")
Back to Journals » Journal of Inflammation Research » Volume 16
An Overview of the Mechanisms Involved in Neuralgia
Authors Zhang BW, Dong H, Wu Z, Jiang X, Zou W
Received 14 June 2023
Accepted for publication 26 August 2023
Published 18 September 2023 Volume 2023:16 Pages 4087—4101
DOI https://doi.org/10.2147/JIR.S425966
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Bai-Wen Zhang,1 Hao Dong,1 Zhe Wu,1 Xi Jiang,2 Wei Zou3
1Heilongjiang University of Chinese Medicine, Harbin, 150040, People’s Republic of China; 2Jinzhou Medical University, Jinzhou, 121001, People’s Republic of China; 3The Third Department of Acupuncture and Moxibustion, First Affiliated Hospital, Heilongjiang University of Chinese Medicine, Harbin, 150040, People’s Republic of China
Correspondence: Wei Zou, The Third Department of Acupuncture and Moxibustion, First Affiliated Hospital, Heilongjiang University of Chinese Medicine, Harbin, 150040, People’s Republic of China, Tel +86 0451-82161746, Email [email protected]
Abstract: Neuralgia is a frequently occurring condition that causes chronic pain and burdens both patients and their families. Earlier research indicated that anti-inflammatory treatment, which was primarily utilized to address conditions like neuralgia, resulted in positive outcomes. However, recent years have witnessed the emergence of various novel mechanisms associated with pain-related disorders. This review provides a concise overview of the inflammatory mechanisms involved in neuralgia. It also examines recent advancements in research, exploring the influence of ion channels and synaptic proteins on neuralgia and its complications. Additionally, the interactions between these mechanisms are discussed with the aim of suggesting innovative therapeutic approaches and research directions for the management of neuralgia.
Keywords: neuralgia, inflammatory mechanisms, innovative therapeutic approaches
Introduction
The International Association for the Study of Pain (IASP) provides a definition for pain as a distressing sensory and emotional experience that is associated with actual or potential damage to tissues, or similar sensations.1 The involvement of the nervous system in transmitting sensations between peripheral receptors and the central nervous system is a crucial aspect of this process.2,3 Statistics indicate that over 30% of the global population experiences chronic pain annually, with approximately 15–25% of chronic pain being neuropathic.4
Trauma, surgery, ischemia, inflammation, viruses, chemotherapy, autoimmune diseases, and channelopathies are all factors that can cause nerve damage, specifically affecting the somatosensory system. The occurrence of thermal hyperalgesia and tactile allodynia (increased sensitivity to heat and pain from touch) in the body can be used to determine the presence of these diseases. In the ICD-11 classification, neuropathic pain is categorized as either central neuropathic pain or peripheral neuropathic pain, depending on the location of the lesion. Central neuropathic pain is caused by damage or disease in the central somatosensory nervous system, which may result from central sensitization (lowered pain threshold or increased response to nociceptive input), including punctate hyperalgesia, ongoing pain, dysesthesia, aftersensations, and temporal summation. Examples of conditions causing central neuropathic pain include stroke, traumatic brain injury, and multiple sclerosis. Spinal cord injury is the most common cause, with approximately 50% of patients experiencing central pain. Peripheral sensitization primarily caused by mechanical insensitivity of C fibers and resulting in pain hypersensitivity and abnormal pain perception is the main cause of peripheral neuropathic pain. This can be seen in conditions such as trigeminal neuralgia, postherpetic neuralgia, and carpal tunnel syndrome (Figure 1).5,6
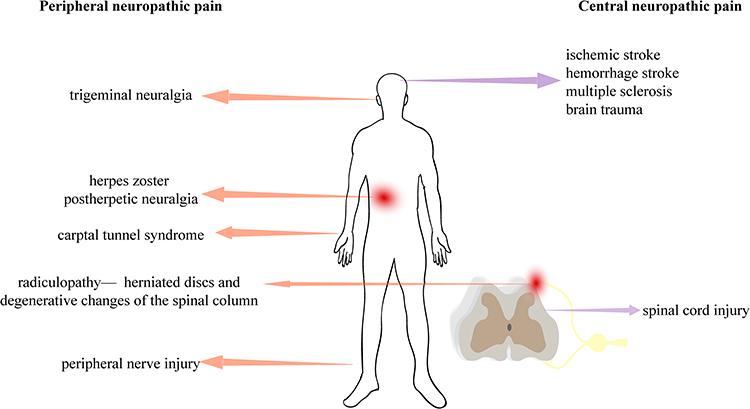 |
Figure 1 The types of neuropathic pain. |
Neuralgia can also lead to complications such as depression, anxiety, and disrupted sleep, which further exacerbate and perpetuate the pain.4 Recent research suggests that while anti-inflammatory mechanisms are important for treatment, exploring the impact of ion channels and synaptic proteins on the condition may offer new therapeutic possibilities.
Effect of Cytokines on the Sensitization of Pain Sense
After nerve injury, various cell factors are upregulated in DRG neurons, increasing neuronal excitability and driving peripheral sensitization. Excitatory synaptic transmission mediated by N-methyl-D-aspartate receptors (NMDAR) and α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptors (AMPAR) reduces inhibitory synaptic transmission mediated by GABA receptors and glycine receptors, leading to central sensitization.7–10
Calcitonin gene-related peptide (CGRP) is a pain-inducing factor that contributes to the development of hyperalgesic responses in nerve injury. It is released from the central terminals of nociceptors, promoting central sensitization,11 and its expression is upregulated in response to inflammatory signaling.12,13 This further exacerbates pain occurrence. Pathological activity of peripheral incoming nerves after injury leads to the release of colony-stimulating factor 1 (CSF1) from damaged afferent neurons, triggering activation of microglia through the activation of CSF receptor 1 on their surface or via the AMPK signaling pathway.14
In the spinal cord, parenchymal microglia are the sole cells expressing colony-stimulating factor 1 receptor (CSF1R). The cross-talk between CSF1-mediated spinal microglia and lymphocytes can either amplify or inhibit pain.15 Substance P (SP) is an 11-amino acid neuropeptide that preferentially activates the neurokinin-1 receptor (NK1R). SP is expressed in several central nervous system regions, including the substantia nigra, amygdala, dorsal root ganglia (DRG), and dorsal horn, and plays a role in transmitting nociceptive signals from primary afferent fibers to secondary neurons in the spinal cord and brainstem.16 SP is also expressed in microglia, T cells, macrophages, and eosinophils. During inflammation, upregulated SP released by primary afferent fibers binds to NK1 receptors on dorsal horn neurons, activating phospholipase C signaling cascade and subsequently modulating membrane depolarization and the function of AMPAR and NMDAR. This regulation affects spinal neuron excitability and plays a crucial role in pain hypersensitivity. Additionally, SP can regulate pathways like nuclear factor kappa-B (NF-κB) and PPAR to alleviate pain.17,18 NMDARs, particularly subtypes NR2A and NR2B, are key regulatory subunits controlling NMDAR channels19 that play a critical role in neuropathic and inflammatory pain. Increased NMDAR expression leads to glial cell activation as well as reduced Kir4.1 expression, resulting in augmented intracellular calcium influx and activation of protein kinases. This initiates cellular cascades that modulate membrane properties and neuronal sensitivity, thereby contributing to central sensitization.20–22
Furthermore, inflammatory mediators such as bradykinin, prostaglandins, nerve growth factor, and pro-inflammatory cytokines like tumor necrosis factor-alpha, interleukin-1beta, and pro-inflammatory chemokines can directly bind to and activate or sensitize G protein-coupled receptors, ion channels, and tyrosine kinase receptors located on the terminals and/or cell bodies of nociceptors. This activation or sensitization ultimately leads to the generation of pain in the body.23–25
A study revealed that the expression of chemokines and their receptors, specifically CX3CL1/CX3CR1, CCL2/CCR2, CXCL1/CXCR2, CXCL12/CXCR4, and CCL3/CCR5, was increased during the onset of neuropathic pain.26
Fractalkine, also known as C-X3-C Motif Chemokine Ligand 1 (CX3CL1), is expressed in primary sensory neurons and spinal neurons and induces activation of microglia through its receptor CX3CR1 (neuron-to-microglia signaling).27 CCL2 and CXCL1 are expressed in spinal astrocytes and act on CCR2 and CXCR2 in spinal neurons, increasing excitatory synaptic transmission.28 CCL2, also called Monocyte chemoattractant protein-1 (MCP-1), is a pain-related chemokine present in substance P (SP) and calcitonin gene-related peptide (CGRP)-positive primary sensory neurons in the superficial dorsal horn. It directly contributes to central sensitization by binding to CCR2. In a rat model of Central post-stroke pain (CPSP), MCP-1 expression levels are elevated in bilateral dorsal horns of the spinal cord, leading to glial activation through neuronal-glia interactions mediated by CCR2.29 Additionally, electrophysiological studies have demonstrated that MCP-1 enhances NMDA and AMPA-induced currents, increasing the frequency of sEPSCs (spontaneous excitatory postsynaptic currents) in spinal second-order neurons and exacerbating inflammatory pain severity.22,30 Conversely, inhibiting MCP-1 signaling can alleviate various types of pain by reducing neuronal excitability, microglial activation, and release of pro-inflammatory cytokines observed in multiple pain models.31,32
CXCL1 is expressed and secreted by activated macrophages, endothelial cells, and fibroblasts. It is expressed in spinal astrocytes and neurons, inducing CXCR2-dependent thermal hyperalgesia. CXCL1 is also involved in various downstream pathways such as c-Raf/MAPK/AP-1, p-PKC-μ/p-ILK/NLRP3, JAK2/STAT3, TAK1/NF-κB.33 Electrophysiological recordings demonstrate that CXCL1 increases NMD-induced currents in dorsal horn neurons through CXCR2.34,35 In a rat model of neuropathic pain following injury, CXCL12 expression is upregulated in the spinal cord and dorsal root ganglia.36 CXCL12 is expressed in neurons, astrocytes, and microglia. Its binding to CXCR4 leads to excessive excitation of glutamatergic neurons, contributing to pain sensitization.37,38 The interaction between CXCL12 and CXCR4 activates downstream signaling pathways such as G protein-mediated pathways (PI3K, ERK1/2, MAPK, and NF-κB), triggers intracellular Ca2+ release, increases neuronal excitability, and promotes pain generation.39,40 Additionally, this signaling axis regulates nav1.8 channels in DRG neurons and is associated with the pathogenesis of mechanical allodynia and small fiber neuropathy observed in painful diabetic neuropathy mouse models.41
CCL3, also known as macrophage inflammatory protein-1α (MIP-1α), has been found to participate in the development of neuropathic pain through its predominant receptors CCR1 and CCR5.42 CCL3 is present in neurons, and studies have discovered that primary cultured microglia produce CCL3 upon ATP stimulation. The activated CCL3 can recruit macrophages/microglia and exacerbate inflammatory neuropathic pain.43–45 CXCL13, also called B lymphocyte chemoattractant (BLC), was originally found to be produced by stromal cells within B-cell follicles. CXCR5 is the sole receptor for CXCL13, also known as cluster of differentiation 185 (CD185) or Burkitt lymphoma receptor 1 (BLR1).46 Both CXCL13 and CXCR5 are expressed in the sensory nervous system, including peripheral sensory ganglia, spinal dorsal horn neurons, and astrocytes. They may contribute to neuropathic pain through neuron-astrocyte interactions. Moreover, CXCR5 can promote the activation of downstream signaling pathways such as NF-κB signaling, excessive activation of spinal cord glial cells, activation of c-Fos protein, and upregulation of pro-inflammatory cytokine IL-6, thereby causing mechanical allodynia.47 Upregulated expression of CXCL13/CXCR5 on DRG neurons can also enhance Nav1.8 channel expression, further contributing to inflammatory pain.48
Additionally, the use of chemokine receptor antagonists was shown to alleviate neuropathic pain, indicating the significance of chemokines in pain initiation. Following peripheral nerve injury (PNI), neuroinflammation occurs in the spinal cord, characterized by the activation of astrocytes and the upregulation of interleukin 6 (IL-6), chemokine (C-C motif) ligand 2 (CCL2), and chemokine (C-X-C motif) ligand 1 (CXCL1). Inhibition of these factors in a rat model of pain demonstrated an improvement in symptoms associated with neuropathic pain.49
Based on the above, it can be concluded that various cell factors may be involved in the mechanisms of neuropathic pain, exerting different effects. These factors not only directly stimulate central or peripheral sensitization but also influence other cells or pathways, leading to the secretion of additional nociceptive factors and exacerbating pain.
Effect of Microglia on the Sensitization of Pain Sense
In 2003, Jin et al50 and Tsuda51 conducted experiments that revealed the significant role of spinal microglia in neuropathic pain. The development of microglia is controlled by the colony stimulating factor 1 receptor (CSF1R)52 and its corresponding ligand interleukin-34 (IL-34).53 Transforming growth factor-β1 (TGF-β1) is also a crucial factor at the molecular level in promoting the differentiation and acquisition of mature properties in microglia.54 However, during the occurrence of pain, TGF-β plays a role in inhibiting the activity of spinal cord glial cells and can reduce the expression of CCL-2, thereby increasing the integrity of the blood-spinal-cord barrier (BSCB) and reducing further infiltration of immune cells into the spinal dorsal horn. It can also suppress inflammation through various pathways such as TGF-β/Smad, NF-κB/ERK1/2,55,56 thereby alleviating neuropathic pain. The inhibitory effects of TGF-β on neuropathic pain can also occur through gene transcription and neuroregulatory pathways in the dorsal root ganglion. TGF-β can rapidly activate TGF-β receptors on neurons, leading to the normalization of heightened excitability in the dorsal root ganglion and synaptic plasticity in the spinal cord induced by nerve injury.57
Microglial activation can be triggered through toll-like receptor-4 (TLR4) or triggering receptor expressed on myeloid cells 2 (TREM2) expressed on trigger receptors, or via Caspase-mediated immune responses, which initiate pro-inflammatory signaling pathways such as the NF-κB and MAPK pathways. Activation of NF-κB pathway can induce the release of pro-inflammatory cytokines, primarily IL-1β and TNF-α, as well as chemokines like MCP1 and CXCL1. On the other hand, activation of the MAPK family including ERK1/2, p38, and JUN-amino-terminal kinase (JNK) pathway can upregulate the expression of pro-inflammatory genes regulated by AP-1 transcription factor, including COX2 and NOS2, further exacerbating inflammatory responses.58 Furthermore, interferon regulatory factor 8 (IRF8) is upregulated in microglia after peripheral nerve injury and regulates cellular genes and responses. This includes purinergic P2 receptors (P2X4R and P2Y12R), TLR2, CX3CR1, diffusion factor IL-1β, histone S, and brain-derived neurotrophic factor (BDNF), all of which collectively mediate the inflammatory response.59
The red nucleus (RN) can bidirectionally regulate the development and maintenance of mononeuropathic pain by secreting both pro- and anti-inflammatory cytokines.60 Li et al61 observed a mononeuropathic pain model in rats with sciatic nerve injury (SNI) and discovered an increased expression of IL-33 and its receptor ST2 in RN neurons, oligodendrocytes, and microglia. This indicates that the activation of microglia induced by pain not only stimulates inflammatory responses around the affected tissues but also affects microglia activation in various regions such as the thalamus, amygdala, ventral tegmental area, anterior cingulate cortex, hippocampus, and periaqueductal gray matter.62–65
It can be understood that microglia play a crucial role in the development, maintenance, and alleviation of neuropathic pain. Following nerve injury, microglia not only act as immune cells to repair the damaged area but can also initiate inflammatory cascades, leading to an increase in the recruitment of inflammatory cytokines and cells within the damaged region. Moreover, microglia can interact with various cell types, ultimately contributing to the onset of pain (Figure 2).
 |
Figure 2 The cytokines in neuropathic pain. |
Effect of Ion Channels on Nociceptive Sensitization
The ion channels associated with chronic pain consist of Nav1.7, a voltage-gated sodium channel, and Kv1.1 and Kv1.2, voltage-gated potassium channels.66 By controlling the levels of proteins associated with calcium (Ca2+), sodium (Na+), potassium (K+), and chloride (Cl−), it is possible to alleviate symptoms of neuropathic pain.67
Potassium Ion Channels
Kv plays a crucial role in regulating the excitability of neurons by controlling the resting membrane potential and repolarization. In rats, the downregulation of Kv1.2 expression in the dorsal root ganglion (DRG) and the dorsal horn of the spinal cord occurred as a result of chronic constrictive injury (CCI). This downregulation led to significant mechanical sensitization and hyperalgesia. Alongside the decrease in Kv1.2 expression, there was an increase in miR-137 expression. However, inhibiting miR-137 reversed these effects by increasing Kv1.2 expression in CCI rats. This restoration of abnormal Kv currents and excitability in dorsal root ganglia (DRG) neurons alleviated mechanical heterotopic pain and hyperalgesia.68 Kv1.2 is highly expressed in large and medium-sized DRG neurons, and its reduced expression can increase the excitability of DRG neurons, leading to mechanical, thermal, and cold hypersensitivity. The decrease in Kv1.2 expression can depolarize the resting membrane potential and lower the current threshold for generating action potentials in injured DRG neurons. It may also increase the release of primary afferent neurotransmitters regulated by mu opioid receptors (MOR), thus promoting the occurrence and maintenance of neuropathic pain hypersensitivity induced by peripheral nerve damage.69
K2p1.1 is abundantly present in DRG neurons and facilitates the outward flow of potassium ions across the cell membrane. Decreased expression of K2p1.1 leads to membrane depolarization and increased neuronal excitability, rendering them sensitive to mechanical and thermal stimuli.70,71 In a study by Jia et al, it was found that systemic administration of paclitaxel resulted in a time-dependent reduction in K2p1.1 mRNA and protein expression in DRG neurons, leading to mechanical and thermal hypersensitivity in rats. Additionally, overexpression of ten-eleven translocation methylcytosine dioxygenase 1 (TET1) in DRG improved the expression of K+ channel 1.1 (K(2p)1.1) and alleviated mechanical heterotopic pain as well as heat and cold pain on the affected side in rats. These findings collectively highlight the significant impact of K+ channel regulation on pain relief.72
Sodium Ion Channels
The sodium leak channel (NALCN) is a channel that allows the leakage of sodium ions and controls the excitability and rhythmicity of neurons. In a study by Zhang et al,73 it was discovered that NALCN is present in the DRG and dorsal spinal cord. When chronic compressive injury (CCI) occurred, NALCN currents and neuronal excitability increased. However, when NALCN was suppressed using NALCN-siRNA or NALCN-shRNA, pain-related symptoms in rats were relieved or eliminated. This suggests that NALCN is involved in neuronal sensitization caused by CCI.
Voltage-gated sodium channels (VGSCs) are crucial for perceiving pain. Nociceptors, which are sensory neurons in the peripheral nervous system, predominantly express different types of Nav channels, including Nav1.1, Nav1.3, Nav1.6, Nav1.7, Nav1.8, and Nav1.9.74 Studies in humans and mice have shown that Nav1.7 is vital for transmitting pain signals.75 Nav1.7 in humans is involved in generating and transmitting neuropathic and nociceptive pain signals.76 It enhances the effectiveness and selectivity of Nav isoforms, which is important for acute and chronic inflammatory pain models and neuropathic pain. Nav1.7 is widely expressed in all sensory neuron subtypes except proprioceptors, as demonstrated by a study.77
Pulsed radiofrequency treatment has been effective for neuropathic pain, and research by Hidaka et al78 on the treatment of resiniferatoxin (RTX)-induced mechanical heterotopic pain suggests that pain relief may be linked to voltage-gated Na+ channels. In a rat model of RTX-induced mechanical heterotopic pain in the DRG, the Nav1.7 channel was upregulated following increased phosphorylation of kinases regulated by extracellular signaling. Early combination therapy of tramadol could inhibit the upregulation of Nav1.7 in the DRG and alleviate pain symptoms. Nav1.7, a subtype of voltage-gated sodium channels, plays a crucial role in transmitting pain signals. Li et al79 discovered that a specific small-molecule inhibitor of CRMP2 SUMOylation, compound 194, selectively reduced Nav1.7 currents in DRG neurons across species and partially reversed mechanical heterotopic pain caused by nerve injury and chemotherapy.
Calcium Channels
Ziconotide is the initial medication that specifically blocks N-type voltage-sensitive calcium channels. By blocking these channels in the spinal cord, ziconotide prevents the release of pain-related neurotransmitters from the central terminals of primary afferent neurons, thus reducing pain.80 Painful diabetic neuropathy (PDN) is a condition characterized by neuropathic pain, where the nociceptors in the DRG become hyperexcitable, leading to excessive calcium levels, degeneration of axons, and loss of cortical innervation. George et al81 discovered that DRG neurons in PDN mice induced by a high-fat diet expressed a significant amount of a protein involved in mitochondrial fission. Examples of proteins involved in mitochondrial dynamics include dynamin-related protein 1 (Drp1), Mitochondrial Fission 1 protein (Fis1), mitochondrial fission factor (Mff), optic atrophy 1 (Opa1), among others. Fis1 and Mff serve as receptors recruiting Drp1 to mitochondria. Mff, a GTP hydrolysis enzyme, catalyzes mitochondrial fission in cells by forming a complex with GTPase Drp1 to promote the process. The normal activity of various pumps and transporters on the membrane of peripheral sensory neurons, as well as long-range transport along the nerve sheath, requires substantial ATP supply. Dysregulation of Drp1, Fis1, Dnm2, and Dnm3 contributes to increased reactive oxygen species (ROS) levels within cells. Furthermore, mitochondrial dysfunction leads to enhanced ROS production, loss of mitochondrial membrane potential, uncoupling of the electron transport chain, and decreased ATP concentration. These factors result in insufficient energy supply and dysfunction of peripheral sensory neurons, ultimately contributing to the occurrence of pain.6,82–84
Furthermore, nociceptive receptors exhibited increased calcium signals in these mice. This particular type of neuropathic pain seems to be associated with axonal degeneration. When there is hyperexcitability of nociceptive receptors and an elevated calcium concentration within the affected cells, excess calcium ions are transported into the mitochondria via the mitochondrial calcium uniporter. This results in increased calcium-dependent mitochondrial fission, ultimately leading to axonal degeneration and neuropathic pain in PDN. Pulsed radiofrequency treatment with high voltage and prolonged duration has proven effective for neuropathic pain. However, the Cav2.2 channel can increase neuronal excitability and neurotransmission, contributing to neuropathic pain. By inhibiting the Cav2.2 channel, the hyperalgesic behaviors caused by NP can be reversed.85 Neuromedin B (Nmb) is involved in the regulation of nociception in sensory neurons. Zhang et al86 found that NmbR, by enhancing Cav3.2 channel currents, increases neuronal excitability in small trigeminal ganglion cells, leading to pain sensitization in a mouse model of inflammatory pain. In another study by Qi et al,87 it was shown that miR-32-5p targeted and inhibited Cav3.2 T-type calcium channels. In a rat model of trigeminal neuralgia, overexpression of miR-32-5p effectively suppressed Cav3.2 expression and relieved mechanical heterotopic pain.
Chloride Channels
Anoctamin-1 (ANO1), a calcium-activated chloride channel, has the potential to be a target for pain relief. ANO1 is activated by harmful stimuli transmitted from sensory neurons in the peripheral nervous system, leading to nerve depolarization. Decreasing the activity of ANO1 alleviates hyperpathia and heterotopic pain resulting from inflammation and nerve damage.88 Reduced glycinergic inhibition could be a spinal mechanism underlying the manifestation of pathological pain symptoms. An increase in sensitivity to mechanical, thermal, and cold stimuli can result from the disruption of glycine receptors, a type of ion channel that permits the passage of chloride ions. Particularly, the glycine receptor α1 subunit in the adult spinal cord is essential for the production of inhibitory neurotransmitters. In a study conducted by Yao et al,89 changes in the glycine receptor α1 subunit were observed in mice experiencing inflammation induced by formalin, and administration of glycine receptor agonists alleviated the aforementioned pain symptoms. These findings suggest that altered expression of glycine receptors or chloride channels may impact pain mechanisms. Liao et al90 demonstrated that using low-intensity focused ultrasound (LIFU) has the ability to hinder calcium/calmodulin-dependent protein kinase type IV (CaMKIV) and activate the potassium chloride co-transporter 2 (KCC2) pathway in neurons located in the spinal dorsal horn. This activation leads to alleviation of neuropathic pain behavior in rats with peripheral nerve injury (PNI). From this, it can be concluded that regulating levels of calcium (Ca2+), potassium (K+), chloride (Cl−), and expression of their associated proteins may provide relief from neuropathic pain.
Lorenzo et al91 discovered that a ligand called L838,417, which targets the benzodiazepine site, plays a significant role in analgesia. However, when administered at high doses, L838,417 failed to induce analgesia due to impaired chloride ion efflux. Conversely, increasing the activity of KCC2 not only enhanced the analgesic effects of L838,417 but also restored its potential to alleviate pain at high doses. This suggests that maintaining chloride ion homeostasis is crucial for analgesia in cases of pathological pain.
Additionally, other proteins related to ion channels, such as P2X7R protein or the transient receptor potential M8 (TRPM8) ion channel, can also influence pain modulation to some extent.
Unlike cell factors, ion channels primarily function by altering the resting membrane potential on the cellular membrane or by facilitating the release of pain-inducing neurotransmitters. Such pathological mechanisms provide avenues for exploring new drugs and therapeutic approaches to address pain.
Effect of Synaptic Proteins on the Sensitization of Pain Sense
Chronic pain is characterized by changes in the dorsal horn of the spinal cord known as synaptic plasticity. This refers to the ability of synapses in this region to undergo long-term potentiation (LTP) and long-term depression (LTD), which are influenced by activity and contribute to central sensitization in chronic pain. The stimulation of the primary afferent tract through burst stimulation (TBS) typically leads to LTP, while tetanic stimulation often results in LTD. The occurrence of synaptic LTP in spinal projection neurons is associated with the development of chronic pain. When primary afferent nerves discharge, closely linked to layer I projection neurons, they can induce synaptic LTP. This, in turn, increases injurious currents in the dorsal horn of the spinal cord, causing intense nociceptive sensations.
Li et al92 discovered that the activation of mGluR5 and intracellular Ca2+ together led to the activation of spinal D1/D5Rs, which facilitated the transition from Hebbian LTP to non-Hebbian LTP. This activation also altered the discharge frequencies of presynaptic and postsynaptic neurons. Primary afferent stimulation alone was sufficient to induce LTP even without action potential discharge in projection neurons. As a result, the sensory synapses on the output neurons that send pain information to the brain were significantly strengthened.
Liu et al93 found that a reduction in glutamatergic synaptic connection strength in the ventral periaqueductal gray (vlPAG) region was associated with increased abdominal sensitivity caused by dibutyltin dichloride (DBTC). This decrease was a result of both pre- and post-synaptic mechanisms. However, they also discovered that activation of AMPA receptors in vlPAG could alleviate DBTC-induced abdominal sensitivity. These findings emphasize the significance of synapses in pain mechanisms and the potential of modulating central mechanisms to influence peripheral pain.
Dong et al94 further demonstrated the importance of NMDARs in pain modulation. They observed a significant upregulation of the α2δ-1 protein encoded by Cacna2d1 in DRG neurons and the spinal dorsal horn after sensory nerve injury. This upregulation enhanced the activity of pre- and post-synaptic NMDARs in spinal dorsal horn neurons, leading to sensitization of pain perception. Xie et al95 conducted a study showing that pre-synaptic NMDARs (PreNMDARs) located at the terminals of spinal pain receptors played a crucial role in activity-dependent pain sensitization. In normal organisms, PreNMDARs inhibit presynaptic transmission, but in injured organisms, they induce presynaptic potentiation.
Interaction of Various Mechanisms
The activation of microglia and their involvement in the development and progression of neuropathic pain can be facilitated by P2X7R. In a study conducted by Wu et al,96 an agonist of P2X7R called 2’(3’)-o-(4-benzoyl)benzoyl ATP (BzATP) was utilized along with electroacupuncture stimulation as a treatment for a rat model of spinal nerve ligation (SNL). The findings demonstrated that electroacupuncture treatment led to a decrease in dendritic spine density, inhibited synaptic remodeling, and reduced the inflammatory response. These effects were consistent with the decrease in P2X7R expression and the improvement of neurobehavioral performance. Conversely, BzATP enhanced abnormal remodeling and inflammation of dendritic spines and synapses. Hence, it was concluded that electroacupuncture may alleviate neuropathic pain by reducing abnormal remodeling of dendritic spines and synapses, as well as improving inflammation through the inhibition of P2X7R expression. Spinal cord injuries result in permanent loss of motor and sensory functions due to complex mechanisms involving the external microenvironment and internal neuro-biochemistry, which limit neuronal plasticity and axonal regeneration. In a study by Li et al,97 it was discovered that the CXCL12 peptide promoted elongation of axons, branch formation, dendrite formation, and synapse formation, leading to alleviation of symptoms associated with spinal cord injuries and enhancement of neurological function recovery. This suggests that chemokines not only influence the inflammatory response but also directly impact synapses, consequently affecting neuropathic pain. Microglia activation encompasses processes such as proliferation, migration, cytokine release, and production of reactive oxygen species which rely on changes in ion homeostasis involving calcium (Ca2+), sodium (Na+), potassium (K+), chloride (Cl−), and hydrogen (H+). These changes are facilitated by ion channels and transporters within microglia.98 High voltage-activated calcium channels that are crucial for synaptic transmission in sensory neurons can be targeted for analgesic purposes. In mice with nerve damage, there is a significant increase in synchronized firing of neurons in the dorsal root ganglion, leading to persistent mechanical, heat, and cold sensitization.
The study conducted by Sun et al99 involved the application of a powerful genetically encoded blocker called CAV-AβLator to inhibit the increase of Ca2+ signaling in DRG neurons following SNI and HVACC currents. This intervention led to the alleviation of SNI-induced mechanical heterotopic pain and sensitization to hot and cold stimuli. In a separate study by Boinon et al,100 it was discovered that collapsin response mediator protein 2 (CRMP2) serves as a dual regulator for both the n-type voltage-gated calcium (Cav2.2) channel and the Nav1.7 voltage-gated sodium channel. By selectively reducing the expression of CRMP2 in neurons, mechanical hyperalgesia in male and female mice caused by selective nerve injury was reversed. This pain mechanism appeared to be related to a decrease in the frequency and amplitude of spontaneous excitatory postsynaptic currents, suggesting that changes in neuronal ion levels might influence synaptic currents and contribute to CRMP2-mediated pain signal transduction.
Complications
Several studies have suggested that a significant percentage of patients (20–30%) with chronic pain experience negative emotions, such as anxiety, depression, aversion, and avoidance.101–103 These negative emotions have been found to potentially increase the perception of pain or reduce the ability to tolerate pain.104 The nucleus accumbens, specifically the medium spiny neurons (MSNs) within it, play a role in reward effects, regulation of motivation, and affective disorders. In a mouse pain model involving spinal nerve ligation (SNL), it was observed that the Ccl2-mediated CCL2/CCR2 signaling pathway in the nucleus accumbens was involved in integrating excitatory and inhibitory synaptic transmission in neuropathic pain. Furthermore, this pathway improved the state of long-term depression (LTD) induced by synapses in MSNs.105 Pain is often associated with poor sleep quality. An inverse relationship exists between the two: pain impairs sleep, which in turn lowers the body’s ability to tolerate pain.106 According to a study by Li et al,107 increased inflammatory response is closely linked to chronic pain symptoms and sleep quality. By regulating the hypothalamus-pituitary-adrenal axis, this inflammatory response can be reduced, thereby alleviating pain and improving sleep quality. Cav3 channels, previously known as T-type low voltage-activated calcium channels, play a role in modulating neuronal excitability, sensory processing, sleep, and the release of hormones and neurotransmitters. Therefore, it can be inferred that regulating ion balance through Cav3 channels may improve sleep and pain symptoms.108 Blum et al109 also demonstrated that modulation of Ca2+ channels contributes to sleep regulation. Sleep deprivation was found to affect the phosphorylation cycle of synaptic neurosomes, which influences synaptic transmission, cytoskeletal reorganization, and the balance between excitation and inhibition.110 Another study by Huang et al111 highlighted the significance of synaptic plasticity in sleep regulation. Thus, it is clear that by modulating inflammatory mechanisms, maintaining ionic balance, and ensuring synaptic function, the complications associated with pain can be improved.
Therapeutic Mechanisms of Analgesic Drugs (Table 1)
Drug resistance or side effects often arise from prolonged use of NSAIDs and opioids, which are commonly prescribed in medical practice to manage pain. Morphine, a widely used painkiller, can develop tolerance with long-term use. With advancements in Traditional Chinese Medicine, certain extracts from Chinese patent medicine have emerged, demonstrating promising results in clinical pain relief. This diversifies the options for analgesic drugs and helps enhance the effectiveness of existing medications or alleviate their resistance to some extent. In a particular study, celastrol was found to reduce mechanical and heat-induced pain in rats with chronic constriction injury (CCI). The mechanism behind this effect was the inhibition of microglia and astrocyte activation in the rats’ spinal cord, subsequently reducing the expression of the toll-like receptor-4/nuclear factor kappa B (TLR4/NF-κB) signaling pathway and levels of pro-inflammatory cytokines such as tumor necrosis factor α (TNF-α), IL-1β, and IL-6 in the spinal cord.112 Corydalis yanhusuo has anti-inflammatory and analgesic properties due to its active ingredient Tetrahydropalmatine (THP). Cheng et al,113 treated rats with DNP using an extract of THP and observed that it promoted a shift from a pro-inflammatory M1 phenotype to an anti-inflammatory M2 phenotype in lipopolysaccharide-induced BV2 microglia. This extract also inhibited the activation of the p38 MAPK signaling pathway, reduced expression levels of inflammatory factors such as NO, IL-1β, IL-6, and TNF-α, increased expression level of anti-inflammatory factor IL-10, and alleviated inflammatory response around the lesions, ultimately improving pain symptoms in the rats. Furthermore, novel medications have been developed to target pain mechanisms. One example is NeuroHeal, a recently developed drug that combines acamprosate and ribavirin. NeuroHeal promotes the regeneration of motor and sensory axons following peripheral nerve injury (PNI), alleviates the distressing sensation of pain caused by diseases, and modulates the P2X4-BDNF-KCC2 axis, a crucial factor in neuropathic pain. Ultimately, NeuroHeal proves effective in alleviating neuropathic pain resulting from PNI.114,115 In summary, recent focus in the field of analgesic drugs has been on exploring Chinese herb extracts and developing new medications that target pain. Additionally, ongoing research aims to improve the utilization of existing drugs, enhance their effectiveness in combination, and mitigate potential toxic side effects they may cause.
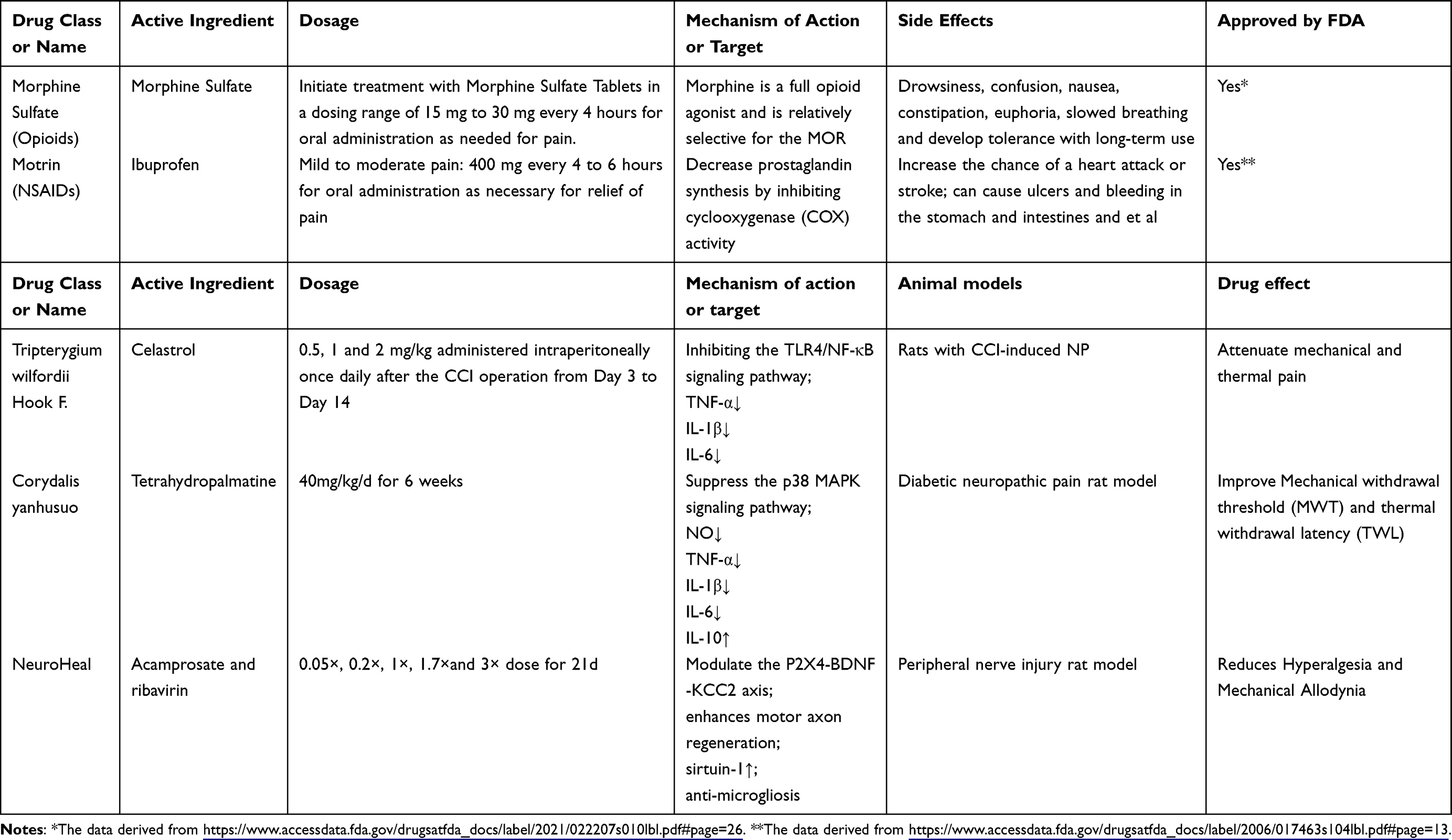 |
Table 1 Pharmacological Profile of Analgesic Medications |
Conclusions
Altering the inflammatory state of the body remains a meaningful approach to pain relief, and various mechanisms of pain can establish a connection between the peripheral and central nervous systems. The abnormal activation of pro-inflammatory substances contributes to the formation, progression, maintenance, and intensification of neuropathic pain. Disturbances in ions and changes in synaptic function are also implicated in pain and its complications (Figure 3). While new drugs with primarily anti-inflammatory mechanisms are emerging in clinical practice, recent findings suggest that altered synaptic function and ionic balance are also associated with pain. Therefore, considering other mechanisms in the development of new drugs could be a promising direction for future drug research and development. However, studies have suggested that nonsteroidal anti-inflammatory drugs may not provide significant relief for neuropathic pain.116 Nevertheless, extensive research on the role of inflammatory mechanisms in pain cannot be ignored, emphasizing the need to examine the effectiveness of multiple inflammatory pathways in neuropathic pain. Identifying specific factors and elucidating the precise mechanisms underlying disease development are crucial for targeted therapies and diagnosing underlying mechanisms. Ion channels have shown potential in improving pain symptoms, and it is also vital to consider the normal activity of cardiac cells and higher central nervous system neurons in the cerebral cortex, as they are sensitive to changes in membrane ion dynamics. These aspects should not be overlooked in future research endeavors.
 |
Figure 3 The mechanisms for pain. |
Abbreviations
IASP, The International Association for the Study of Pain; NP, neuropathic pain; CX3CL1, chemokine C-X3-C motif ligand 1; CSF1R, colony stimulating factor 1 receptor; IL-34, interleukin-34; TGF-β1, transforming growth factor β1; TLR4, Toll- like receptor 4; TREM2, triggering receptor expressed on myeloid cells 2; IRF8, interferon regulator factor 8; BDNF, brain-derived neurotrophic factor; SNI, sciatic nerve injury; CCI, chronic constrictive injury; DRG, dorsal root ganglion; VGSCs, voltage gated sodium channels; NAc, nucleus accumbens; Nmb, neuromedin B; TET1, ten-eleven translocation methylcytosine dioxygenase 1; Kv, voltage-gated potassium channel; MSNs, medium spiny neurons; CRMP2, collapsin response mediator protein 2; LTP, long-term potentiation; LTD, long-term depression; RTX, resiniferatoxin; NALCN, sodium leak channel; PNI, peripheral nerves injuries; LIFU, Low-intensity focused ultrasound; CaMKIV, calmodulin-dependent protein kinase IV.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Raja SN, Carr DB, Cohen M, et al. The revised international association for the study of pain definition of pain: concepts, challenges, and compromises. Pain. 2020;161(9):1976–1982. doi:10.1097/j.pain.0000000000001939
2. Kuner R, Flor H. Structural plasticity and reorganisation in chronic pain. Nat Rev Neurosci. 2016;18(1):20–30. doi:10.1038/nrn.2016.162
3. Peirs C, Seal RP. Neural circuits for pain: recent advances and current views. Science. 2016;354(6312):578–584. doi:10.1126/science.aaf8933
4. Cohen SP, Vase L, Hooten WM. Chronic pain: an update on burden, best practices, and new advances. Lancet. 2021;397(10289):2082–2097. doi:10.1016/S0140-6736(21)00393-7
5. Scholz J, Finnerup NB, Attal N, et al. Classification committee of the neuropathic pain special interest G. The IASP classification of chronic pain for ICD-11: chronic neuropathic pain. Pain. 2019;160:53–59. doi:10.1097/j.pain.0000000000001365
6. Finnerup NB, Kuner R, Jensen TS. Neuropathic pain: from mechanisms to treatment. Physiol Rev. 2021;101(1):259–301. doi:10.1152/physrev.00045.2019
7. Xie RG, Gao YJ, Park CK, et al. Spinal CCL2 promotes central sensitization, long-term potentiation, and inflammatory pain via CCR2: further insights into molecular, synaptic, and cellular mechanisms. Neurosci Bull. 2018;34(1):13–21. doi:10.1007/s12264-017-0106-5
8. Atta AA, Ibrahim WW, Mohamed AF, Abdelkader NF. Microglia polarization in nociplastic pain: mechanisms and perspectives. Inflammopharmacology. 2023;31(3):1053–1067. doi:10.1007/s10787-023-01216-x
9. Sun JL, Dai WJ, Shen XY, Lü N, Zhang YQ. Interleukin-17 is involved in neuropathic pain and spinal synapse plasticity on mice. J Neuroimmunol. 2023;377:578068. doi:10.1016/j.jneuroim.2023.578068
10. Kwiatkowski K, Mika J. The importance of chemokines in neuropathic pain development and opioid analgesic potency. Pharmacol Rep. 2018;70(4):821–830. doi:10.1016/j.pharep.2018.01.006
11. Iyengar S, Ossipov MH, Johnson KW. The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain. 2017;158:543–559. doi:10.1097/j.pain.0000000000000831
12. Zhao Z, Zhang Z, Li J, et al. Sustained TNF-α stimulation leads to transcriptional memory that greatly enhances signal sensitivity and robustness. Elife. 2020;9:e61965. doi:10.7554/eLife.61965
13. Greco R, Demartini C, Francavilla M, Zanaboni AM, Tassorelli C. Antagonism of CGRP receptor: central and peripheral mechanisms and mediators in an animal model of chronic migraine. Cells. 2022;11(19):3092. doi:10.3390/cells11193092
14. Yang G, Tan Q, Li Z, et al. The AMPK pathway triggers autophagy during CSF1-induced microglial activation and may be implicated in inducing neuropathic pain. J Neuroimmunol. 2020;345:577261. doi:10.1016/j.jneuroim.2020.577261
15. Kuhn JA, Vainchtein ID, Braz J, et al. Regulatory T-cells inhibit microglia-induced pain hypersensitivity in female mice. Elife. 2021;10:e69056. doi:10.7554/eLife.69056
16. Cao YQ, Mantyh PW, Carlson EJ, Gillespie AM, Epstein CJ, Basbaum AI. Primary afferent tachykinins are required to experience moderate to intense pain. Nature. 1998;392(6674):390–394. doi:10.1038/32897
17. Zieglgänsberger W. Substance P and pain chronicity. Cell Tissue Res. 2019;375(1):227–241. doi:10.1007/s00441-018-2922-y
18. Ebrahimi S, Erfani B, Alalikhan A, et al. The in vitro pro-inflammatory functions of the SP/NK1R System in prostate cancer: a focus on nuclear factor-kappa B (NF-κB) and its pro-inflammatory target genes. Biochem Biotechnol. 2023:1. doi:10.1007/s12010-023-04495-w
19. Franchini L, Carrano N, Di Luca M, Gardoni F. Synaptic GluN2A-Containing NMDA receptors: from physiology to pathological synaptic plasticity. Int J Mol Sci. 2020;21(4):1538. doi:10.3390/ijms21041538
20. Zhang YY, Liu F, Fang ZH, et al. Differential roles of NMDAR subunits 2A and 2B in mediating peripheral and central sensitization contributing to orofacial neuropathic pain. Brain Behav Immun. 2022;106:129–146. doi:10.1016/j.bbi.2022.08.010
21. Zhang YY, Liu F, Lin J, et al. Activation of the N-methyl-D-aspartate receptor contributes to orofacial neuropathic and inflammatory allodynia by facilitating calcium-calmodulin-dependent protein kinase II phosphorylation in mice. Brain Res Bull. 2022;185:174–192. doi:10.1016/j.brainresbull.2022.05.003
22. Zhang H, Ma S-B, Gao Y-J, et al. Spinal CCL2 promotes pain sensitization by rapid enhancement of NMDA-induced currents through the ERK-GluN2B pathway in mouse lamina II neurons. Neurosci Bull. 2020;36(11):1344–1354 doi:10.1007/s12264-020-00557-9.
23. Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi:10.1016/j.cell.2009.09.028
24. Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. 2010;16:1248–1257. doi:10.1038/nm.2235
25. Liu JA, Yu J, Cheung CW. Immune actions on the peripheral nervous system in pain. Int J Mol Sci. 2021;22(3). doi:10.3390/ijms22031448
26. Zhou L, Ao L, Yan Y, et al. The therapeutic potential of chemokines in the treatment of chemotherapy- induced peripheral neuropathy. Curr Drug Targets. 2020;21(3):288–301. doi:10.2174/1389450120666190906153652
27. Subbarayan MS, Joly-Amado A, Bickford PC, Nash KR. CX3CL1/CX3CR1 signaling targets for the treatment of neurodegenerative diseases. Pharmacol Ther. 2022;231:107989. doi:10.1016/j.pharmthera.2021.107989
28. Zhang ZJ, Jiang BC, Gao YJ. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cell Mol Life Sci. 2017;74(18):3275–3291. doi:10.1007/s00018-017-2513-1
29. Yang F, Jing JJ, Fu SY, et al. Spinal MCP-1 contributes to central post-stroke pain by inducing central sensitization in rats. Mol Neurobiol. 2023;60(4):2086–2098. doi:10.1007/s12035-022-03184-9
30. Gao YJ, Zhang L, Samad OA, et al. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29(13):4096–4108. doi:10.1523/JNEUROSCI.3623-08.2009
31. Yan Y, Liang Y, Ding T, Chu H. PI3K/Akt signaling pathway may be involved in MCP-1-induced P2X4R expression in cultured microglia and cancer-induced bone pain rats. Neurosci Lett. 2019;701:100–105. doi:10.1016/j.neulet.2019.02.024
32. Zhu X, Xie W, Zhang J, Strong JA, Zhang JM. Sympathectomy decreases pain behaviors and nerve regeneration by downregulating monocyte chemokine CCL2 in dorsal root ganglia in the rat tibial nerve crush model. Pain. 2022;163(1):e106–e120. doi:10.1097/j.pain.0000000000002321
33. Jiang S, Liang J, Li W, et al. The role of CXCL1/CXCR2 axis in neurological diseases. Int Immunopharmacol. 2023;120:110330. doi:10.1016/j.intimp.2023.110330
34. Zhang ZJ, Cao DL, Zhang X, Ji RR, Gao YJ. Chemokine contribution to neuropathic pain: respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain. 2013;154(10):2185–2197. doi:10.1016/j.pain.2013.07.002
35. Cao DL, Zhang ZJ, Xie RG, Jiang BC, Ji RR, Gao YJ. Chemokine CXCL1 enhances inflammatory pain and increases NMDA receptor activity and COX-2 expression in spinal cord neurons via activation of CXCR2. Exp Neurol. 2014;261:328–336. doi:10.1016/j.expneurol.2014.05.014
36. Knerlich-Lukoschus F, von der Ropp-Brenner B, Lucius R, Mehdorn HM, HeldFeindt J. Spatiotemporal CCR1, CCL3(MIP-1α), CXCR4, CXCL12(SDF-1α) expression patterns in a rat spinal cord injury model of posttraumatic neuropathic pain. J Neurosurg Spine. 2011;14:583–597. doi:10.3171/2010.12.SPINE10480
37. Song ZH, Song XJ, Yang CL, et al. Up-regulation of microglial chemokine CXCL12 in anterior cingulate cortex mediates neuropathic pain in diabetic mice. Acta Pharmacol Sin. 2023;44(7):1337–1349. doi:10.1038/s41401-022-01046-7
38. Liu ZY, Song ZW, Guo SW, et al. CXCL12/CXCR4 signaling contributes to neuropathic pain via central sensitization mechanisms in a rat spinal nerve ligation model. CNS Neurosci Ther. 2019;25(9):922–936. doi:10.1111/cns.13128
39. Li M, Ransohoff RM. Multiple roles of chemokine CXCL12 in the central nervous system: a migration from immunology to neurobiology. Prog Neurobiol. 2008;84:116–131. doi:10.1016/j.pneurobio.2007.11.003
40. Xing F, Kong C, Bai L, et al. CXCL12/CXCR4 signaling mediated ERK1/2 activation in spinal cord contributes to the pathogenesis of postsurgical pain in rats. Mol Pain. 2017;13:1744806917718753. doi:10.1177/1744806917718753
41. Mines MA, Goodwin JS, Limbird LE, Cui FF, Fan GH. Deubiquitination of CXCR4 by USP14 is critical for both CXCL12-induced CXCR4 degradation and chemotaxis but not ERK ativation. J Biol Chem. 2009;284:5742–5752. doi:10.1074/jbc.M808507200
42. Kiguchi N, Kobayashi Y, Kishioka S. Chemokines and cytokines in neuroinflammation leading to neuropathic pain. Curr Opin Pharmacol. 2012;12(1):55–61. doi:10.1016/j.coph.2011.10.007
43. Rojewska E, Zychowska M, Piotrowska A, Kreiner G, Nalepa I, Mika J. Involvement of macrophage inflammatory protein-1 family members in the development of diabetic neuropathy and their contribution to effectiveness of morphine. Front Immunol. 2018;9:494. doi:10.3389/fimmu.2018.00494
44. Kataoka A, Tozaki-Saitoh H, Koga Y, Tsuda M, Inoue K. Activation of P2X7 receptors induces CCL3 production in microglial cells through transcription factor NFAT. J Neurochem. 2009;108:115–125. doi:10.1111/j.1471-4159.2008.05744.x
45. Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of monocyte chemoattractant protein-1 and other beta-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J Neuroimmunol. 1998;84:238–249. doi:10.1016/S0165-5728(97)00208-7
46. Ansel KM, Ngo VN, Hyman PL, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406(6793):309–314. doi:10.1038/35018581
47. Wang J, Yin C, Pan Y, et al. CXCL13 contributes to chronic pain of a mouse model of CRPS-I via CXCR5-mediated NF-κB activation and pro-inflammatory cytokine production in spinal cord dorsal horn. J Neuroinflammation. 2023;20(1):109. doi:10.1186/s12974-023-02778-x
48. Wu XB, Cao DL, Zhang X, et al. CXCL13/CXCR5 enhances sodium channel Nav1.8 current density via p38 MAP kinase in primary sensory neurons following inflammatory pain. Sci Rep. 2016;6:34836. doi:10.1038/srep34836
49. Tsymbalyuk O, Gerzanich V, Mumtaz A, et al. SUR1, newly expressed in astrocytes, mediates neuropathic pain in a mouse model of peripheral nerve injury. Mol Pain. 2021;17:17448069211006603. doi:10.1177/17448069211006603
50. Jin SX, Zhuang ZY, Woolf CJ, et al. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23(10):4017–4022. doi:10.1523/JNEUROSCI.23-10-04017.2003
51. Tsuda M, Shigemoto-Mogami Y, Koizumi S, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424(6950):778–783. doi:10.1038/nature01786
52. Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi:10.1126/science.1194637
53. Wang Y, Szretter KJ, Vermi W, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. 2012;13(8):753–760. doi:10.1038/ni.2360
54. Butovsky O, Jedrychowski MP, Moore CS, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17(1):131–143. doi:10.1038/nn.3599
55. Chen X, Yao J, Lai J, et al. ADAM17 aggravates the inflammatory response by modulating microglia polarization through the TGF-β1/smad pathway following experimental traumatic brain injury. J Neurotrauma. 2023;40(13–14):1495–1509. doi:10.1089/neu.2022.0373
56. Xie Y, Chen X, Li Y, et al. Transforming growth factor-β1 protects against LPC-induced cognitive deficit by attenuating pyroptosis of microglia via NF-κB/ERK1/2 pathways. J Neuroinflammation. 2022;19(1):194. doi:10.1186/s12974-022-02557-0
57. Priyanto B, Islam AA, Hatta M, Bukhari A, Rosyidi RM. Effect of MLC901 on MIR30C-5P expression, TGF-Β expression, VEGF receptor expression, degree of axon demyelination and changes in neuropathic pain behaviour in experimental animals experiencing neuropathic pain with circumferential spinal stenosis method. Ann Med Surg. 2022;81:104489. doi:10.1016/j.amsu.2022.104489
58. Rodríguez-Gómez JA, Kavanagh E, Engskog-Vlachos P, et al. Microglia: agents of the CNS pro-inflammatory response. Cells. 2020;9(7):1717. doi:10.3390/cells9071717
59. Masuda T, Tsuda M, Yoshinaga R, et al. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep. 2012;1(4):334–340. doi:10.1016/j.celrep.2012.02.014
60. Matsushita T, Otani K, Oto Y, et al. Sustained microglial activation in the area postrema of collagen-induced arthritis mice. Arthritis Res Ther. 2021;23(1):273. doi:10.1186/s13075-021-02657-x
61. Li HN, Yang QQ, Wang WT, et al. Red nucleus IL-33 facilitates the early development of mononeuropathic pain in male rats by inducing TNF-α through activating ERK, p38 MAPK, and JAK2/STAT3. J Neuroinflammation. 2021;18(1):150. doi:10.1186/s12974-021-02198-9
62. Liu Y, Zhou LJ, Wang J, et al. TNF-α differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J Neurosci. 2017;37(4):871–881. doi:10.1523/JNEUROSCI.2235-16.2016
63. Miyamoto K, Kume K, Ohsawa M. Role of microglia in mechanical allodynia in the anterior cingulate cortex. J Pharmacol Sci. 2017;134(3):158–165. doi:10.1016/j.jphs.2017.05.010
64. Ni HD, Yao M, Huang B, et al. Glial activation in the periaqueductal gray promotes descending facilitation of neuropathic pain through the p38 MAPK signaling pathway. J Neurosci Res. 2016;94(1):50–61. doi:10.1002/jnr.23672
65. Taylor AM, Mehrabani S, Liu S, et al. Topography of microglial activation in sensory- and affect-related brain regions in chronic pain. J Neurosci Res. 2017;95(6):1330–1335. doi:10.1002/jnr.23883
66. Goins AE, Gomez K, Ran D, et al. Neuronal allodynic mechanisms of Slc7a5 (LAT1) in the spared nerve injury rodent model of neuropathic pain. Pflugers Arch. 2022;474(4):397–403. doi:10.1007/s00424-021-02653-9
67. Tilley DM, Cedeño DL, Vetri F, et al. Differential target multiplexed spinal cord stimulation programming modulates proteins involved in ion regulation in an animal model of neuropathic pain. Mol Pain. 2022;18:17448069211060181. doi:10.1177/17448069211060181
68. Zhang J, Rong L, Shao J, et al. Epigenetic restoration of voltage-gated potassium channel Kv1.2 alleviates nerve injury-induced neuropathic pain. J Neurochem. 2021;156(3):367–378. doi:10.1111/jnc.15117
69. Zhang Z, Zheng B, Du S, et al. Eukaryotic initiation factor 4 gamma 2 contributes to neuropathic pain through down-regulation of Kv1.2 and the mu opioid receptor in mouse primary sensory neurones. Br J Anaesth. 2021;126(3):706–719. doi:10.1016/j.bja.2020.10.032
70. Kim SS, Park J, Kim E, Hwang EM, Park JY. β-COP suppresses the surface expression of the TREK2. Cells. 2023;12(11):1500. doi:10.3390/cells12111500
71. Mao Q, Wu S, Gu X, et al. DNMT3a-triggered downregulation of K 2p 1.1 gene in primary sensory neurons contributes to paclitaxel-induced neuropathic pain. Int J Cancer. 2019;145:2122–2134. doi:10.1002/ijc.32155
72. Jia S, Wei G, Bono J, et al. TET1 overexpression attenuates paclitaxel-induced neuropathic pain through rescuing K(2p)1.1 expression in primary sensory neurons of male rats. Life Sci. 2022;297:120486. doi:10.1016/j.lfs.2022.120486
73. Zhang D, Zhao W, Liu J, et al. Sodium leak channel contributes to neuronal sensitization in neuropathic pain. Prog Neurobiol. 2021;202:102041. doi:10.1016/j.pneurobio.2021.102041
74. Bennett DL, Clark AJ, Huang J, et al. The role of voltage-gated sodium channels in pain signaling. Physiol Rev. 2019;99(2):1079–1151. doi:10.1152/physrev.00052.2017
75. Lu W, Cheng X, Chen J, et al. A buthus martensii Karsch scorpion sting targets Nav1.7 in mice and mimics a phenotype of human chronic pain. Pain. 2022;163(2):e202–e214. doi:10.1097/j.pain.0000000000002397
76. Zhang Y, Wang L, Peng D, et al. Engineering of highly potent and selective HNTX-III mutant against hNa(v)1.7 sodium channel for treatment of pain. J Biol Chem. 2021;296:100326. doi:10.1016/j.jbc.2021.100326
77. Zeisel A, Hochgerner H, Lönnerberg P, et al. Molecular architecture of the mouse nervous system. Cell. 2018;174(4):999–1014.e1022. doi:10.1016/j.cell.2018.06.021
78. Hidaka K, Maruta T, Koshida T, et al. Extracellular signal-regulated kinase phosphorylation enhancement and Na(V)1.7 sodium channel upregulation in rat dorsal root ganglia neurons contribute to resiniferatoxin-induced neuropathic pain: the efficacy and mechanism of pulsed radiofrequency therapy. Mol Pain. 2022;18:17448069221089784. doi:10.1177/17448069221089784
79. Li J, Stratton HJ, Lorca SA, et al. Small molecule targeting NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces pain in chronic constriction injury (CCI) rats. Channels. 2022;16(1):1–8. doi:10.1080/19336950.2021.2023383
80. Schmidtko A, Lötsch J, Freynhagen R, et al. Ziconotide for treatment of severe chronic pain. Lancet. 2010;375(9725):1569–1577. doi:10.1016/S0140-6736(10)60354-6
81. George DS, Hackelberg S, Jayaraj ND, et al. Mitochondrial calcium uniporter deletion prevents painful diabetic neuropathy by restoring mitochondrial morphology and dynamics. Pain. 2022;163(3):560–578. doi:10.1097/j.pain.0000000000002391
82. Rios L, Pokhrel S, Li SJ, Heo G, Haileselassie B, Mochly-Rosen D. Targeting an allosteric site in dynamin-related protein 1 to inhibit Fis1-mediated mitochondrial dysfunction. Nat Commun. 2023;14(1):4356. doi:10.1038/s41467-023-40043-0
83. Liu X, Rothe K, Yen R, et al. A novel AHI-1-BCR-ABL-DNM2 complex regulates leukemic properties of primitive CML cells through enhanced cellular endocytosis and ROS-mediated autophagy. Leukemia. 2017;31(11):2376–2387. doi:10.1038/leu.2017.108
84. Gu C, Yao J, Sun P. Dynamin 3 suppresses growth and induces apoptosis of hepatocellular carcinoma cells by activating inducible nitric oxide synthase production. Oncol Lett. 2017;13(6):4776–4784. doi:10.3892/ol.2017.6057
85. Cai Z, Quan L, Chang X, et al. High-voltage long-duration pulsed radiofrequency attenuates neuropathic pain in CCI rats by inhibiting Cav2.2 in spinal dorsal horn and dorsal root ganglion. Brain Res. 2022;1785:147892. doi:10.1016/j.brainres.2022.147892
86. Zhang Y, Qian Z, Jiang D, et al. Neuromedin B receptor stimulation of Cav3.2 T-type Ca(2+) channels in primary sensory neurons mediates peripheral pain hypersensitivity. Theranostics. 2021;11(19):9342–9357. doi:10.7150/thno.62255
87. Qi R, Cao J, Sun Y, et al. Histone methylation-mediated microRNA-32-5p down-regulation in sensory neurons regulates pain behaviors via targeting Cav3.2 channels. Proc Natl Acad Sci USA. 2022;119(14):e2117209119. doi:10.1073/pnas.2117209119
88. Wang Y, Hu X, Huang H, et al. Optimization of 4-arylthiophene-3-carboxylic acid derivatives as inhibitors of ANO1: lead optimization studies toward their analgesic efficacy for inflammatory pain. Eur J Med Chem. 2022;237:114413. doi:10.1016/j.ejmech.2022.114413
89. Yao L, Zhang TY, Diao XT, et al. Functional expression of glycine receptors in DRG neurons of mice. Eur J Pharmacol. 2021;899:174034. doi:10.1016/j.ejphar.2021.174034
90. Liao YH, Wang B, Chen MX, et al. LIFU alleviates neuropathic pain by improving the KCC(2) expression and inhibiting the CaMKIV-KCC(2) pathway in the L4-L5 section of the spinal cord. Neural Plast. 2021;2021:6659668. doi:10.1155/2021/6659668
91. Lorenzo LE, Godin AG, Ferrini F, et al. Enhancing neuronal chloride extrusion rescues α2/α3 GABA(A)-mediated analgesia in neuropathic pain. Nat Commun. 2020;11(1):869. doi:10.1038/s41467-019-14154-6
92. Li J, Price TJ, Baccei ML. D1/D5 dopamine receptors and mGluR5 jointly enable non-hebbian long-term potentiation at sensory synapses onto lamina I spinoparabrachial neurons. J Neurosci. 2022;42(3):350–361. doi:10.1523/JNEUROSCI.1793-21.2021
93. Liu Q, Ko CY, Zheng C, et al. Decreased glutamatergic synaptic strength in the periaqueductal gray contributes to maintenance of visceral pain in male rats with experimental pancreatitis. Neuroscience. 2020;428:60–69. doi:10.1016/j.neuroscience.2019.12.004
94. Dong L, Zhang Y, Chen J. 新型镇痛靶点α2δ-1-NMDAR稳定表达细胞株构建及镇痛药物筛选的应用 [A novel cell tool for α2δ-1-NMDAR target-based analgesic drug discovery]. Shengwu Gongcheng Xuebao. 2022;38(3):1149–1158. Chinese. doi:10.13345/j.cjb.210162
95. Xie RG, Chu WG, Liu DL, et al. Presynaptic NMDARs on spinal nociceptor terminals state-dependently modulate synaptic transmission and pain. Nat Commun. 2022;13(1):728. doi:10.1038/s41467-022-28429-y
96. Wu Q, Yue J, Lin L, et al. Electroacupuncture may alleviate neuropathic pain via suppressing P2X7R expression. Mol Pain. 2021;17:1744806921997654. doi:10.1177/1744806921997654
97. Li J, Wu Y, Chen P, et al. CXCL12 promotes spinal nerve regeneration and functional recovery after spinal cord injury. Neuroreport. 2021;32(6):450–457. doi:10.1097/WNR.0000000000001613
98. Luo L, Song S, Ezenwukwa CC, et al. Ion channels and transporters in microglial function in physiology and brain diseases. Neurochem Int. 2021;142:104925. doi:10.1016/j.neuint.2020.104925
99. Sun L, Tong CK, Morgenstern TJ, et al. Targeted ubiquitination of sensory neuron calcium channels reduces the development of neuropathic pain. Proc Natl Acad Sci USA. 2022;119(20):e2118129119. doi:10.1073/pnas.2118129119
100. Boinon L, Yu J, Madura CL, et al. Conditional knockout of CRMP2 in neurons, but not astrocytes, disrupts spinal nociceptive neurotransmission to control the initiation and maintenance of chronic neuropathic pain. Pain. 2022;163(2):e368–e381. doi:10.1097/j.pain.0000000000002344
101. Edwards RR, Dworkin RH, Sullivan MD, et al. The role of psychosocial processes in the development and maintenance of chronic pain. J Pain. 2016;17(9 Suppl):T70–92. doi:10.1016/j.jpain.2016.01.001
102. Ahn H, Weaver M, Lyon D, et al. Depression and pain in asian and white Americans with knee osteoarthritis. J Pain. 2017;18(10):1229–1236. doi:10.1016/j.jpain.2017.05.007
103. Gerrits M, van Oppen P, van Marwijk HWJ, et al. Pain and the onset of depressive and anxiety disorders. Pain. 2014;155(1):53–59. doi:10.1016/j.pain.2013.09.005
104. Becker S, Navratilova E, Nees F, et al. Shared mechanisms of chronic pain and emotional-motivational problems: from basic science to the clinics. Pain Res Manag. 2018;2018:9305026. doi:10.1155/2018/9305026
105. Wu XB, Zhu Q, Gao YJ. CCL2/CCR2 contributes to the altered excitatory-inhibitory synaptic balance in the nucleus accumbens shell following peripheral nerve injury-induced neuropathic pain. Neurosci Bull. 2021;37(7):921–933. doi:10.1007/s12264-021-00697-6
106. Haack M, Simpson N, Sethna N, et al. Sleep deficiency and chronic pain: potential underlying mechanisms and clinical implications. Neuropsychopharmacology. 2020;45(1):205–216. doi:10.1038/s41386-019-0439-z
107. Li MT, Robinson CL, Ruan QZ, et al. The influence of sleep disturbance on chronic pain. Curr Pain Headache Rep. 2022;26(10):795–804. doi:10.1007/s11916-022-01074-2
108. Lory P, Nicole S, Monteil A. Neuronal Cav3 channelopathies: recent progress and perspectives. Pflugers Arch. 2020;472(7):831–844. doi:10.1007/s00424-020-02429-7
109. Blum ID, Keleş MF, Baz ES, et al. Astroglial calcium signaling encodes sleep need in drosophila. Curr Biol. 2021;31(1):150–162.e157. doi:10.1016/j.cub.2020.10.012
110. Brüning F, Noya SB, Bange T, et al. Sleep-wake cycles drive daily dynamics of synaptic phosphorylation. Science. 2019;366(6462). doi:10.1126/science.aav3617
111. Huang S, Sigrist SJ. Presynaptic and postsynaptic long-term plasticity in sleep homeostasis. Curr Opin Neurobiol. 2021;69:1–10. doi:10.1016/j.conb.2020.11.010
112. Jin GJ, Peng X, Chen ZG, et al. Celastrol attenuates chronic constrictive injury-induced neuropathic pain and inhibits the TLR4/NF-κB signaling pathway in the spinal cord. J Nat Med. 2022;76(1):268–275. doi:10.1007/s11418-021-01564-4
113. Cheng LZ, Zhou JM, Ma JL, et al. 延胡索乙素通过抑制p38 MAPK信号通路介导的小胶质细胞活化改善糖尿病大鼠神经病理性疼痛[Tetrahydropalmatine alleviated diabetic neuropathic pain by inhibiting activation of microglia via p38 MAPK signaling pathway]. Zhongguo Zhong yao za zhi. 2022;47(9):2533–2540. Chinese. doi:10.19540/j.cnki.cjcmm.20220119.702
114. Romeo-Guitart D, Casas C. NeuroHeal treatment alleviates neuropathic pain and enhances sensory axon regeneration. Cells. 2020;9(4):808. doi:10.3390/cells9040808
115. Liu Q, Li R, Yang W, et al. Role of neuroglia in neuropathic pain and depression. Pharmacol Res. 2021;174:105957. doi:10.1016/j.phrs.2021.105957
116. Petroianu GA, Aloum L, Adem A. Neuropathic pain: mechanisms and therapeutic strategies. Front Cell Dev Biol. 2023;11:1072629. doi:10.3389/fcell.2023.1072629
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.