Supporting Information). In addition, a signal at δ 5.89 and 3.42 showed broad singlet peaks attributed to phenolic hydroxyl proton. The remaining 12 protons were displayed in the aromatic region.
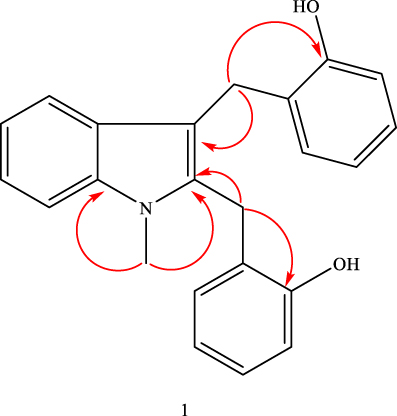 |
Figure 2 Important HMBC correlations of compound 1 from root extract of U. scheffleri. |
The 13C NMR spectrum showed twelve aromatic methines, two sp3 methylene carbons at δ 42.8 (C-2a) and 40.8 (C-3a), two oxygenated sp2 quaternary carbons at δ 158.9 and 151.2 along with six sp2 quaternary carbons at δ 143.0, 140.7, 134.5, 132.9, 119.8 and 122.9. Placement of two 2-hydroxybenzyl substituents at C-2 and C-3 positions were done with the help of 2J and 3J HMBC correlations of methylene protons (Figure 2). Based on the above spectroscopic evidence and comparison with spectral data of uvarindole-C,24 compound (1) is found to be a new compound named N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol (Figure 1, 1), reported herein for the first time.
Compound (2) was obtained as deep gray needles. It’s ESI-MS m/z showed 378.12. The 1H NMR spectrum displayed a singlet peak at δ 4.90 (2H, Ar-CH2) attributed to methylene linked with two phenyl rings and two signals with A2B2 spin pattern observed at δ 2.85 and 3.15 suggesting a β-propiophenone moiety. Two singlets at δ 4.37 and δ 11.33 attributed to methoxy and hydroxyl group with peri effect to the carbonyl group, respectively. The presence of a mono substituted aromatic ring was clearly evident from peaks at δ 7.77 (d, J = 8.2, 1H, H-2), 7.81 (dd, J = 8.1, 1.7 Hz, 1H, H-3), 7.56 (t, 1H, H-4), 7.30 (m, 1H, H-5) and 7.49 (dd, J = 7.5, 1.7 Hz, 1H, H-6). This substitution pattern coupled with the appearance of a singlet aromatic proton at δ 7.23 (s, 1H) displayed by H-5ʹ suggest a tetra substituted ring A and mono substituted ring B pattern of dihydrochalcone. The 13C NMR spectrum supported by DEPT-135 displayed 22 carbon signals. Ten sp2 methines were observed at δ 112.1, 116.6, 119.5, 119.9, 126.8, 128.4, 128.5, 131.3, and 136.8. Four sp2 oxygenated quaternary carbons and one carbonyl carbon were observed at δ 162.9 (C-2ʹ), 155.4 (C-4ʹ), 155.0 (C-6ʹ), 154.0 (C-2”) and δ 203.3, respectively. In addition, four sp2 quaternary carbons were observed at δ 118.2 (C-1ʹ), 121.2 (C-3ʹ), 122.0 (C-1”), and 140.6 (C-1). Furthermore, three sp3 methylenes were observed at δ 28.2 (C-3ʹa), 30.6 (α-CH2) and 41.0 (β-CH2). Thus, based on the above spectroscopic evidence and comparison with literature,24 compound 2 was identified as uvaretin (Figure 1, 2) previously reported from various Uvaria species,24–28 reported herein for the first time from the species.
Compound (3) was obtained from the root extract of C. burgensis as a yellow amorphous crystal. Its HRMS displayed [M+H]+ m/z at 419.3363 (calculated 419.3156 for the chemical formula C26H42O4). Its 1H NMR spectrum showed a typical AA’BB’ system at δ 7.64 (2H, dd, J = 8.8, 2.2 Hz, H-3, 6), and 7.46 (2H, dd, J = 8.8, 2.2 Hz, H-4, 5) which implied an ortho-substituted benzene ring.18 Twelve methylene protons were observed at δ 4.21 (4H, t, CH2-1, 1ʹ), 1.65 (4H, m, CH2-2, 2ʹ), 1.25 (4H, m, CH2 −3, 3ʹ), 1.29 (8H, m, H CH2 - 4, 4ʹ, 5, 5ʹ), 1.31 (4H, m, CH2 −6, 6ʹ), 1.40 (4H, m, CH2 −8, 8ʹ), along with four methyl protons at δ 0.90 (6H, m, CH3 −9, 9ʹ) and 0.89 (6H, m, CH3-7, 7ʹ). The 13C NMR, assigned with the aid of DEPT-135 and HSQC spectra, confirmed the presence of ester carbonyl carbon at δ 167.7 (C-1, 8), two sp2 methines at δ 130.9 (C-3, 6), 128.8 (C-4, 5) one sp2 quaternary carbon at δ 132.2 (C-2, 7), two methyls at δ 14.0 and 10.9 (C-7ʹ,7” and C-9ʹ,9”), O-bearing methylene group at δ 68.1 (C-1ʹ,1”) and five methylene groups at δ 23.7, 30.3, 28.9, 29.7, and 22.9. Finally, based on the above spectral evidence and comparison with literature,29 the structure of compound 3 was assigned to bis(2-ethylheptyl) phthalate (3, Figure 1) previously isolated from Cynodon dactylon,29 reported herein for the first time from Clematis species.
Compound (4) was obtained as a white powder, and its ESI-MS [M]+ m/z appeared at 426. The 1H NMR spectrum showed oxygenated sp3 methine proton at δ 3.18 (dd, J = 10.9, 5.3 Hz, 1H, H-3) and seven methyl signals at δ 1.66, 1.05, 0.96, 0.85, 0.85, 0.83 and 0.78. One terminal methylene was observed at δ 4.68 and 4.56 (dd, J = 2.6, 1.4 Hz, 2H, H-29a, 29b). Its 13C NMR and DEPT-135 spectra suggested a triterpenoid skeleton with characteristic peaks at δ 151.2 (C-20) and 109.5 (C-29) attributed to sp2 quaternary carbon and terminal sp2 methine, respectively, along with oxygenated sp3 methine peaks at δ 79.2 attributed to C-3. Consequently, the above spectroscopic evidence and comparison with literature,30,31 compound 4 was identified to be lupeol (Figure 1, 4), reported herein for the first time from the Clematis species.
Compound (5) was obtained as yellow needles with ESI-MS [M]+ m/z appeared at 246.1. Its 1H NMR spectrum showed two doublets at δ 6.26 and 7.61 (J = 9.5 Hz), and two singlets at δ 6.84 (H-8) and 6.91 (H-5) corresponding to 6,7-disubstituted coumarin.32 The presence of a doublet methyl signals at δ 0.88 (d, J = 5.3 Hz, 6H) coupled with methine at 2.02 (1H, m) and methylene protons at δ 2.02 (m, 2H) and 2.31 (m, 2H) suggested the presence of a reduced prenyl moiety. The singlet peak at δ 3.95 belongs to the methoxyl group. Its 13C NMR spectra, displayed 15 carbon signals assigned with the aid of DEPT-135. One carbonyl appeared at δ 161. 5, two oxygenated sp2 quaternary carbons at δ 150.2 and 149.7, and two sp2 quaternary carbon at δ 114.0 and 103.2. In addition, two aromatic methines at δ 111.4 and 107.5 and two olefinic methines at 143.3 and 113.3 were observed. Methoxy signal was observed at δ 56.4. Two methylene groups at δ 31.9 and 31.6, one sp3 methine at δ 31.6, and two methyl groups at δ 22.7 and 14.8 support the presence of reduced prenyl moiety. Based on the above spectroscopic data and comparison with spectral data of suberosin,32,33 compound 5 was identified as suberosin derivative (5, Figure 1), which was previously reported from Citrus grandis by Wu (1988)33 but reported herein for the first time from the species.
Antimicrobial Study
The antimicrobial activity (Table 1) of dichloromethane root extract of U. scheffleri and isolated compounds 1 and 4–5 were tested in vitro against S. aureus and E. coli as well as a fungal strain (C. albicans). The results displayed dichloromethane extract displayed potential antibacterial activity against S. aureus (MIC 6.25 µg/mL) compared with gentamicin (5 µg/mL against S. aureus). The antibacterial activity of dichloromethane root extract of U. scheffleri showed strong activity; whereas, the isolated compound N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol (1) was inactive, which might be accounted by synergic effects of compounds within the extract or bioactive constituents which are not identified by the present study might be responsible for the observed activity. Antimicrobial assay of rest compounds (4) and (5) showed that (4) and (5) displayed weak activity with respect to gentamicin and fluconazole. However, the crude extract and isolated compound were not shown any effect on the tested fungal strain. The findings of this study support the traditional uses of the root decoction of U. scheffleri for the treatment of infectious diseases caused by gram-positive bacteria strains such as pneumonia, asthma, and sore throat.8
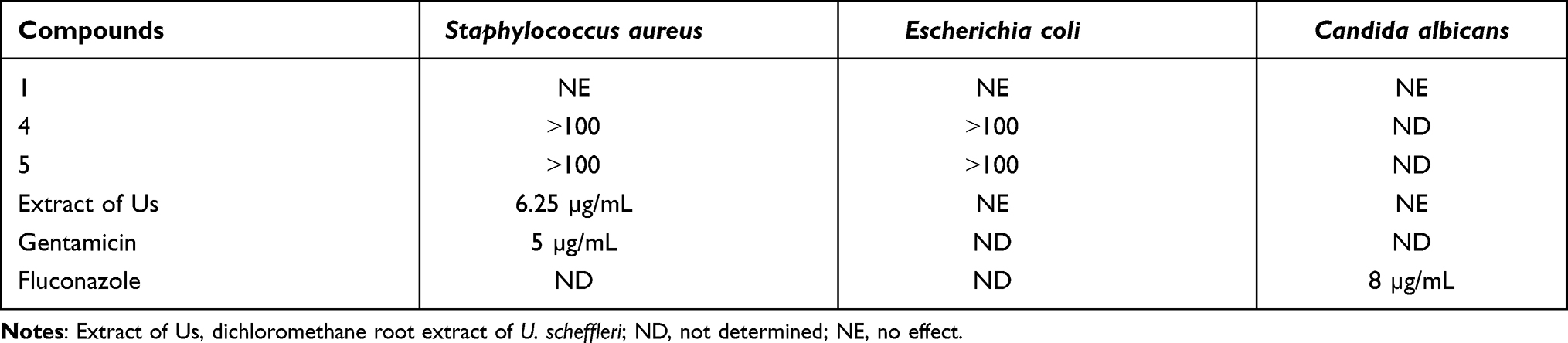 |
Table 1 Antimicrobial Activity of Compounds Against Selected Microorganisms (MIC in µg/mL) |
Molecular Docking Studies
Results obtained from the molecular docking study demonstrated that among the isolated N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol (1), lupeol (4), and coumarin (5) showed a strong binding affinity towards E. coli DNA gyrase B, with binding energy value ranging from −6.9 to −6.0 kcal/mol, with respect to ciprofloxacin (−7.2 kcal/mol) (Table 2, Figure 3). Compounds (1, 4, and 5) showed two hydrogen bond interactions with (Arg-76, Glu-50), (Ans-46, Glu-50), and (Asp-73, Thr-156) protein residue, respectively. Compounds 3 showed one hydrogen bond interaction with active site amino acid residue Asn-46 at a distance of 1.5 Å. Hydrophobic interactions were observed for compound 1 with Ile-78, Pro-79 and Ile-94, compound 3 with Asp-49, Val-43, Val-71, Val-167, Ile-78, and Ile-94, lupeol (4) with Pro-79, and compound 5 with Val-43, Ile-78, and Ile-167, suggesting the compounds may act as inhibitors of DNA gyrase B enzyme, from the docked ligands, lupeol (4) and coumarin (5) showed better binding energy and amino acid interactions (−6.6 and −6.9 kcal/mol) compared to ciprofloxacin (−7.2 kcal/mol).
 |
Table 2 Molecular Docking Value of Compounds 1, 3–5 Against E. coli DNA gyrase B (PDB ID: 6F86) |
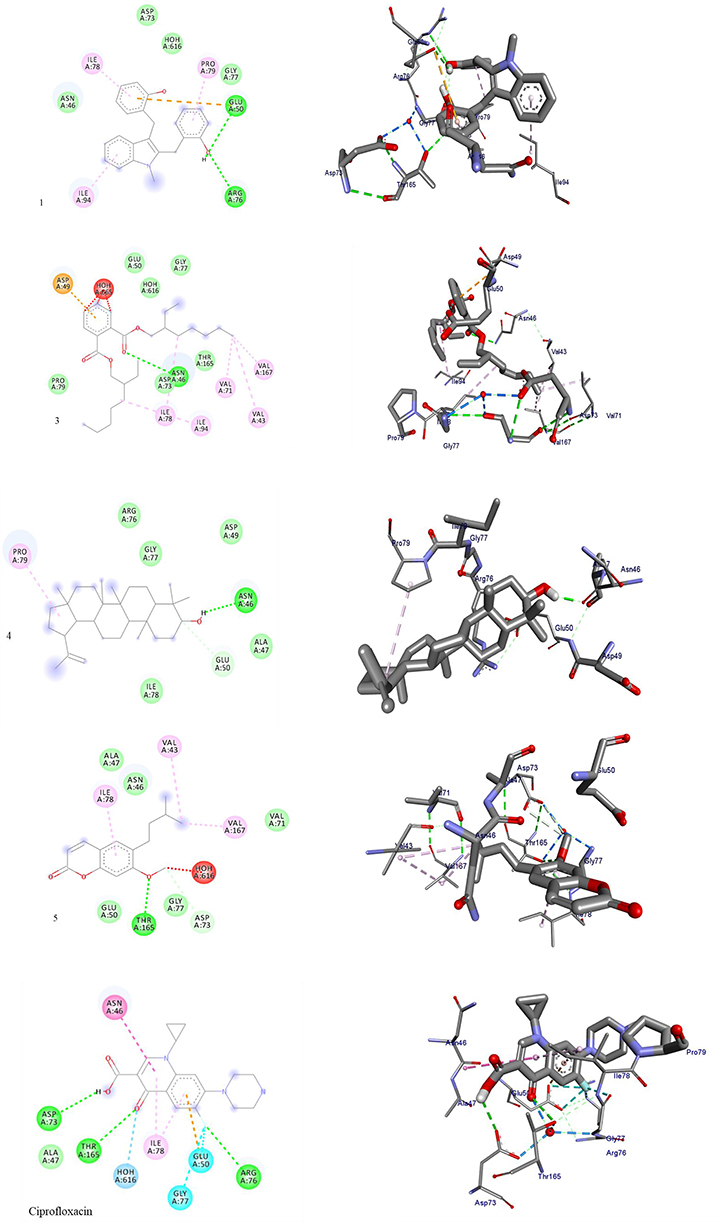 |
Figure 3 The 2D and 3D binding interactions compounds (1, 3–5) and standard ciprofloxacin against DNA gyrase B (PDB ID: 6F86), respectively. Hydrogen bonds between compounds and amino acids are shown as green dash lines, and hydrophobic interactions are shown as pink lines. |
On the other hand, compounds (1, 3–5) displayed a strong binding affinity for target protein topoisomerase IIα (Table 3, Figure 4) ranging from −5.9 to −5.3 kcal/mol compared to vosaroxin (−6.2 kcal/mol). N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol (1) and coumarin (5) showed higher binding affinity −5.4 and −5.9 kcal/mol, similar residual and DNA interaction profile with amino acid residues Ser-480, Glu-477, Asp-577, and Asp-479, and nucleic acid residues DC-8, DT-9, and DG-10 compared to vosaroxin (−6.2 kcal/mol). The results obtained suggest that N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol (1) and coumarin (5) are potential topoisomerase II α inhibitors and might be used as anticancer agents.
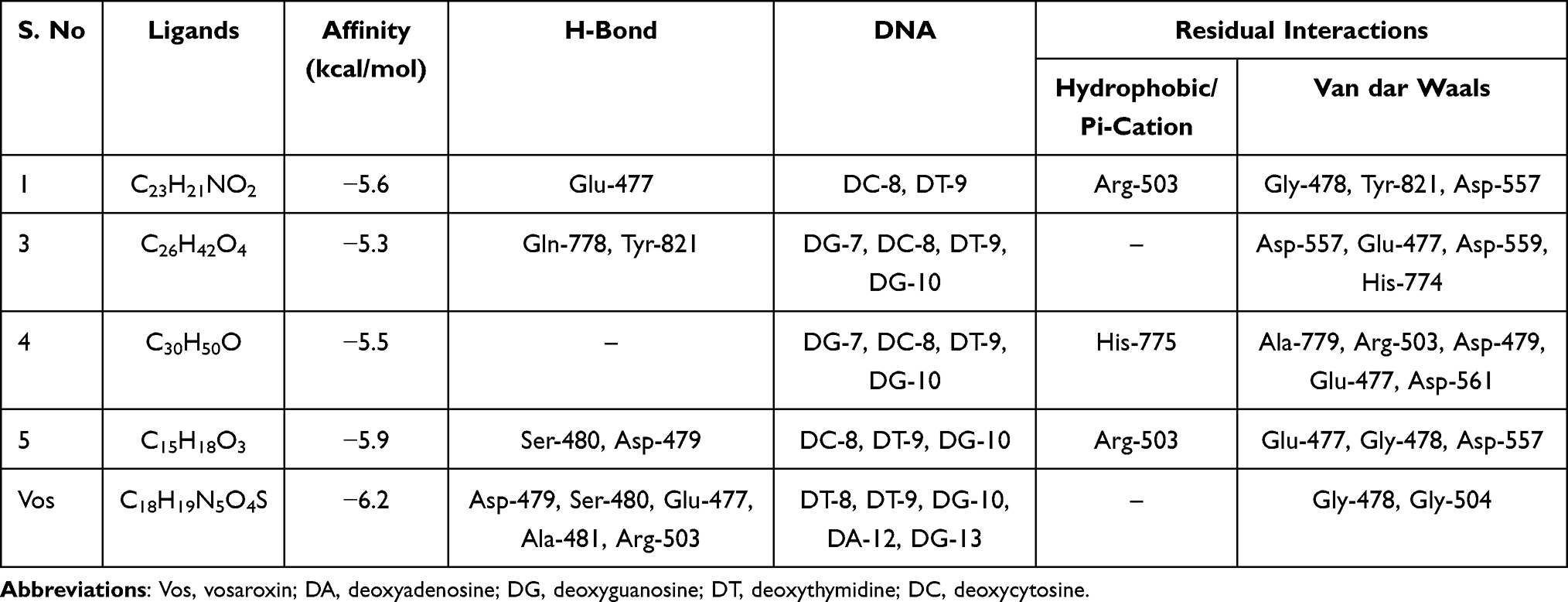 |
Table 3 Molecular Docking Value of Compounds 1, 3–5 Against Human DNA Topoisomerase II α (PDB ID 3QX3) |
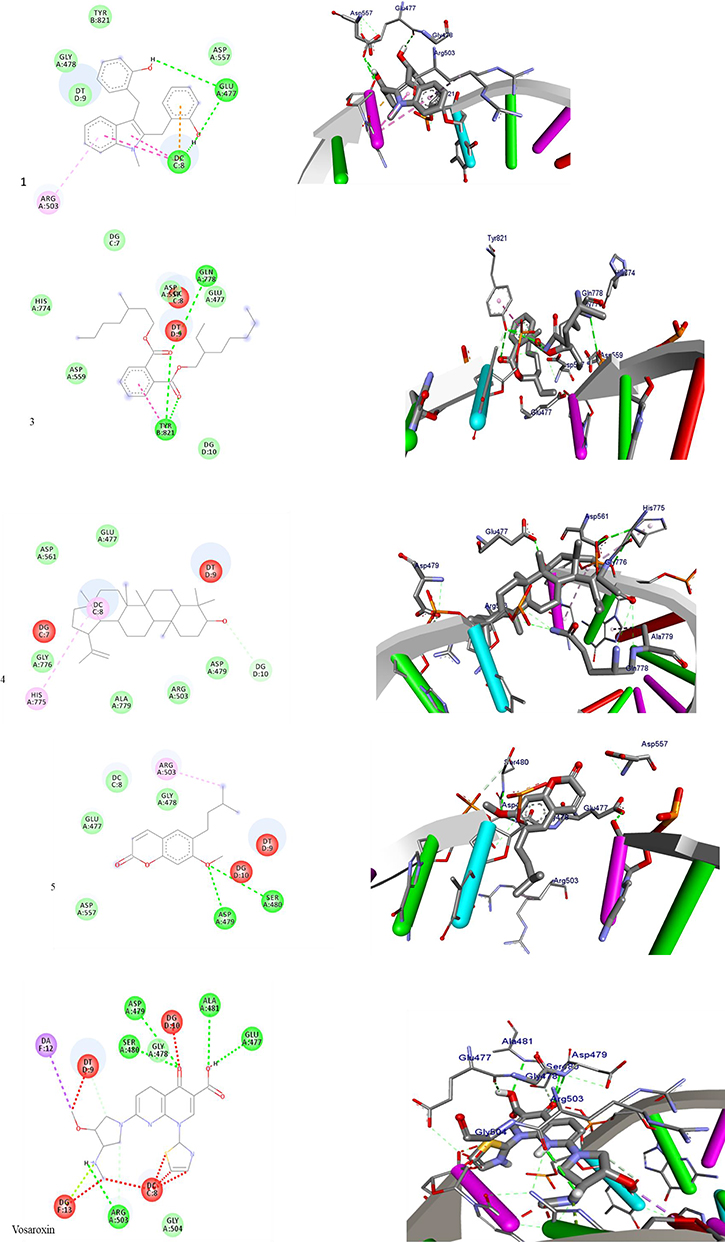 |
Figure 4 The 2D and 3D binding interactions of compound 1, 3–5 and standard vosaroxin against human topoisomerase II α (PDB ID: 3QX3), respectively. Hydrogen bonds between compounds and amino acids are shown as green dash lines, and hydrophobic interaction is shown as pink lines. |
In silico Pharmacokinetics (Drug-Likeness) and Toxicity Analysis
The structures of isolated compounds (1, 3–5) were converted to their canonical simplified molecular-input line-entry system (SMILE) and submitted to the SwissADME tool to estimate in silico pharmacokinetic parameters (drug-likeness properties) according to ‘Lipinski’s rule of five.19 Lipinski’s rule of five implies that the drugs and/or candidates should obey the five-parameter rule, which states that hydrogen-bond donors (HBDs) should be less than 5, hydrogen-bond acceptors (HBAs) should be less than 10, molecular mass should be less than 500 Da, log P should not be less than 5, and total polar surface area (TPSA) should not be greater than 140Å. Drug-likeness is a prediction that screens whether a particular organic molecule has properties consistent with being an orally active drug.19 In the present study, the SwissADME prediction revealed the studied compounds obeyed Lipinski’s rule of five and are likely to be orally active except compound 3 (Table 4). The hydrogen bonding potential and bioavailability of molecules are closely correlated to the TPSA value. Thus, the TPSA value of the studied compounds was noticed in the range from 20.23 to 82.69 Å and is well below the limit of 140 Å. The calculated number of rotatable bonds (NRB) values for the isolated N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol (1), lupeol (4), and coumarin (5) are less than 10, which indicated the compounds are conformationally stable.34
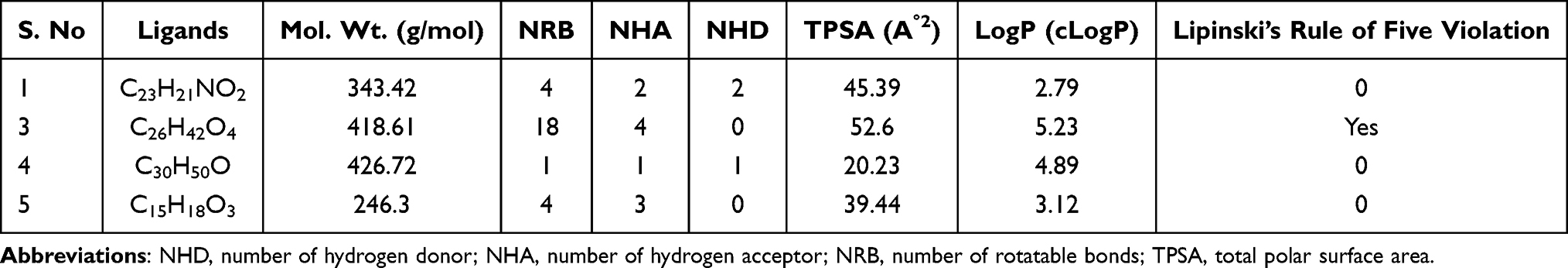 |
Table 4 Drug-Likeness Predictions of Compounds 1, 3–5, Computed by SwissADME |
ADMET Properties
The absorption, distribution, metabolism, excretion, and toxicity (ADMET) studies of isolated compounds (1, 3–5) were predicted using Swiss ADMET. The skin permeability value (Kp) in cm/s indicates the skin absorption of molecules. In silico, the skin permeability, Kp, values of all compounds ranged from −1.90 to –4.96 cm/s suggesting low skin permeability and are within the range of broad-spectrum antibiotic ciprofloxacin (−9.09 cm/s) and under the clinical trial anticancer agent vosoroxin (−9.43 cm/s). Additionally, gastrointestinal (GI) and blood–brain barrier (BBB) permeation indicate the absorption and distribution of drug molecules.34,35 The in-silico prediction results of absorption, distribution, metabolism and excretion (ADME) of the compounds (1, 3–5) studied are presented in (Table 5). The Swiss ADME prediction parameters indicated that N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol (1) and coumarin (5) showed high gastrointestinal (GI) absorption, whereas (3 and 4) displayed low absorption. In the same way, Swiss ADME prediction showed that compounds 3 and 4 showed blood–brain barrier (BBB) permeation, but compounds (1 and 5) were not. Moreover, a range of cytochromes (CYP’s) regulates the drug metabolism, in which CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 are vital for the biotransformation of drug molecules.36 Thus, in silico SwissADME prediction, compound (1) inhibited all cytochromes except CYP2C9 and substrate of permeability glycoprotein (P-gp). Compound 3 inhibited CYP2C9 and CYP3A4 whereas compound 5 inhibited CYP1A2, CYP2C19, CYP2C9, and CYP2D6 but both are not substrate of permeability glycoprotein (P-gp). However, compounds (4) neither cytochromes inhibitor nor a substrate of permeability glycoprotein (P-gp).
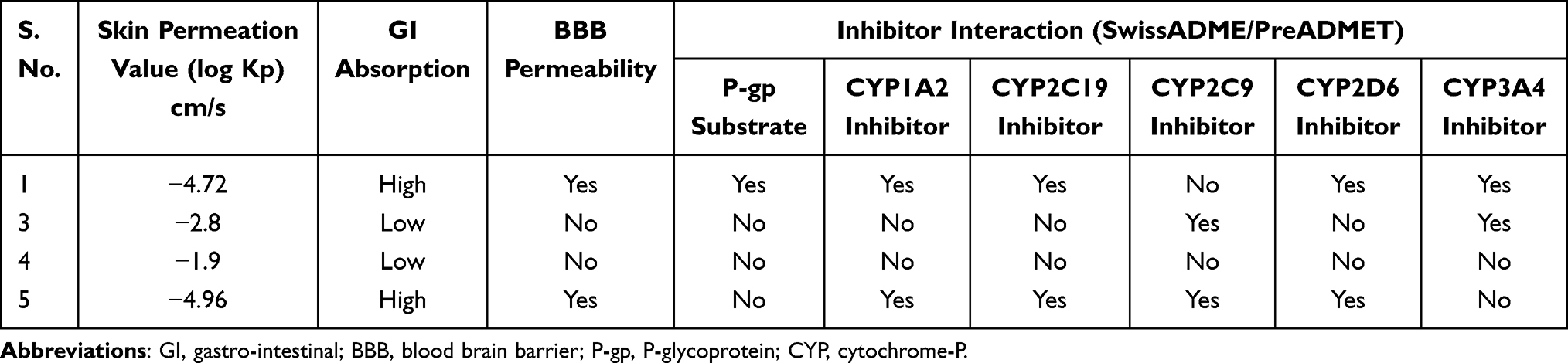 |
Table 5 ADME Predictions of Compounds 1, 3–5, Computed by SwissADME and PreADMET |
Acute toxicity prediction results, such as toxicity class classification and LD50 values, predict that none of the isolated compounds has acute toxicity. The toxicological prediction gives results of endpoints such as hepatotoxicity, carcinogenicity, mutagenicity, and cytotoxicity. The studied compounds were predicted to be non-carcinogenic and non-irritant. However, all compounds were inactive to hepatotoxicity, mutagenicity, and cytotoxicity. Pre-ADMET and OSIRIS property explorer prediction analyses are shown in Table 6. Hence, based on ADMET prediction analysis, isolated compounds may be good candidates in this investigation except compound (3).
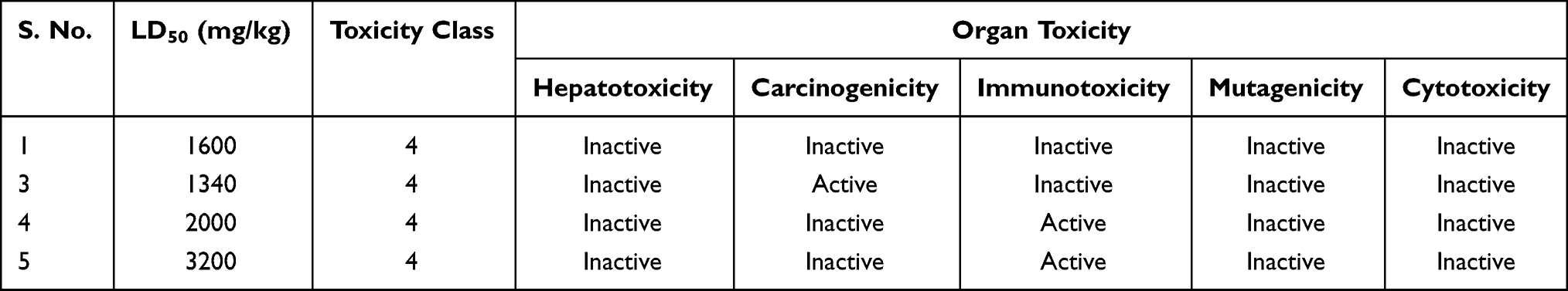 |
Table 6 Toxicity Prediction of Compounds 1, 3–5 Computed by PreADMET and OSIRIS Property Explorer |
DFT Analysis
The DFT (density functional theory) study is an important approach to explore the relationship between geometry and electronic properties of chemical compounds.37 Therefore, we report herein the DFT calculations with the basis sets B3LYP/6-31 G (d,p) involving optimized geometries (Figure 5), molecular electrostatic potential (MEP), Mulliken’s atomic charges, highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO) and energy gap for isolated compounds (1, 3–5). The determination of the MEP region is the best fit for identifying sites for intra- and intermolecular interactions (Figure 6).37 Red/Yellow regions indicate negative electrostatic potentials and the blue region shows positive, and the green color designates potential neutral region. The Mulliken’s atomic charges (Figure 7) of the DFT calculation revealed a charge distribution in individual atoms. The charges on carbon atoms exhibited either positive or negative values. All hydrogen atoms displayed a net positive charge and act as acceptor atoms. All oxygen atoms of the optimized compounds were shown to have a negative charge, which acts as donor atoms. The HOMO, LUMO, and energy gap (∆E) of the studied compounds are presented in Table 7. The results show that all studied compounds exhibited the least energy gap (∆E), suggesting high chemical reactivity and considerable intramolecular charge transfer from an electron donor (HOMO) to electron acceptor (LUMO) groups. Additionally, the isolated compounds (1, 3–5) have large electronegativity (χ, eV)), global softness (σ, eV-1), and global electrophilicity (ω, eV) (Table 7). Based on the results, compounds (1, 3–5) have good bioactivity.
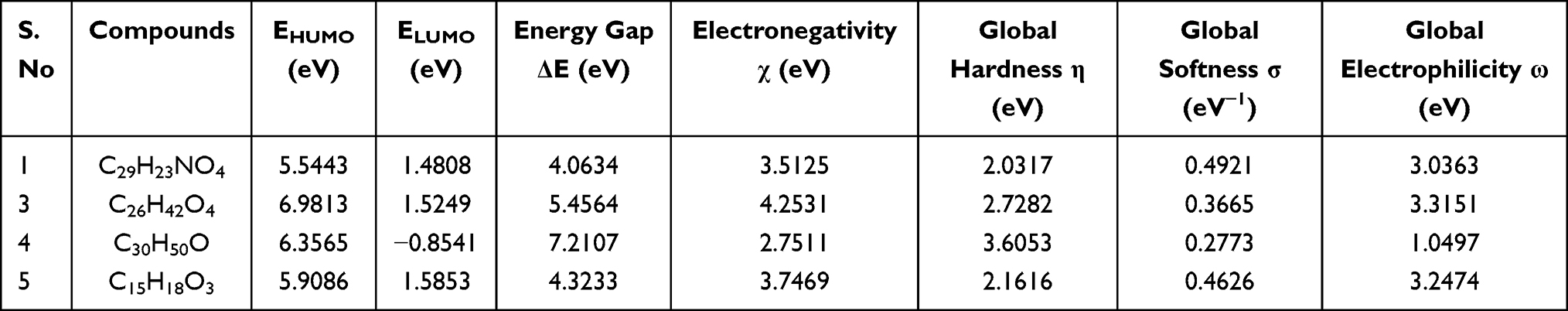 |
Table 7 The Various Quantum Chemical Parameters of Isolated Compounds |
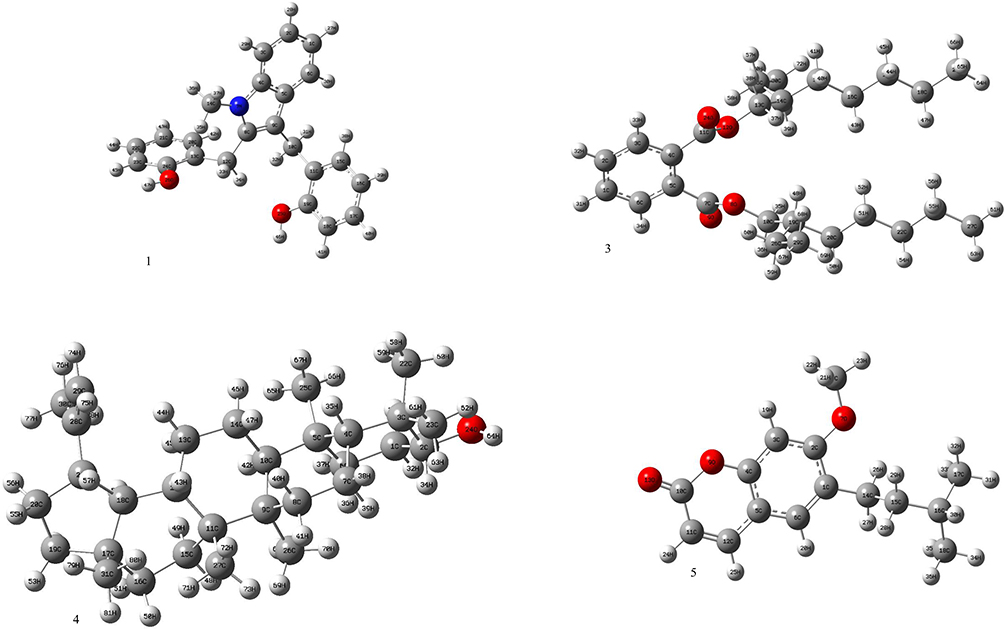 |
Figure 5 The optimized structures of compounds 1, 3–5. |
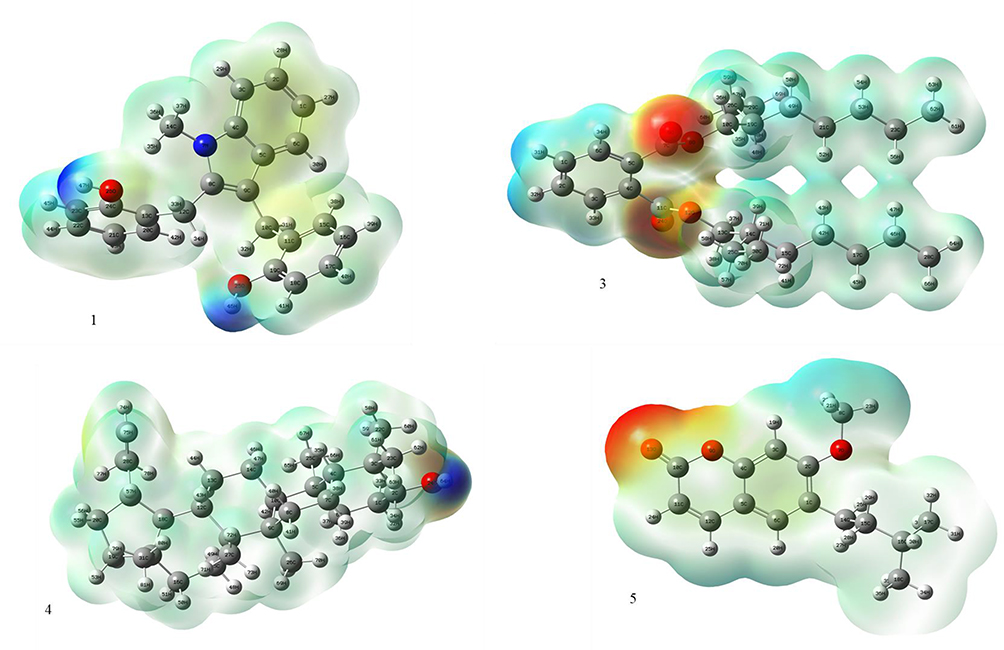 |
Figure 6 Molecular electrostatic potential surface of compounds 1, 3–5. |
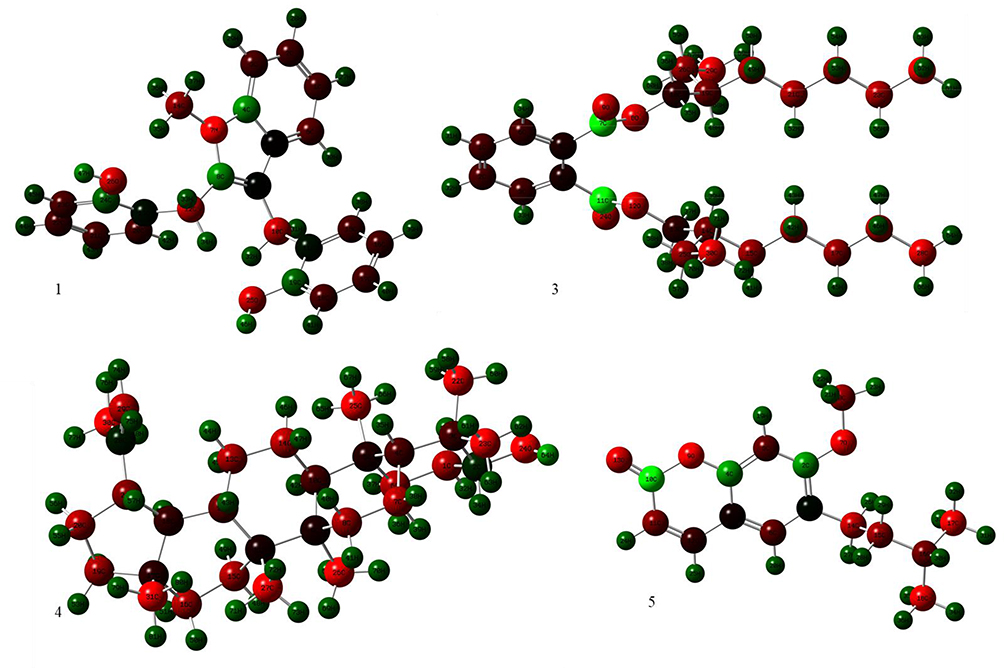 |
Figure 7 The DFT calculated Mulliken’s atomic charges of compounds 1, 3–5. |
Conclusion
U. scheffleri, C. burgensis, and E. schimperiana are traditionally used to treat various ailments in Ethiopia such as cough, tuberculosis, asthma, sore throat, fever, and skin infections. The present study identified five compounds from the root extracts aforementioned three medicinal plants of which N-methyl-2,3-bis(2-hydroxybenzyl)-1Н-indol (1) isolated herein for the first time. Dichloromethane root extract of U. scheffleri showed potent antibacterial activity against S. aureus (MIC = 6.25 µg/mL) compared with gentamicin (5 µg/mL against S. aureus) and fluconazole (8 µg/mL against C. albicans).
In silico molecular docking analysis of isolated compounds (1, 3–5) against E. coli DNA gyrase B and human topoisomerase II α revealed promising scoring pose (lowest energy) with a value ranging from −6.9 to −6.0 kcal/mol and −5.3 to −5.9 kcal/mol, compared to ciprofloxacin (−7.2 kcal/mol) and vosaroxin (−6.2 kcal/mol), respectively. Lupeol (4) and prenylated coumarin (5) showed better binding energy (−6.6 and −6.8 kcal/mol, respectively) with that of ciprofloxacin (−7.2 kcal/mol), suggesting this compound could be considered as antibacterial drug lead against E. coli. Coumarin (5) displayed comparable binding affinity (- 5.9 kcal/mol) with that of vosaroxin (−6.2 kcal/mol) suggesting it can be potential topoisomerase II α inhibitor and might be used as an anticancer agent. The findings of the present work support previous reports that revealed suberosin coumarin (close derivative compound 5) is a potential candidate for killing MCF-7 breast cancer cells as well as increasing the therapeutic efficiency of radiotherapy or hyperthermia.38
DFT calculations suggested that studied compounds showed the lowest gap energy and chemically reactive. In addition, ADMET (drug-likeness) studies showed the highest drug-likeness properties of the studied compounds, except compound 3, which suggests these compounds can act as a drug and exhibit remarkable biological activities. The results obtained from molecular docking, drug-likeness properties, ADMET analysis, and DFT calculation are in good agreement with those obtained from experimental studies suggesting the potential use of the isolated compounds as potential drug leads, which corroborate with the traditional uses of the roots of U. scheffleri, C. burgensis, and E. schimperiana.
Acknowledgments
The authors are thankful for the facilities provided by the University of Valencia, Spain, during spectroscopic analysis and antimicrobial study. Adama Science and Technology University and Wolaita Sodo University are gratefully acknowledged for giving Ph.D. study opportunity and leave of absence, respectively, for Mr. Mathewos Anza.
Disclosure
The authors declare that they have no conflicts of interest regarding this work or the publication of this paper.
References
1. Jamshidi-Kia F, Lorigooini Z, Amini-Khoei H. Medicinal plants: past history and future perspective. J HerbMed Pharmacol. 2018;7:1–7. doi:10.15171/jhp.2018.01
2. Schenone M, Vlado D, Bridget K, et al. Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol. 2013;9:232–240. doi:10.1038/nchembio.1199
3. Gajdács M, Urbán E, Stájer A, et al. Antimicrobial resistance in the context of the sustainable development goals: a brief review. Eur J Investig Heal Psychol Educ. 2021;11:71–82.
4. Jones KE, Patel NG, Levy MA, et al. Global trends in emerging infectious diseases. Nature. 2008;451:990–993. doi:10.1038/nature06536
5. Cassell GH, Mekalanos J. Development of antimicrobial agents in the era of new and reemerging infectious diseases and increasing antibiotic resistance. JAMA. 2001;285:601–605. doi:10.1001/jama.285.5.601
6. Blaser MJ. Who are we? Indigenous microbes and the ecology of human diseases. EMBO Rep. 2006;7(10):956–960. doi:10.1038/sj.embor.7400812
7. Seo M, Hyun KS, Yoochan M, et al. Development of natural compound molecular fingerprint (NC-MFP) with the dictionary of natural products (DNP) for natural product-based drug development. J Cheminformatic. 2020;12:1–17. doi:10.1186/s13321-020-0410-3
8. Wanzala W, Marcellina SS, Joseck OA. A survey of the applications and use of ethnomedicinal plants and plant products for healthcare from the Ukambani Region in Eastern Kenya. Indian J Ethnophyopharmaceutical. 2016;2:6–58.
9. Kuma M, Shibru S. Floristic composition, vegetation structure and regeneration status of kimphe lafa natural forest. J Botany. 2015;5:19–32.
10. Hawaze S, Deti H, Suleman S. In vitro antimicrobial activity and phytochemical screening of clematis species indigenous to Ethiopia. Indian J Pharm Sci. 2012;74:29–35. doi:10.4103/0250-474X.102540
11. Shaker KH, Al Shehri BM, Oteef MDY, Mahmoud MF. Antioxidant compounds from Euphorbia schimperiana scheele in Aseer region, Saudi Arabia. Int J Pharm Sci Rev Res. 2015;32:117–122.
12. Valgas C, Simone MDS, Elza F, et al. Screening methods to determine antibacterial activity of natural products. Braz J Microbiol. 2007;38:369–380. doi:10.1590/S1517-83822007000200034
13. Trott O, Arthur JO. Software news and update autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461.
14. Seeliger D, Bert L, De G. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. 2010. J Comput Aided Mol Des. 2010;24:417–422. doi:10.1007/s10822-010-9352-6
15. Matin MM, Uzzaman M, Chowdhury SA. et al. In vitro antimicrobial, physicochemical, pharmacokinetics and molecular docking studies of benzoyl uridine esters against SARS-CoV-2 main protease. J Biomol Struct Dyn; 2020:1–13. doi:10.1080/07391102.2020.1850358
16. Matin MM, Chakraborty P, Alam MS, et al. Novel mannopyranoside esters as sterol 14alpha-demethylase inhibitors: synthesis, PASS predication, molecular docking, and pharmacokinetic studies. Carbohydr Res. 2020;496:108130. doi:10.1016/j.carres.2020.108130
17. Matin MM, Bhattacharjee SC, Chakraborty P, et al. Synthesis, PASS predication, in vitro antimicrobial evaluation and pharmacokinetic study of novel n-octyl glucopyranoside esters. Carbohydr Res. 2019;485:1–29. doi:10.1016/j.carres.2019.107812
18. Narramore S, Clare EM, Stevenson AM, et al. New insights into the binding mode of pyridine-3-carboxamide inhibitors of E. coli DNA gyrase. Bioorg Med Chem. 2019;27:3546–3550. doi:10.1016/j.bmc.2019.06.015
19. Lipinski CA, Franco L, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. doi:10.1016/S0169-409X(96)00423-1
20. Oduselu GO, Olayinka OA, Yvonne UA, et al. Homology modelling and molecular docking studies of selected substituted benzo [d] Imidazol-1-Yl) Methyl) benzimidamide scaffolds on plasmodium falciparum adenylosuccinate lyase receptor. Bioinform Biol Insights. 2019;13:1–10. doi:10.1177/1177932219865533
21. Behrouz S, Navid MSR, Taghavi BS, et al. Design, synthesis, and in silico studies of novel eugenyloxy propanol azole derivatives having potent antinociceptive activity and evaluation of their β -adrenoceptor blocking property. Mol Divers. 2019;23:147–164. doi:10.1007/s11030-018-9867-7
22. Sulpizi M, Folkers G, Rothlisberger U, et al. Applications of density functional theory-based methods in medicinal chemistry. Quant Struct -Act Relat. 2002;21:173–181. doi:10.1002/1521-3838(200207)21:2<173::AID-QSAR173>3.0.CO;2-B
23. Abu-melha S. Design, synthesis and DFT/DNP modeling study of new 2-amino-5-arylazothiazole derivatives as potential antibacterial agents. Molecules. 2018;23:434. doi:10.3390/molecules23020434
24. Muhammad I, Waterman PG. Chemistry of the Annonaceae, part 18. benzylated indoles and dihydrochalcones in Uvaria angolensis from Tanzania. J Nat Prod. 1985;51:571–580. doi:10.1021/np50040a009
25. Cole JR, Torrance SJ, Wiedhopf RM, et al. Uvaretin, a new antitumor agent from Uvaria acuminata (Annonaceae). J Org Chem. 1976;41:1852–1855. doi:10.1021/jo00872a037
26. Lasswell WL, Hufford CD. Cytotoxic C-benzylated Flavonoids from Uvaria chamae. J Org Chem. 1977;42:1295–1302. doi:10.1021/jo00428a006
27. Parmar VS, Malhotra A, Singh SK, et al. Novel constituents of Uvaria species. Nat Prod Rep. 1994;11:219–224. doi:10.1039/np9941100219
28. Ichimaru M, Nakatani N, Takahashi T, et al. Cytotoxic C-benzylated dihydrochalcones from Uvaria acuminata. Chem Pharm Bull. 2004;52:138–141. doi:10.1248/cpb.52.138
29. Allahresani A, Fatemeh G, Mohammad AN, et al. Isolation and characterization of bis(2-ethylheptyl) phthalate from Cynodon dactylon (L.) and studies on the catalytic activity of its Cu(II) complex in the green preparation of 1,8-Dioxo-Octahydroxanthenes. Iran Chem Commun. 2020;8:212–227.
30. Jain PS, Bari BS. Isolation of lupeol, stigmasterol, and campesterol from petroleum ether extract of woody stem of Wrightia Tinctoria. Asian J Plant Sci. 2010;9:163–167. doi:10.3923/ajps.2010.163.167
31. Laghari AH, Shahabuddin M, Aisha N, et al. Alhagi Maurorum: a convenient source of lupeol. Ind Crops Prod. 2011;34:1141–1145. doi:10.1016/j.indcrop.2011.03.031
32. El-Shafae AM, Ibrahim MA. Bioactive kaurane diterpenes and coumarins from Fortunella margarita. Pharmazie. 2003;58:143–147.
33. Wu TS. Alkaloids and coumarins of Citrus Grandis. Phytochemistry. 1988;27:3717–3718. doi:10.1016/0031-9422(88)80815-X
34. Abdelrheem DA, Rahman AA, Elsayed KNM, et al. Isolation, characterization, in vitro anticancer activity, DFT calculations, molecular docking, bioactivity score, drug-likeness and ADMET studies of eight phytoconstituents from brown alga sargassum platycarpum. J Mol Struct. 2021;1225:12245. doi:10.1016/j.molstruc.2020.129245
35. Eswaramoorthy R, Hailekiros H, Kedir F, et al. In silico molecular docking, DFT analysis and ADMET studies of carbazole alkaloid and coumarins from roots of clausena anisata: a potent inhibitor for quorum sensing. Adv Appl Bioinforma Chem. 2021;14:13–24.
36. Das P, Majumder R, Mahitosh M, et al. In-silico approach for identification of effective and stable inhibitors for COVID-19 main protease (Mpro) from flavonoid based phytochemical constituents of Calendula officinalis. J Biomol Struct Dyn. 2020;38:1–16.
37. Mumit MA, Pal TK, Alam MA, et al. DFT studies on vibrational and electronic spectra, HOMO–LUMO, MEP, HOMA, NBO and molecular docking analysis of benzyl-3-N-(2,4,5-trimethoxyphenylmethylene) hydrazinecarbodithioate. J Mol Struct. 2020;1220:128715. doi:10.1016/j.molstruc.2020.128715
38. Nodooshan SJ, Amini P, Ashrafizadeh M, et al. Suberosin attenuates the proliferation of MCF-7 breast cancer cells in combination with radiotherapy or hyperthermi. Curr Drug Res Rev. 2020. doi:10.2174/2589977512666201228104528
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.