Back to Journals » Cancer Management and Research » Volume 11
Analyzing biological and molecular characteristics and genomic damage induced by exposure to asbestos
Authors Ospina D, Villegas VE ![]() , Rodríguez-Leguizamón G
, Rodríguez-Leguizamón G ![]() , Rondón-Lagos M
, Rondón-Lagos M ![]()
Received 17 February 2019
Accepted for publication 19 April 2019
Published 30 May 2019 Volume 2019:11 Pages 4997—5012
DOI https://doi.org/10.2147/CMAR.S205723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Diana Ospina,1 Victoria Eugenia Villegas,1 Giovanni Rodríguez-Leguizamón,2 Milena Rondón-Lagos3
1Biology Program, Faculty of Natural Sciences and Mathematics, Universidad del Rosario, Bogotá 111221, Colombia; 2Hospital Universitario Mayor Méderi – Universidad del Rosario. School of Medicine and Health Sciences, Universidad del Rosario, Bogotá, 111221, Colombia; 3School of Biological Sciences, Universidad Pedagógica y Tecnológica de Colombia, Tunja 150003, Colombia
Abstract: Asbestos is one of the most important occupational carcinogens. Currently, about 125 million people worldwide are exposed to asbestos in the workplace. According to global estimates, at least 107,000 people die each year from lung cancer, mesothelioma, and asbestosis as a result of occupational exposure to asbestos. The high pathogenicity of this material is currently known, being associated with the development of pulmonary diseases, of which lung cancer is the main cause of death due to exposure to this mineral. Pulmonary diseases related to asbestos are a common clinical problem and a major health concern worldwide. Extensive research has identified many important pathogenic mechanisms; however, the precise molecular mechanisms involved, and the generated genomic damage that lead to the development of these diseases, are not completely understood. The modes of action that underlie this type of disease seem to differ depending on the type of fiber, lung clearance, and genetics. This evidences the need to increase our knowledge about these effects on human health. This review focuses on the characteristics of asbestos and the cellular and genomic damage generated in humans via exposure.
Keywords: occupational exposure, cellular damage, genomic damage, cancer
Introduction
The term asbestos, or earth flax, was assigned generically to a group of fibrous minerals characterized by their resistance to high temperatures and isolation of heat and noise.
Due to their physical characteristics, their properties of tension and resistance to heat, the chemical structure, and their lower cost, asbestos has been used since antiquity, especially in twentieth-century industry.1 Since the beginning of the twentiethcentury, the relationship between exposure to asbestos and lung damage has been known. Since then, several studies have been conducted that have demonstrated the degree of danger represented by the constant use of this fiber for human health.2 In 1987, the International Agency for Research in Cancer (IARC) classified asbestos as a Group 1 human carcinogen (defined) by direct, indirect, and domestic exposure.3,4 Exposure to asbestos occurs through the inhalation of fibers, mainly from contaminated air in the work environment, as well as from ambient air in the neighborhood of point sources, or air inside homes and buildings containing friable asbestos materials.5
The high pathogenicity of this material is currently known, being associated with the development of pulmonary diseases of which lung cancer is the main cause of death due to exposure to this mineral. Pulmonary diseases related to asbestos are a common clinical problem and a major concern for health worldwide. Epidemiological studies have established that exposure to asbestos fibers causes pleural abnormalities (effusions and plaques), pulmonary fibrosis (asbestosis), and malignancies (bronchogenic carcinoma and mesothelioma).6–9 Extensive research has identified many important pathogenic mechanisms; however, the precise molecular mechanisms involved and the generated genomic damage that lead to the development of these diseases are not completely understood. The modes of action that underlie this type of disease seem to differ depending on the type of fiber, lung clearance, and genetics. This evidences the need to deepen and increase our knowledge about the effects of asbestos on human health.
Asbestos: general considerations
The term asbestos, or earth flax, was generically assigned to a group of fibrous minerals characterized by their resistance to high temperatures and isolation of heat and noise. This mineral is found naturally in rocks and soils, is extracted from mines, and its processing is cheap. Asbestos is used to make clothing (gloves, anti-flame jogging suit, aprons, mittens, and ropes), in construction (fiber cement, tiles, slabs, etc.), in rubber, and in some household appliances (irons, toasters, hair dryers, and coffee makers).
Asbestos is one of the most important occupational carcinogens. Currently, about 125 million people worldwide are exposed to asbestos in the workplace.5 According to global estimates, at least 107,000 people die each year from lung cancer, mesothelioma, and asbestosis as a result of occupational exposure to asbestos.5 Almost 400 deaths have been attributed to non-professional exposure to asbestos. The number of asbestos-related diseases continues to rise, even in countries that banned its use in the early 1990s. Due to the long latency periods associated with the diseases in question, suspending from now the use of asbestos, will result in a decrease in the number of deaths related to asbestos alone after several decades.5
According to the IARC, asbestos and earth flax were classified as Group 1 human carcinogens by direct, indirect, and domestic exposure.3,4 Many industrialized countries have introduced legislation that prevents factories from using compounds such as asbestos in production due to its high carcinogenic risk. Additionally, it has been proven that joint exposure to tobacco smoke and asbestos fibers increases the risk of lung cancer – the more one smokes, the greater this risk.
Exposure to asbestos occurs by inhalation of fibers dispersed in the air and can be of three types: occupational (people who manipulate asbestos or who work in places of exploitation or its use), domestic (people living with workers exposed to asbestos, also those living in houses or buildings built with materials based on it), or environmental (people who live or have lived in the proximity of sites that use asbestos).10 Nevertheless, the occupational exposure has always been and remains the most likely source of human exposure. Asbestos is dispersed in air due to the extraction of the mineral, its production, the inadequate disposal of the material, and the repair of facilities containing asbestos. Taking into account that asbestos is harmful to health in the phases in which it is dispersed in the air, this mineral has been considered a carcinogen by the IARC.3,4
Two groups of asbestos are distinguished: serpentines, which include chrysotile; and amphiboles, among which are crocidolite, amosite, tremolite, anthophyllite, and actinolite.11 The first type consists of curled, wavy, flexible, long, and easily breakable fibers, soluble in the tissues, with diameters of 0.02–0.03 microns.6 The second type, amphibole, are rigid, short, sharp, and highly resistant fibers to chemical and biological solutions, and have a greater biological persistence compared to chrysotile.6,12
Etiopathology related to asbestos
The determinants of the toxicity of asbestos fibers depend on multiple factors, including dose, dimension, biopersistence, surface reactivity, and genetic history of those exposed. The dose of asbestos is a crucial factor triggering inflammation: high doses during short periods promote a predominant acute inflammation characterized by neutrophil accumulation, whereas low doses during prolonged exposure periods promote a chronic inflammation linked to accumulation of alveolar macrophages (AMs).6 The dimensions of the fibers and their chemical characteristics seem to determine the biological potency of fibrogenesis. It is thought that these characteristics, together with the surface properties, are also important for carcinogenesis. Thin and long fibers are more active than short fibers and amphiboles are more active than chrysotile – a property attributed to its greater biological persistence.
The ability of the inhaled fibers to penetrate into the lung spaces depends on their size, so fibers with aerodynamic diameters equal to or <5 μm show a penetration of more than 80%, but also a lower retention (10–20%).13 The dimensions of the fiber are important because only the very thin fibers (diameter <0.4 μm and length <10 μm) are respirable in the distal alveolar space; the long fibers cannot be swallowed by the AM because they are biodurable. Phagocytosis of the fibers is limited by the size of the AMs (generally 14–21 μm). In general, although fibers longer than 20 μm in length are associated with asbestosis, fibers longer than 10 μm are the most carcinogenic. However, the carcinogenicity of amphiboles is two orders of magnitude greater than that of chrysotile.6 Additionally, it has been reported that fibers <5 μm in length can also promote pulmonary fibrosis and malignancy, especially when administered as a pulmonary overload condition, as can occur in dust clouds.7 When the fibers are too long to be completely phagocytosed, the AMs try to swallow them, which results in their death when their membrane is crossed, which is called “frustrated phagocytosis”. This process results in the release of digestive enzymes, reactive oxygen species (ROS), reactive nitrogen species (RNS), proteases and cytokines that affect the lungs and other tissues.7 Considering that frustrated phagocytosis by phagocytic cells is associated with an increase in the release of ROS and RNS, long and thin fibers are considered more genotoxic and mutagenic, which has been related to the alteration of mitosis by interfering with cytokinesis8 by breaking the mitotic spindle.14 These fibers can penetrate deep into the lungs, unlike short fibers that are completely wrapped by AMs and are eliminated as any particle.7 However, the smaller-diameter fibers are likely deposited in the alveoli.5
The biopersistence of the fibers depends on the site and speed of deposition, the rates of elimination by AMs or mucociliary transport, their solubility in pulmonary fluids, their rate of rupture, and transport through the biological membranes.7 The biopersistence of chrysotile fibers is greater than that of amphibole fibers (months vs years, respectively), but chrysotile has a smaller surface area (27 vs ~8 m2g−1, respectively).12 For fibers whose chemical composition makes them totally or partially soluble inside the lung, it is possible that they completely dissolve or weaken sufficiently to be broken into shorter fibers, which can be eliminated through macrophage-mediated phagocytosis and mucociliary transport7 through the nasal and tracheo-bronchial region.5 The relatively low biopersistence of chrysotile could be explained by the fact that the leached fibers break into shorter fibers that are eliminated more easily. The leaching of chrysotile occurs in acidic or strong chelating conditions, which produces the elimination of magnesium (Mg), as in phagocytosis by AMs, thereby decreasing its biological potential.5
Cellular damage induced by exposure to asbestos
Exposure to asbestos has been shown to cause damage at both the cellular and genomic levels.8 Notably, several studies have shown that the damage to the organism caused by asbestos differs depending on the concentration, the exposure, and the type of fiber – chrysotile is the most pathogenic fiber, followed by crocidolite.15 At the cellular level, the accumulation of fibers causes, among other effects, oxidative stress, fibrosis, and chronic inflammation (Figure 1).8
 | Figure 1 Cellular damage induced by exposure to asbestos. High doses of asbestos during short periods promote acute inflammation of neutrophils. Low doses during prolonged exposure periods promote neutrophil accumulation and thus acute inflammation. Free radicals, ROS and RNS result from the chronic inflammation generated by the prolonged phagocytic activity by macrophages. This condition causes DNA damage inducing the activation of proto-oncogenes, cell proliferation, and susceptibility to mutations. Abbreviations: ROS, reactive oxygen species; RNS, reactive nitrogen species. |
Oxidative stress
Asbestos fibers tend to accumulate on the pleural surface and interact with the mesothelial cell layer, leading to the generation of ROS and RNS, and the formation of free radicals.5 The formation of ROS and RNS results from the chronic inflammation generated by the prolonged phagocytic activity by macrophages that function in the elimination of biopersistent fibers (Figure 1).7 The catalytic iron (Fe) associated with asbestos fibers is one of the main sources of ROS production.14 The amphiboles have a higher Fe content than serpentines, which are more mutagenic. This difference in harm potential may be due to the variation in the activity of the superficial Fe. Importantly, the valence and mobility status of Fe are determining factors in the mutagenic potential of the fiber.14 It is estimated that free radicals derived from Fe produce other effects such as lipid peroxidation, the release of tumor necrosis factor, cellular apoptosis, adhesion, and an increase in the absorption of fibers by epithelial cells.5 It has been suggested that the generation of free radicals, ROS, RNS, growth factors, and the induction of inflammatory cytokines secondary to fiber accumulation cause desoxyribonucleic acid (DNA) damage and induce the activation of proto-oncogenes, cell proliferation, and susceptibility to mutations.
Fibrosis
The mode of action of the long fiber is mechanical and not chemical; its length is essential in the production of fibrosis since the short fibers are unable to produce such reaction. The retention time determines its development.7 The target lung cells that work in the elimination of fibers, especially AMs and epithelial cells, produce cytokines, proteases, and growth factors that promote cell proliferation and tissue repair, their prolonged activation by chronic exposure leads to pulmonary fibrosis.8
An early event of fibrogenesis is the injury of Type I epithelial cells, followed by the hypertrophy of Type II epithelial cells. Increases in epithelial cells proliferation and that of fibroblasts are determining factors in the repair and regeneration of tissue that, when not controlled, can lead to fibrosis.16 Therefore, this non-mutagenic process can be related to excessive apoptosis caused by the genotoxicity of ROS that results in lung tissue injury and uncontrolled and prolonged cell proliferation.8
Chronic inflammation
Chronic inflammation is a recognized risk factor for human cancer17–19 that can promote all stages of tumorigenesis, including DNA damage, continuous replication, evasion of apoptosis, prolonged angiogenesis, resistance to signaling of anti-growth, and invasion/metastases. Furthermore, crocidolite asbestos fibers are capable of inducing cell proliferation, cell cycle detention, and apoptosis in diverse populations of mesothelial cells20 and epithelial cells of the lung.21 The inflammatory response and the release of ROS and RNS are triggered by the frustrated phagocytosis of the long asbestos fibers by the AMs.22,23 ROS and RNS recruit more macrophages and other inflammatory cells to the lung (Figure 1). Therefore, the persistence of asbestos fibers in the lungs can trigger the prolonged production of free radicals and chronic inflammation at the sites of fiber deposition.
Signaling pathways involved
In addition to inducing direct DNA damage and mutagenesis, chronic inflammation (induced by asbestos, ROS, and RNS) activates multiple signaling cascades,24–26 including the signaling pathway of the mitogen-activated protein kinase (MAPK) (Figure 2). A rapid increase in signaling of the MAPK pathway subsequently activates transcription factors, such as the activator protein-1 (AP-1) and the nuclear transcription factor kappa-B (NFκB), in target cells exposed to asbestos (Figure 2). AP-1 is a family of transcription factors comprised of homo- and heterodimers of the JUN and FOS early response protooncogenes. It is a redox-sensitive transcription factor classically associated with the development of cell proliferation and tumor promotion.27 NF-κB is a critical transcription factor in inflammation and responses in target cells of asbestos-related diseases, since its activation is crucial in the upregulation of many genes related to proliferation and apoptosis.28 Moreover, asbestos fibers caused transcriptional activation of a number of NFκB dependent genes, including c-MYC, through an oxidant-dependent pathway.29
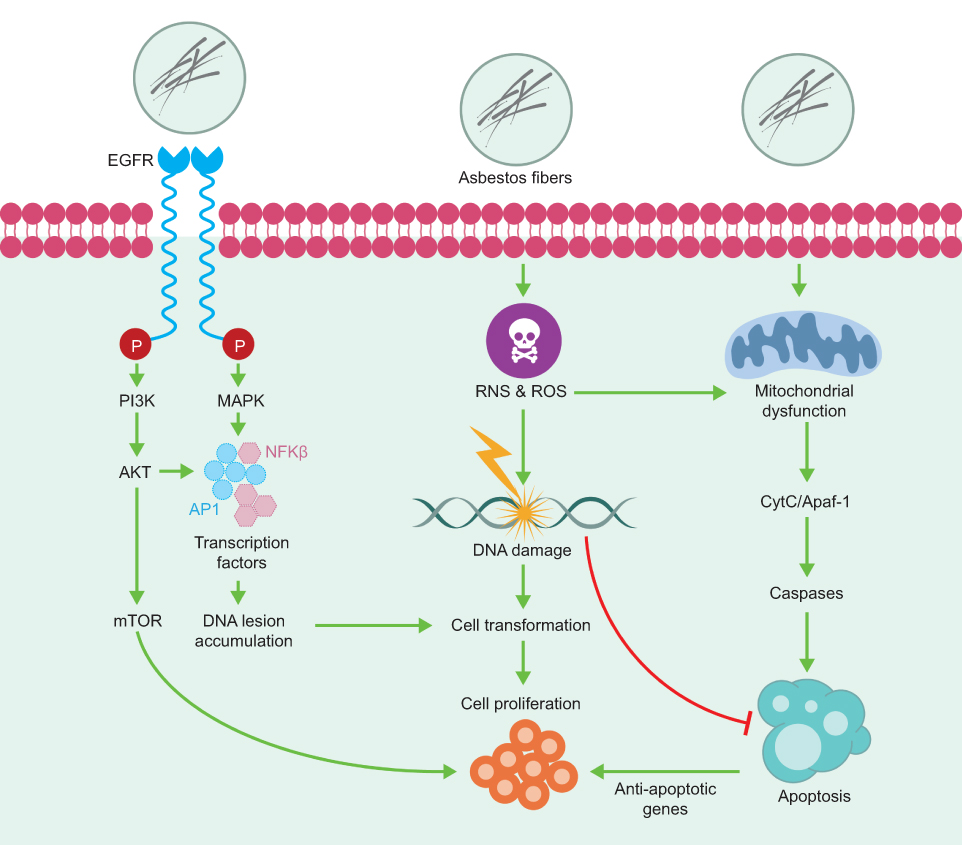 | Figure 2 Chronic inflammation activates multiple signaling cascades. The signaling pathway of the mitogen-activated protein kinase (MAPK) and the signaling pathway regulated by the epidermal growth factor receptor (EGFR) are some of the cellular signaling pathways involved in the response to asbestos inhalation. Abbreviations: PI3K, phosphatidylinositol 3-kinase; AKT, AKT serine/threonine kinase; mTOR, mechanistic target of rapamycin kinase; AP-1, activator protein-1; NFκB, nuclear transcription factor kappa-B; ROS, reactive oxygen species; RNS, reactive nitrogen species; CytC, cytochrome C; Apaf-1, apoptotic peptidase activating factor 1. |
The properties of asbestos fibers eliciting these cell signaling cascades and the consequences of asbestos-induced AP-1 and NFκB dependent gene expression may be related to the initiation of asbestos-associated cell responses and lung/pleural diseases. Cross-talk between these cell signaling pathways also exists, and may be relevant to asbestos-induced inflammation and proliferation.30,31
Extensive studies by Mossman et al7,28 identified additional cellular signaling pathways involved in the response to asbestos inhalation, especially the Epidermal growth factor receptor (EGFR) regulated signaling pathway (Figure 2). In 2010, Heintz et al28 showed that asbestos, chrysotile, and crocidolite fibers activate the EGFR in mesothelial cells - an event related to the activation of extracellular signal regulated protein kinases (ERK) 1 and 2 (ERK1/2).32 ERK1/2 partially regulates the transcriptional activity of FOS, and the mRNA levels of both FOS and JUN induced by earth flax in the distal bronchial epithelium are reduced in mice without EGFR.33 These studies provide a link between the activation of EGFR and ERK1/2. Additional studies have shown that phosphorylation of EGFR also occurs with other types of cancer fibers and may be related to the generation of oxidants after an incomplete phagocytosis of long fibers.34 The activation of these signaling pathways promotes the proliferation of fibroblasts and epithelial cells of the lung as a result of lung inflammation after chronic inhalation of earth flax (chrysotile and crocidolite).
Genomic damage induced by exposure to asbestos
At the genomic level, asbestos fibers can directly induce mutagenicity and genotoxicity through physical interaction with the mitotic machinery of dividing cells after they are phagocytosed by the target cells, or indirectly as a result of DNA damage (genetic damage) and to chromosomes (chromosomal damage) (Figure 3) by ROS and RNS.19,23,24,26 ROS and RNS are responsible for producing a wide variety of DNA and chromosome damage and generate single chain breaks, chromosomal fragments, and 8-hydroxy-2ʹ-deoxyguanosine (8-OHdG), which is a product of DNA oxidation.8,35
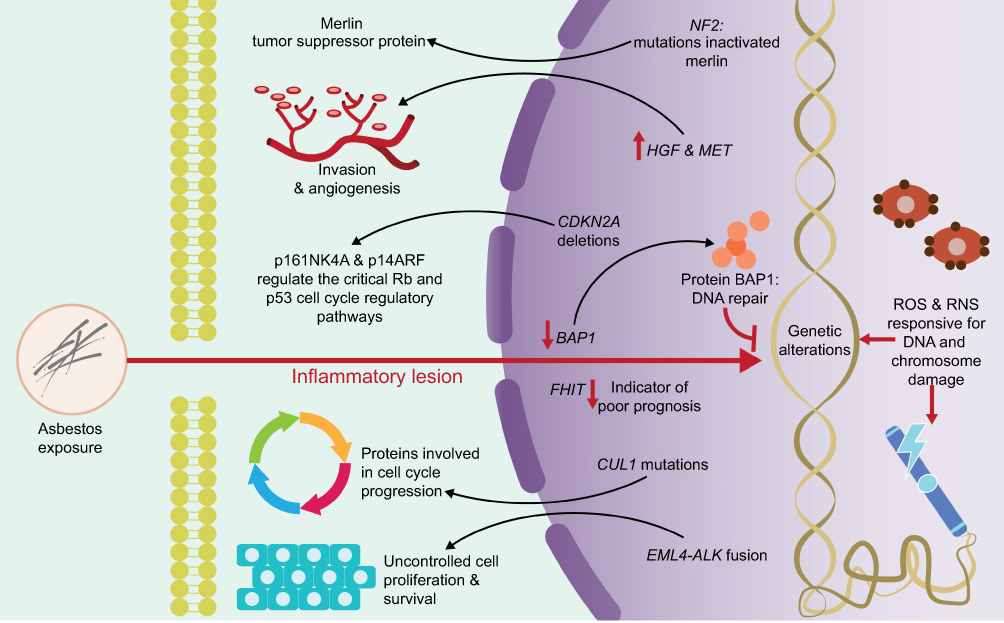 | Figure 3 Genomic damage induced by exposure to asbestos. ROS and RNS are responsible for producing a wide variety of DNA and chromosome damage. The activation or deactivation of certain genes has been associated with the progressive development and treatment of pleuro-pulmonary diseases. Abbreviations: RNS, reactive nitrogen species; p16INK4A and p14ARF, cyclin-dependent kinase Inhibitor 2A; p53, protein p53; Rb, retinoblastoma protein; NF2, neurobrimonin 2 gene; HGF, hepatocyte growth factor; MET, mesenchymal-epithelial transition factor; CDKN2A, cyclin-dependent kinase inhibitor 2A; BAP1, BRCA1 associated protein 1 gene; FHIT, fragile histidine triad gene; CUL1, cullin 1 gene; EML4, echinoderm gene associated with microtubules 4 gene; ALK, anaplastic lymphoma kinase gene. |
Genetic damage
The activation or deactivation of certain genes by amplification or structural rearrangements (deletions or inversions) has been associated with the progressive development and treatment of pleuro-pulmonary diseases. Genes, such as BRCA1 Associated Protein 1 (BAP1), anaplastic lymphoma kinase (ALK), and mesenchymal-epithelial transition (MET) factor, are highly related to these diseases and play well-established roles within them (Figure 3).
BAP1 gene has been proposed as a tumor suppressor gene, with important functions in cell proliferation and growth inhibition.36 This gene is located on the short arm of chromosome 3 (3p21.1), a region that harbors germline mutations associated with an inherited multicancer syndrome with an autosomal dominant transmission37 (Table 1). BAP1 is the first and only gene that has been proposed to influence environmental carcinogenesis, such that a germinal mutation in the BAP1 gene leads to a greater susceptibility to asbestos, which favors the clinical onset of Malignant Mesothelioma (MM).36,38–40 This gene encodes the protein BAP1, a nuclear deubiquitinate enzyme,41 which plays important roles in the ubiquitin-proteasome pathway, in the deubiquitination of histones, in the regulation of cell cycle progression, and DNA repair (Figure 3). The loss of BAP1, independent of the mechanism that leads to such loss, including deletion or point mutation (detected with a high incidence in MM),42 translates into nuclear negativity for the expression of BAP1 assessed by immunohistochemistry.43–45 The loss of the expression of the BAP1 nuclear protein is useful for differentiating both MM and malignant pleural imitators (lung and ovarian cancers) and malignant vs reactive mesothelial proliferation with a high specificity despite the variable sensitivity.45,46
 | Table 1 Genes commonly altered in lung and pleural diseases associated with asbestos exposure |
ALK gene, located on the short arm of chromosome 2 (2p23), encodes for a receptor with tyrosine kinase activity. ALK regulates several signaling pathways including mitogen-activated protein kinase RAS (MAPK), phosphatidyl-inositol 3-kinase (PI3K)-AKT, and the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathways. Within the rearrangements that involve the ALK gene, the most frequently observed in lung cancer is the fusion of the ALK gene with the echinoderm gene associated with microtubules 4 (EML4) (Figure 3). The EML4-ALK fusion results in a paracentric inversion within the 2p chromosomal region that fuses different parts of the EML4 gene with a portion of the ALK gene47 (Table 1 and Figure 4). Such rearrangement leads to fusion of the 5ʹ end of EML4 with the intracellular tyrosine kinase domain of ALK, leading to the constitutive activation of the ALK kinase and its downstream signaling pathways, and hence to uncontrolled cell proliferation and survival.
 | Figure 4 ALK Break-Apart FISH Probe for testing for the presence of EML4 (2p21) - ALK (2p23) fusion gene (ALK rearrangement) in lung cancers. The ALK break-apart probe is typically designed by labeling the 3ʹ (telomeric) part of the fusion breakpoint with one fluorochrome (orange signal) and the 5ʹ (centromeric) part with another fluorochrome (green signal). (A) In normal cells, the genomic areas homologous to the 3ʹ and 5ʹ probes are molecularly very close and these signals are seen as fused or adjacent. In contrast, (B) in abnormal cells, as result of the paracentric inversion on short arm of chromosome 2 (inv(2)(p21p23)), a gene fusion occurs between the AML and ALK genes. When the EML4-ALK fusion gene is present, the 5ʹ ALK green signal becomes far removed from the 3ʹ ALK red signal (by approximately 12.5 Mb), and the signals are seen as being split. The inv(2)(p21p23) is present when a green/orange fusion signal, specific for ALK, splits into separate green and orange signals. Abbreviations: ALK, anaplastic lymphoma kinase gene; EML4, echinoderm gene associated with microtubules 4 gene; inv, chromosomal inversion. |
Considering that this gene rearrangement involves large chromosomal inversion and translocation, fluorescence in situ hybridization (FISH) has become the method of choice for detecting all forms of ALK gene rearrangement, so that a cell is considered normal (ALK negative) when the 5' and 3ʹ signals are fused, whereas a cell is considered positive when 5' and 3ʹ signals are separated (ALK positive) (Figures 4 and 5). EML4-ALK is the predominant ALK fusion in lung cancer, with several studies demonstrating that ALK fusion proteins are oncogenic and enough to induce pulmonary tumorigenesis in vivo.48 Thus, the presence of EML4-ALK gene fusion (ALK positive) (Figure 5) has not only been associated with several distinctive clinicopathological features in lung diseases, including the absence of a history of smoking, but is considered an important therapeutic target, sensitive to treatment with small-molecule ALK kinase inhibitors, such as crizotinib.47,49 The early performance of FISH tests at the time of diagnosis of diseases such as MM, adenocarcinomas and large cell lung carcinomas, can determine the appropriate treatment directed to ALK.
 | Figure 5 Signal patterns in lung tumor nuclei hybridized with ALK break-apart FISH. (A) A cell is interpreted as having a normal pattern (ALK negative) when the 5ʹ and 3ʹ signals are fused (indicated by arrows); (B) A cell is interpreted as having a split pattern (ALK positive) when the 5ʹ and 3ʹ signals are separated (indicated by arrows), regardless of the number of actual isolated signals. Abbreviation: ALK, anaplastic lymphoma kinase gene. |
MET gene, located on the long arm of chromosome 7 (7q31.2), encodes for a high-affinity receptor for hepatocyte growth factor (HGF), also known as a dispersion factor, and is involved in cell growth and differentiation, neovascularization, and tissue repair in normal tissues (Table 1).50 The deregulation of MET and HGF has been implicated in tumor development, invasion, and angiogenesis for a variety of malignancies51 (Figure 3). Such deregulation can be caused by different mechanisms, including overexpression of the MET protein, amplification of the MET gene, mutations, or rearrangements. Amplification of the MET gene has been associated with poor prognosis, tumor development, invasion, and angiogenesis in a variety of malignancies including ovarian, breast,52 lung,53 thyroid, stomach, and colon cancer.51 Also, amplification of this gene has been associated with secondary resistance to the tyrosine kinase inhibitor and with aggressive anatomopathological features in lung adenocarcinoma, such as increased tumor size, pleural invasion, and invasion of lymphatic vessels.54,55 Additionally, studies in lung cancer cell lines with MET gene amplification have shown a significantly higher sensitivity to MET inhibitors, suggesting that patients with tumors harboring amplified MET may present clinical responses to MET inhibitors.56 Considering the above findings, the MET gene has been postulated as a poor independent prognostic marker in lung adenocarcinoma,54 and as a promising target for the treatment of lung cancer.57,58
Asbestos has been indicated to act as a tumor promotor and facilitates the mutagenic effects in synergy with other carcinogens,59 such as cigarette smoke, generating significant DNA damage that increases in proportion to the dose of exposure to earth flax, size, and biodurability of the fibers. There are two possible mechanisms by which cigarette smoke act: absorbing the carcinogens on the surface of the fibers, favoring their retention time; and increasing the penetration of the target cells due to the chemical carcinogens of the smoke.7 In addition, it has been established that asbestos can inactivate the p53 gene in epithelial, mesothelial,8 and alveolar5 cells in the lungs, stopping the cell cycle and apoptosis to allow time for DNA repair.
Chromosomal damage
Some studies have shown that the direct interaction between asbestos fibers with the mitotic spindle and chromosomes during mitosis in vitro60 can lead to the induction of chromosomal instability (CIN)23,61,62 (Figure 3). The main chromosomal alterations reported as a result of exposure to asbestos, include numerical (aneuploidy, polyploidy, and hyperploidy) and structural alterations (deletions, translocations, inversions, duplications, chromosomal ruptures, and exchange of sister chromatids).15 However, these alterations were observed in rat embryos,15 with information in humans being scarce.
CIN, defined as the rate of gain or loss of complete chromosomes or fractions of chromosomes, has been recognized as a hallmark of cancer and a source of genetic variation that favors the adaptation of the tumor to stressful environments. CIN favors the simultaneous growth of various tumor subpopulations, leading to inter- and intra-tumor genomic heterogeneity (clonal heterogeneity).63–65 CIN and clonal heterogeneity lead to gene regulatory interactions and variable concentrations of proteins, which could affect the cellular response to drug treatment.66
Although several studies in humans have been aimed at determining the induction of CIN by exposure to asbestos, such studies have been limited to the identification of micronucleus (MN) and sister chromatid exchange (SCE), demonstrating an increase in the frequency of the same.35,67–71 In general, the results of these studies were extremely heterogeneous in terms of type of exposure (occupational, domestic, and environmental), type of fibers, and duration of exposure. Only a few of the investigated populations showed significantly higher levels of MN. These results suggest the existence of differences in the level of DNA damage and repair between the different types of asbestos fibers. These findings suggest that the damage induced by chrysotile (white asbestos) could be repaired more easily than asbestos,71,72 causing minor damage. However, few studies have described the type and frequency of specific chromosomal alterations induced by exposure to asbestos.
For instance, in MM, a rare aggressive neoplasm arising from the pleural, peritoneal, or pericardial lining, 40% to 70% of both pleural and peritoneal mesotheliomas harbor loss of 9p including loss of the cyclin-dependent kinase inhibitor 2A (CDKN2A) gene, or 22q, including loss of the neurobrimonin 2 (NF2) gen.73,74 Specifically, in Malignant Pleural Mesothelioma (MPM), losses of chromosome arms 1p, 3p, 4q, 6q, 9p, 13q, 14q, and 22q and gains of chromosome arms 1q, 5p, 7p, 8q, and 17q,75 deletions in CDKN2A, cyclin-dependent kinase inhibitor 2B (CDKN2B), and NF2 genes76,77 and mutations in BAP1 and Cullin 1 (CUL1) genes78 have been reported (Table 1 and Figure 3). Additional studies have demonstrated losses in chromosomal regions 3p14–p21, 8p12-pter, and 17p12-pter or gain in 7q.79 Losses of the short arm of chromosome 3 (3p) have been reported as an early common aberration in lung cancer, observed more frequently in tumors of patients exposed to asbestos than in unexposed patients.80 This chromosomal region (3p14), contains the fragile histidine triad (FHIT) gene, has also been associated with exposure to asbestos and smoking (Table 1).81 In this disease (MPM), significant correlations have been described between high contents of asbestos fibers in lung tissue and partial or total losses of chromosomes 1, 4, and 9, and chromosomal rearrangements involving a breakpoint at 1p11–p22.82,83 Comparison between recurrent altered regions in asbestos-exposed and unexposed patients showed a significant difference in the 14q11.2–q21 region, which is also lost in fiber-induced murine mesothelioma.84 Chromosomal regions and genes altered in MPM are indicated in Table 1.
In Malignant Peritoneal Mesothelioma, analysis by comparative genomic hybridization showed the presence of CIN, which was characterized by losses of the chromosomal regions 3p21, 9p21, and 22q12. Interestingly, Chirac et al (2016)85 reported that in patients with malignant peritoneal mesothelioma exposed to asbestos, the proportion of chromosomal losses and gains was higher than that observed in patients without asbestos exposure. Additional studies have been performed on cell lines, including V79 lung fibroblasts. Such studies demonstrated that the chrysotile and rock wool fibers cause chromosome aberrations, as indicated by a dose-dependent increase in MN frequency.86 However, studies that describe the type and frequency of specific chromosomal alterations induced by exposure to asbestos are limited.
Asbestos and disease
Prolonged exposure to asbestos fibers, the accumulation of these in the lungs, and the sum of other risk factors, such as smoking, lead to the development of various diseases, which are mainly pulmonary.3–5,87 Usually, people who have diseases related to the mineral do not show signs of the disease until a long time after the first exposure. It can take 10 to 40 years or more for the symptoms of an asbestos-related condition to appear.5,6 Among the diseases generated by exposure to asbestos fibers are pulmonary fibrosis (asbestosis) and malignant tumors (lung cancer and mesothelioma),6–9 among others.
Asbestosis
Asbestosis is a chronic lung disease caused by the inhalation exposure to asbestos after a latent period of more than 20 years.88 Prolonged exposure to these fibers and their deposition in the lungs triggers an inflammatory process that can lead to the formation of scars (fibrosis) inside the lung. In this process, the fibrosis is of an interstitial and diffuse type, tends to affect primarily the lower lobes and the peripheral areas, and, in advanced cases, is associated with the obliteration of the normal architecture of the lung. Fibrosis of the adjacent pleura is common. None of the histological features of asbestosis differentiates it from interstitial fibrosis due to other causes, except for the presence of earth flax in the lung in the form of asbestos bodies, visible under an optical microscope, or uncoated fibers, most of which are too thin to be visualized except by electron microscopy.13
Sometimes fibrosis can be limited to relatively few areas, mainly affecting the peribronchiolar regions, causing disease of the small airways related to earth flax. In this case, none of the histological changes of this process distinguishes it from disease of the small airways due to other causes (such as tobacco use or exposure to other mineral powders), except for the presence of earth flax in the lung. The disease of the small airways may be the only manifestation of asbestos-related pulmonary fibrosis, or it may coexist with varying degrees of interstitial fibrosis.13
The initial stage of asbestosis is characterized by discrete foci of fibrosis within the respiratory bronchiole walls and alveolar duct bifurcations associated with the accumulation of earth flax bodies.7,89,90 Asbestos triggers the accumulation of AMs and an inflammatory reaction, followed by a more diffuse pulmonary involvement characterized by 1) loss of the alveolar epithelium type I and Alveolar Type II (AT2) cells, 2) proliferation of fibroblasts, and 3) collagen deposition. Pulmonary fibrosis of asbestosis is associated with fibrosis of the walls of the respiratory bronchioles and alveolar ducts. The site of asbestos-induced inflammation occurs in the area of fiber deposition along the airways and in the alveolar spaces.89,90
The ingestion of asbestos fibers by macrophages triggers a fibrogenic response of fibroblasts by the release of growth factors, such as TGF-β and the platelet-derived growth factor, as well as cytokines, such as the TNF-α and IL-1β, which collectively promote collagen deposition.16,91
Indicative characteristics for the diagnosis of asbestosis include reliable exposure to asbestos; an appropriate latency period, typically >20 years; where exertional dyspnea and dry cough together with the late inspiratory crackles are the most frequent symptoms and signs; abnormal chest images showing subpleural reticular abnormalities with basal predominance, typically with pleural plaques (80–90%); and restrictive pulmonary physiology with reduced gas exchange.90 In cases of occupational exposure, a chest X-ray with reading is used with applying the radiological classification of the International Labor Organization.13
Lung cancer
Lung cancer remains the leading cause of incidence and mortality due to cancer worldwide (with 2.1 million new cases of lung cancer and 1.8 million deaths expected in 2018).92 Even if mesothelioma is commonly known as the primary type of cancer related to asbestos, it has been estimated that asbestos results in an equal or greater number of lung cancer cases compared to mesothelioma.80 The IARC concluded that there is sufficient evidence of carcinogenicity in humans for all types of asbestos, including chrysotile.5
Asbestos fibers have been indicated to interact with other genotoxic agents, such as tobacco smoke, increasing not only CIN93 but also the risk of lung cancer.94
Malignant Mesothelioma (MM)
MM is an aggressive and fatal tumor strongly associated with asbestos exposure. MM is responsible for ~3,000 deaths per year in the United States and 5,000 deaths in Western Europe.95 According to Montanaro et al,91 MM incidence or mortality predicts a steady growth in the number of cases among industrialized countries, following a plateau or decline as a consequence of the restriction on the use of asbestos. In addition, the demography of MM has changed; the age of MM patients has decreased and there is an increased incidence in women, likely reflecting exposure from non-occupational sources.96 MM is a highly aggressive, fast-growing type of cancer, associated with a low rate of patient survival, poor prognosis, and low overall survival, relatively resistant to chemotherapy and radiotherapy, with limited therapeutic options.97 The median overall survival for MM following frontline chemotherapy with pemetrexed and cisplatin is only ~12 months.98
This type of neoplasm results from the uncontrolled proliferation of mesothelial cells lining pleural, pericardial, and peritoneal cavities. According to the IARC,5 MM has been related to occupational, domestic, and environmental exposure to asbestos. Thus, in at least 376 cases of MM, the causative agent was non-occupational (domestic) exposure to asbestos.99
The populations most exposed to the development of this type of neoplasm are those who work in the automobile industry, fiber cement products factories, and construction, in combination causing 70–80% of cases of mesothelioma. However, the development of diseases due to exposure to asbestos is not only occupational, but also domestic or even environmental. It has been reported that the families of these aforementioned workers and the communities surrounding the factories can also develop harmful symptoms and diseases.100
Although it has been observed that 10% of those who died due to MM did not present asbestos or earth flax residues in their biopsy, 90% of patients who have MM attribute it to exposure to these compounds. The rare cases of MM without exposure to asbestos have been related to exposure to factors such as ionizing radiation, other fibrous minerals, and genetic predisposition.101 The time from exposure to asbestos to the diagnosis is considerably long, but the time from the onset of the disease to the malignancy is short. In addition, the affected organism shows symptoms soon after the initial growth.
At the genetic level, the activation or deactivation of certain genes allows the progressive development of MM. Genes, such as CDKN2A, NF2, and BAP1, are highly related to this disease and play well-established roles within it. The next-generation sequencing data indicate that NF2 and BAP1 genes are the most frequently mutated genes in MM.73,78,102
Biomarkers of asbestos exposure in the evaluation of cancer risk
The identification of biomarkers for the evaluation of the carcinogenic risk in populations exposed to asbestos and also for an early diagnosis of malignant diseases, has been the topic of research of several studies. Among the most studied biomarkers of asbestos exposure are: the soluble mesothelin-related protein,103,104 osteopontin,105 fibulin-3 (Fb-3),106,107 high mobility group box 1 protein (HMGB1),40,108 aquaporin 1 (AQP1),109 fibronectin,110 (IL-6,111 and IL-8112 (Table 2). However, according to Ledda et al (2018),113 none of the markers available today are sufficiently reliable to be used in the surveillance of subjects exposed to asbestos. Of note that, new biomarkers such as miRNAs have been recently introduced, which could be useful to monitor sensitivity to therapy and for prognostic purposes. Some examples of such miRNAs include miRNA-16-5p, miRNA-126-3p, miRNA-143-3p, miRNA-145-5p, miRNA-192-5p, miRNA-193a-3p, miRNA-200b-3p, miRNA-203a-3p, and miRNA-652-3p.114–118
 | Table 2 Biomarkers of asbestos exposure in the evaluation of cancer risk |
Epidemiology
According to global estimates, at least 107,000 people die every year from lung cancer, mesothelioma, and asbestosis as a result of occupational exposure to asbestos.5 Almost 400 deaths have been attributed to non-professional exposure to asbestos. The number of asbestos-related diseases continues to rise, even in countries that banned the use of asbestos in the early 1990s. Due to the long latency periods associated with the diseases in question, suspending from now the use of asbestos will result in a decrease in the number of deaths related to asbestos alone after several decades.5
Even though asbestos has been banned in several countries around the world, in Colombia, limitations on the use of asbestos are few. In Colombia, asbestos consumption in 2015 was 5960 metric tons according to data published by the United States Geological Survey in 2018.119 According to the Ministry of Social Protection, in Colombia there is only one exploitation of chrysotile asbestos, with an approximate production of 9,000 tons per year in recent years and 270,000 tons per year of asbestos-cement (10% asbestos+90% cement) registered in the 1980s.120 There are no exact data of the other economic activities in which there is exposure to asbestos. However, the study group that, together with the Ministry, carried out the National Plan for the Prevention of Silicosis, Pneumoconiosis of the Coal Miner and Asbestosis 2010–2030, managed to detect – by surveying professional risk insurance companies (ARP) – 256 companies that develop 25 economic activities with asbestos use. In these companies, it was calculated that 7% of the workers (688 of 15,170) are exposed to asbestos.120
Conclusions
Asbestos is one of the most important occupational carcinogens used in many industries around the world. The high pathogenicity of this mineral fiber is currently known and has been shown that exposure to it causes oxidative stress, fibrosis, chronic inflammation, direct damage to DNA, and mutagenesis, all of the above associated with the development of lung diseases. Additionally, due to the long latency periods associated with diseases generated by asbestos exposure, the diagnosis of these is delayed, leading to a high percentage of deaths in people exposed. Taking into account the above, improving our knowledge about the mechanisms of cellular and molecular response to asbestos, could have a significant impact on our ability to determine susceptibility to exposure and to establish early diagnoses and more effective treatments.
Acknowledgments
Graphic designer Elizabeth Cruz Tapias is acknowledged for the illustrations. The authors also thank MDPI English editing group for editing a draft of this manuscript. This research was funded by Corporación Hospitalaria Juan Ciudad – Hospital Universitario Mayor – Méderi.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zurbriggen R, Capone L. [Pulmonary disease due to asbestos in steel industry workers]. Medicina (B Aires). 2013;73(3):224–230.
2. Corfiati M, Scarselli A, Binazzi A, et al. Epidemiological patterns of asbestos exposure and spatial clusters of incident cases of malignant mesothelioma from the Italian national registry. BMC Cancer. 2015;15:286. doi:10.1186/s12885-015-1584-3
3.
4.
5.
6. Case BW, Abraham JL, Meeker G, Pooley FD, Pinkerton KE. Applying definitions of “asbestos” to environmental and “low-dose” exposure levels and health effects, particularly malignant mesothelioma. J Toxicol Environ Health B Crit Rev. 2011;14(1–4):3–39. doi:10.1080/10937404.2011.556045
7. Mossman BT, Lippmann M, Hesterberg TW, Kelsey KT, Barchowsky A, Bonner JC. Pulmonary endpoints (lung carcinomas and asbestosis) following inhalation exposure to asbestos. J Toxicol Environ Health B Crit Rev. 2011;14(1–4):76–121. doi:10.1080/10937404.2011.556047
8. Huang SX, Jaurand MC, Kamp DW, Whysner J, Hei TK. Role of mutagenicity in asbestos fiber-induced carcinogenicity and other diseases. J Toxicol Environ Health B Crit Rev. 2011;14(1–4):179–245. doi:10.1080/10937404.2011.556051
9. Kamp DW. Asbestos-induced lung diseases: an update. Transl Res. 2009;153(4):143–152. doi:10.1016/j.trsl.2009.01.004
10. Ossa A, Gomez D, Espinal C. Asbestos in Colombia: A silent enemy. Latreia. 2014;27(1):9.
11. Liu G, Cheresh P, Kamp DW. Molecular basis of asbestos-induced lung disease. Annu Rev Pathol. 2013;8:161–187. doi:10.1146/annurev-pathol-020712-163942
12. Yao S, DellaVentura G, Petibois C. Analytical characterization of cell-asbestos fiber interactions in lung pathogenesis. Anal Bioanal Chem. 2010;397(6):2079–2089. doi:10.1007/s00216-010-3773-x
13.
14. Betti M, Casalone E, Ferrante D, et al. Germline mutations in DNA repair genes predispose asbestos-exposed patients to malignant pleural mesothelioma. Cancer Lett. 2017;405:38–45. doi:10.1016/j.canlet.2017.06.028
15. Libbus BL, Illenye SA, Craighead JE. Induction of DNA strand breaks in cultured rat embryo cells by crocidolite asbestos as assessed by nick translation. Cancer Res. 1989;49(20):5713–5718.
16. Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1666–1680. doi:10.1164/ajrccm.157.5.9707141
17. Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer. 2007;121(11):2381–2386. doi:10.1002/ijc.23192
18. Kundu JK, Surh YJ. Inflammation: gearing the journey to cancer. Mutat Res. 2008;659(1–2):15–30. doi:10.1016/j.mrrev.2008.03.002
19. Weitzman SA, Gordon LI. Inflammation and cancer: role of phagocyte-generated oxidants in carcinogenesis. Blood. 1990;76(4):655–663.
20. Goldberg JL, Zanella CL, Janssen YM, et al. Novel cell imaging techniques show induction of apoptosis and proliferation in mesothelial cells by asbestos. Am J Respir Cell Mol Biol. 1997;17(3):265–271. doi:10.1165/ajrcmb.17.3.2991
21. Buder-Hoffmann S, Palmer C, Vacek P, Taatjes D, Mossman B. Different accumulation of activated extracellular signal-regulated kinases (ERK 1/2) and role in cell-cycle alterations by epidermal growth factor, hydrogen peroxide, or asbestos in pulmonary epithelial cells. Am J Respir Cell Mol Biol. 2001;24(4):405–413. doi:10.1165/ajrcmb.24.4.4290
22. Branchaud RM, Garant LJ, Kane AB. Pathogenesis of mesothelial reactions to asbestos fibers. Monocyte recruitment and macrophage activation. Pathobiology. 1993;61(3–4):154–163. doi:10.1159/000163784
23. Kane AB. Mechanisms of mineral fibre carcinogenesis. IARC Sci Publ. 1996;140:11–34.
24. Kamp DW, Weitzman SA. The molecular basis of asbestos induced lung injury. Thorax. 1999;54(7):638–652.
25. Manning CB, Vallyathan V, Mossman BT. Diseases caused by asbestos: mechanisms of injury and disease development. Int Immunopharmacol. 2002;2(2–3):191–200.
26. Shukla A, Gulumian M, Hei TK, Kamp D, Rahman Q, Mossman BT. Multiple roles of oxidants in the pathogenesis of asbestos-induced diseases. Free Radic Biol Med. 2003;34(9):1117–1129.
27. Su B, Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr Opin Immunol. 1996;8(3):402–411.
28. Heintz NH, Janssen-Heininger YM, Mossman BT. Asbestos, lung cancers, and mesotheliomas: from molecular approaches to targeting tumor survival pathways. Am J Respir Cell Mol Biol. 2010;42(2):133–139. doi:10.1165/rcmb.2009-0206TR
29. Janssen YM, Barchowsky A, Treadwell M, Driscoll KE, Mossman BT. Asbestos induces nuclear factor kappa B (NF-kappa B) DNA-binding activity and NF-kappa B-dependent gene expression in tracheal epithelial cells. Proc Natl Acad Sci USA. 1995;92(18):8458–8462. doi:10.1073/pnas.92.18.8458
30. Janssen-Heininger YM, Macara I, Mossman BT. Cooperativity between oxidants and tumor necrosis factor in the activation of nuclear factor (NF)-kappaB: requirement of Ras/mitogen-activated protein kinases in the activation of NF-kappaB by oxidants. Am J Respir Cell Mol Biol. 1999;20(5):942–952. doi:10.1165/ajrcmb.20.5.3452
31. Mathas S, Hinz M, Anagnostopoulos I, et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. Embo J. 2002;21(15):4104–4113.
32. Zanella CL, Posada J, Tritton TR, Mossman BT. Asbestos causes stimulation of the extracellular signal-regulated kinase 1 mitogen-activated protein kinase cascade after phosphorylation of the epidermal growth factor receptor. Cancer Res. 1996;56(23):5334–5338.
33. Manning CB, Cummins AB, Jung MW, et al. A mutant epidermal growth factor receptor targeted to lung epithelium inhibits asbestos-induced proliferation and proto-oncogene expression. Cancer Res. 2002;62(15):4169–4175.
34. Zanella CL, Timblin CR, Cummins A, et al. Asbestos-induced phosphorylation of epidermal growth factor receptor is linked to c-fos and apoptosis. Am J Physiol. 1999;277(4 Pt 1):L684–L693. doi:10.1152/ajplung.1999.277.4.L684
35. Dusinska M, Collins A, Kazimirova A, et al. Genotoxic effects of asbestos in humans. Mutat Res. 2004;553(1–2):91–102. doi:10.1016/j.mrfmmm.2004.06.027
36. Testa JR, Cheung M, Pei J, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43(10):1022–1025. doi:10.1038/ng.912
37. Betti M, Aspesi A, Biasi A, et al. CDKN2A and BAP1 germline mutations predispose to melanoma and mesothelioma. Cancer Lett. 2016;378(2):120–130. doi:10.1016/j.canlet.2016.05.011
38. Baumann F, Flores E, Napolitano A, et al. Mesothelioma patients with germline BAP1 mutations have 7-fold improved long-term survival. Carcinogenesis. 2015;36(1):76–81. doi:10.1093/carcin/bgu227
39. Carbone M, Flores EG, Emi M, et al. Combined genetic and genealogic studies uncover a large BAP1 cancer syndrome kindred tracing back nine generations to a common ancestor from the 1700s. PLoS Genet. 2015;11(12):e1005633. doi:10.1371/journal.pgen.1005633
40. Napolitano A, Pellegrini L, Dey A, et al. Minimal asbestos exposure in germline BAP1 heterozygous mice is associated with deregulated inflammatory response and increased risk of mesothelioma. Oncogene. 2016;35(15):1996–2002. doi:10.1038/onc.2015.243
41. Ventii KH, Devi NS, Friedrich KL, et al. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res. 2008;68(17):6953–6962. doi:10.1158/0008-5472.CAN-08-0365
42. Assis LV, Isoldi MC. Overview of the biochemical and genetic processes in malignant mesothelioma. J Bras Pneumol. 2014;40(4):429–442.
43. Nasu M, Emi M, Pastorino S, et al. High incidence of somatic BAP1 alterations in sporadic malignant mesothelioma. J Thorac Oncol. 2015;10(4):565–576. doi:10.1097/JTO.0000000000'000471
44. Bott M, Brevet M, Taylor BS, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet. 2011;43(7):668–672. doi:10.1038/ng.855
45. McGregor SM, Dunning R, Hyjek E, Vigneswaran W, Husain AN, Krausz T. BAP1 facilitates diagnostic objectivity, classification, and prognostication in malignant pleural mesothelioma. Hum Pathol. 2015;46(11):1670–1678. doi:10.1016/j.humpath.2015.06.024
46. Cigognetti M, Lonardi S, Fisogni S, et al. BAP1 (BRCA1-associated protein 1) is a highly specific marker for differentiating mesothelioma from reactive mesothelial proliferations. Mod Pathol. 2015;28(8):1043–1057. doi:10.1038/modpathol.2015.65
47. Wong DW, Leung EL, So KK, et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009;115(8):1723–1733. doi:10.1002/cncr.24181
48. Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol. 2013;31(8):1105–1111. doi:10.1200/JCO.2012.44.5353
49. Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253. doi:10.1200/JCO.2009.22.6993
50. Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003;22(4):309–325.
51. Eder JP, Vande Woude GF, Boerner SA, LoRusso PM. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009;15(7):2207–2214. doi:10.1158/1078-0432.CCR-08-1306
52. Carracedo A, Egervari K, Salido M, et al. FISH and immunohistochemical status of the hepatocyte growth factor receptor (c-Met) in 184 invasive breast tumors. Breast Cancer Res. 2009;11(2):402. doi:10.1186/bcr2239
53. Zucali PA, Ruiz MG, Giovannetti E, et al. Role of cMET expression in non-small-cell lung cancer patients treated with EGFR tyrosine kinase inhibitors. Ann Oncol. 2008;19(9):1605–1612. doi:10.1093/annonc/mdn240
54. Jin Y, Sun PL, Kim H, et al. MET gene copy number gain is an independent poor prognostic marker in Korean stage I lung adenocarcinomas. Ann Surg Oncol. 2014;21(2):621–628. doi:10.1245/s10434-013-3355-1
55. Toschi L, Cappuzzo F. Clinical implications of MET gene copy number in lung cancer. Future Oncol. 2010;6(2):239–247. doi:10.2217/fon.09.164
56. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi:10.1126/science.1141478
57. Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol. 2013;31(8):1089–1096. doi:10.1200/JCO.2012.43.9422
58. Goldman JW, Laux I, Chai F, et al. Phase 1 dose-escalation trial evaluating the combination of the selective MET (mesenchymal-epithelial transition factor) inhibitor tivantinib (ARQ 197) plus erlotinib. Cancer. 2012;118(23):5903–5911. doi:10.1002/cncr.27575
59. Hicks J. Biologic, cytogenetic, and molecular factors in mesothelial proliferations. Ultrastruct Pathol. 2006;30(1):19–30. doi:10.1080/01913120500313168
60. Hesterberg TW, Barrett JC. Induction by asbestos fibers of anaphase abnormalities: mechanism for aneuploidy induction and possibly carcinogenesis. Carcinogenesis. 1985;6(3):473–475.
61. Dopp E, Saedler J, Stopper H, Weiss DG, Schiffmann D. Mitotic disturbances and micronucleus induction in Syrian hamster embryo fibroblast cells caused by asbestos fibers. Environ Health Perspect. 1995;103(3):268–271. doi:10.1289/ehp.95103268
62. Ault JG, Cole RW, Jensen CG, Jensen LC, Bachert LA, Rieder CL. Behavior of crocidolite asbestos during mitosis in living vertebrate lung epithelial cells. Cancer Res. 1995;55(4):792–798.
63. Tanaka K, Hirota T. Chromosomal instability: A common feature and a therapeutic target of cancer. Biochim Biophys Acta. 2016;1866(1):64–75. doi:10.1016/j.bbcan.2016.06.002
64. Geigl JB, Obenauf AC, Schwarzbraun T, Speicher MR. Defining ‘chromosomal instability’. Trends Genet. 2008;24(2):64–69. doi:10.1016/j.tig.2007.11.006
65. Gagos S, Irminger-Finger I. Chromosome instability in neoplasia: chaotic roots to continuous growth. Int J Biochem Cell Biol. 2005;37(5):1014–1033. doi:10.1016/j.biocel.2005.01.003
66. Dayal J, Albergant L, Newman T, South A. Quantitation of multiclonality in control and drug-treated tumour populations using high-throughput analysis of karyotypic heterogeneity. Converg Sci Phys Oncol. 2015;1:2. doi:10.1088/2057-1739/1/2/025001
67. Marini V, Michelazzi L, Cioe A, Fucile C, Spigno F, Robbiano L. Exposure to asbestos: correlation between blood levels of mesothelin and frequency of micronuclei in peripheral blood lymphocytes. Mutat Res. 2011;721(1):114–117. doi:10.1016/j.mrgentox.2010.12.014
68. Horska A, Kazimirova A, Barancokova M, Wsolova L, Tulinska J, Dusinska M. Genetic predisposition and health effect of occupational exposure to asbestos. Neuro Endocrinol Lett. 2006;27(Suppl 2):100–103.
69. Kawami M, Ebihara I. Cytogenetic damage and cell-mediated immunity in pneumoconiosis. J Environ Pathol Toxicol Oncol. 2000;19(1–2):103–108.
70. Govercin M, Tomatir AG, Evyapan F, Acikbas I, Coskun G, Akdag B. Elevated micronucleus frequencies in patients with pleural plaque secondary to environmental exposure to asbestos. Genet Mol Res. 2014;13(1):598–604. doi:10.4238/2014.January.28.5
71. Donmez-Altuntas H, Baran M, Oymak FS, et al. Investigation of micronucleus frequencies in lymphocytes of inhabitants environmentally exposed to chrysotile asbestos. Int J Environ Health Res. 2007;17(1):45–51. doi:10.1080/09603120601124231
72. Bolognesi C, Martini F, Tognon M, et al. A molecular epidemiology case control study on pleural malignant mesothelioma. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1741–1746. doi:10.1158/1055-9965.EPI-04-0903
73. Bueno R, Stawiski EW, Goldstein LD, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. 2016;48(4):407–416. doi:10.1038/ng.3520
74. Singhi AD, Krasinskas AM, Choudry HA, et al. The prognostic significance of BAP1, NF2, and CDKN2A in malignant peritoneal mesothelioma. Mod Pathol. 2016;29(1):14–24. doi:10.1038/modpathol.2015.121
75. Jean D, Daubriac J, Le Pimpec-Barthes F, Galateau-Salle F, Jaurand MC. Molecular changes in mesothelioma with an impact on prognosis and treatment. Arch Pathol Lab Med. 2012;136(3):277–293. doi:10.5858/arpa.2011-0215-RA
76. Sekido Y, Pass HI, Bader S, et al. Neurofibromatosis type 2 (NF2) gene is somatically mutated in mesothelioma but not in lung cancer. Cancer Res. 1995;55(6):1227–1231.
77. Hirao T, Bueno R, Chen CJ, Gordon GJ, Heilig E, Kelsey KT. Alterations of the p16(INK4) locus in human malignant mesothelial tumors. Carcinogenesis. 2002;23(7):1127–1130.
78. Guo G, Chmielecki J, Goparaju C, et al. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res. 2015;75(2):264–269. doi:10.1158/0008-5472.CAN-14-1008
79. Krismann M, Muller KM, Jaworska M, Johnen G. Molecular cytogenetic differences between histological subtypes of malignant mesotheliomas: DNA cytometry and comparative genomic hybridization of 90 cases. J Pathol. 2002;197(3):363–371. doi:10.1002/path.1128
80. Nymark P, Wikman H, Hienonen-Kempas T, Anttila S. Molecular and genetic changes in asbestos-related lung cancer. Cancer Lett. 2008;265(1):1–15. doi:10.1016/j.canlet.2008.02.043
81. Nelson HH, Wiencke JK, Gunn L, Wain JC, Christiani DC, Kelsey KT. Chromosome 3p14 alterations in lung cancer: evidence that FHIT exon deletion is a target of tobacco carcinogens and asbestos. Cancer Res. 1998;58(9):1804–1807.
82. Tiainen M, Tammilehto L, Rautonen J, Tuomi T, Mattson K, Knuutila S. Chromosomal abnormalities and their correlations with asbestos exposure and survival in patients with mesothelioma. Br J Cancer. 1989;60(4):618–626.
83. Tammilehto L, Tuomi T, Tiainen M, et al. Malignant mesothelioma: clinical characteristics, asbestos mineralogy and chromosomal abnormalities of 41 patients. Eur J Cancer. 1992;28A(8–9):1373–1379.
84. Jean D, Thomas E, Manie E, et al. Syntenic relationships between genomic profiles of fiber-induced murine and human malignant mesothelioma. Am J Pathol. 2011;178(2):881–894. doi:10.1016/j.ajpath.2010.10.039
85. Chirac P, Maillet D, Lepretre F, et al. Genomic copy number alterations in 33 malignant peritoneal mesothelioma analyzed by comparative genomic hybridization array. Hum Pathol. 2016;55:72–82. doi:10.1016/j.humpath.2016.04.015
86. Cui Y, Ma J, Ye W, et al. Chrysotile and rock wool fibers induce chromosome aberrations and DNA damage in V79 lung fibroblast cells. Environ Sci Pollut Res Int. 2018;25(23):22328–22333. doi:10.1007/s11356-017-9403-9
87. IARC working group on the evaluation of carcinogenic risks to humans: Asbestos. International Agency for Research on Cancer. 1987;Supplement 7:106–116.
88. Algranti EM. S Parkes’ Occupational Lung Disorders.
89. Roggli VL, Gibbs AR, Attanoos R, et al. Pathology of asbestosis- An update of the diagnostic criteria: report of the asbestosis committee of the college of american pathologists and pulmonary pathology society. Arch Pathol Lab Med. 2010;134(3):462–480. doi:10.1043/1543-2165-134.3.462
90. American Thoracic S. Diagnosis and initial management of nonmalignant diseases related to asbestos. Am J Respir Crit Care Med. 2004;170(6):691–715. doi:10.1164/rccm.200310-1436ST
91. Montanaro F, Bray F, Gennaro V, et al. Pleural mesothelioma incidence in Europe: evidence of some deceleration in the increasing trends. Cancer Causes Control. 2003;14(8):791–803.
92. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018. doi:10.3322/caac.21492
93. Nelson HH, Kelsey KT. The molecular epidemiology of asbestos and tobacco in lung cancer. Oncogene. 2002;21(48):7284–7288. doi:10.1038/sj.onc.1205804
94. Kjuus H, Skjaerven R, Langard S, Lien JT, Aamodt T. A case-referent study of lung cancer, occupational exposures and smoking. II. Role of asbestos exposure. Scand J Work Environ Health. 1986;12(3):203–209.
95. Carbone M, Ly BH, Dodson RF, et al. Malignant mesothelioma: facts, myths, and hypotheses. J Cell Physiol. 2012;227(1):44–58. doi:10.1002/jcp.22724
96. Goldberg M, Luce D. The health impact of nonoccupational exposure to asbestos: what do we know? Eur J Cancer Prev. 2009;18(6):489–503. doi:10.1097/CEJ.0b013e32832f9bee
97. Damhuis RA, Khakwani A, De Schutter H, Rich AL, Burgers JA, van Meerbeeck JP. Treatment patterns and survival analysis in 9014 patients with malignant pleural mesothelioma from Belgium, the Netherlands and England. Lung Cancer. 2015;89(2):212–217. doi:10.1016/j.lungcan.2015.05.014
98. Vogelzang NJ, Rusthoven JJ, Symanowski J, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21(14):2636–2644. doi:10.1200/JCO.2003.11.136
99. Donovan EP, Donovan BL, McKinley MA, Cowan DM, Paustenbach DJ. Evaluation of take home (para-occupational) exposure to asbestos and disease: a review of the literature. Crit Rev Toxicol. 2012;42(9):703–731. doi:10.3109/10408444.2012.709821
100. Espinosa Munoz E, Ramirez Ocana D, Gutierrez Cardo AL. Malignant pleural mesothelioma in a young adult with no known exposure to asbestos. Arch Bronconeumol. 2016;52(12):615–616. doi:10.1016/j.arbres.2016.03.008
101. Carmona E, María R. Asbestosis and malignant pleural mesothelioma. Revista De La Facultad De Medicina (México). 2013;52(2):5–17.
102. Lo Iacono M, Monica V, Righi L, et al. Targeted next-generation sequencing of cancer genes in advanced stage malignant pleural mesothelioma: a retrospective study. J Thorac Oncol. 2015;10(3):492–499. doi:10.1097/JTO.0000000000000436
103. Tian L, Zeng R, Wang X, et al. Prognostic significance of soluble mesothelin in malignant pleural mesothelioma: a meta-analysis. Oncotarget. 2017;8(28):46425–46435. doi:10.18632/oncotarget.17436
104. Creaney J, Robinson BW. Detection of malignant mesothelioma in asbestos-exposed individuals: the potential role of soluble mesothelin-related protein. Hematol Oncol Clin North Am. 2005;19(6):1025–1040. doi:10.1016/j.hoc.2005.09.007
105. Creaney J, Yeoman D, Demelker Y, et al. Comparison of osteopontin, megakaryocyte potentiating factor, and mesothelin proteins as markers in the serum of patients with malignant mesothelioma. J Thorac Oncol. 2008;3(8):851–857. doi:10.1097/JTO.0b013e318180477b
106. Pass HI, Levin SM, Harbut MR, et al. Fibulin-3 as a blood and effusion biomarker for pleural mesothelioma. N Engl J Med. 2012;367(15):1417–1427. doi:10.1056/NEJMoa1115050
107. Pei D, Li Y, Liu X, et al. Diagnostic and prognostic utilities of humoral fibulin-3 in malignant pleural mesothelioma: evidence from a meta-analysis. Oncotarget. 2017;8(8):13030–13038. doi:10.18632/oncotarget.14712
108. Yang H, Rivera Z, Jube S, et al. Programmed necrosis induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein release and resultant inflammation. Proc Natl Acad Sci US A. 2010;107(28):12611–12616. doi:10.1073/pnas.1006542107
109. Lopez-Campos JL, Sanchez Silva R, Gomez Izquierdo L, et al. Overexpression of Aquaporin-1 in lung adenocarcinomas and pleural mesotheliomas. Histol Histopathol. 2011;26(4):451–459. doi:10.14670/HH-26.451
110. Begin R, Martel M, Desmarais Y, et al. Fibronectin and procollagen 3 levels in bronchoalveolar lavage of asbestos-exposed human subjects and sheep. Chest. 1986;89(2):237–243.
111. Ilavska S, Jahnova E, Tulinska J, et al. Immunological monitoring in workers occupationally exposed to asbestos. Toxicology. 2005;206(2):299–308. doi:10.1016/j.tox.2004.09.004
112. Tulinska J, Jahnova E, Dusinska M, et al. Immunomodulatory effects of mineral fibres in occupationally exposed workers. Mutat Res. 2004;553(1–2):111–124. doi:10.1016/j.mrfmmm.2004.06.030
113. Ledda C, Senia P, Rapisarda V. Biomarkers for early diagnosis and prognosis of malignant pleural mesothelioma: the quest goes on. Cancers (Basel). 2018;10:6. doi:10.3390/cancers10110400
114. Ledda C, Rapisarda V. Malignant pleural mesothelioma: the need to move from research to clinical practice. Arch Med Res. 2016;47(5):407. doi:10.1016/j.arcmed.2016.08.009
115. Weber DG, Johnen G, Bryk O, Jockel KH, Bruning T. Identification of miRNA-103 in the cellular fraction of human peripheral blood as a potential biomarker for malignant mesothelioma–a pilot study. PLoS One. 2012;7(1):e30221. doi:10.1371/journal.pone.0030221
116. Mairinger FD, Werner R, Flom E, et al. miRNA regulation is important for DNA damage repair and recognition in malignant pleural mesothelioma. Virchows Arch. 2017;470(6):627–637. doi:10.1007/s00428-017-2133-z
117. Gayosso-Gomez LV, Zarraga-Granados G, Paredes-Garcia P, et al. Identification of circulating miRNAs profiles that distinguish malignant pleural mesothelioma from lung adenocarcinoma. Excli J. 2014;13:740–750.
118. Cavalleri T, Angelici L, Favero C, et al. Plasmatic extracellular vesicle microRNAs in malignant pleural mesothelioma and asbestos-exposed subjects suggest a 2-miRNA signature as potential biomarker of disease. PLoS One. 2017;12(5):e0176680. doi:10.1371/journal.pone.0176680
119. Flanagan DM. Asbestos - 2016 [Advance Release]. Washington, DC: U.S. Geological survey minerals yearbook; 2018.
120.
121. Pylkkanen L, Sainio M, Ollikainen T, et al. Concurrent LOH at multiple loci in human malignant mesothelioma with preferential loss of NF2 gene region. Oncol Rep. 2002;9(5):955–959.
122. Pylkkanen L, Wolff H, Stjernvall T, Knuuttila A, Anttila S, Husgafvel-Pursiainen K. Reduced Fhit protein expression in human malignant mesothelioma. Virchows Arch. 2004;444(1):43–48. doi:10.1007/s00428-003-0902-3
123. Thurneysen C, Opitz I, Kurtz S, Weder W, Stahel RA, Felley-Bosco E. Functional inactivation of NF2/merlin in human mesothelioma. Lung Cancer. 2009;64(2):140–147. doi:10.1016/j.lungcan.2008.08.014
124. Napolitano A, Antoine DJ, Pellegrini L, et al. HMGB1 and its hyperacetylated isoform are sensitive and specific serum biomarkers to detect asbestos exposure and to identify mesothelioma patients. Clin Cancer Res. 2016;22(12):3087–3096. doi:10.1158/1078-0432.CCR-15-1130
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.