Back to Journals » ImmunoTargets and Therapy » Volume 4
Antigen-based immunotherapy for autoimmune disease: current status
Received 22 August 2014
Accepted for publication 26 September 2014
Published 16 December 2014 Volume 2015:4 Pages 1—11
DOI https://doi.org/10.2147/ITT.S49656
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Darren Lowell Hirsch, Punita Ponda
Division of Allergy and Immunology, North Shore-Long Island Jewish Health System/Hofstra North Shore-LIJ School of Medicine, New Hyde Park, NY, USA
Abstract: Autoimmune diseases are common chronic disorders that not only have a major impact on the quality of life but are also potentially life-threatening. Treatment modalities that are currently favored have conferred significant clinical benefits, but they may have considerable side effects. An optimal treatment strategy for autoimmune disease would specifically target disease-associated antigens and limit systemic side effects. Similar to allergen-specific immunotherapy for allergic rhinitis, antigen-specific immunotherapy for autoimmune disease aims to induce immune deviation and promote tolerance to specific antigens. In this review, we present the current status of studies and clinical trials in both human and animal hosts that use antigen-based immunotherapy for autoimmune disease.
Keywords: autoimmune disease, immunotherapy, antigen, diabetes, rheumatoid arthritis, multiple sclerosis
Introduction
Autoimmune disorders affect over 23 million Americans, with an estimated prevalence of 7.6%–9.4% of the population.1 These diseases can be devastating, as they are chronic and potentially life-threatening. Autoimmune disease is one of the top ten leading causes of death in women younger than 64 years. The National Institutes of Health estimates the direct health care costs for autoimmune diseases to be approximately $100 billion annually.2
Treatment for autoimmune diseases has improved over the past several decades, but the optimal treatment for these conditions remains a work in progress. Type 1 diabetes (T1D), caused by autoimmune destruction of pancreatic β-cells, has been increasing in prevalence in recent years3 and can lead to many complications. Although intensive insulin therapy reduces the risks of complications from T1D, these risks are not eliminated. The current treatment options for rheumatoid arthritis (RA), systemic lupus erythematosis (SLE), and multiple sclerosis (MS), which include physical therapy, nonsteroidal anti-inflammatory drugs, corticosteroids, disease-modifying anti-inflammatory drugs, anti-cytokine therapies, monoclonal antibodies, biological inhibitors of T-cell function, and B-cell inhibition,4 have had a significant impact on the quality of life of millions of patients but may have considerable drawbacks. Current treatment options are generally nonspecific immunosuppressants, and medications ranging from cyclophosphamide, glucocorticoids, and azathioprine to biologic therapies have been associated with an increased risk for infections5,6 as well as several other adverse effects including hepatotoxicity, gastrointestinal perforation, nausea, diarrhea, and fatigue.7 Autoimmune diseases typically require lifelong therapy, as current drugs do not induce the restoration of immune tolerance to self-antigens.8 The ideal treatment would target disease-associated antigens rather than act as a global immunosuppressant, thereby limiting side effects as well as focusing on the underlying cause of the disease.
Autoimmune disorders are caused by physiologic immune responses to autoantigens (Table 1).1,9–12 In diseases where the pathophysiology is understood and the culprit autoantigens are recognized, these pathways can theoretically be manipulated to induce immune tolerance to self-antigens. There have been considerable efforts to use autoantigen-based immunotherapy to modify the immune response,13 and studies in several animal models that simulate chronic inflammatory conditions have found that controlled administration of autoantigens can provide protection from autoimmune disease.14 Antigen-specific immunotherapy (ASI) for autoimmune disease has the potential to control the disease much like allergen-specific immunotherapy has been used to treat allergic diseases. However, there are fundamental differences between allergen-specific immunotherapy and ASI, including that allergic diseases consist of Th2 dominant responses whereas autoimmune diseases consist of Th1 and Th17 dominant responses. While the promising animal studies of ASI have not yet been translated into clinical efficacy, there have been encouraging advances.
 | Table 1 Representative autoantigens involved in autoimmune disease |
Immunological changes induced by immunotherapy
ASI for autoimmune disease is conceptually similar to allergen-specific immunotherapy, which has been used with good (and potentially curative) effect for >100 years. ASI is thought to work through repeatedly exposing the immune system to increasing amounts of an allergen, which results in immune deviation (alteration in cytokine production) upon exposure to allergens from a Th2 response to a Th1 response as well as the induction of FOXP3+CD4+CD25+ regulatory T-cells (Tregs) that secrete interleukin (IL)-10 and transforming growth factor (TGF)-β.15,16 Antigen-specific therapy for autoimmune disease similarly aims to take advantage of immune deviation and the induction of Tregs17 in order to promote autoantigen-specific tolerance. The long-term disease modification and safety profile that is seen with allergen immunotherapy provides hope that a similar therapeutic modality could be effective for autoimmune diseases with known autoantigens.
In contrast to allergic diseases that are typically dominated by Th2 responses, autoimmune disorders are usually associated with Th1 and Th17 responses targeted against self-antigens.18 However, Th1, Th2, Th9, and Th17 cells all secrete IL-10 in response to chronic exposure to an antigen.19 Autoimmune diseases could potentially be treated by eliminating pathogenic Th1 and Th17 cells that are specific for autoantigens or by blocking the immune response directed by autoantigen-specific T-cells. Through repeated exposure to antigens, both allergen immunotherapy and autoantigen specific-immunotherapy aim to manipulate this phenomenon to promote tolerance.
Studies in animals have demonstrated the induction of Tregs and immune deviation with increased production of IL-4, IL-10, and TGF-β after administration of autoantigenic peptides.14 In humans, some studies have shown immune deviation consistent with Treg generation, peptide-specific IL-10, and increased levels of IFNγ, IL-5, IL-13, IL-17, IL-6, tumor necrosis factor-α (TNFα), and FoxP3 after administration of autoantigens.20,21 Yet, other studies demonstrated no clear biological effects after ASI.22
Another method to induce immunological changes is via manipulation of dendritic cells (DCs). DCs are essential to the induction phase of the immune response and are therefore critically important in determining whether a response toward an antigen will be inflammation or tolerance. DCs can influence if naïve T-cells will undergo deletion, anergy, or differentiation.23 Deletion and anergy of T-cells can occur when DCs present the antigen without costimulation. DC responses to a specific antigen are influenced by the tissue environment and innate stimuli associated with that antigen. Emerging therapies are beginning to target DCs to induce tolerance.
In this manuscript, we review the available data on autoantigen immunotherapy and DC immunotherapy.
Safety concerns of ASI
One of the most important elements in designing novel therapeutic modalities is the safety profile. The major areas of concern for the use of ASI are inciting the progression of disease, inducing hypersensitivity reactions, and precipitating other autoimmune disorders. This concern is reviewed in a disease-specific way in this manuscript.
ASI for type 1 diabetes
T1D is one of the most studied autoimmune diseases, as autoantigens are well defined and mouse models (non-obese diabetic [NOD] mice) are available. In T1D, pancreatic β cells (which normally secrete insulin) are damaged by cytotoxic lymphocytes as well as proinflammatory helper T-cells (Th1 cells) that are primed against autoantigens such as GAD65 or preproinsulin by inflammatory DCs. The goal of ASI for T1D is to induce tolerance to known autoantigens in order to prevent the immune response against these antigens and avoid destruction of pancreatic β cells. ASI can achieve this goal through deletion of harmful T-cells and induction of regulation through either Tregs or immune deviation. Treg induction may also result in linked suppression, in which Tregs induced to one autoantigen also regulate response to other autoantigens presented by the same DC.
One complicating factor in potentially treating T1D with immunotherapy is that T1D does not typically manifest itself until <10%–20% of functional β cells remain in the pancreas.24 At this point, the path to organ damage may already be under way. Therefore, strategies for ASI have not only been attempted immediately after diagnosis of T1D but also prior to clinical onset but after the appearance of islet autoantibodies (secondary prevention). Prevention of T1D with immunotherapy is clearly the more desirable path. However, this strategy relies on an accurate means of identifying at-risk patients as well as a safe intervention that would not cause any harm to patients who otherwise may not have developed the disease. Models have been developed to identify patients at risk of developing T1D based on screening for islet autoantibodies and genetic markers in the human leucocyte antigen (HLA) region, as well as metabolic assessments.24 Studies have also been conducted on patients with recent onset T1D in the hope of preventing progression of the disease or even possibly reversing the disease. While this approach does not have the potential upside of disease prevention, it may be more feasible, as patients are easily identified and there has been evidence that it may be possible to reverse clinically established T1D.24
A variety of approaches have been evaluated for immunotherapy for T1D, some of which have shown promising results in mouse models and Phase I human studies. Unfortunately, few of these approaches have demonstrated efficacy in Phase II trials, raising significant concern that ASI for treatment of T1D may not be possible. However, Phase III studies are under way.
The practice of ASI in treatment of T1D is confounded by the presence of several known autoantigens. Below, we outline the approaches that have been taken using these autoantigens.
ASI using insulin as the target antigen
Insulin and proinsulin molecules are thought to be important autoantigens in the initiation of the autoimmune-mediated destruction of β cells. Insulin antibodies typically precede the clinical onset of T1D and therefore represent a potentially useful target for ASI. Models of immunotherapy (intravenous, subcutaneous, oral, or intranasal) using insulin, proinsulin, or insulin peptides in NOD mice have yielded promising results in protecting against diabetes,25 but these results have not been consistently replicated in human subjects.
The results of studies performed using insulin are summarized in Table 2. Ultimately, these studies failed to show any disease modification in the treatment groups.
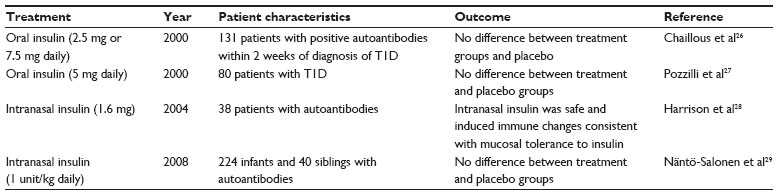 | Table 2 Antigen-specific immunotherapy using insulin as the target antigen |
Results from the Diabetes Prevention Trial-Type 1 (DPT-1), a randomized, double-blind, placebo-controlled trial studying the effects of administration of intravenous, subcutaneous, or oral insulin on preventing T1D in nondiabetic relatives at risk for diabetes, were released in 2005. The results are summarized in Table 3. Initially, low-dose systemic insulin (annual intravenous insulin infusions and daily subcutaneous injections) was given to a high-risk group of individuals, but this treatment had no effect on diabetes incidence (15.1% of the 69 subjects in the intervention group progressed annually to diabetes compared to 14.6% of the 70 subjects in the observation group).30 In the second phase of the trial, oral insulin administration was studied. Screening was carried out on 103,391 first- and second-degree relatives of individuals with T1D. Of these, 3,483 were antibody positive and 2,523 underwent genetic, immunological, and metabolic testing to stage their risk of developing diabetes. Three-hundred and eighty-eight individuals were found to have a 26%–50% 5-year risk projection of developing T1D, and 372 of these individuals were randomly assigned to oral insulin (7.5 mg/day) or placebo. Ultimately, the annual rate of development of diabetes was similar in both groups (6.4% [n=44] with oral insulin and 8.2% [n=53] with placebo). However, a subgroup analysis suggested a possible benefit in subjects with insulin autoantibody levels >80 nU/mL (rate of diabetes development in this group was 6.2% with oral insulin and 10.4% with placebo).22 Other intervention trials studying the use of insulin in ASI have also failed to show clinical efficacy in slowing the loss of β-cell function even though laboratory evidence was initially suggestive.31
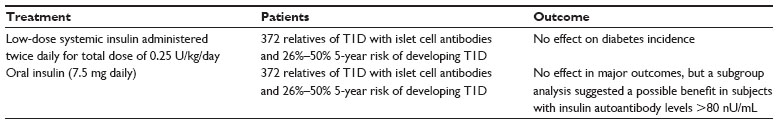 | Table 3 Diabetes Prevention Trial-Type 1 |
ASI with altered peptide ligands (APL) of insulin has also been attempted. Although promising in a Phase 1a clinical study using an APL, NBI-6024 (corresponding to the insulin B chain that is recognized by Th1 cells in T1D),32 a Phase II study released in 2009 determined that NBI-6024 did not improve or maintain islet cell function.33
However, there are several ongoing studies such as the Pre-POINT (Primary Oral INsulin Trial) trial which aims to determine whether oral insulin ASI can prevent or delay the onset of diabetes in children who are at high risk for developing T1D (at least two first-degree family members with T1D but no pancreatic autoantibodies).34 Another is the Intranasal Insulin (INIT II) Phase II trial in Australia, New Zealand, and Germany, treating patients who have autoantibodies without clinical diabetes with intranasal insulin. Finally, the Oral Insulin Trial is an American study to assess whether oral insulin can delay the onset of T1D in at-risk individuals.35
ASI using GAD65 as the target antigen
GAD65, an enzyme that is involved in the production of γ-aminobutyric acid (GABA), is another major autoantigen involved in the pathogenesis of T1D and may also be involved in the initiation of the autoimmune process. The presence of anti-GAD65 antibodies may predict conversion to insulin dependence in patients with latent autoimmune diabetes in the adult.36 In NOD mice, administration of GAD65 prevents and blocks autoimmune destruction of β cells.37–39 NOD mice that received oral administration of GAD65 and cholera toxin B demonstrated a significant reduction in pancreatic islet inflammation and the development of diabetes.40
From 2005 to 2007, a 30-month randomized, double-blind, placebo-controlled Phase II study of recombinant human GAD65 (Diamyd®) was conducted involving 70 Swedish children with T1D. The treatment was well tolerated with no serious side effects reported, and significant long-term efficacy was demonstrated in preserving β-cell function.21 A Phase II study of 47 patients with latent autoimmune diabetes in the adult showed that patients who received two injections of Diamyd® were less likely to require insulin treatment after 5 years of follow-up (14% of patients receiving Diamyd® vs 64% of patients receiving insulin).41
However, an American Phase II study published in 2011 randomly assigned patients who had recently been diagnosed with T1D within 100 days to receive Diamyd® or placebo and found no difference in loss of insulin secretion.42 Also in 2011, a European Phase III study of 334 patients who were diagnosed with T1D within 3 months of entering the study showed that, while Diamyd® was safe and well tolerated, there was no significant difference in β-cell function (determined by C-peptide levels) after 15 months of follow-up compared with placebo.43
A research group at Lund University in Sweden is currently conducting a double-blind, randomized study of Diamyd® in children at risk for developing T1D (positive GAD antibodies prior to onset of clinical diabetes) called Diabetes Prevention-Immune Tolerance (DIAPREV-IT). The initial results of this study should be compiled in 2015. Another Swedish study, DIABGAD-1, is currently recruiting patients to determine the effects of combining Diamyd® with Vitamin D and ibuprofen on C-peptide levels.
ASI using HSP60 as the target antigen
Heatshock protein 60 (HSP60), a ubiquitous intracellular chaperone that is involved in the regulation of innate immunity and is found in mature insulin-secretory granules of pancreatic β cells, may be an important autoantigen in the development of T1D. DiaPep277 is a synthetic peptide derived from human HSP60 that activates the T-cell receptor (TCR) and T-cell Toll-like receptor 2 (TLR2). Unlike HSP60, Diapep277 does not affect TLR4 receptors on macrophages and therefore does not induce a proinflammatory effect on innate immune cells. The TCR interaction ensures an antigen-specific response and the interaction with TLR2 receptors triggers anti-inflammatory cytokine secretion such as IL-10, which promotes Treg responses.
Again, mouse models have shown promising results with the use of DiaPep277 in preventing T1D development.44 Clinical studies in humans have also been encouraging. The DiaPep277-Arresting Immune Diabetes (DIA-AID 2) study, an international, Phase III, randomized, double-blind, placebo-controlled, parallel group study of 457 patients newly diagnosed with T1D who received either subcutaneous injections of DiaPep277 or placebo, found that DiaPep277 was not only safe and well tolerated but also showed clinical efficacy. There was significant preservation of C-peptide secretion in the treatment group (relative treatment effect of 23.4% higher C-peptide levels in the treatment group). More patients in the treatment group maintained target HbA1c (56% vs 44% in the placebo group) and entered partial remission (38.4% in the treatment group compared with 29.3% in the placebo group).45 Further phase III studies are currently under way to confirm these results.
Safety
In at least 100 published studies involving administration of peptide autoantigens to NOD mice, it has been extremely rare for studies to demonstrate acceleration of disease after autoantigen administration.9 This phenomenon seems to be similarly unusual in human studies as well. However, in one human study of administration of oral insulin to individuals with insulin autoantibodies and first-degree relatives with diabetes, it was noted that a subset of patients (subjects with insulin autoantibodies that were not >80 nU/mL) developed diabetes at a greater rate than the subjects receiving placebo.22 Also, fatal anaphylaxis was reported in NOD mice after injections of insulin peptides, when using large doses and repeated injections.46
ASI for MS
MS, a potentially debilitating, chronic inflammatory demyelinating disease of the central nervous system, results from the autoimmune destruction of myelin, which can cause irreversible deterioration of nerves.
While MS is a heterogeneous disease, there are several identified autoantigens that are considered important in the pathogenesis of the disease. The established antigens that are thought to be most significant are myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), and proteolipid protein (PLP).47 These autoantigens can induce experimental autoimmune encephalomyelitis (EAE) in mouse models. However, given the different subtypes, multiple possible auto antigens, and the possibility of CD4+ cells to respond to multiple epitopes within the myelin protein, the concept of ASI for MS is inherently difficult.
Some animal studies of EAE demonstrated modest efficacy of oral or nasal administration of soluble myelin peptides in preventing EAE but not in treating EAE after onset. Other animal studies found that an oligomerized T-cell epitope of myelin PLP (PLP139-151) is effective in both preventing and treating EAE. Treatment with this protein increased the production of IL-10 and decreased the levels of TNFα.48–50 However, human clinical trials with oral bovine MBP have been unsuccessful, and the oral route appears limited in its ability to induce tolerance in ongoing disease.51
Intravenous trials using an epitope of MBP in chronic progressive disease seemed to induce tolerance but the trials were largely abandoned due to safety concerns of anaphylaxis.52,53
As early as the mid-1990s, an APL of MBP, where the native peptide is modified at key amino acid residues to ensure contact with the TCR, was shown to treat EAE in rodents.54 Because APLs compete with the naïve peptide for TCR binding and bind to the TCR with lower affinity, they can function as antagonists or partial agonists. Antagonists induce T-cell anergy, and partial agonists incompletely activate T-cells and can induce immune deviation.55 Administration of this APL was found to decrease the production of TNFα and IFNγ. Phase I studies on humans demonstrated that APL was generally well tolerated and seemed to induce a Th2-like response. However, two Phase II studies were stopped because of adverse events. One study was stopped because it was found that APL caused exacerbation of the disease in several patients.56 Another Phase II study involving an APL was stopped after a 9% incidence of hypersensitivity reactions among trial patients was noted.57 However, these trials did show that APL treatment induces a shift from a Th1 response to a Th2 response.
In September 2013, a study described 30 patients with relapsing-remitting MS who were treated with a skin patch delivering a mixture of three myelin peptides (MBP85-99, MOG35-55, and PLP139-155). Individuals treated with the myelin peptide skin patch benefited from significantly reduced radiographic (66.5% reduction in gadolinium-enhanced lesions per scan) and clinical disease activity (0.43% annual relapse rate in the treatment group compared to 1.4% in the placebo group). The patch was safe and well tolerated.58 An earlier study by the same group reported that the myelin peptide skin patch caused immunologic tolerance to myelin antigens by activating dendritic Langerhans cells in the skin, induced a unique population of granular DCs in local lymph nodes, generated IL-10 producing Tregs in the periphery, and suppressed IFNγ and TGF-β production.59
Additionally, one of the currently FDA-approved medications for MS, glatiramer acetate, may represent a type of immunotherapy. Glatiramer acetate is a random mixture of glutamine, lysine, alanine, and tyrosine, and acts as an APL by stimulating MBP-reactive T-cells.57 Glatiramer acetate may induce immune deviation from a Th1/Th17 cell-type response to a Th2 cell-type response.60
Safety
Some studies using mouse models of MS have shown not only a lack of efficacy but also high rates of anaphylaxis.61 Earlier studies using an APL of MBP for MS raised concern after one had to be suspended after 9% of patients experienced hypersensitivity reactions.57 Human studies using APLs to MBP resulted in exacerbations of MS.57
ASI for rheumatoid arthritis
RA is a systemic inflammatory disease caused by autoimmune inflammation of synovial tissue and subsequent cartilage and bone erosion.
Antigen-specific therapies for RA have been limited, partly because of an incomplete understanding of the pathogenesis of autoimmunity.7 Several antigenic targets have been identified in RA. Humans predominantly have antibodies targeting citrullinated peptides, which include type II collagen (CII), vimentin, α-enolase, clusterin, histones, and peptidyl arginine deiminase-4,8 as well as rheumatoid factor. However, the ideal antigen to target for ASI remains unclear.62
CII is the primary protein in cartilage, is crucial to joint health and function, and is thought to be an important autoantigen in the pathogenesis of RA.63 Studies in the 1980s and 1990s demonstrated that oral administration of CII could reduce the incidence of collagen-induced arthritis (the animal model used to study RA) in mice,64 reduce immunoglobulin G (IgG)-collagen specific antibodies, and generate a T-cell population that was capable of transferring tolerance to naïve mice.65 However, these effects were noted only when the therapy was performed prior to disease onset. There was no therapeutic effect noted if CII was administered orally after disease onset.66 Further studies demonstrated that adjusting the method of administration (conjugating with a biodegradable polymer) can induce protection from collagen-induced arthritis after disease onset.67 However, a clinical study looking at the effects of switching RA patients from treatment with methotrexate to oral CII revealed clinical deterioration in the group that was switched to CII.68
Attempts at ASI for RA then progressed to the use of epitope-specific antigens. It has been hypothesized that RA is secondary to an interaction between HLA and dnaJ-derived peptides that promotes T-cells which ultimately result in autoimmune inflammation.69 A pilot study treated a group of patients recently diagnosed with RA with dnaJP1 orally for 6 months and found that treatment with this peptide induced T-cell production of IL-4 and IL-10 and decreased production of IL-2, IFNγ, and TNFα.69 A Phase II trial released in 2009 studied 160 patients with RA and found that the dnaJP1 peptide was safe and well tolerated and again demonstrated evidence for immune deviation with an increase in IL-10 production and a decrease in TNFα. While the primary endpoints for clinical efficacy (20% improvement criteria) were not met, there was a trend toward clinical effect. Post hoc analysis showed a potentially synergistic effect between epitope-specific therapy and hydroxychloroquine.70
A group in Cuba has been studying a new peptide called APL-1 which is an APL of HSP60 that has an altered amino acid residue to increase its affinity to bind to HLA class II molecule. In animal models, this APL was found to increase Tregs and inhibit progression of the disease.71 APL-1 was also found to modulate inflammatory immune responses in peripheral blood mononuclear cells from human RA patients by inducing Tregs and apoptosis of activated CD4+ cells.72 It remains to be seen whether the promising effects seen in animal models and in vitro human studies can translate into clinical success.
Safety
There has been no evidence for disease acceleration in ASI models for RA.9
ASI for SLE
SLE is a complex disease characterized by diverse clinical manifestations and multiorgan involvement.73 Most of the studies of peptide-based immunotherapy for SLE have focused on epitopes from nucleosomal histones, ribonucleoproteins, and autoantibodies. In mouse models, studies using peptides from histone regions and studies using a phosphorylated analog (peptide p140) of a small nuclear ribonucleoprotein U1-70K have been associated with laboratory and clinical evidence of disease improvement.74,75 The precise mode of action of the P140 peptide is not fully understood, but repeated administration of P140 has been found to transiently eliminate T-cell responses to spliceosomal proteins,75 perhaps through its ability to affect endogenous autoantigen processing and peptide loading to class II major histocompatibility complex molecules and thereby induce tolerance.76
A Phase IIa study conducted in 2007 in Bulgaria of 20 patients with SLE who received three administrations of P140 (IPP-201101) demonstrated safety and tolerability and achieved its primary efficacy endpoint of a significant reduction in anti-double-stranded DNA antibody titers.77 A Phase IIb study conducted in 2008 in Europe and Latin America again found that IPP-201101(now marketed as Lupuzor™ through ImmuPharma plc, London, UK) was well tolerated and resulted in a significant decrease in the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) score.78 A Phase III trial is currently under way.
In a mouse model of lupus, monthly injections of an artificial peptide modeled on the variable heavy chain region of anti-DNA antibodies (pConsensus or pCons) significantly delayed the onset of nephritis, induced Tregs, decreased autoantibody levels, and prolonged survival.73 Oral pCons was also found to have similar beneficial effects.79 However, clinical trials with another synthetic peptide, hCDR1 (Edratide™) in humans passed Phase I clinical studies but did not meet primary endpoints in a Phase II study.80 Further studies are needed to investigate the potential role of ASI for SLE.
ASI for celiac disease
Celiac disease, or gluten-sensitive enteropathy, is an increasingly diagnosed autoimmune disease that is induced by exposure to wheat gliadans and is characterized by IgA antibodies to tissue transglutaminase. ImmusanT has demonstrated that the majority of gluten-specific T-cells circulating in the blood after wheat, barley, or rye challenge recognize one of three peptides derived from wheat α-gliadin (NPL001), wheat ω-gliadin/barley C-hordein (NPL002), and barley B-hordein (NPL003). These three peptides include five distinct HLA DQ2-restricted epitopes that are recognized by almost all of the patients with celiac disease who have HLA DQ2 (80% of celiac patients).81 Nexvax2®, developed by ImmusanT, contains NPL001, NPL002, and NPL003. Phase I trials have shown that Nexvax2® is well tolerated and a Phase Ib trial is currently under way in Australia and New Zealand and a second Phase Ib trial is enrolling patients in the United States.82
Future directions: DC therapy
DCs are professional antigen-presenting cells that process antigens and present them to T-cells. They are capable of directing the immune response and influencing the differentiation of naïve T-cells into Th1, Th2, Th17, or Treg cells. Specialized DCs can also produce inflammatory or suppressive mediators and promote tolerance. Because DCs play a critical role in self-tolerance, they have recently have become targets of therapeutics in autoimmune disease. While there are potential drawbacks of presenting tolerogenic antigens through mucosal delivery to mucosal DCs (including the possibility of inciting disease flares, the altered gastrointestinal tract flora in patients with autoimmune disease, instead, promoting inflammatory conditions, and other unpredictable effects of activated DCs in inflammatory sites in patients with autoimmune conditions), tolerogenic DCs could be generated ex vivo and modified to optimize their ability to delete antigen-specific T-cells or to induce antigen-specific Tregs (with the use of NF-κB inhibitors or incubation with TNF, IL-6, or low-dose endotoxin to induce tolerant DCs). These modified DCs could then be delivered with the appropriate autoantigen to the patient to manipulate the induction of Tregs in an antigen-specific manner that induces tolerance and avoids generalized immune suppression.8
Preclinical experiments in an animal model of methylated bovine serum albumin antigen (mBSA)-induced arthritis demonstrated that animals treated with a single dose of DCs exposed to mBSA (along with an inhibitor of NF-κB in order to promote Treg induction and prevent immune activation) benefited from suppressed arthritis and that anti-mBSA antibodies were switched from Th1 to Th2 and regulatory isotypes.83 In a mouse model of autoimmunity using EAE, DCs were targeted to MOG and were found to delay onset and reduce the severity of EAE.84 In mouse models of SLE, mycophenolate nanogels (without antigen) were found to target DCs in vivo and suppress inflammatory cytokines.85 In vitro studies have shown that tolerogenic DCs pulsed with PLP-induced anergy in CD4+ T-cells from MS patients.86 Another study used a single infusion of autologous peripheral blood mononuclear cells coupled with seven myelin peptides and found this treatment to be safe and well tolerated and resulted in a decrease of antigen-specific T-cell responses.87 Another recent in vitro study loaded DCs with insulin or GAD65 in the presence of IL-10 and TGF-β1 (to create tolerogenic DCs) and cultured these cells with T lymphocytes. The T-cells were then rechallenged with insulin or GAD65, and it was found that the tolerogenic DCs induced antigen-specific T-cell hyporesponsiveness.88
Two Phase I studies using tolerogenic DCs in RA are ongoing. Thomas et al89 at the University of Queensland are evaluating a potential treatment known as Rheumavax®. In this approach, DCs are modified with an NF-κB inhibitor and pulsed with a mixture of four citrullinated peptides. The antigen-pulsed tolerogenic DCs are then intradermally administered to patients. Thus far, Rheumavax® has been well tolerated with no major adverse events.90
This targeted therapy should have fewer systemic side effects and has the potential for even greater therapeutic advantage. DCs can be loaded with immunosuppressive drugs and antigen to deliver the drug to T-cells during antigen presentation.91 This would allow elimination of T-cells in an antigen-specific manner. Further studies need to be conducted in order to analyze DC vaccination for autoimmune disease including identifying the optimal timing of vaccination, the appropriate antigen to induce antigen-specific tolerance, the optimal route and frequency of tolerogenic DC administration, and appropriate ways of monitoring biological effects of therapy.92
Discussion
While ASI has conceptually been an attractive therapeutic modality for years, it has not been successfully translated into clinical practice the way allergen immunotherapy has been used to treat allergic disease. There are several possible reasons for the difficulty in developing an effective therapy with ASI. The allergens responsible for allergic diseases are well classified. After a patient with allergic symptoms undergoes diagnostic skin testing, the allergens that elicit a positive response on skin testing can then be incorporated into an immunotherapy regimen. However, the antigens responsible for initiating autoimmune disease (ideal targets for immunotherapy) are not always as obvious. Autoimmune disorders are often characterized by autoantibodies against several different antigens, and choosing the proper antigen to target with immunotherapy has proven to be both critically important and difficult. Complicating matters even further is the fact that, at the time of diagnosis for several autoimmune diseases, substantial tissue damage has already occurred.12 Once inflammation has occurred, the ability of Tregs to suppress pathologic cytokine production may be decreased.7 Therefore, the timing of administration of ASI to optimize tolerance must be clarified.
Furthermore, allergen immunotherapy elicits a shift in the immunological response to an allergen from a Th2-dominated response to a Th1-dominated response with an associated decrease in allergen-specific IgE and increase in allergen-specific IgG1 and IgG4. While studies with ASI for autoimmune disease have shown a shift toward tolerance with several biological markers, it is important to note that IgG antibodies are responsible for autoimmune disease, as opposed to IgE antibodies in allergic disease. The beneficial effects of allergen immunotherapy may be mediated by the decrease in allergen-specific IgE levels that are associated with allergen immunotherapy. In contrast, ASI for autoimmune disease aims to alter primarily Th1- or Th17-dominated conditions, which may prove to be more difficult to manipulate. This fundamental difference between these two sets of diseases may explain the vastly different clinical experience with ASI.
It remains both discouraging and unclear as to why the promising results of ASI that are seen in mouse models have not been replicated in human studies. Induction of antigen-specific tolerance may not affect the underlying disease process without eliminating the pathogenic effector cells. The optimal route of delivery may be an important variable that has not yet fully been elucidated. The optimal dosage is still being tested in different studies and has been difficult to determine. Again, the timing of administration of ASI is critical, and it may be most effective before the onset of subclinical disease. In future studies, it would be tremendously beneficial for human trials to monitor the immune response and measure relevant immune biomarkers after autoantigen administration.13
ASI for autoimmune diseases is not ready to be translated into the clinical realm. Further studies are needed as there is still much more to be learned.
Disclosure
The authors report no conflicts of interest in this work.
References
Cooper GS, Bynam ML, Somers EC. Recent insights in the epidemiology of autoimmune diseases: improved prevalence estimates and understanding of clustering of diseases. J Autoimmun. 2009;33(3–4):197–207. | |
American Autoimmune Related Disease Association, Inc. c2004–2014. Available from: http://www.aarda.org/autoimmune-information/autoimmune-statistics/. Accessed January 17, 2014. | |
Lipman TH, Levitt Katz LE, Ratcliffe SJ, et al. Increasing incidence of Type 1 diabetes in youth. Diabetes Care. 2013;36(6):1597–1603. | |
Rosata E, Pisarri S, Salsano F. Current strategies for the treatment of autoimmune diseases. J Biol Regul Homeost Agents. 2010;24(3):251–259. | |
Bernatsky S, Hudson M, Suissa S. Anti-rheumatic drug use and risk of serious infections in rheumatoid arthritis. Rheumatology. 2010;46(7):1157–1160. | |
Atzeni F, Sarzi-Puttini P, Botsios C, et al. Long-term anti-TNF therapy and the risk of serious infections in a cohort of patients with rheumatoid arthritis: comparison of adalimumab, etanercept, and infliximab in the GISEA registry. Autoimmun Rev. 2012;12:225–229. | |
Rosman Z, Shoenfeld Y, Zandman-Goddard G. Biologic therapy for autoimmune diseases: an update. BMC Med. 2013;11:88. | |
Thomas R. Dendritic cells and the promise of antigen-specific therapy in rheumatoid arthritis. Arthritis Res Ther. 2013;15:204. | |
Roep BO, Peakmann M. Antigen targets of Type 1 diabetes autoimmunity. Cold Spring Harb Perspect Med. 2012;2(4):a007781. | |
Mirshafiey A, Kianiasinni M. Autoantigens and autoantibodies in multiple sclerosis. Iran J Allergy Asthma Immunol. 2013;12(4):292–303. | |
Riemekasten G, Hahn BH. Key autoantigens in SLE. Rheumatology. 2005;44:975–982. | |
Biswas S, Sharma S, Saroha A, et al. Identification of novel autoantigen in the synovial fluid of rheumatoid arthritis patients using an immunoproteomics approach. PLoS One. 2013;8(2):e56246. | |
Harrison LC, Wentworth JM, Zhang Y, et al. Antigen-based vaccination and prevention of Type 1 diabetes. Curr Diab Rep. 2013;13(5):616–623. | |
Peakman M, von Herrath M. Antigen-specific immunotherapy for Type 1 diabetes: maximizing the potential. Diabetes. 2010;59(9):2087–2093. | |
Hochfelder JL, Ponda P. Allergen immunotherapy: routes, safety, efficacy, and mode of action. Immunotargets Ther. 2013;2:61–71. | |
Fujita H, Soyka MB, Akdis M, Akdis CA. Mechanisms of allergen-specific immunotherapy. Clin Transl Allergy. 2012;2:2. | |
Sabatos-Peyton CA, Verhagen J, Wraith DC. Antigen-specific immunotherapy of autoimmune and allergic diseases. Curr Opin Immunol. 2010;22(5):609–615. | |
Anderson RP, Jabri B. Vaccine against autoimmune disease: antigen-specific immunotherapy. Curr Opin Immunol. 2013;25(3):410–417. | |
Jankovic D, Kugler DG, Sher A. IL-10 production by CD4+ effector T cells: a mechanism for self-regulation. Mucosal Immunol. 2010;3:239–246. | |
Thrower SL, James L, Peakman M. Proinsulin peptide immunotherapy in Type 1 diabetes: report of a first-in-man Phase I safety study. Clin Exp Immunol. 2009;155(2):156–165. | |
Ludvigsson J, Faresjö M, Hjorth M, et al. GAD treatment and insulin secretion in recent-onset Type 1 diabetes. N Engl J Med. 2008;359:1909–1920. | |
The Diabetes Prevention Trial-Type I Study Group. Effects of oral insulin in relatives of patients with Type I diabetes. Diabetes Care. 2005;28(5):1068–1076. | |
Kalinski P, Hilkens CMU, Wierenga EA, Kapsenberg ML. T-cell priming by type-1 and type-2 polarized dendritic cells: the concept of a third signal. Immunol Today. 1999;20(12):561–567. | |
Staeva-Vieira T, Peakman M, von Herrath MV. Translational mini-review series on Type 1 diabetes: immune-based therapeutic approaches for Type 1 diabetes. Clin Exp Immunol. 2007;148(1):17–31. | |
Chatneoud L. Immune therapy for Type 1 diabetes mellitus – what is unique about anti-CD3 antibodies? Nat Rev Endocrinol. 2010;6(3):149–157. | |
Chaillous L, Lefèvre H, Thivolet C, et al. Oral insulin administration and residual beta-cell function in recent-onset Type 1 diabetes: a multicentre randomized controlled trial. Diabete Insuline Orale Group. Lancet. 2000;356(9229):545–549. | |
Pozzilli P, Pitocco D, Visalli N, et al. No effect of oral insulin on residual beta-cell function in recent-onset Type 1 diabetes (the IMDIAB VII). IMDIAB Group. Diabetologia. 2000;43(8):1000–1004. | |
Harrison LC, Honeyman MC, Steele CE. Pancreatic beta-cell function and immune responses to insulin after administration of intranasal insulin to humans at risk for Type 1 diabetes. Diabetes Care. 2004;27(10):2348–2355. | |
Näntö-Salonen K, Kupila A, Simell S, et al. Nasal insulin to prevent Type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind randomized controlled trial. Lancet. 2008;372(9651):1746–1755. | |
Diabetes Prevention Trial – Type 1 Diabetes Study Group. Effects of insulin in relatives of patients with Type I diabetes mellitus. N Engl J Med. 2002;346:1685–1691. | |
Fourlanos S, Perry C, Harrison LC. Evidence that nasal insulin induces immune tolerance to adults with autoimmune diabetes. Diabetes. 2011;60(4):1237–1245. | |
Alleva DG, Maki RA, Putnam AL, et al. Immunomodulation in Type 1 diabetes by NBI-6024, an altered peptide ligand of the insulin B (9-23) epitope. Scand J Immunol. 2006;63:59–69. | |
Walter M, Philotheou A, Bonnici F, Ziegler AG, Jimenez R. No effect of the altered peptide ligand NBI-6024 on beta-cell residual function and insulin needs in new-onset Type 1 diabetes. Diabetes Care. 2009;32(11):2036–2040. | |
Achenbach P, Barker J, Bonifacio E. Modulating the natural history of Type 1 diabetes in children at high genetic risk by mucosal insulin immunization. Curr Diab Rep. 2008;8(2):87–93. | |
Staeva TP, Chatenoud L, Insel R, Atkinson MA. Recent lessons learned from prevention and recent-onset Type 1 diabetes immunotherapy trials. Diabetes. 2013;62(1):9–17. | |
Uibo R, Lernmark A. GAD65 autoimmunity-clinical studies. Adv Immunol. 2008;100:39–78. | |
Wang H, Yang J, Jin L, et al. Immunotherapy of autoimmune diabetes by nasal administration of tandem glutamic acid decarboxylase 65 peptides. Immunol Invest. 2009;38(8):690–703. | |
Tian J, Clare-Salzler M, Herschenfeld A, et al. Modulating autoimmune responses to GAD inhibits disease progression and prolongs islet graft survival in diabetes-prone mice. Nat Med. 1996;2(12):1348–1353. | |
Tisch R, Liblau RS, Yang XD, Liblau P, McDevitt HO. Induction of GAD65-specific regulatory T-cells inhibits autoimmune diabetes in nonobese diabetic mice. Diabetes. 1998;47(6):894–899. | |
Gong Z, Pan L, Le Y, et al. Gluatmic acid decarboxylase epitope protects against autoimmune diabetes through activation of Th2 immune response and induction of possible regulatory mechanism. Vaccine. 2010;28(24):4052–4058. | |
Diamyd.com (homepage on the Internet). Stockholm: Diamyd Medical AB; c2009–2013. Available from: http://www.diamyd.com/docs/trialsDiabetes.aspx?section=trials. Accessed May 21, 2014. | |
Wherrett DK, Bundy B, Becker DJ, et al; Type 1 Diabetes TrialNet GAD Study Group. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset Type 1 diabetes: a randomized double-blind trial. Lancet. 2011;378(9788):319–327. | |
Ludvigsson J, Krisky D, Casas R, et al. Gad65 antigen therapy in recently diagnosed Type 1 diabetes mellitus. N Engl J Med. 2012;366(5):433–442. | |
Fierabracci A. Peptide immunotherapies in Type 1 diabetes: lessons from animal models. Curr Med Chem. 2011;18(4):577–586. | |
Raz I, Ziegler AG, Linn T, et al; DIA-AID 1 Writing Group. Treatment of recent-onset Type 1 diabetic patients with DiaPep277: results of a double-blind, placebo-controlled, randomized phase 3 trial. Diabetes Care. 2014;37(5):1392–1400. | |
Liu E, Moriyama H, Eisenbarth GS. Anti-peptide autoantibodies and fatal anaphylaxis in NOD mice in response to insulin self-peptides B:9-23 and B:13-23. J Clin Invest. 2002;110(7):1021–1027. | |
Hohlfeld R, Wekerle H. Autoimmune concepts of multiple sclerosis as a basis for selective immunotherapy: from pipe dreams to (therapeutic) pipelines. Proc Natl Acad Sci U S A. 2004;101(2):14599–14606. | |
Puentes F, Dickhaut K, Hofstatter M, Falk K, Rotzschke O. Active suppression induced by repetitive self-epitopes protects against EAE development. PLoS One. 2013;8(5):e64888. | |
Metzler B, Wraith DC. Inhibition of experimental autoimmune encephalomyelitis by inhalation but not oral administration of the encephalitogenic peptide: influence of MHC binding affinity. Int Immunol. 1993;5(9):1159–1165. | |
Bai XF, Li HL, Link H. Complexities of applying nasal tolerance induction as a therapy for ongoing relapsing experimental autoimmune encephalomyelitis (EAE) in DA rats. Clin Exp Immunol. 1998;111(1):205–210. | |
Weiner HL, Mackin GA, Matsui M, et al. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science. 1993;259(5099):1321–1324. | |
Warren KG, Catz I, Wucherpfennig KW. Tolerance induction to myelin basic protein by intravenous synthetic peptides containing epitope P85 VVHFFKNIVTP96 in chronic progressive multiple sclerosis. J Neurol Sci. 1997;152(1):31–38. | |
Smith CE, Eagar TN, Strominger JL, Miller SD. Differential induction of IgE-mediated anaphylaxis after soluble vs cell-bound tolerogenic peptide therapy of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2005;102(27):9595–9600. | |
Karin N, Mitchell DJ, Brocke S, Ling N, Steinman L. Reversal of experimental autoimmune encephalomyelitis by a soluble peptide variant of a myelin basic protein epitope: T cell receptor antagonism and reduction of interferon gamma and tumor necrosis factor alpha production. J Exp Med. 1994;180(6):2227–2237. | |
Turley DM, Miller SD. Prospects for antigen-specific tolerance based therapies for the treatment of multiple sclerosis. Results Probl Cell Differ. 2010;51:217–235. | |
Bielekova B, Goodwin B, Richert N, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6(10):1167–1175. | |
Kappos L, Comi G, Panitch H, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo controlled, randomized phase II trial. Nat Med. 2000;6(10):1176–1182. | |
Walczak A, Siger M, Ciach A, Szczepanik M, Selmaj K. Transdermal application of myelin peptides in multiple sclerosis treatment. JAMA Neurol. 2013;70(9):1105–1109. | |
Jurynczyk M, Walczak A, Jurewicz A, Jesionek-Kupnicka D, Szczepanik M, Selmaj K. Immune regulation of multiple sclerosis by transdermally applied myelin peptides. Ann Neurol. 2010;68(5):593–601. | |
Aharoni R, Teitelbaum D, Arnon R, Sela M. Copolymer 1 acts against the immunodominant epitope 82-100 of myelin basic protein by T cell receptor antagonism in addition to major histocompatibility complex blocking. Proc Natl Acad Sci U S A. 1999;96(2):634–639. | |
Smith CE, Eagar TN, Miller SD. Differential induction of IgE-mediated anaphylaxis after soluble vs cell-bound tolerogenic peptide therapy of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2005;102(27):9595–9600. | |
Ichim TE, Zheng X, Suzuki M, et al. Antigen-specific therapy of rheumatoid arthritis. Expert Opin Biol Ther. 2008;8(2):191–199. | |
Nissim A, Winyard PG, Corrigall V, et al. Generation of neoantigenic epitopes after posttranslational modification of Type II collagen by factors present within the inflamed joint. Arthritis Rheum. 2005;52(12):3829–3838. | |
Nagler-Anderson C, Bober LA, Robinson ME, Siskind GW, Thorbecke GJ. Suppression of Type II collagen-induced arthritis by intragastric administration of soluble Type II collagen. Proc Natl Acad Sci U S A. 1986;83(19):7443–7446. | |
Thompson HS, Harper N, Bevan DJ, Staines NA. Suppression of collagen induced arthritis by oral administration of Type II collagen: changes in immune and arthritic responses mediated by active peripheral suppression. Autoimmunity. 1993;16(3):189–199. | |
Yoshino S. Antigen-induced arthritis in rats is suppressed by the inducing antigen administered orally before, but not after immunization. Cell Immunol. 1995;163(1):55–58. | |
Kim WU, Lee WK, Ryoo JW, et al. Suppression of collagen-induced arthritis by single administration of poly(lactic-co-glycolic acid) nanoparticles entrapping Type II collagen: a novel treatment strategy for induction of oral tolerance. Arthritis Rheum. 2002;46(4):1109–1120. | |
Häuselmann HJ, Caravatti M, Seifert B, et al. Can collagen Type II sustain a methotrexate-induced therapeutic effect in patients with long-standing rheumatoid arthritis? A double-blind, randomized trial. Br J Rheumatol. 1998;37(10):1110–1117. | |
Prakken BJ, Samodal R, Le TD, et al. Epitope-specific immunotherapy induces immune deviation of proinflammatory T cells in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2004;101(12):4228–4233. | |
Koffeman EC, Genovese M, Amox D, et al. Epitope-specific immunotherapy of rheumatoid arthritis. Arthritis Rheum. 2009;60(11):3207–3216. | |
Dominguez MDC, Lorenzo N, Barbera A, et al. An altered peptide ligand corresponding to a novel epitope from heat-shock protein 60 induces regulatory T cells and suppresses pathogenic response in an animal model of adjuvant-induced arthritis. Autoimmunity. 2011;44(6):471–482. | |
Barbera A, Lorenzo N, Garrido G. APL-1, an altered peptide ligand derived from human heat-shock protein 60, selectively induces apoptosis in activated CD4+CD25+ T cells from peripheral blood of rheumatoid arthritis patients. Int Immunopharmacol. 2013;17:1075–1083. | |
Iikuni N, Hahn BH, La Cava A. Potential for anti-DNA immunoglobulin peptide therapy in systemic lupus erythematosus. Expert Opin Biol Ther. 2009;9(2):201–206. | |
Monneaux F, Lozano JM, Patarroyo ME, Briand JP, Muller S. T cell recognition and therapeutic effect of a phosphorylated synthetic peptide of the 70K snRNP protein administered in MR/lpr mice. Euro J Immunol. 2003;33:287–296. | |
Kaliyaperumal A, Michaels MA, Datta SK. Antigen-specific therapy of murine lupus nephritis using nucleosomal peptides: tolerance spreading impairs pathogenic function of autoimmune T and B cells. J Immunol. 1999;162:5775–5783. | |
Muller S, Wallace DJ. The importance of implementing proper selection of excipients in lupus clinical trials. Lupus. 2014;2:609–614. | |
Muller S, Monneaux F, Schall N, et al. Spliceosomal peptide P140 for immunotherapy of systemic lupus erythematosus. Arthritis Rheum. 2008;58(12):3873–3883. | |
ImmuPharma.org (homepage on the Internet). ImmuPharma PLC 2005–2014. Available from: http://www.immupharma.org/lupuzor. Accessed May 12, 2014. | |
Skaggs BJ, Lourenco EV, Hahn BH. Oral administration of different forms of a tolerogenic peptide to define the preparations and doses that delay anti-DNA antibody production and nephritis and prolong survival in SLE-prone mice. Lupus. 2011;20:912–920. | |
Tevapharm.com (homepage on the Internet). Available from: http://ir.tevapharm.com/phoenix.zhtml?c=73925&p=irol-newsArticle_Print&ID=1554647&highlight=. Accessed May 12, 2014. | |
Tye-Din JA, Stewart JA, Dromey JA, et al. Comprehensive, quantitative mapping of T cell epitopes in gluten in celiac disease. Sci Transl Med. 2010;2:41ra51. | |
Immusant.com (homepage on the Internet). ImmusanT, Inc.; c2011. Available from: http://www.immusant.com/clinical-trial/. Accessed May 12, 2014. | |
Martin E, Capini C, Duggan E, et al. Antigen-specific suppression of established arthritis in mice by dendritic cells deficient in NF-KB. Arthritis Rheum. 2007;56:2255–2266. | |
Loschko J, Heink S, Hackl D, et al. Antigen targeting to plasmacytoid dendritic cells via Siglec-H inhibits Th cell-dependent autoimmunity. J Immunol. 2011;187:6346–6356. | |
Look M, Stern E, Wang QA, et al. Nanogel-based delivery of mycophenolic acid ameliorates systemic lupus erythematosus in mice. J Clin Invest. 2013;123:1741–1749. | |
Raϊch-Regué D, Grau-López L, Naranjo-Gómez M, et al. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. Eur J Immunol. 2012;42:771–782. | |
Lutterotti A, Yousef S, Sputtek A, et al. Induction of immune tolerance by autologous peptide-coupled cells: a phase I trial in relapsing-remitting and secondary-progressive multiple sclerosis patients. Mult Scler J. 2012;18:509. | |
Segovia-Gamboa N, Rodríguez-Arellano ME, Rangel-Cruz R, et al. Tolerogenic dendritic cells induce antigen-specific hyporesponsiveness in insulin- and glutamic acid decarboxylase 65-autoreactive T lymphocytes from Type 1 diabetic patients. Clin Immunol. 2014;154(1):72–83. | |
Thomas R, White D, Vecchio P. Phase I trial of Rheumavax to induce antigen specific tolerance in patients with rheumatoid arthritis. University of Queensland. Available from http://www.di.uq.edu.au/clinicaltrialra. Accessed May 12, 2014. | |
Van Brussel I, Lee WP, Rombouts M, et al. Tolerogenic dendritic cell vaccines to treat autoimmune diseases: can the unattainable dream turn into reality? Autoimmun Rev. 2014;13:138–150. | |
Orange DE, Blachere NE, Fak J, et al. Dendritic cells loaded with FK506 kill T cells in an antigen-specific manner and prevent autoimmunity in vivo. Elife. 2013;2:e00105. | |
Gross CC, Wiendl H. Dendritic cell vaccination in autoimmune disease. Curr Opin Rheumatol. 2013;25:268–274. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.