Back to Journals » Journal of Inflammation Research » Volume 16
Anti-Inflammatory and Therapeutic Effects of a Novel Small-Molecule Inhibitor of Inflammation in a Male C57BL/6J Mouse Model of Obesity-Induced NAFLD/MAFLD
Authors McCall KD ![]() , Walter D, Patton A, Thuma JR, Courreges MC, Palczewski G, Goetz DJ, Bergmeier S, Schwartz FL
, Walter D, Patton A, Thuma JR, Courreges MC, Palczewski G, Goetz DJ, Bergmeier S, Schwartz FL
Received 8 April 2023
Accepted for publication 31 October 2023
Published 16 November 2023 Volume 2023:16 Pages 5339—5366
DOI https://doi.org/10.2147/JIR.S413565
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Adam Bachstetter
Kelly D McCall,1– 6 Debra Walter,1,2 Ashley Patton,1,2 Jean R Thuma,3 Maria C Courreges,3 Grzegorz Palczewski,7 Douglas J Goetz,1,6,8 Stephen Bergmeier,1,6,9 Frank L Schwartz3,5,6
1Molecular and Cellular Biology Program, Ohio University College of Arts & Sciences, Athens, OH, USA; 2Department of Biological Sciences, Ohio University College of Arts & Sciences, Athens, OH, USA; 3Department of Specialty Medicine, Ohio University Heritage College of Osteopathic Medicine, Athens, OH, USA; 4Department of Biomedical Sciences, Ohio University Heritage College of Osteopathic Medicine, Athens, OH, USA; 5Diabetes Institute, Ohio University Heritage College of Osteopathic Medicine, Athens, OH, USA; 6Biomedical Engineering Program, Ohio University Russ College of Engineering and Technology, Athens, OH, USA; 7Metabolon Inc., Durham, NC, USA; 8Department of Chemical & Biomolecular Engineering, Ohio University Russ College of Engineering and Technology, Athens, OH, USA; 9Department of Chemistry & Biochemistry, Ohio University College of Arts & Sciences, Athens, OH, USA
Correspondence: Kelly D McCall, Department of Specialty Medicine, Ohio University Heritage College of Osteopathic Medicine, Academic & Research Center 302B, Athens, OH, 45701, USA, Tel +1 740 593 0926, Fax +1 740 597 1371, Email [email protected]
Purpose: Non-alcoholic fatty liver disease (NAFLD), recently renamed metabolic (dysfunction) associated fatty liver disease (MAFLD), is the most common chronic liver disease in the United States. Presently, there is an intense and ongoing effort to identify and develop novel therapeutics for this disease. In this study, we explored the anti-inflammatory activity of a new compound, termed IOI-214, and its therapeutic potential to ameliorate NAFLD/MAFLD in male C57BL/6J mice fed a high fat (HF) diet.
Methods: Murine macrophages and hepatocytes in culture were treated with lipopolysaccharide (LPS) ± IOI-214 or DMSO (vehicle), and RT-qPCR analyses of inflammatory cytokine gene expression were used to assess IOI-214’s anti-inflammatory properties in vitro. Male C57BL/6J mice were also placed on a HF diet and treated once daily with IOI-214 or DMSO for 16 weeks. Tissues were collected and analyzed to determine the effects of IOI-214 on HF diet-induced NAFL D/MAFLD. Measurements such as weight, blood glucose, serum cholesterol, liver/serum triglyceride, insulin, and glucose tolerance tests, ELISAs, metabolomics, Western blots, histology, gut microbiome, and serum LPS binding protein analyses were conducted.
Results: IOI-214 inhibited LPS-induced inflammation in macrophages and hepatocytes in culture and abrogated HF diet-induced mesenteric fat accumulation, hepatic inflammation and steatosis/hepatocellular ballooning, as well as fasting hyperglycemia without affecting insulin resistance or fasting insulin, cholesterol or TG levels despite overall obesity in vivo in male C57BL/6J mice. IOI-214 also decreased systemic inflammation in vivo and improved gut microbiota dysbiosis and leaky gut.
Conclusion: Combined, these data indicate that IOI-214 works at multiple levels in parallel to inhibit the inflammation that drives HF diet-induced NAFLD/MAFLD, suggesting that it may have therapeutic potential for NAFLD/MAFLD.
Keywords: high fat diet, toll-like receptor 4, IOI-214, metabolomics, gut microbiome, lipopolysaccharide
Introduction
NAFLD/MAFLD is the hepatic manifestation of the metabolic syndrome that is linked to acquisition of visceral obesity (a systemic inflammatory state), dyslipidemia, insulin resistance (IR), and type 2 diabetes mellitus (T2DM); all of which have reached epidemic proportions.1–5 The pathogenesis of NAFLD/MAFLD is multifactorial and complex, leading to diagnostic and therapeutic challenges. In short, fatty liver results from ectopic accumulation of a variety of lipids due to systemic dysregulation of lipid homeostasis. This occurs as a consequence of 1) increased consumption of dietary fat and/or carbohydrate, 2) increased circulating free fatty acid (FFA) levels derived primarily from increased adipose tissue lipolysis and/or 3) increased hepatic de novo lipogenesis, 4) decreased FFA oxidation, and 5) decreased hepatic very low-density lipoprotein (VLDL)-triglyceride (TG) secretion.6,7
Obesity induces a chronic inflammatory state, and chronic inflammation is a main contributor to the development of NAFLD/MAFLD.8–28 A pathologic shift (ie, an increased Firmicutes to Bacteroidetes ratio) in gut-derived bacteria from obesity (ie, gut microbiota dysbiosis) contributes via additionally increased levels of LPS,27–29 which triggers inflammation in the gut (at least in part via TLR4 signaling) (reviewed in29) leading to downregulation of tight junction proteins [occluding (OCLN), zona-occludin (ZO-1), etc.], disrupting the integrity of the gut barrier30,31(reviewed in32,33), and leading to “leaky gut syndrome” (reviewed in29,32,33). Bacterial LPS, FFAs, other inflammatory molecules and their metabolites are then released into the bloodstream from the leaky gut, causing inflammation systemically in other tissues (such as adipose tissue) contributing to a state of chronic inflammation/metabolic endotoxemia7,34,35(reviewed in29). In addition, itaconate is an anti-inflammatory metabolite36–39 that has been shown to be significantly less abundant in obesity-associated steatotic liver,19 and genetic deletion of the gene that encodes the protein (ie, immune response gene 1, Irg1) exacerbates HF diet-induced NAFLD/MAFLD in C57BL/6J mice.18 It has been reported that the induction of NAFLD/MAFLD is often accompanied by an increase in circulating tryptophan and a decrease in circulating levels of the tryptophan catabolite, kynurenine (a marker of immune system activation),40 and that this change also contributes to a pro-inflammatory status.41 A schematic diagram of how each of the aforementioned work together to contribute to HF diet-induced chronic inflammation and the development of NAFLD/MAFLD is provided in Figure 1.
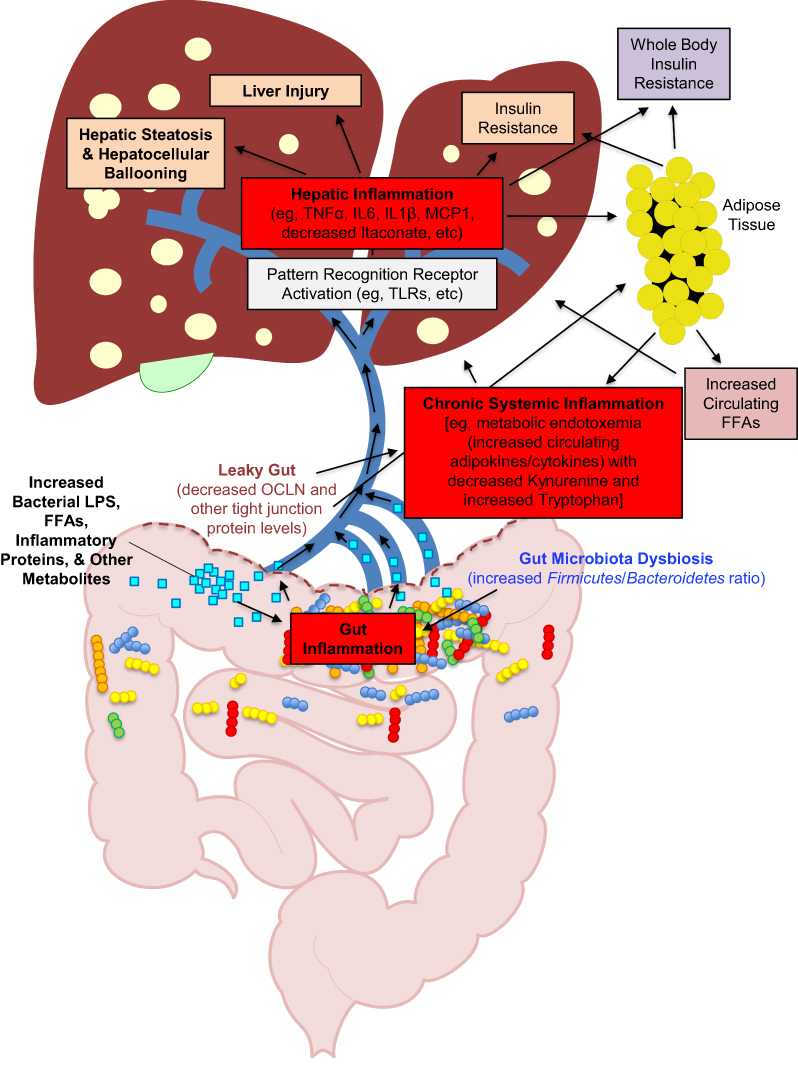 |
Figure 1 Simplified schematic diagram of the pathologic cycle in obesity-induced NAFLD/MAFLD resulting from multiple parallel hits. Notes: In a state of obesity, there is an over-abundance of circulating free fatty acids (FFAs) that are released from adipose tissue and absorbed in the gut. There is also a pathologic shift in the gut microbiome (ie, gut microbiome dysbiosis) that leads to an inflammatory state in the gut (in part via activation of TLR4 signaling by gut derived FFAs and bacterial LPS). Gut inflammation then leads to a decrease in gut tight junction proteins (such as OCLN), which causes a disruption in the gut protective barrier (ie, leaky gut). The leaky gut then leads to the release of increased levels of LPS, FFAs, inflammatory proteins (ie, cytokines and chemokines), and other metabolites into the portal vein, where it then goes to the liver. In the liver, these substances cause hepatic inflammation (in part via activation of TLR4 signaling in both hepatocytes and resident immune cells). This hepatic inflammation then leads to an increase in hepatic steatosis (via modulation of lipid metabolism in the liver), hepatocellular ballooning, hepatic insulin resistance, and other liver injury that is associated with NAFLD/MAFLD. In parallel, the increase in chronic, systemic circulating levels of LPS, FFAs, inflammatory proteins, and other metabolites also leads to the activation of inflammation (again, in part, via activation of TLR4 signaling) in adipose tissue (in both adipocytes and associated macrophages/immune cells). This also contributes to the overall state of chronic inflammation systemically in the obese individual. This chronic systemic inflammation is the cause of whole-body insulin resistance that occurs in NAFLD/MAFLD. This is certainly not a complete, comprehensive schematic, but encompasses the main overall processes in the pathogenesis of obesity-induced NAFLD/MAFLD that are relevant to this study. |
Presently, there is a dearth of Food and Drug Administration approved drugs for the treatment of NAFLD/MAFLD and nonalcoholic steatohepatitis (NASH).42–45 The current management of NAFLD/MAFLD includes lifestyle modifications (ie, weight loss, exercise) and the treatment of co-morbidities (ie, diabetes, dyslipidemia, hypertension and atherosclerosis). However, there is poor patient adherence to lifestyle modifications, efficacy studies of metformin in improving liver histology have led to mixed results,46–48 and insulin-sensitizing drugs such as pioglitazone have shown some beneficial effects,49 but cause increased water retention and weight gain,50,51 while glucagon-like peptide 1 (GLP-1) agonists and other “dual and triple” incretin analogs combinations are being developed.52 Moreover, less than half of NASH patients will respond to current therapeutic regimens,53 underscoring the critical need for continued research aimed at the development of novel therapeutics for the treatment of this rapidly expanding, life-threatening liver disease.
In the current study, we evaluated the ability of a promising, non-toxic (Supplemental Table 1) and novel small molecule, IOI-214 (Figure 2), to inhibit LPS-induced inflammation and its efficacy in preventing HF diet-induced NAFLD/MAFLD in male C57BL/6J mice. Our group previously studied a phenyl derivative of methimazole termed phenylmethimazole (ie, C10).11,54–60 In an attempt to find more effective compounds, we generated a library of small organic molecules derived from C10 in which a large number of aryl groups (Ar), substituted alkyl groups (R), and different heteroatoms (X, Y) were inserted into the ring system of C10 (see scheme in Figure 2). The pharmacological properties of these compounds were investigated in a series of assays. These assays revealed a compound’s ability to reduce cytokine expression in in vitro models of pathological inflammation. This effort led to the identification of the compound used in this study (IOI-214) that was more potent than C10 in the screening assays. We hypothesized that IOI-214, a novel compound that is similar to, yet structurally distinct from phenylmethimazole (C10) which we have shown previously to be a potent inhibitor of TLR3 and TLR4-mediated inflammation,11,55–59,61,62 would block LPS-induced inflammatory cytokine expression, and would therefore ameliorate HF diet-induced inflammation and hepatic steatosis in C57BL/6J male mice. Herein, we show that IOI-214 blocks inflammation and abates HF diet-induced NAFLD/MAFLD and begin to explore its therapeutic mechanism(s) of action.
 |
Figure 2 Derivation and structure of IOI-214 from C10. Notes: IOI-214 is a small organic compound that was derived from a phenyl derivative of methimazole termed C10. In an attempt to generate compounds that had more potent anti-inflammatory activity than C10, we generated a library of small organic molecules derived from C10 (see scheme above) in which a large number of aryl groups (Ar), substituted alkyl groups (R), and different heteroatoms (X, Y) were inserted into the ring system of C10. The molecular structures of C10 and IOI-214 are shown here and were drawn using the ChemDraw program (Perkin Elmer Informatics, Waltham, MA, USA). The structure of IOI-214 used in these studies was verified by both 1H-NMR and 13C-NMR and was shown to be 98% pure by HPLC. |
Materials and Methods
IOI-214 Synthesis
IOI-214 was prepared from commercially available a-halo ketone using the general method of Gan.63 A schematic describing the synthesis of 3-propyl-4-(pyridin-3-yl)thiazole-2(3H)-thione (IOI-214) can be found in Figure 3. In brief, to a stirred solution of the amine 1 (0.22 mL, 2.67 mmol) in a 1:1 mixture of H2O/EtOH (9 mL) was added CS2 (0.32mL, 5.34mmol) and K2CO3 (0.495 g, 3.56 mmol). The reaction was stirred for 5 min and 3-(2-bromoacetyl) pyridine hydrobromide (1) (0.503 g, 1.78 mmol) was added and stirred for an additional 3 h. The yellow solid was filtered and dried under vacuum overnight to provide 0.36 g, (80%) of 4-hydroxy-3-propyl-4-(pyridin-3-yl)thiazolidine-2-thione (2), Rf = 0.28 (2% CH3OH in CH2Cl2) mp 129–131 °C; 1H NMR (CD3OD, 500 MHz) δ 8.68 (s, 1H) 8.54 (d, J = 4.9, 1H) 7.91 (d, J = 7.8, 1H) 7.51 (m, 1H) 3.67 (s, 1H) 3.11 (s, 1H), 1.60 (m, 2H) 0.79 (t, J = 7.3, 3H).
 |
Figure 3 Synthesis of 3-propyl-4-(pyridin-3-yl)thiazole-2(3H)-thione (IOI-214). Notes: To a stirred solution of the amine 1 (0.22 mL, 2.67 mmol) in a 1:1 mixture of H2O/EtOH (9 mL) was added CS2 (0.32mL, 5.34mmol) and K2CO3 (0.495 g, 3.56 mmol). The reaction was stirred for 5 min and 3-(2-bromoacetyl) pyridine hydrobromide (1) (0.503 g, 1.78 mmol) was added and stirred for an additional 3 h. The yellow solid was filtered and dried under vacuum overnight to provide 0.36 g, (80%) of 4-hydroxy-3-propyl-4-(pyridin-3-yl)thiazolidine-2-thione (2), Rf = 0.28 (2% CH3OH in CH2Cl2) mp 129–131 °C; 1H NMR (CD3OD, 500 MHz) δ 8.68 (s, 1H) 8.54 (d, J = 4.9, 1H) 7.91 (d, J = 7.8, 1H) 7.51 (m, 1H) 3.67 (s, 1H) 3.11 (s, 1H), 1.60 (m, 2H) 0.79 (t, J = 7.3, 3H). To the crude alcohol (2) (200 mg, 0.78 mmol) in a dry flask was added a freshly prepared solution of anhydrous HCl (1 M in EtOAc, 2.0 mL) and dry EtOH (1.6 mL). The reaction mixture was refluxed for 24 h. TLC confirmed reaction completion and the solvent was removed under vacuum to provide 190 mg (100%) of IOI-214 as a dark yellow solid, Rf = 0.62 (2% CH3OH in CH2Cl2) mp 175–177 °C; 1H NMR (CD3OD, 500 MHz) δ 9.16 (s, 1H), 9.05 (d, J = 4.0, 1H), 8.79 (d, J = 7.4, 1H), 8.26 (t, J = 6.9, 1H), 7.20 (s, 1H) 4.15 (t, J = 7.8, 2H), 1.64 (m, 2H), 0.80 (t, J = 7.6, 3H); 13C NMR (CD3OD, 125 MHz) δ 189.0, 147.0, 142.9, 142.57, 136.9, 130.7, 127.5, 114.0, 48.9, 20.6, 9.7; HPLC (30–80%- Acetonitrile/H2O-20min), TR 4.98min, 98.1%. The sample was run on a Hypersil Gold C8 Column (5µm, 4.6 x 150mm) with solvent (30–80%- Acetonitrile/H2O) gradually increasing the percentage of Acetonitrile from 30 to 80% over 20 mins. |
To the crude alcohol (2) (200 mg, 0.78 mmol) in a dry flask was added a freshly prepared solution of anhydrous HCl (1 M in EtOAc, 2.0 mL) and dry EtOH (1.6 mL). The reaction mixture was refluxed for 24 h. TLC confirmed reaction completion and the solvent was removed under vacuum to provide 190 mg (100%) of IOI-214 as a dark yellow solid, Rf = 0.62 (2% CH3OH in CH2Cl2) mp 175–177 °C; 1H NMR (CD3OD, 500 MHz) δ 9.16 (s, 1H), 9.05 (d, J = 4.0, 1H), 8.79 (d, J = 7.4, 1H), 8.26 (t, J = 6.9, 1H), 7.20 (s, 1H) 4.15 (t, J = 7.8, 2H), 1.64 (m, 2H), 0.80 (t, J = 7.6, 3H); 13C NMR (CD3OD, 125 MHz) δ 189.0, 147.0, 142.9, 142.57, 136.9, 130.7, 127.5, 114.0, 48.9, 20.6, 9.7; HPLC (30–80%- Acetonitrile/H2O-20min), TR 4.98min, 98.1%. The sample was run on a Hypersil Gold C8 Column (5µm, 4.6 x 150mm) with solvent (30–80%- Acetonitrile/H2O) gradually increasing the percentage of Acetonitrile from 30 to 80% over 20 mins.
IOI-214 Solutions
Dr. Stephen Bergmeier (Ohio University, Athens, OH, USA) prepared IOI-214 for these studies as a 200 mM stock solution using 100% (v/v) dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO, USA). This stock solution was then further diluted using sterile 1X phosphate-buffered saline (PBS) (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) to a 10% working solution.
Cell Culture
To culture the murine macrophage cell line, RAW 264.7 (ATCC, Manassas, VA, USA), Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) was prepared using 10% (v/v) fetal bovine serum. To culture the murine hepatocyte cell line, AML-12 (ATCC, Manassas, VA, USA), DMEM/Nutrient Mixture F-12 (DMEM-F12) (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) was prepared with insulin (0.005 mg/mL), transferrin (0.005 mg/mL), selenium (5 ng/mL), dexamethasone (40 ng/mL), fetal bovine serum [10% (v/v)] (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), and penicillin/streptomycin [1% (v/v)] (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). The conditions used to grow all cell lines were 37°C in 5% carbon dioxide. A working solution of IOI-214 (20 μM) was prepared in 0.25% DMSO (Sigma-Aldrich, St. Louis, MO, USA). LPS treatment of cells (passaged less than 20 times) was done using 10 ng/mL LPS (Sigma-Aldrich, St. Louis, MO, USA) in complete culture media.
Mice and Experimental Design
All mouse studies were approved by the Ohio University Institutional Animal Care and Use Committee (Approval Number 13-H-035) and conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Experimental Design
Male C57BL/6J mice were purchased from The Jackson Laboratories (Bar Harbor, ME, USA) at 6 weeks of age and acclimated for one week prior to the initiation of the experiment (ie, diet introduction and initiation of compound treatment). These mice were housed in a temperature- and humidity-controlled environment (18°C–22°C) that is maintained on a 14:10 hour light/dark cycle.
Male mice were used in these studies since it has recently been shown that C57BL/6J male mice, and to a much lesser extent female C57BL/6J mice,64–68 fed a HF diet develop obesity, IR, and NAFLD/MAFLD.8,69–78 In fact, female C57BL/6J mice have even been shown to be protected from HF diet-induced metabolic syndrome, adipose tissue inflammation, and hepatic steatosis.67,68,79 More specifically, estrogens have been shown to attenuate metabolic dysfunction and NAFLD due to their effects on hepatic lipogenic and inflammatory signaling and energy balance (eg, regulation of food intake, energy expenditure, direct effects on the physiology of energy metabolism-regulating organs such as liver, muscle, adipose tissue and pancreas).80–88
At the initiation of the experiment, the mice were randomly divided into the following groups: Chow Diet, HF DMSO (10% v/v), and HF IOI-214 (1 mg/kg) in 10% DMSO. The mice were then placed on either a chow diet consisting of 10% fat, 20% protein, 70% carbohydrate [(#D12450B, Research Diets Inc., New Brunswick, NJ, USA), or a HF diet consisting of 60% fat, 20% protein, 20% carbohydrate [(#D12492, Research Diets Inc., New Brunswick, NJ, USA)]. We chose this diet since it is a highly used model for studying NAFLD/MAFLD89–93 and one in which our laboratory has experience using.11 On the same day, mice were placed on their respective diets, the mice on the HF diet began receiving daily intraperitoneal injections. The diets and treatments continued for 16 weeks. Body weights of the mice were measured weekly. Multiple cohorts of mice were used to generate all of the data.
Intraperitoneal Glucose Tolerance Tests
For intraperitoneal glucose tolerance tests (IPGTT), mice were fasted for 4–5 hours. Blood glucose (Freestyle Freedom Lite Blood Glucose Monitoring System, Abbott Laboratories, Lake Bluff, IL, USA) and body weight were then measured. Next, glucose (1 g/kg body weight) (Sigma-Aldrich, St. Louis, MO, USA) was delivered via intraperitoneal injection. Blood glucose measurements were then measured at time 0, 15, 30, 60, 90, 120, and 180-minutes following the glucose injection.
Hepatic Histological Analysis
To evaluate liver morphology and steatosis microscopically, 10% buffered formalin was used to fix the liver tissue for a period of 12–24 hours. After fixation in formalin, the tissues were dehydrated in ethanol, embedded in paraffin, and cut to 5 µm using a microtome. The liver tissues were then subjected to hematoxylin and eosin (H&E) staining. The Ohio University Heritage College of Osteopathic Medicine Histological Core Services conducted the tissue preparation and staining.
For Oil Red O (ORO) staining, frozen liver samples were cut to 10 µm using a cryostat. The sections were then dried onto the slides, fixed in 10% buffered formalin, and then briefly washed with running tap water. The slides were then rinsed with 60% isopropanol and stained with freshly prepared ORO working solution. After rinsing again with 60% isopropanol, the slides were lightly stained with haematoxylin, rinsed with distilled water, and mounted with aqueous mountant (CC/Mount, C9368-30ML, Sigma-Aldrich, St. Louis, MO, USA).
Hepatic Triglyceride Quantification
A protocol based on the Salmon and Flatt method of lipid saponification was used to measure hepatic triglyceride content.94 Triglycerides concentration in the samples was determined using Triglycerides Liquid reagent set (Pointe Scientific, Canton, MI, USA) following the manufacturer’s instructions and corrected using a dilution factor and amount of tissue used. Final triglyceride concentration is expressed as mg/g of liver tissue.
Untargeted Global Metabolomics Analysis
Serum and liver samples were collected from HF DMSO and HF IOI-214 mice and sent to Metabolon Inc. (Durham, NC, USA) (N = 8/group) for nontargeted metabolite profiling as previously described.95 Briefly, samples were extracted and split into equal parts for by liquid chromatography/tandem mass spectrometry (LC/MS/MS). Proprietary software was used to match ions to an in-house library of standards for metabolite identification and for metabolite quantitation by peak area integration. For all analyses, following normalization to volume extracted for serum, missing values, if any, were imputed with the observed minimum for that particular compound. The statistical analyses were performed on natural log-transformed data.
Serum Insulin, Cholesterol, and Triglyceride Analyses
Serum was collected from 4–6-hour fasted mice at experimental termination (end of the 16-week experiment). Insulin (80-INSMSU-E01, ALPCO, Salem, NH, USA), cholesterol (ab65359, ABCAM, Waltham, MA, USA), and triglyceride (10,010,303, Cayman Chemical, Ann Arbor, MI, USA) levels were obtained according to the manufacturer’s protocols.
Fasting Blood Glucose Measurements
Fasting blood glucose was measured on mice fasted for 4–6 hours using Freestyle Freedom Lite Blood Glucose Monitoring System (Abbott Laboratories, Lake Bluff, IL, USA) via the tail vein just prior to euthanasia.
Quantitative Real Time-PCR Analysis
TRIzol reagent (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) was used to isolate total RNA from frozen liver tissue. Total RNA was isolated from cell lines (RAW 264.7 and AML-12) using the RNeasy kit (Qiagen, Germantown, MD, USA). cDNA was synthesized using the high-capacity cDNA reverse transcription kit (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) with the inclusion of RNase inhibitor. Gene expression was then quantified via qPCR using SYBR Green biochemistry (iTaq Universal SYBR Green Supermix, Bio-Rad Laboratories, Inc., Hercules, CA, USA). Murine Taqman (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) gene expression assays that were used as qPCR primers include: tumor necrosis factor (Tnf) (Mm00443258_m1), interferon beta 1 (Ifnb1) (Mm00439546_s1), C-C motif chemokine ligand 2/monocyte chemoattractant protein 1 (Mcp1) (Mm00441242_m1), interleukin 1 beta (Il1b) (Mm00434228_m1), interleukin 6 (Il6) (Mm00446190_m1), and perilipin 5 (Plin5) (Mm00508852_m1). Murine housekeeping Taqman gene expression assays that were used as qPCR primers include: Beta actin (Actb) (Mm02619580_g1) and glyceraldehyde 3-phosphate dehydrogenase (Gapdh) (Mm99999915_g1). Cycling parameters were: 40 cycles of 95°C for 3 seconds, followed by 60°C for 30 seconds. Melting curves were run for each qPCR. The delta delta Ct Method was used to evaluate relative fold change in gene expression.96
ELISAs
Protein was isolated from the liver using a standard protein lysis buffer (10mM Tris HCL, pH 7.5, 150 mM NaCl, 1% NP-40) supplemented with protease inhibitors (Calbiochem, San Diego, CA, USA) followed by manual homogenization using zirconium oxide beads (Next Advance Inc., Troy, NY, USA). The same amount of protein from each sample was then subjected to Enzyme-Linked Immunosorbent Assays (ELISAs) specific for detecting TNFα (ELM-TNFα-CL-1), MCP1 (ELM-MCP1-CL-1), IL6 (ELM-IL6-CL-1), and IL1β (ELM-IL1β-CL-1) (RayBiotech Life, Peachtree Corners, GA, USA) according to the manufacturer’s protocol.
Western Blot
For the detection of colonic OCLN, 60 µg of total protein isolated from the colon of the mice at the end of the experiment was subjected to SDS-PAGE and Western blot analysis using an antibody specific for the detection of OCLN (91131S, Cell Signaling Technologies, Danvers, MA, USA) at a dilution of 1:500. Blots were also stripped and re-probed with an antibody specific for the detection of βActin (4967S, Cell Signaling Technologies, Danvers, MA, USA) at a dilution of 1:1000. Goat anti-rabbit IgG-HRP conjugated secondary antibody (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) at a dilution of 1:20,000 was used. For visualization of signals, the ECL Western Blotting Analysis System (Amersham/Cytiva Life Sciences, Marlborough, MA, USA) was used. Western blots were read using the ChemiDoc MP Imaging System (BioRad Laboratories, Hercules, CA, USA). OCLN and βActin band intensities were measured using GelQuant.NET software provided by biochemlabsolutions.com (University of California, San Francisco, San Francisco, CA, USA).
Microbiome Analysis
The microbiome data was generated using University of California (UC) Davis Mouse Metabolic Phenotyping Center (MMPC, Davis, CA, USA) services. Fecal samples from the colon of odd numbered mice (chosen for randomization) in each group were collected at the end of the 16-week study (before euthanasia), stored at −80°C, and sent to the UC Davis MMPC for microbiome analysis according to their protocol (https://www.mmpc.org/shared/protocols.aspx).
LPS Binding Protein Analysis
Serum samples from the mice collected at the end of the 16-week experiment were sent to the UC Davis MMPC (Davis, CA, USA) for LPS binding protein enzyme-linked immunosorbent assay (ELISA) analysis. The LPS binding protein (LBP) kit (Biometec GmbH, Greifswald, Germany) was used to determine serum LPS binding protein levels according to the manufacturer’s instructions.
Statistical Analysis
Statistical analyses were performed using NCSS (V8.0.24, Kaysville, UT, USA). Statistical differences in RT-qPCR for cell culture studies were determined using a one-way analysis of variance (ANOVA) followed by a Tukey honestly significant difference (HSD) test for post hoc comparison with alpha set to 0.05. Statistical differences in animal study data were determined using either a one-way ANOVA followed by a Tukey HSD test for post hoc comparison with alpha set to 0.05, or Independent Samples t-tests with alpha set to 0.05 (LBP) as appropriate. Pearson’s Correlations were calculated, and linear regression and correlation analyses were conducted to evaluate relationships between hepatic TG and cytokine, Plin5 expression, and LBP levels with alpha set to 0.05. Statistical differences in metabolites from the metabolomics study were determined using Welch’s two-sample t-tests with alpha set to 0.05. Outliers were identified in NCSS to be >1.5X the inter-quartile range.
Results
IOI-214 is a Potent Inhibitor of LPS-Induced Inflammatory Cytokine Production in Murine Macrophages and Hepatocytes in Culture
Obesity induces a chronic inflammatory state, in large part, via toll-like receptor 4 (TLR4)-dependent mechanisms.9 Hepatocytes and macrophages in the liver express TLR4 which is necessary for both FFA/lipid- and LPS-induced inflammation in HF diet-induced NAFLD.8,97 Although recent evidence indicates that TLR4 is not a receptor for saturated FFAs, TLR4 is required for saturated FFA/lipid-induced inflammation through TLR4-dependent priming and altered cellular metabolism.10 Thus, we evaluated the effects of our novel small molecule, IOI-214, to prevent LPS-induced inflammation in murine macrophages (RAW264.7) and hepatocytes (AML-12) in culture. Treatment with IOI-214 potently attenuated the robust LPS-induced upregulation of proinflammatory cytokine gene expression (Tnf, Ifnb1, Ccl2, Il1b, and Il6) in the cultured murine macrophages (Figure 4A). The induction of Tnf in the murine hepatocytes (AML-12 cells) by LPS, as expected, was not as robust as in the RAW264.7 cells, however IOI-214 was also effective at blocking this induction of Tnf expression in the AML-12 cells in culture (Figure 4B).
 |
Figure 4 IOI-214 prevents LPS-induced inflammation in murine macrophages and hepatocytes in culture. Notes: (A) The murine macrophage cell line, RAW264.7 (A), and the murine hepatocyte cell line, AML-12 (B) were treated with 20 μM IOI-214 and DMSO (control) to determine if IOI-214 could block the upregulation of Tnf, Ifnb1, Ccl2, Il1b, and Il6 expression in the presence of 10 ng/mL LPS. All cells were pretreated with IOI-214 or DMSO for 1 hour prior to LPS treatment for 4 hours. Gene expression was determined via RT-qPCR. Error bars in all bar graphs represent ±SEM. Significance was determined using a one-way ANOVA followed by Tukey’s HSD post hoc analysis for multiple comparison. Bars indicate differences between indicated groups. #Indicates difference from all other groups, p < 0.05. *Indicates p < 0.05. ***Indicates p < 0.001. *****Indicates p < 0.00001. |
IOI-214 Impairs HF Diet-Induced Hepatic Steatosis and Hepatocyte Damage, as Well as Mesenteric Fat Accumulation and Fasting Blood Glucose Levels, Without Affecting Other Metabolic and Lipid Parameters (Serum Cholesterol, Triglyceride, or Insulin Levels or Glucose Tolerance) in Obese Mice
Given the potent anti-inflammatory activity of IOI-214 in vitro, we questioned the effectiveness of IOI-214 to prevent hepatic steatosis/NAFLD/MAFLD induced by a HF diet in vivo. To this end, we fed C57BL/6J male mice either a chow diet (10% fat) or a HF diet (60% fat) and treated them once daily for 16-weeks with DMSO (vehicle) or IOI-214 (1 mg/kg). We chose this diet since it is a frequently used model for studying NAFLD/MAFLD89–93 and one in which our laboratory has experience using.11
There was an increase in total weight after HF diet feeding in both the IOI-214 and comparably fed DMSO control group compared to the chow diet-fed group after 16-weeks (Figure 5A and B). Treatment with IOI-214 did not alter weight gain or impede the development of overall obesity (Figure 5A and B). Mice on the HF diet and treated with IOI-214 (HF IOI-214) gained the same amount of weight as the HF diet-fed DMSO (HF DMSO) group (Figure 5A and B).
 |
Figure 5 IOI-214 blocks HF diet-induced mesenteric fat accumulation without affecting HF diet-induced weight gain or subcutaneous fat accumulation. Notes: Seven-week-old male C57BL/6J mice were fed a HF diet and treated once daily with 1mg/kg IOI-214 or DMSO (10% in sterile saline) via intraperitoneal injection for 16 weeks. Weights of the mice were measured weekly and weight over the course of the experiment was plotted (A) and body weight gain was calculated (B). At the end of the 16-week experiment, mice were euthanized and mesenteric (C) and subcutaneous (D) fat was extracted from the mice and weighed. (B–D) Black squares (HF DMSO), gray triangles (HF IOI-214), and green diamonds (Chow Diet) represent data points from individual mice in each group. Error bars in all line and bar graphs represent ±SEM. Significance was determined using a one-way ANOVA followed by Tukey’s HSD post hoc analysis for multiple comparison. Bars in bar graphs indicate differences between indicated groups. Asterisks in the line graph indicate differences of HF DMSO and HF IOI-214 groups from the Chow Diet group at each time point. ***Indicates p < 0.001. *****Indicates p < 0.00001. ******Indicates p < 0.000001. Outliers are indicated by pink circles. |
The amount of both mesenteric and subcutaneous adipose tissue in the HF diet-fed mice was significantly higher than in the chow diet-fed control mice (Figure 5C and D). While the amount of subcutaneous adipose tissue in HF IOI-214 mice was no different than that in the HF DMSO control mice (Figure 5D), HF IOI-214 mice had significantly less mesenteric adipose tissue than HF DMSO control mice (Figure 5C).
Oil Red O staining of liver sections revealed the presence of hepatic steatosis in the HF diet-fed mice compared to the chow diet-fed mice. The HF IOI-214 mice also had less hepatic steatosis than the HF DMSO control mice (Figure 6A). H&E staining of liver sections also revealed the presence of some hepatocellular ballooning (Figure 6B, lower left panel marked with arrowhead) and widespread evidence of the presence of early stages of hepatocellular ballooning in HF DMSO mice. However, there was no hepatocellular ballooning observed in the HF IOI-214 mice (Figure 6B, lower middle panel) or chow diet-fed mice (Figure 6B, lower right panel). To confirm the histological findings related to hepatic steatosis, and to further quantify the IOI-214 inhibition of HF diet-induced hepatic steatosis, we quantified TG content biochemically using the Salmon and Flatt method.94 In line with what was observed histologically (Figure 6A), the HF diet-induced increase in hepatic TG content (ie, hepatic steatosis) was significantly attenuated with IOI-214 treatment (Figure 6C). There was no significant difference in the amount of hepatic TG content between the HF IOI-214 and the chow diet-fed mice (Figure 6C). To lend additional support to this finding, we evaluated the hepatic expression of the lipid-associated marker, perilipin 5 (Plin5). We found that Plin5 expression in the HF DMSO mice was significantly higher than in the HF IOI-214 mice and the chow diet-fed mice (Figure 6D). There was no significant difference in the hepatic expression of Plin5 between the HF IOI-214 mice and the chow diet-fed mice (Figure 6D). Moreover, we found a significant (P < 0.001) positive correlation (r = 0.56) between hepatic Plin5 expression and hepatic TG levels (Figure 6E).
Figure 6 Continued. Figure 6 IOI-214 ameliorates HF diet-induced hepatic steatosis. Notes: Seven-week-old male C57BL/6J mice were fed a HF diet and treated once daily with 1mg/kg IOI-214 or DMSO (10% in sterile saline) via intraperitoneal injection for 16 weeks. (A) Oil Red O (ORO) staining and (B) Hematoxylin and eosin (H&E) staining was performed on liver tissue sections prepared from the mice after 16 weeks of HF diet feeding and treatment. All images in (A, top row) were taken at 40X magnification and images in (A, bottom row) were taken at 100X. All images in (B, top row) were taken at 20X and images in (B, bottom row) were taken at 200X. The black arrow in B represents an example of hepatocellular ballooning. Scale bars are as indicated in A&B. These are representative images from mice in each group. (C) Liver triglyceride content was quantified biochemically in liver tissue extracted from the mice after 16 weeks of treatment. (D) RNA was isolated from the liver of the HF IOI-214 and HF DMSO mice from the 16-week study and Plin5 gene expression was determined via RT-qPCR. (E) Pearson’s correlation (r) analysis of hepatic TG content and relative fold change in Plin5 expression in HF DMSO, HF IOI-214 and Chow Diet mice. (C–E) Black squares (HF DMSO), gray triangles (HF IOI-214), and green diamonds (Chow Diet) represent data points from individual mice in each group. Error bars in all bar graphs represent ±SEM. Significance was determined using a one-way ANOVA followed by Tukey’s HSD post hoc analysis for multiple comparison. Bars indicate differences between indicated groups. *Indicates p < 0.05. ****Indicates p < 0.0001. Outliers are indicated by pink circles.

Fasting serum cholesterol, TG, insulin, and glucose levels were measured biochemically. Serum cholesterol levels were significantly elevated in the HF DMSO and HF IOI-214 mice compared to the chow diet-fed mice. There was no difference in the amount of serum cholesterol between the HF DMSO and HF IOI-214 groups (Figure 7A). There was also no difference in serum TG levels between HF and chow diet-fed mice. This is an observation that is consistent with our findings in previous studies using this mouse model.11 IOI-214 had no effect on serum TG levels (Figure 7B). There were also no observed differences in the amount of fasting serum insulin among the three experimental groups (Figure 7C). Fasting blood glucose levels in the HF DMSO mice were significantly elevated compared to the chow diet-fed and HF IOI-214 mice (Figure 7D). The fasting blood glucose levels in the HF IOI-214 mice were not statistically different from the chow diet-fed mice (Figure 7D). Moreover, intraperitoneal glucose tolerance tests revealed decreased glucose tolerance in the HF DMSO and HF IOI-214 groups compared to the chow diet-fed group, however there was no difference in glucose tolerance in the HF IOI-214 mice compared to the HF DMSO mice (Figure 7E). Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were not measured in this study as it has been previously demonstrated that AST and ALT levels are not elevated in this mouse model until 34–36 weeks on the HF diet.98
 |
Figure 7 IOI-214 mitigates HF diet-induced fasting hyperglycemia, without affecting fasting serum insulin, cholesterol or triglyceride levels, or glucose intolerance. Notes: Blood was collected from the mice at the end of the 16-week study and fasting serum cholesterol (A), TG (B), insulin (C), and blood glucose (D) levels were measured. A 3-hour intraperitoneal glucose tolerance test (E) was performed at week 15. Black squares (HF DMSO), gray triangles (HF IOI-214), and green diamonds (Chow Diet) represent data points from individual mice in each group. Error bars in all line and bar graphs represent ±SEM. Significance was determined using a one-way ANOVA followed by Tukey’s HSD post hoc analysis for multiple comparison or Independent Samples t-tests as appropriate. Bars indicate differences between indicated groups in bar graphs. Asterisks in the line graph indicate differences of HF DMSO and/or HF IOI-214 groups from the Chow Diet group at each time point. *Indicates p < 0.05. **Indicates p < 0.01. ****Indicates p < 0.0001. Outliers are indicated by pink circles. |
IOI-214 Reduces HF Diet-Induced Inflammation in Obese Male C57BL/6J Mice
Inflammation is a multifaceted biological response that incorporates numerous signaling pathways for the local or systemic activation of the immune system. As such, changes in multiple pathways, both at the tissue level and in circulation, may implicate a mechanism by which IOI-214 regulates inflammation either directly or indirectly. Since inflammation is associated with enhanced hepatic steatosis,12–16 and exogenous TNF in mice and rats is known to cause increased TG production and storage in the liver,12,15 we first evaluated TNFα protein levels in the liver of our mice. HF IOI-214 and chow diet-fed mice had significantly lower levels of hepatic TNFα than the HF IOI-214 DMSO mice (Figure 8A). There was no significant difference in TNFα levels between chow diet-fed mice and HF IOI-214 mice (Figure 8A). The hepatic protein levels of other pro-inflammatory cytokines known to participate in the pathogenesis of HF diet-induced NAFLD/MAFLD (MCP1, IL6, and IL1β)73 were also assessed in the liver of our mice. Hepatic MCP1, IL6 and IL1β protein levels were also significantly reduced in HF IOI-214 mice compared to HF DMSO mice (Figure 8B–D), although hepatic MCP1 and IL6 protein levels in HF IOI-214 mice were still significantly elevated compared to chow diet-fed mice (Figure 8B and D). There was no significant difference in hepatic IL1β levels between chow diet-fed mice and HF IOI-214 mice (Figure 8C). To lend further support to the relationship between hepatic inflammation and TG levels in the liver, there were highly significant positive correlations between hepatic TG levels and TNFα (P < 0.00001), MCP1 (P = 0.0001), IL6 (P < 0.001) and IL1β (P < 0.00001) protein levels. These positive correlations corresponded with clear separation of treatment groups (as indicated by corresponding circles) (Figure 9A–D).
 |
Figure 8 IOI-214 blocks HF diet-induced hepatic inflammatory cytokine and chemokine protein levels in vivo. Notes: Protein levels of inflammatory cytokines [TNFα (A), IL6 (B) and IL1β(C)] and the chemokine MCP1 (D) were evaluated in the liver of the mice at the end of the 16-week experiment via cytokine and chemokine-specific ELISAs. Black squares (HF DMSO), gray triangles (HF IOI-214), and green diamonds (Chow Diet) represent data points from individual mice in each group. Error bars in all graphs represent ±SEM. Significance was determined using a one-way ANOVA followed by Tukey’s HSD post hoc analysis for multiple comparison. Bars indicate differences between indicated groups. *Indicates p < 0.05. **Indicates p < 0.01. |
 |
Figure 9 Correlations between hepatic TG content and inflammatory cytokine and chemokine protein levels in the liver of HF DMSO, HF IOI-214 and Chow Diet mice. Notes: Pearson’s correlation (r) analyses of hepatic TG content and hepatic protein levels of TNFα (A), MCP1 (B), IL6 (C) and IL1β (D). Hepatic TG content was significantly and highly correlated with the hepatic protein levels of each of the cytokines and chemokine as indicated. The HF IOI-214 mice, in all cases, clearly had decreased levels of hepatic TG content compared to the HF DMSO mice that was correlated with decreased levels of the cytokines and chemokine evaluated. |
In humans and mice PLIN5/Plin5 expression has recently been shown to be involved in the regulation of pro-inflammatory cytokines, endoplasmic reticulum stress, and mitochondrial damage, which has made it an important target of studies focused on NAFLD/MAFLD.17 Plin5 depletion has also recently been shown to be protective in the pathogenesis of liver injury, which supports that pathways associated with PLIN5 are important.17 Thus, we evaluated hepatic Plin5 expression and found that IOI-214 significantly mitigated HF diet-induced hepatic Plin5 expression (Figure 6D).
Additional markers of systemic inflammation that have significance in NAFLD/MAFLD were assessed via metabolomics. It has been shown that in NAFLD/MAFLD, where there is increased inflammation, hepatic itaconate levels are low, and circulating levels of tryptophan are high and kynurenine low.40,41 Metabolomics revealed that hepatic itaconate and circulating kynurenine levels were significantly elevated in the HF IOI-214 mice in comparison to the HF DMSO mice (Table 1). In addition, circulating tryptophan levels were significantly lower in HF IOI-214 mice compared to the HF DMSO mice (Table 1).
 |
Table 1 Effects of IOI-214 on Key Metabolites Related to Inflammation |
Given the significant contribution of the gut microbiome to inflammation that contributes to NAFLD/MAFLD,20–26 changes in the gut microbiome composition by IOI-214 may help to explain IOI-214’s therapeutic effects on NAFLD/MAFLD in this HF diet-induced mouse model. To evaluate the effects of IOI-214 on the gut microbiome in HF diet-fed mice, we collected colonic fecal samples from HF DMSO and HF IOI-214 mice after 16 weeks of HF diet feeding and sent them to UC Davis MMPC and their Host Microbe Systems Biology Core for microbiome analysis. Partial Least Squares-Discriminant Analysis (PLS-DA) using Genus level abundances mostly separated populations of HF IOI-214 and HF DMSO groups (Figure 10A). Moreover, there was a greater abundance of Bacteroidetes in HF IOI-214 mice when compared to the HF DMSO mice, as well as a smaller abundance of Firmicutes in the HF IOI-214 mice when compared to the HF DMSO mice (Figure 10B). There was also a larger abundance of Verrucomicrobia, Proteobacteria, and Actinobacteria in the HF IOI-214 mice compared to the HF DMSO mice (Figure 10B).
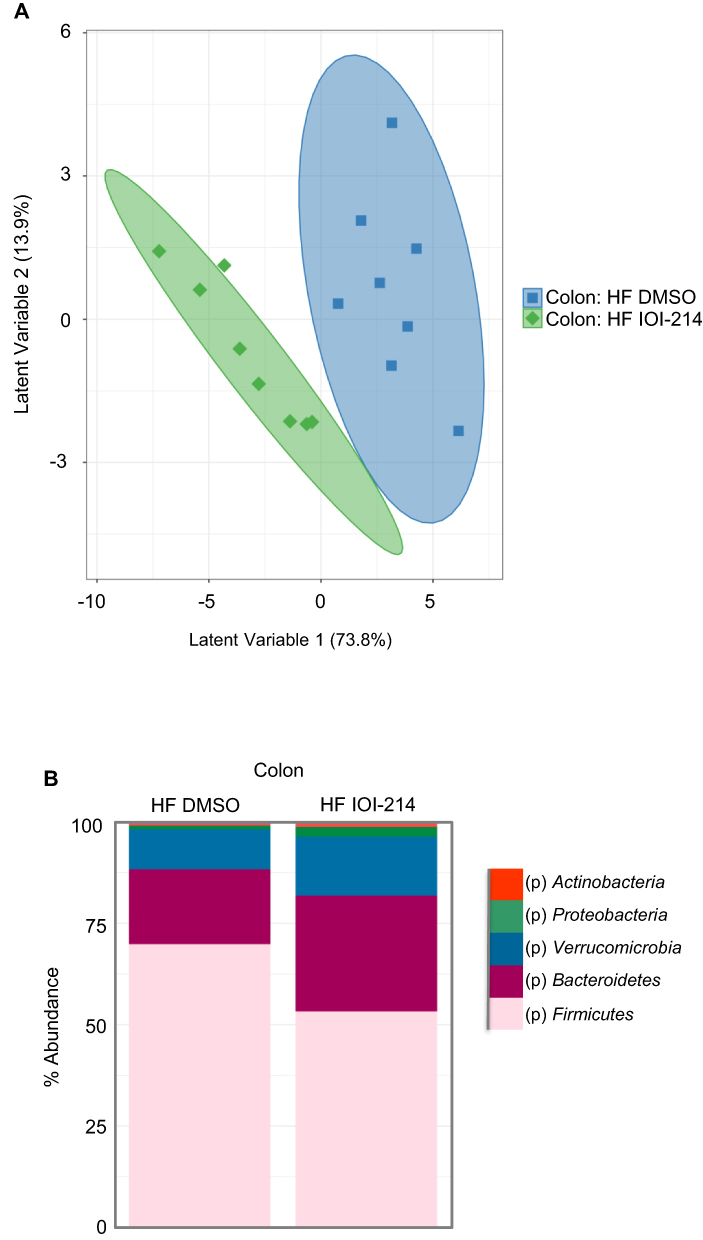 |
Figure 10 IOI-214 improves gut microbiota dysbiosis in HF diet-fed mice. Notes: Fecal samples from the colon of HF IOI-214 and HF DMSO mice from the 16-week study were collected and sent to the UC Davis Mouse Metabolic Phenotyping Center (MMPC) for microbiome analysis. (A) The principal component analysis was generated using genus-level abundance data. (B) Phylum abundance levels were measured among fecal samples collected from HF-fed mice. |
It is well known that in humans and mice, the pathologic shift in the gut microbiome that occurs in response to HF diet feeding (ie, gut microbiota dysbiosis) leads to an increase in intestinal permeability (ie, “leaky gut”), wherein bacterial LPS, and other inflammatory molecules, are released into the circulation.27 This consequently triggers inflammation in the liver, as the liver gets ~70% of its blood supply through the portal vein from the intestines.28,78 Lipopolysaccharide binding protein (LBP) binds to bacterial LPS and is important for presenting LPS to its cluster of differentiation 14 (CD14)-TLR4 complex to elicit immune responses.79 LBP levels are used as a surrogate of LPS. Thus, an increased level of serum LBP is indicative of increased circulating levels of LPS (ie, endotoxemia), which leads to increased hepatic inflammation. To evaluate the effects of IOI-214 on circulating levels of LBP, as a measure of systemic inflammation, in the HF diet-fed mice treated with DMSO or IOI-214, we collected serum samples from HF DMSO and HF IOI-214 mice after 16 weeks of HF diet feeding and sent them to the UC Davis MMPC and their Host Microbe Systems Biology Core for analysis. Circulating LBP levels in the HF IOI-214 mice were found to be significantly lower than in the HF DMSO mice (Figure 11A). To lend additional support to the relationship between circulating LPS/LBP levels and TG levels in the liver, there was a significant positive correlation (r = 0.58, P < 0.05) between hepatic TG levels and circulating LBP levels, which corresponded with clear separation of treatment groups (as indicated by the color-coded corresponding circles) (Figure 11B).
 |
Figure 11 IOI-214 decreases gut permeability in HF diet-fed mice. Notes: Serum samples from HF DMSO and HF IOI-214 collected at the end of the 16-week experiment and sent to the UC Davis Mouse Metabolic Phenotyping Center (MMPC) for analysis of LBP levels (A). (B) Pearson’s correlation (r) analysis of hepatic TG content and serum LBP levels in HF DMSO and HF IOI-214 mice. (C) Colon samples were collected from some of the mice at random at the end of the 16-week experiment, total protein was isolated, and Western blots were done to evaluate colonic OCLN protein levels. Densitometry was then conducted to evaluate the relative levels of OCLN protein relative to the amount of βActin. The average amount of OCLN per group was then calculated for the mice in each group wherein OCLN was present in the colon (n = 4 mice/group). Black squares (HF DMSO) and gray triangles (HF IOI-214) represent data points from individual mice in each group. Error bars represent ±SEM. Significance was determined using Independent Samples t-tests. Bars indicate differences between the groups. *Indicates p < 0.05. Outliers are indicated by pink circles. |
In addition to increased circulating levels of LPS as a marker of increased gut permeability, Occludin (OCLN) protein levels in the gut are known to decrease in obese mice with leaky gut syndrome30,31(reviewed in32,33). Thus, as a measure of gut permeability, we assessed OCLN levels in the colon of our mice that were fed the HF diet. As can be seen in Figure 10C, colonic OCLN protein levels in the majority of the HF diet-fed mice treated with IOI-214 were much higher than OCLN protein levels in the HF diet-fed DMSO mice (Figure 11C, last 5 lanes compared to the first 5 lanes, respectively). In the mice containing OCLN protein (n = 4 out of 5 in each group), the OCLN levels were significantly higher in the HF IOI-214 mice compared to the HF DMSO mice (Figure 11C, bottom panel).
Discussion
The liver plays a central role in metabolic homeostasis and is a major site for synthesis, metabolism, storage and redistribution of carbohydrates, lipids and proteins. Due to the rapid and pervasive increase in obesity world-wide, NAFLD/MAFLD remains among the most prevalent liver diseases and is a leading cause of liver-related morbidity and mortality. Risk factors for NAFLD are well established and contribute to our understanding of the etiology and pathogenesis for this multisystem disease. Obesity and metabolic derangements promote alterations in hepatic glucose and lipid metabolism and these alterations are linked to the pathophysiologies of NAFLD/ MAFLD.
Inflammation is widely recognized to be a key component in the pathogenesis of NAFLD/MAFLD. Obesity is considered the chief risk factor for the development of NAFLD/MAFLD and is the key foundation of the inflammation that is responsible for its progression.99,100 Currently, there are no therapies to prevent the inflammation associated with NAFLD/MAFLD. Given that NAFLD/MAFLD is associated with metabolic disease, pharmacological agents that target lipid accumulation or insulin resistance, such as certain anti-diabetic therapies including metformin,101 rosiglitazone,102 statins,103 and more recently GLP-1 agonists52 are being used as front-line therapies. Consequently, there is presently an intense and ongoing effort to identify and develop other novel therapeutics for this disease.
Obesity is considered to be a state of chronic, low-grade inflammation characterized by an abundance of pro-inflammatory cytokines in tissues (eg, liver and adipose tissue) and in the circulation, and this elevated inflammatory state contributes to insulin resistance and metabolic disorders.25,104–106 Adipocytes and associated macrophages produce and release cytokines systemically that interfere with peripheral insulin signaling. In addition, ectopic lipid deposition in insulin target tissues leads to “lipotoxicity”, an event that allows release of cytokines from the ectopically deposited adipocytes and associated macrophages to blunt insulin action (ie, insulin resistance). The combination of both local lipotoxicity and increases in circulating cytokines in obese individuals disrupts normal glucose homeostasis (ie, increased blood glucose levels).107–111 Moreover, the liver is central to maintaining whole-body glucose and lipid metabolism, and accumulating evidence suggests that hepatocyte-initiated inflammation also plays an important role in the development of insulin resistance. Normally, there is little to no production of cytokines in the liver, however in states of obesity where there is an increase in FFAs, LPS and other pathological stimuli, hepatic cells (ie, hepatocytes and immune cells) are stimulated to increase their production of inflammatory molecules (ie, pro-inflammatory cytokines and chemokines), which will trigger additional hepatic inflammation that leads to hepatocellular injury/death and fibrosis. Notably, TNFα, a pro-inflammatory adipokine/cytokine, that is produced by multiple types of inflammatory cells (monocytes/macrophages, neutrophils, and T cells) and tissues such as adipose and endothelial tissue, is produced directly by hepatocytes and Kupffer cells, or indirectly, by abdominal fat in the liver.112
TLR4 is a major inflammatory signaling pathway that mediates chronic inflammation in states of obesity.8,113–115 Tlr4 is abundantly expressed not only in immune cells, but also in adipose tissue, liver, the gut, and skeletal muscle, and is an important modulator of cellular insulin action.116–123 TLR4 is known to be a critical mediator of obesity-induced insulin resistance and T2DM.124 Non-immune ligands for TLR4 include saturated FFAs (sFFAs) (ie, palmitate and stearate,115,125,126 as well as increased circulating bacterial LPS, a phenomenon termed “metabolic endotoxemia”,25,26,127–133 all of which are in excess in obese individuals. Saturated FFA- and LPS-induced activation of TLR4 (and TLR2) signaling in insulin target cells (hepatocytes, adipocytes, skeletal myocytes, etc.) blunts insulin action8,113,118,121,122,126,134–140 because activation of TLR4 signaling results in the generation of pro-inflammatory cytokines (TNFα, IL6, etc.) and chemokines [CXCL10, MCP1 (CCL2), etc.] that contribute to insulin resistance.116–122,126,141,142 Consistently, mice lacking TLR4 display a marked decrease in diet-induced inflammation and IR.8,122,126,143–147 In fact, LPS derived from the gut (ie, leaky gut) stimulates TLR4 in obesity,148–150 and although TLR4 is not a receptor for saturated FFAs derived from the HF diet, TLR4 is required for saturated FFA/lipid-induced inflammation through TLR4-dependent priming and altered cellular metabolism.10 TLR4 signaling in hepatocytes,8 and hepatic macrophages, particularly Kupffer cells, is a critical source of inflammatory cytokines and chemokines (eg, TNF, IL6, MCP1, IL1B, etc.) that are key players in the pathogenesis of NAFLD/MAFLD.97
In the current study, we evaluated a promising novel small molecule, IOI-214, to inhibit LPS-induced inflammation in vitro and its efficacy to prevent HF diet-induced NAFLD/MAFLD in male C57BL/6J mice in vivo. We hypothesized that IOI-214, a novel compound that is similar to, yet structurally distinct from phenylmethimazole (C10), which we have shown previously to be a potent inhibitor of TLR3 and TLR4-mediated inflammation,11,55–59,61,62 would block LPS-induced inflammatory cytokine expression, and would therefore ameliorate HF diet-induced inflammation and hepatic steatosis in C57BL/6J male mice. In vitro, IOI-214 exhibited potent anti-inflammatory properties by inhibiting LPS-induced pro-inflammatory cytokine gene expression in murine macrophages (Tnf, Ifnb1, Mcp1, Il1b, Il6) and hepatocytes (Tnf) in culture. In vivo, we observed a significant decrease in hepatic steatosis and hepatocellular ballooning (ie, NAFLD/MAFLD) in HF IOI-214-treated mice compared to HF DMSO mice. Similar anti-inflammatory properties of IOI-214 were observed in vivo as HF diet-induced pro-inflammatory cytokine (TNFα, MCP1, IL6, IL1β) levels were significantly reduced in the liver of the mice fed a HF diet and treated with IOI-214. We also found that the levels of these inflammatory cytokines in the liver were significantly highly correlated with the amount of TG in the liver (ie, hepatic steatosis). In this context, it is worth noting again that IOI-214 mitigated HF diet-induced hepatic Plin5 expression because, as mentioned earlier, in humans and mice PLIN5/Plin5 expression is involved in lipid droplet formation and degradation,151 pro-inflammatory cytokine regulation and direct mitochondrial damage,152 as well as endoplasmic reticulum stress, which has made it a key target of NAFLD/MAFLD studies.17 Moreover, serum LBP levels were reduced in HF IOI-214 mice compared to HF DMSO mice, also providing evidence of IOI-214’s anti-inflammatory activity. The decrease in LBP levels was also significantly highly correlated with hepatic TG levels. The decrease in circulating LBP levels (and likely circulating inflammatory cytokines/chemokines) is presumably due to the positive effects of IOI-214 on the gut microbiota dysbiosis (as determined by the observed decrease in the Firmicutes/Bacteroidetes ratio in the gut of IOI-214-treated mice compared to DMSO-treated controls) and decreased gut permeability (as determined by the observed higher levels of OCLN in the gut of the IOI-214-treated mice compared to DMSO-treated controls) induced by the HF diet. The decreased Firmicutes/Bacteroidetes ratio is indicative of decreased gut inflammation, and the increased levels of OCLN in the gut are indicative of a decrease in leaky gut (which leads to systemic and hepatic inflammation). Both of these improvements by IOI-214 in the HF diet-fed mice suggest that part of the therapeutic mechanism(s) (ie, its ability to decrease hepatic steatosis) of IOI-214 is via its positive, anti-inflammatory, effects on the gut.
Another rather interesting finding that is relevant to inflammation is the accumulation of hepatic itaconate within the HF IOI-214 mice in comparison to the HF DMSO mice. Itaconate is an anti-inflammatory metabolite36–39 that has been shown to be significantly less abundant in obesity-associated steatotic liver,19 and genetic deletion of the gene that encodes the protein (ie, immune response gene 1, Irg1) exacerbates HF diet-induced NAFLD/MAFLD in C57BL/6J mice.18 As itaconate is primarily produced by macrophages and is an anti-inflammatory metabolite, this change may indicate a change in infiltrating/resident immune cells and promote inflammation resolution.37–39 Moreover, there was a significant drop in circulating tryptophan levels and substantial increase in circulating kynurenine (a catabolite of tryptophan) levels in HF IOI-214 mice compared to HF DMSO mice. Kynurenine is also a marker of immune system activation and it has been reported that the induction of NAFLD/MAFLD is often accompanied by a decrease in circulating kynurenine (and increase in tryptophan)40 and that this change may contribute to a pro-inflammatory status.41 As such, this change may be a marker of reduced systemic inflammation and an improvement in liver function. Hence, the increase in hepatic itaconate levels and the increase in circulating levels of kynurenine and decrease in circulating levels of tryptophan by IOI-214 in the HF diet-fed mice supports the notion that IOI-214 decreases systemic inflammation. We propose that this decrease in inflammation is part of the mechanism by which IOI-214 protects from HF diet-induced NAFLD/MAFLD. Exactly how IOI-214 modulates the levels of hepatic itaconate and circulating levels of kynurenine and tryptophan will be the focus of future studies.
As mentioned above, it is widely accepted that mesenteric adipose tissue is a significant source of inflammation in obesity that contributes to the lipotoxicity present in metabolic diseases.153 Thus, the fact that there was significantly less mesenteric adipose tissue in the HF IOI-214 mice than in the HF DMSO mice lends additional evidence to support the decrease in overall inflammation in the IOI-214-treated mice.
Given the clear anti-inflammatory effects of IOI-214 in the HF diet-fed mice, and the known role of inflammation in driving hepatic steatosis in NAFLD/MAFLD,8–28 it is not surprising that IOI-214 treatment significantly abated hepatic steatosis induced by the HF diet, a finding confirmed independently in this study by histological (ORO and H&E staining), biochemical (direct measurements of hepatic TG levels) and molecular analyses (RT-qPCR measures of Plin5 expression). It is also noteworthy to mention that the anti-inflammatory effects of IOI-214 also offers an explanation for how IOI-214 can reduce visceral fat accumulation, and glucose levels, in addition to hepatic steatosis in these mice since obesity-induced inflammation (at least in part by blocking TLR4 signaling) in the liver, gut, adipose tissue, and systemically drives not only hepatic steatosis but also lipid accumulation and insulin resistance.
What is surprising, given the overall decreased state of inflammation in the HF IOI-214 mice, is that there was no observed statistical decrease in systemic insulin resistance (as measured by an oral GTT) with IOI-214 treatment. We can only speculate at this point that the large effects on attenuating hepatic steatosis, in the absence of such an effect on overall systemic insulin resistance in the IOI-214-treated mice, may be the result of the largest effects of IOI-214 on gut microbiota dysbiosis and gut barrier integrity. In states of obesity, there is a pathologic shift in the gut microbiota (ie, gut microbiota dysbiosis) that leads to leaky gut syndrome, wherein bacterial LPS, and other inflammatory molecules, are released directly into the portal circulation,27 and thereby trigger inflammation in the liver, as the liver gets ~70% of its blood supply through the portal vein from the intestines.28,29 Because of the large amount of LPS and other inflammatory molecules produced in the gut, directly and continuously being delivered to the liver in high quantities in HF diet-fed mice, it is logical that by IOI-214 inhibiting the inflammation derived in the gut AND the inflammation that is triggered in the liver directly under these pathological conditions, that the most dramatic effects observed would be in the liver first and foremost. It is possible that the current experiment may have been too short, or the IOI-214 dosage too small to observe additional possible effects of IOI-214 on overall systemic insulin resistance. Thus, future studies will address these issues. However, although we did not see significant effects of IOI-214 on HF diet-induced glucose intolerance in our GTTs, we did observe that HF diet feeding caused fasting hyperglycemia, which can be the first phase of impaired GTT/insulin resistance. The HF diet-induced fasting hyperglycemia was significantly decreased by IOI-214 treatment. The exact mechanism(s) by which IOI-214 reverses HF diet-induced fasting hyperglycemia is uncertain at present; it could be due to IOI-214 lowering TG content in the liver since TGs in the liver contribute to hepatic gluconeogenesis in the fasting state of insulin-resistant patients and decreased hepatic fat content could reverse glucose synthesis in the fasting state of IOI-214-treated mice.154
Conclusion
Chronic inflammation, as a consequence of obesity-induced steatosis, leads to NAFLD/MAFLD, which can progress to more severe stages of the disease (ie, NASH, cirrhosis, or hepatocellular carcinoma). Herein, we report for the first time that IOI-214 is a potent inhibitor of LPS/TLR4-mediated inflammation in murine macrophages and hepatocytes in culture and is highly effective at mitigating HF diet-induced NAFLD/MAFLD in male C57BL/6J mice by ameliorating HF diet-induced inflammation (both in the liver and systemically), abating mesenteric fat accumulation, improving gut microbiota dysbiosis, decreasing gut permeability (by preventing HF diet-induced decreases in OCLN levels in the gut), and preventing release of gut-derived LPS into the circulation (Figure 12, green boxes). Thus, IOI-214 has the potential to have a profound clinical impact and an exciting novel therapeutic option for the treatment of NAFLD/MAFLD; especially NASH and the more progressive forms of the disease.
 |
Figure 12 Hypothesized working model of IOI-214 activity to ameliorate HF diet-induced NAFLD/MAFLD in vivo. Notes: Green boxes represent IOI-214 targets in vivo. We present data in this manuscript that supports the hypothesis that IOI-214 ameliorates HF diet-induced NAFLD/MAFLD in vivo by improving gut microbiota dysbiosis (Figure 9), decreasing systemic inflammation (Table 1) and mesenteric fat accumulation (Figure 4C), decreasing gut permeability, and circulating levels of LPS (Figure 10), and decreasing hepatic inflammation (Figure 7). Thus, collectively, IOI-214 works at multiple levels in parallel to ameliorate the effects of the obesity-induced inflammation that drives NAFLD/MAFLD. |
Abbreviations
NAFLD, non-alcoholic fatty liver disease; MAFLD, metabolic (dysfunction) associated fatty liver disease; IOI-214, inhibitor of inflammation-214; HF, high fat; RT-qPCR, reverse transcription-quantitative polymerase-chain reaction; LPS, lipopolysaccharide; TG, triglyceride; LPB, LPS binding protein; IR, insulin resistance; T2DM, type 2 diabetes mellitus; FFA, free fatty acid; VLDL, very low-density lipoprotein; NASH, nonalcoholic steatohepatitis; DMSO, dimethyl sulfoxide; PBS, phosphate-buffered saline; DMEM, Dulbecco’s Modified Eagle Medium; DMEM-F12, DMEM/Nutrient Mixture F-12; IPGTT, intraperitoneal glucose tolerance test; IPITT, intraperitoneal insulin tolerance test; HOMA-IR, homeostatic model assessment of insulin resistance; H&E, hematoxylin and eosin; LC/MS/MS, liquid chromatography/tandem mass spectrometry; UC, University of California; MMPC, Mouse Metabolic Phenotyping Center; PCA, Principal Components Analysis; PLS-DA, partial least squares-discriminant analysis; ELISA, enzyme-linked immunosorbent assay; ANOVA, analysis of variance; HSD, honestly significant difference; TLR4, toll-like receptor 4; Tnf/TNFα, tumor necrosis factor alpha; Ifnb1, interferon beta 1; Mcp1/MCP1, monocyte chemoattractant protein 1; Il1b/IL1β, interleukin 1 beta; Il6/IL6, interleukin 6; Plin5, perilipin 5; Actb/βActin, beta actin; Gapdh, glyceraldehyde 3-phosphate dehydrogenase; AST, aspartate aminotransferase; ALT, alanine aminotransferase; CD14, cluster of differentiation 14; Irg1, immune response gene 1; ORO, Oil Red O; OCLN, occludin; ZO1, zona-occludin; GLP-1, glucagon-like peptide-1.
Acknowledgments
The authors would like to thank Metabolon Inc. (Durham, NC, USA) for the generation of the metabolomics data and the UC Davis MMPC (Davis, CA, USA) for the generation of the gut microbiome data. The authors would also like to thank Macie Matta for her assistance with the cell culture experiments involving IOI-214 and Samantha Shaw for her assistance with the gut microbiome study. The authors would also like to thank the Ohio University Heritage College of Osteopathic Medicine Histological Core Services for their assistance with the liver tissue preparation and staining. University Research was supported by NIH grant U24-DK092993 [UC Davis Mouse Metabolic Phenotyping Center (RRID:SCR 015361)]. This research was also supported by the Ohio University Heritage College of Osteopathic Medicine. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Disclosure
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Ohio University owns two patents on IOI-214. Kelly D McCall, Frank L Schwartz, Stephen C Bergmeier and Douglas J Goetz are inventors on the patents. Kelly D McCall, Douglas J Goetz and Jean R Thuma report grants from UC Davis Mouse Metabolic Phenotyping Center, during the conduct of the study. All other authors declare that they have no conflicts of interest regarding the publication of this paper.
References
1. Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol. 2009;40(1):S5–S10.
2. Zelber-Sagi S, Nitzan-Kaluski D, Halpern Z, Oren R. Prevalence of primary non-alcoholic fatty liver disease in a population-based study and its association with biochemical and anthropometric measures. Liver Int. 2006;26(7):856–863. doi:10.1111/j.1478-3231.2006.01311.x
3. Younossi Z, Anstee QM, Marietti M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11–20. doi:10.1038/nrgastro.2017.109
4. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. doi:10.1002/hep.28431
5. El-Serag HB, Kanwal F. Epidemiology of hepatocellular carcinoma in the United States: where are we? Where do we go? Hepatology. 2014;60(5):1767–1775. doi:10.1002/hep.27222
6. Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008;134(2):424–431. doi:10.1053/j.gastro.2007.11.038
7. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol. 2018;68(2):280–295. doi:10.1016/j.jhep.2017.11.014
8. Jia L, Vianna CR, Fukuda M, et al. Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat Commun. 2014;5(1):3878. doi:10.1038/ncomms4878
9. Rogero MM, Calder PC. Obesity, inflammation, toll-like receptor 4 and fatty acids. Nutrients. 2018;10(4):432. doi:10.3390/nu10040432
10. Lancaster GI, Langley KG, Berglund NA, et al. Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab. 2018;27(5):1096–110 e5. doi:10.1016/j.cmet.2018.03.014
11. Patton A, Church T, Wilson C, et al. Phenylmethimazole abrogates diet-induced inflammation, glucose intolerance and NAFLD. J Endocrinol. 2018;237(3):337–351. doi:10.1530/JOE-18-0078
12. Feingold KR, Grunfeld C. Tumor necrosis factor-alpha stimulates hepatic lipogenesis in the rat in vivo. J Clin Invest. 1987;80(1):184–190. doi:10.1172/JCI113046
13. Grunfeld C, Verdier JA, Neese R, Moser AH, Feingold KR. Mechanisms by which tumor necrosis factor stimulates hepatic fatty acid synthesis in vivo. J Lipid Res. 1988;29(10):1327–1335. doi:10.1016/S0022-2275(20)38435-2
14. Grunfeld C, Dinarello CA, Feingold KR. Tumor necrosis factor-alpha, interleukin-1, and interferon alpha stimulate triglyceride synthesis in HepG2 cells. Metabolism. 1991;40(9):894–898. doi:10.1016/0026-0495(91)90062-2
15. Feingold KR, Adi S, Staprans I, et al. Diet affects the mechanisms by which TNF stimulates hepatic triglyceride production. Am J Physiol. 1990;259(2 Pt 1):E177–E184. doi:10.1152/ajpendo.1990.259.2.E177
16. Feingold KR, Staprans I, Memon RA, et al. Endotoxin rapidly induces changes in lipid metabolism that produce hypertriglyceridemia: low doses stimulate hepatic triglyceride production while high doses inhibit clearance. J Lipid Res. 1992;33(12):1765–1776. doi:10.1016/S0022-2275(20)41334-3
17. Mass Sanchez PB, Krizanac M, Weiskirchen R, Asimakopoulos A. Understanding the role of perilipin 5 in non-alcoholic fatty liver disease and its role in hepatocellular carcinoma: a review of novel insights. Int J Mol Sci. 2021;22(10):5284. doi:10.3390/ijms22105284
18. Zhang X, Zhi Y, Zan X, et al. Immune response gene 1 deficiency aggravates high fat diet-induced nonalcoholic fatty liver disease via promotion of redox-sensitive AKT suppression. Biochim Biophys Acta Mol Basis Dis. 2023;1869(4):166656. doi:10.1016/j.bbadis.2023.166656
19. Azzimato V, Chen P, Barreby E, et al. Hepatic miR-144 Drives Fumarase Activity Preventing NRF2 Activation During Obesity. Gastroenterology. 2021;161(6):1982–97 e11. doi:10.1053/j.gastro.2021.08.030
20. Moschen AR, Kaser S, Tilg H. Non-alcoholic steatohepatitis: a microbiota-driven disease. Trends Endocrinol Metab. 2013;24(11):537–545. doi:10.1016/j.tem.2013.05.009
21. Kirpich IA, Parajuli D, McClain CJ. Microbiome in NAFLD and ALD. Clin Liver Dis. 2015;6(3):55–58. doi:10.1002/cld.494
22. Gangarapu V, Yildiz K, Ince AT, Baysal B. Role of gut microbiota: obesity and NAFLD. Turk J Gastroenterol. 2014;25(2):133–140. doi:10.5152/tjg.2014.7886
23. Kim KA, Gu W, Lee IA, Joh EH, Kim DH. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS One. 2012;7(10):e47713. doi:10.1371/journal.pone.0047713
24. Jin C, Flavell RA. Innate sensors of pathogen and stress: linking inflammation to obesity. J Allergy Clin Immunol. 2013;132(2):287–294. doi:10.1016/j.jaci.2013.06.022
25. Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. doi:10.2337/db06-1491
26. Cani PD, Bibiloni R, Knauf C, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–1481. doi:10.2337/db07-1403
27. Harte AL, da Silva NF, Creely SJ, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm. 2010;7(1):15. doi:10.1186/1476-9255-7-15
28. Compare D, Coccoli P, Rocco A, et al. Gut--liver axis: the impact of gut microbiota on non alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2012;22(6):471–476. doi:10.1016/j.numecd.2012.02.007
29. Aron-Wisnewsky J, Gaborit B, Dutour A, Clement K. Gut microbiota and non-alcoholic fatty liver disease: new insights. Clin Microbiol Infect. 2013;19(4):338–348. doi:10.1111/1469-0691.12140
30. Han X, Fink MP, Yang R, Delude RL. Increased iNOS activity is essential for intestinal epithelial tight junction dysfunction in endotoxemic mice. Shock. 2004;21(3):261–270. doi:10.1097/01.shk.0000112346.38599.10
31. Chin AC, Flynn AN, Fedwick JP, Buret AG. The role of caspase-3 in lipopolysaccharide-mediated disruption of intestinal epithelial tight junctions. Can J Physiol Pharmacol. 2006;84(10):1043–1050. doi:10.1139/y06-056
32. Lee B, Moon KM, Kim CY. Tight junction in the intestinal epithelium: its association with diseases and regulation by phytochemicals. J Immunol Res. 2018;2018:2645465. doi:10.1155/2018/2645465
33. Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. 2018;50(8):1–9. doi:10.1038/s12276-018-0126-x
34. Liangpunsakul S, Chalasani N. Lipid mediators of liver injury in nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol. 2019;316(1):G75–G81. doi:10.1152/ajpgi.00170.2018
35. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52(5):1836–1846. doi:10.1002/hep.24001
36. Chen LL, Morcelle C, Cheng ZL, et al. Itaconate inhibits TET DNA dioxygenases to dampen inflammatory responses. Nat Cell Biol. 2022;24(3):353–363. doi:10.1038/s41556-022-00853-8
37. Mills EL, Ryan DG, Prag HA, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556(7699):113–117. doi:10.1038/nature25986
38. O’Neill LAJ, Artyomov MN. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol. 2019;19(5):273–281. doi:10.1038/s41577-019-0128-5
39. Oates JR, McKell MC, Moreno-Fernandez ME, et al. Macrophage function in the pathogenesis of non-alcoholic fatty liver disease: the mac attack. Front Immunol. 2019;10:2893. doi:10.3389/fimmu.2019.02893
40. Berge RK, Cacabelos D, Senaris R, et al. Hepatic steatosis induced in C57BL/6 mice by a non-ss oxidizable fatty acid analogue is associated with reduced plasma kynurenine metabolites and a modified hepatic NAD(+)/NADH ratio. Lipids Health Dis. 2020;19(1):94. doi:10.1186/s12944-020-01271-1
41. Yeung AW, Terentis AC, King NJ, Thomas SR. Role of indoleamine 2,3-dioxygenase in health and disease. Clin Sci. 2015;129(7):601–672.
42. Sumida Y, Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterol. 2018;53(3):362–376. doi:10.1007/s00535-017-1415-1
43. Sarwar R, Pierce N, Koppe S. Obesity and nonalcoholic fatty liver disease: current perspectives. Diabetes Metab Syndr Obes. 2018;11:533–542. doi:10.2147/DMSO.S146339
44. Wong VW, Chitturi S, Wong GL, Yu J, Chan HL, Farrell GC. Pathogenesis and novel treatment options for non-alcoholic steatohepatitis. Lancet Gastroenterol Hepatol. 2016;1(1):56–67. doi:10.1016/S2468-1253(16)30011-5
45. Sharma M, Premkumar M, Kulkarni AV, Kumar P, Reddy DN, Rao NP. Drugs for Non-alcoholic Steatohepatitis (NASH): quest for the Holy Grail. J Clin Transl Hepatol. 2021;9(1):40–50. doi:10.14218/JCTH.2020.00055
46. Lavine JE, Schwimmer JB, Van Natta ML, et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA. 2011;305(16):1659–1668. doi:10.1001/jama.2011.520
47. Loomba R, Lutchman G, Kleiner DE, et al. Clinical trial: pilot study of metformin for the treatment of non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2009;29(2):172–182. doi:10.1111/j.1365-2036.2008.03869.x
48. Nair S, Diehl AM, Wiseman M, Farr GH, Perrillo RP. Metformin in the treatment of non-alcoholic steatohepatitis: a pilot open label trial. Aliment Pharmacol Ther. 2004;20(1):23–28. doi:10.1111/j.1365-2036.2004.02025.x
49. Cusi K, Orsak B, Bril F, et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med. 2016;165(5):305–315. doi:10.7326/M15-1774
50. Belfort R, Harrison SA, Brown K, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355(22):2297–2307. doi:10.1056/NEJMoa060326
51. Aithal GP, Thomas JA, Kaye PV, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135(4):1176–1184. doi:10.1053/j.gastro.2008.06.047
52. Jastreboff AM, Kaplan LM, Frias JP, et al. Triple-hormone-receptor agonist retatrutide for obesity - a Phase 2 trial. N Engl J Med. 2023;389(6):514–526. doi:10.1056/NEJMoa2301972
53. Spengler EK, Loomba R. Recommendations for diagnosis, referral for liver biopsy, and treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mayo Clin Proc. 2015;90(9):1233–1246. doi:10.1016/j.mayocp.2015.06.013
54. Dagia NM, Harii N, Meli AE, et al. Phenyl methimazole inhibits TNF-alpha-induced VCAM-1 expression in an IFN regulatory factor-1-dependent manner and reduces monocytic cell adhesion to endothelial cells. J Immunol. 2004;173(3):2041–2049. doi:10.4049/jimmunol.173.3.2041
55. McCall KD, Harii N, Lewis CJ, et al. High basal levels of functional Toll-Like Receptor 3 (TLR3) and non-cannonical wnt5a are expressed in Papillary Thyroid Cancer (PTC) and are coordinately decreased by phenylmethimazole together with cell proliferation and migration. Endocrinology. 2007;148(9):4226–4237. doi:10.1210/en.2007-0459
56. McCall KD, Holliday D, Dickerson E, et al. Phenylmethimazole blocks palmitate-mediated induction of inflammatory cytokine pathways in 3T3L1 adipocytes and RAW 264.7 macrophages. J Endocrinol. 2010;207(3):343–353. doi:10.1677/JOE-09-0370
57. McCall KD, Schmerr MJ, Thuma JR, et al. Phenylmethimazole suppresses dsRNA-induced cytotoxicity and inflammatory cytokines in murine pancreatic beta cells and blocks viral acceleration of type 1 diabetes in NOD mice. Molecules. 2013;18(4):3841–3858. doi:10.3390/molecules18043841
58. Schwartz AL, Malgor R, Dickerson E, et al. Phenylmethimazole decreases Toll-like receptor 3 and noncanonical Wnt5a expression in pancreatic cancer and melanoma together with tumor cell growth and migration. Clin Cancer Res. 2009;15(12):4114–4122. doi:10.1158/1078-0432.CCR-09-0005
59. Deosarkar SP, Bhatt P, Gillespie J, Goetz DJ, McCall KD. Inhibition of LPS-induced TLR4 signaling products in murine macrophages by phenylmethimazole: an assay methodology for screening potential phenylmethimazole analogs. Drug Dev Res. 2014;75(8):497–509. doi:10.1002/ddr.21231
60. Courreges MC, Kantake N, Goetz DJ, Schwartz FL, McCall KD. Phenylmethimazole blocks dsRNA-induced IRF3 nuclear translocation and homodimerization. Molecules. 2012;17(10):12365–12377. doi:10.3390/molecules171012365
61. Harii N, Lewis C, Vasko V, et al. Thyrocytes express a functional toll-like receptor 3(TLR3): overexpression can be induced by viral infection, reversed by Phenylmethimazole, and is associated with Hashimoto’s autoimmune thyroiditis. Mol Endocrinol. 2005;19(5):1231–1250. doi:10.1210/me.2004-0100
62. Schwartz AL, Dickerson E, Dagia N, Malgor R, McCall KD. TLR signaling inhibitor, phenylmethimazole, in combination with tamoxifen inhibits human breast cancer cell viability and migration. Oncotarget. 2017;8(69):113295–113302. doi:10.18632/oncotarget.10358
63. Gan SF, Wan JP, Pan YJ, Sun CR. Water-mediated multicomponent reaction: a facile and efficient synthesis of multisubstituted thiazolidine-2-thiones. Synlett. 2010;6:973–975.
64. Hwang LL, Wang CH, Li TL, et al. Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity. 2010;18(3):463–469. doi:10.1038/oby.2009.273
65. Benz V, Bloch M, Wardat S, et al. Sexual dimorphic regulation of body weight dynamics and adipose tissue lipolysis. PLoS One. 2012;7(5):e37794. doi:10.1371/journal.pone.0037794
66. Yang Y, Smith DL, Keating KD, Allison DB, Nagy TR. Variations in body weight, food intake and body composition after long-term high-fat diet feeding in C57BL/6J mice. Obesity. 2014;22(10):2147–2155. doi:10.1002/oby.20811
67. Pettersson US, Walden TB, Carlsson PO, Jansson L, Phillipson M, Maedler K. Female mice are protected against high-fat diet induced metabolic syndrome and increase the regulatory T cell population in adipose tissue. PLoS One. 2012;7(9):e46057. doi:10.1371/journal.pone.0046057
68. Stubbins RE, Najjar K, Holcomb VB, Hong J, Nunez NP. Oestrogen alters adipocyte biology and protects female mice from adipocyte inflammation and insulin resistance. Diabetes Obes Metab. 2012;14(1):58–66. doi:10.1111/j.1463-1326.2011.01488.x
69. Hill-Baskin AE, Markiewski MM, Buchner DA, et al. Diet-induced hepatocellular carcinoma in genetically predisposed mice. Hum Mol Genet. 2009;18(16):2975–2988. doi:10.1093/hmg/ddp236
70. VanSaun MN, Lee IK, Washington MK, Matrisian L, Gorden DL. High fat diet induced hepatic steatosis establishes a permissive microenvironment for colorectal metastases and promotes primary dysplasia in a murine model. Am J Pathol. 2009;175(1):355–364. doi:10.2353/ajpath.2009.080703
71. Nakamura A, Tajima K, Zolzaya K, et al. Protection from non-alcoholic steatohepatitis and liver tumourigenesis in high fat-fed insulin receptor substrate-1-knockout mice despite insulin resistance. Diabetologia. 2012;55(12):3382–3391. doi:10.1007/s00125-012-2703-1
72. Pan X, Wang P, Luo J, et al. Adipogenic changes of hepatocytes in a high-fat diet-induced fatty liver mice model and non-alcoholic fatty liver disease patients. Endocrine. 2014;48:834–847.
73. Fraulob JC, Ogg-Diamantino R, Fernandes-Santos C, Aguila MB, Mandarim-de-Lacerda CA. A mouse model of metabolic syndrome: insulin resistance, fatty liver and Non-Alcoholic Fatty Pancreas Disease (NAFPD) in C57BL/6 mice fed a high fat diet. J Clin Biochem Nutr. 2010;46(3):212–223. doi:10.3164/jcbn.09-83
74. Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37(9):1163–1167. doi:10.2337/diab.37.9.1163
75. Surwit RS, Feinglos MN, Rodin J, et al. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism. 1995;44(5):645–651. doi:10.1016/0026-0495(95)90123-X
76. Qiu L, List EO, Kopchick JJ. Differentially expressed proteins in the pancreas of diet-induced diabetic mice. Mol Cell Proteomics. 2005;4(9):1311–1318. doi:10.1074/mcp.M500016-MCP200
77. List EO, Berryman DE, Palmer AJ, et al. Analysis of mouse skin reveals proteins that are altered in a diet-induced diabetic state: a new method for detection of type 2 diabetes. Proteomics. 2007;7(7):1140–1149. doi:10.1002/pmic.200600641
78. Sun G, Jackson CV, Zimmerman K, et al. The FATZO mouse, a next generation model of type 2 diabetes, develops NAFLD and NASH when fed a Western diet supplemented with fructose. BMC Gastroenterol. 2019;19(1):41. doi:10.1186/s12876-019-0958-4
79. Stubbins RE, Holcomb VB, Hong J, Nunez NP. Estrogen modulates abdominal adiposity and protects female mice from obesity and impaired glucose tolerance. Eur J Nutr. 2012;51(7):861–870. doi:10.1007/s00394-011-0266-4
80. Fu X, Xing L, Xu W, Shu J. Treatment with estrogen protects against ovariectomy-induced hepatic steatosis by increasing AQP7 expression. Mol Med Rep. 2016;14(1):425–431. doi:10.3892/mmr.2016.5236
81. Xin G, Qin S, Wang S, Wang X, Zhang Y, Wang J. Sex hormone affects the severity of non-alcoholic steatohepatitis through the MyD88-dependent IL-6 signaling pathway. Exp Biol Med. 2015;240(10):1279–1286. doi:10.1177/1535370215570189
82. Gao H, Bryzgalova G, Hedman E, et al. Long-term administration of estradiol decreases expression of hepatic lipogenic genes and improves insulin sensitivity in ob/ob mice: a possible mechanism is through direct regulation of signal transducer and activator of transcription 3. Mol Endocrinol. 2006;20(6):1287–1299. doi:10.1210/me.2006-0012
83. Chow JD, Jones ME, Prelle K, Simpson ER, Boon WC. A selective estrogen receptor alpha agonist ameliorates hepatic steatosis in the male aromatase knockout mouse. J Endocrinol. 2011;210(3):323–334. doi:10.1530/JOE-10-0462
84. Paquette A, Wang D, Jankowski M, Gutkowska J, Lavoie JM. Effects of ovariectomy on PPAR alpha, SREBP-1c, and SCD-1 gene expression in the rat liver. Menopause. 2008;15(6):1169–1175. doi:10.1097/gme.0b013e31817b8159
85. Geary N, Asarian L, Korach KS, Pfaff DW, Ogawa S. Deficits in E2-dependent control of feeding, weight gain, and cholecystokinin satiation in ER-alpha null mice. Endocrinology. 2001;142(11):4751–4757. doi:10.1210/endo.142.11.8504
86. Jones ME, Thorburn AW, Britt KL, et al. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci U S A. 2000;97(23):12735–12740. doi:10.1073/pnas.97.23.12735
87. Cooke PS, Naaz A. Role of estrogens in adipocyte development and function. Exp Biol Med. 2004;229(11):1127–1135. doi:10.1177/153537020422901107
88. Monteiro R, Teixeira D, Calhau C. Estrogen signaling in metabolic inflammation. Mediators Inflamm. 2014;2014:615917. doi:10.1155/2014/615917
89. Lai YS, Chen WC, Kuo TC, et al. Mass-spectrometry-based serum metabolomics of a C57BL/6J mouse model of high-fat-diet-induced non-alcoholic fatty liver disease development. J Agric Food Chem. 2015;63(35):7873–7884. doi:10.1021/acs.jafc.5b02830
90. Gopal SS, Sukhdeo SV, Vallikannan B, Ponesakki G. Lutein ameliorates high-fat diet-induced obesity, fatty liver, and glucose intolerance in C57BL/6J mice. Phytother Res. 2023;37(1):329–341. doi:10.1002/ptr.7615
91. List EO, Palmer AJ, Berryman DE, Bower B, Kelder B, Kopchick JJ. Growth hormone improves body composition, fasting blood glucose, glucose tolerance and liver triacylglycerol in a mouse model of diet-induced obesity and type 2 diabetes. Diabetologia. 2009;52(8):1647–1655. doi:10.1007/s00125-009-1402-z
92. Mukai T, Egawa M, Takeuchi T, Yamashita H, Kusudo T. Silencing of FABP1 ameliorates hepatic steatosis, inflammation, and oxidative stress in mice with nonalcoholic fatty liver disease. FEBS Open Bio. 2017;7(7):1009–1016. doi:10.1002/2211-5463.12240
93. Ni X, Wang H. Silymarin attenuated hepatic steatosis through regulation of lipid metabolism and oxidative stress in a mouse model of nonalcoholic fatty liver disease (NAFLD). Am J Transl Res. 2016;8(2):1073–1081.
94. Salmon DM, Flatt JP. Effect of dietary fat content on the incidence of obesity among ad libitum fed mice. Int J Obes. 1985;9(6):443–449.
95. Zeybel M, Altay O, Arif M, et al. Combined metabolic activators therapy ameliorates liver fat in nonalcoholic fatty liver disease patients. Mol Syst Biol. 2021;17(10):e10459. doi:10.15252/msb.202110459
96. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
97. Xu L, Liu W, Bai F, et al. Hepatic macrophage as a key player in fatty liver disease. Front Immunol. 2021;12:708978. doi:10.3389/fimmu.2021.708978
98. Ito M, Suzuki J, Tsujioka S, et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol Res. 2007;37(1):50–57. doi:10.1111/j.1872-034X.2007.00008.x
99. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–1053. doi:10.2337/diacare.27.5.1047
100. Imes CC, Burke LE. The obesity epidemic: the United States as a cautionary tale for the rest of the world. Curr Epidemiol Rep. 2014;1(2):82–88. doi:10.1007/s40471-014-0012-6
101. Haukeland JW, Konopski Z, Eggesbo HB, et al. Metformin in patients with non-alcoholic fatty liver disease: a randomized, controlled trial. Scand J Gastroenterol. 2009;44(7):853–860. doi:10.1080/00365520902845268
102. Ratziu V, Giral P, Jacqueminet S, et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 2008;135(1):100–110. doi:10.1053/j.gastro.2008.03.078
103. Eslami L, Merat S, Malekzadeh R, Nasseri-Moghaddam S, Aramin H. Statins for non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Cochrane Database Syst Rev. 2013;12:CD008623.
104. Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–1830. doi:10.1172/JCI200319451
105. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi:10.1172/JCI200319246
106. Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012;15(5):635–645. doi:10.1016/j.cmet.2012.04.001
107. Davi G, Guagnano MT, Ciabattoni G, et al. Platelet activation in obese women: role of inflammation and oxidant stress. JAMA. 2002;288(16):2008–2014. doi:10.1001/jama.288.16.2008
108. Kriketos AD, Greenfield JR, Peake PW, et al. Inflammation, insulin resistance, and adiposity: a study of first-degree relatives of type 2 diabetic subjects. Diabetes Care. 2004;27(8):2033–2040. doi:10.2337/diacare.27.8.2033
109. Lemieux I, Pascot A, Prud’homme D, et al. Elevated C-reactive protein: another component of the atherothrombotic profile of abdominal obesity. Arterioscler Thromb Vasc Biol. 2001;21(6):961–967. doi:10.1161/01.ATV.21.6.961
110. Poitou C, Coussieu C, Rouault C, et al. Serum amyloid A: a marker of adiposity-induced low-grade inflammation but not of metabolic status. Obesity. 2006;14(2):309–318. doi:10.1038/oby.2006.40
111. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–1119. doi:10.1172/JCI25102
112. Targher G, Arcaro G. Non-alcoholic fatty liver disease and increased risk of cardiovascular disease. Atherosclerosis. 2007;191(2):235–240. doi:10.1016/j.atherosclerosis.2006.08.021
113. Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity. 2008;16(6):1248–1255. doi:10.1038/oby.2008.210
114. Pierre N, Deldicque L, Barbe C, Naslain D, Cani PD, Francaux M. Toll-like receptor 4 knockout mice are protected against endoplasmic reticulum stress induced by a high-fat diet. PLoS One. 2013;8(5):e65061. doi:10.1371/journal.pone.0065061
115. Sawada K, Ohtake T, Hasebe T, et al. Augmented hepatic Toll-like receptors by fatty acids trigger the pro-inflammatory state of non-alcoholic fatty liver disease in mice. Hepatol Res. 2014;44(8):920–934. doi:10.1111/hepr.12199
116. Frost RA, Nystrom GJ, Lang CH. Lipopolysaccharide regulates proinflammatory cytokine expression in mouse myoblasts and skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2002;283(3):R698–R709. doi:10.1152/ajpregu.00039.2002
117. Gustot T, Lemmers A, Moreno C, et al. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology. 2006;43(5):989–1000. doi:10.1002/hep.21138
118. Song MJ, Kim KH, Yoon JM, Kim JB. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem Biophys Res Commun. 2006;346(3):739–745. doi:10.1016/j.bbrc.2006.05.170
119. Frost RA, Nystrom GJ, Lang CH. Multiple Toll-like receptor ligands induce an IL-6 transcriptional response in skeletal myocytes. Am J Physiol Regul Integr Comp Physiol. 2006;290(3):R773–R784. doi:10.1152/ajpregu.00490.2005
120. Lang CH, Silvis C, Deshpande N, Nystrom G, Frost RA. Endotoxin stimulates in vivo expression of inflammatory cytokines tumor necrosis factor alpha, interleukin-1beta, −6, and high-mobility-group protein-1 in skeletal muscle. Shock. 2003;19(6):538–546. doi:10.1097/01.shk.0000055237.25446.80
121. Reyna SM, Ghosh S, Tantiwong P, et al. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes. 2008;57(10):2595–2602. doi:10.2337/db08-0038
122. Radin MS, Sinha S, Bhatt BA, Dedousis N, O’Doherty RM. Inhibition or deletion of the lipopolysaccharide receptor Toll-like receptor-4 confers partial protection against lipid-induced insulin resistance in rodent skeletal muscle. Diabetologia. 2008;51(2):336–346. doi:10.1007/s00125-007-0861-3
123. Burgueno JF, Abreu MT. Epithelial Toll-like receptors and their role in gut homeostasis and disease. Nat Rev Gastroenterol Hepatol. 2020;17(5):263–278. doi:10.1038/s41575-019-0261-4
124. Jialal I, Kaur H, Devaraj S. Toll-like receptor status in obesity and metabolic syndrome: a translational perspective. J Clin Endocrinol Metab. 2014;99(1):39–48. doi:10.1210/jc.2013-3092
125. Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology. 2013;57(2):577–589. doi:10.1002/hep.26081
126. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025. doi:10.1172/JCI28898
127. Erridge C, Attina T, Spickett CM, Webb DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr. 2007;86(5):1286–1292. doi:10.1093/ajcn/86.5.1286
128. Creely SJ, McTernan PG, Kusminski CM, et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;292(3):E740–E747. doi:10.1152/ajpendo.00302.2006
129. Mehta NN, McGillicuddy FC, Anderson PD, et al. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes. 2009;59(1):172–181. doi:10.2337/db09-0367
130. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi:10.1038/4441022a
131. Bajzer M, Seeley RJ. Physiology: obesity and gut flora. Nature. 2006;444(7122):1009–1010. doi:10.1038/4441009a
132. de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol. 2010;299(2):G440–G448. doi:10.1152/ajpgi.00098.2010
133. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi:10.1038/nature05414
134. Kopp A, Buechler C, Neumeier M, et al. Innate immunity and adipocyte function: ligand-specific activation of multiple toll-like receptors modulates cytokine, adipokine, and chemokine secretion in adipocytes. Obesity. 2009;17(4):648–656. doi:10.1038/oby.2008.607
135. Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. The c-Jun N-terminal kinase mediates the induction of oxidative stress and insulin resistance by palmitate and toll-like receptor 2 and 4 ligands in 3T3-L1 adipocytes. Horm Metab Res. 2009;41(7):523–530. doi:10.1055/s-0029-1202852
136. Kopp A, Gross P, Falk W, et al. Fatty acids as metabolic mediators in innate immunity. Eur J Clin Invest. 2009;39(10):924–933. doi:10.1111/j.1365-2362.2009.02185.x
137. Poulain-Godefroy O, Le Bacquer O, Plancq P, et al. Inflammatory role of Toll-like receptors in human and murine adipose tissue. Mediators Inflamm. 2010;2010:823486. doi:10.1155/2010/823486
138. Lee JY, Zhao L, Youn HS, et al. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem. 2004;279(17):16971–16979. doi:10.1074/jbc.M312990200
139. Holland WL, Bikman BT, Wang LP, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest. 2011;121(5):1858–1870. doi:10.1172/JCI43378
140. Uchimura K, Hayata M, Mizumoto T, et al. The serine protease prostasin regulates hepatic insulin sensitivity by modulating TLR4 signalling. Nat Commun. 2014;5(1):3428. doi:10.1038/ncomms4428
141. Underhill DM. Toll-like receptors: networking for success. Eur J Immunol. 2003;33(7):1767–1775. doi:10.1002/eji.200324037
142. Lin Y, Lee H, Berg AH, Lisanti MP, Shapiro L, Scherer PE. The lipopolysaccharide-activated toll-like receptor (TLR)-4 induces synthesis of the closely related receptor TLR-2 in adipocytes. J Biol Chem. 2000;275(32):24255–24263. doi:10.1074/jbc.M002137200
143. Kim F, Pham M, Luttrell I, et al. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res. 2007;100(11):1589–1596. doi:10.1161/CIRCRESAHA.106.142851
144. Poggi M, Bastelica D, Gual P, et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia. 2007;50(6):1267–1276. doi:10.1007/s00125-007-0654-8
145. Suganami T, Mieda T, Itoh M, Shimoda Y, Kamei Y, Ogawa Y. Attenuation of obesity-induced adipose tissue inflammation in C3H/HeJ mice carrying a Toll-like receptor 4 mutation. Biochem Biophys Res Commun. 2007;354(1):45–49. doi:10.1016/j.bbrc.2006.12.190
146. Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56(8):1986–1998. doi:10.2337/db06-1595
147. Li L, Chen L, Hu L, et al. Nuclear factor high-mobility group box 1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. 2011;54(5):1620–1630. doi:10.1002/hep.24552
148. Lassenius MI, Pietilainen KH, Kaartinen K, et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care. 2011;34(8):1809–1815. doi:10.2337/dc10-2197
149. Jayashree B, Bibin YS, Prabhu D, et al. Increased circulatory levels of lipopolysaccharide (LPS) and zonulin signify novel biomarkers of proinflammation in patients with type 2 diabetes. Mol Cell Biochem. 2014;388(1–2):203–210. doi:10.1007/s11010-013-1911-4
150. Velloso LA, Folli F, Saad MJ. TLR4 at the crossroads of nutrients, gut microbiota, and metabolic inflammation. Endocr Rev. 2015;36(3):245–271. doi:10.1210/er.2014-1100
151. Hashani M, Witzel HR, Pawella LM, et al. Widespread expression of perilipin 5 in normal human tissues and in diseases is restricted to distinct lipid droplet subpopulations. Cell Tissue Res. 2018;374(1):121–136. doi:10.1007/s00441-018-2845-7
152. Kimmel AR, Sztalryd C. Perilipin 5, a lipid droplet protein adapted to mitochondrial energy utilization. Curr Opin Lipidol. 2014;25(2):110–117. doi:10.1097/MOL.0000000000000057
153. Kahn DE, Bergman BC. Keeping it local in metabolic disease: adipose tissue paracrine signaling and insulin resistance. Diabetes. 2022;71(4):599–609. doi:10.2337/dbi21-0020
154. Jin ES, Szuszkiewicz-Garcia M, Browning JD, Baxter JD, Abate N, Malloy CR. Influence of liver triglycerides on suppression of glucose production by insulin in men. J Clin Endocrinol Metab. 2015;100(1):235–243. doi:10.1210/jc.2014-2404
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Association of Lipopolysaccharide-Toll-Like Receptor 4 Signaling and Microalbuminuria in Patients with Type 2 Diabetes Mellitus
Zhang L, Zhang Y, Liu J, Li Y, Quan J
Diabetes, Metabolic Syndrome and Obesity 2022, 15:3143-3152
Published Date: 17 October 2022
Calcium-Sensitive Receptors Alters Intestinal Microbiota Metabolites Especially SCFAs and Ameliorates Intestinal Barrier Damage in Neonatal Rat Endotoxemia
Sun Y, Song J, Lan X, Ma F, Jiang M, Jiang C
Infection and Drug Resistance 2023, 16:5707-5717
Published Date: 30 August 2023
Metabolomics Analysis Reveals Gut Microbiota-Associated Sakuranin Modulates Endometrial Stem Cell Differentiation and Inflammation to Alleviate Pain in Endometriosis
Shi W, Wang M, Jin Z, Chen X, Li J, Lai H, Li X, Zhong Q, Chen Y, Chen S
Journal of Pain Research 2026, 19:557430
Published Date: 17 February 2026