Back to Journals » Journal of Hepatocellular Carcinoma » Volume 11
An Engineered Influenza a Virus Expressing the Co-Stimulator OX40L as an Oncolytic Agent Against Hepatocellular Carcinoma
Authors Yang H ![]() , Lei G, Deng Z
, Lei G, Deng Z ![]() , Sun F, Tian Y, Cheng J
, Sun F, Tian Y, Cheng J ![]() , Yu H, Li C
, Yu H, Li C ![]() , Bai C, Zhang S, An G, Yang P
, Bai C, Zhang S, An G, Yang P
Received 3 March 2023
Accepted for publication 24 October 2023
Published 9 January 2024 Volume 2024:11 Pages 1—13
DOI https://doi.org/10.2147/JHC.S410703
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ahmed Kaseb
Hao Yang,1– 3,* Guanglin Lei,2,* Zhuoya Deng,1 Fang Sun,1 Yuying Tian,1 Jinxia Cheng,1 Hongyu Yu,1 Cong Li,1 Changqing Bai,4 Shaogeng Zhang,2 Guangwen An,5 Penghui Yang1
1Faculty of Hepato-Pancreato-Biliary Surgery, Institute of Hepatobiliary Surgery, the First Medical Center, Chinese PLA General Hospital, Beijing, People’s Republic of China; 2Department of Hepatological Surgery, the Fifth Medical Center of Chinese PLA General Hospital, Beijing, People’s Republic of China; 3Department of Surgery, Taian City Central Hospital, Taian, People’s Republic of China; 4Department of Respiratory, Shenzhen University General Hospital, Shenzhen University Clinical Medical Academy, Guangdong, People’s Republic of China; 5Department of Pharmacy, No. 984 Hospital of the PLA, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Penghui Yang; Guangwen An, Email [email protected]; [email protected]
Background: Oncolytic virus (OV) therapy has emerged as a promising novel form of immunotherapy. Moreover, an increasing number of studies have shown that the therapeutic efficacy of OV can be further improved by arming OVs with immune-stimulating molecules.
Methods: In this study, we used reverse genetics to produce a novel influenza A virus, termed IAV-OX40L, which contained the immune-stimulating molecule OX40L gene in the influenza virus nonstructural (NS1) protein gene. The oncolytic effect of IAV-OX40L was explored on hepatocellular carcinoma (HCC)HCC cells in vitro and in vivo.
Results: Hemagglutination titers of the IAV-OX40L virus were stably 27– 28 in specific-pathogen-free chicken embryos. The morphology and size distribution of IAV-OX40L are similar to those of the wild-type influenza. Expression of OX40L protein was confirmed by Western blot and immunofluorescence. MTS assays showed that the cytotoxicity of IAV-OX40L was higher in HCC cells (HepG2 and Huh7) than in normal liver cells (MIHA) in a time- and dose-dependent manner in vitro. We found that intratumoral injection of IAV-OX40L reduced tumor growth and increased the survival rate of mice compared with PR8-treated controls in vivo. In addition, the pathological results showed that IAV-OX40L selectively destroyed tumor tissues without harming liver and lung tissues. CD4+ and CD8+ T cells of the IAV-OX40L group were significantly increased in the splenic lymphocytes of mice. Further validation confirmed that IAV-OX40L enhanced the immune response mainly by activating Th1-dominant immune cells, releasing interferon-γ and interleukin-2.
Conclusion: Taken together, our findings demonstrate the novel chimeric influenza OV could provide a potential therapeutic strategy for combating HCC and improve the effectiveness of virotherapy for cancer therapy.
Keywords: OVs, OX40L, IAV, HCC
Introduction
Hepatocellular carcinoma (HCC) is one of the most prevalent malignant tumors worldwide, accounting for a large proportion of cancer mortality.1 Considerable progress has been made in the treatment of HCC, but the therapeutic efficacy is still not ideal.2,3 Previous studies have shown that HCC is mainly caused by immune escape and proliferation of tumor cells.4,5 Simultaneously, tumor cells create an immune microenvironment with the participation of various cell types, allowing the tumor cells to escape from immune supervision, survival, and proliferation.6 Cancer immunotherapy relies on the recognition of tumor-associated antigens, which can restore the patient’s immune response against tumor cells.7 In recent years, many researchers have been interested in methods of improve the effectiveness of immunotherapy for HCC.8,9
Oncolytic virus (OV) therapy is a revolutionary tool for cancer immunotherapy. Tumor cells have defective damage- or pathogen-recognition responses, which make them more susceptible to viral infection. OVs can replicate and destroy tumor cells without affecting normal cells.10,11 The mechanisms underlying the antitumor effects of OVs include targeted oncolytic effects and activation of the host’s innate and adaptive immune responses.12,13 OVs kill tumor cells directly and alter the tumor microenvironment to result in tumor cell destruction.14,15 With the development of genetic engineering techniques, a variety of viral vectors can be engineered to express immunoregulatory genes for cancer treatment, these viral vectors include influenza virus, herpes virus, adenovirus, and so on. Such methods can be used to develop safer and more powerful tumor-specific OVs that can be used in combination with chimeric immune checkpoint inhibitors (ICIs) and adoptive T cell therapy (ACT) to promote sustained antitumor immune responses.16,17 Several investigations are in progress to determine the optimal combinations of OVs and ICIs.18
OX40L (CD252, TNFSF4) is a type II transmembrane protein, which is a class of proteins involved in the costimulation and differentiation of T cells with a positive role in the immune response.19,20 OX40L is predominantly expressed by antigen-presenting cells (APCs), including activated dendritic cells (DCs), B cells, macrophages, Langerhans cells, T cells, and endothelial cells.21 Previous studies have shown that OX40L can enhance the antitumor activity of T cells by delivering effective costimulatory signals to CD4+ and CD8 T+ cells, which is critical for the activation and survival of T cells and the generation of memory T cells from activated effector T cells.22,23 Plasmacytoid DCs can regulate T helper responses by balancing OX40 and type I interferon (IFN) expression.24,25 Another study reported that OX40–OX40L interactions play a critical role in key immunomodulatory checkpoints that may be involved in the development of autoimmune disease.26 A cancer mRNA vaccine delivering the OX40L gene has been developed by the pharmaceutical and biotechnology company Moderna and is under investigation for colon adenocarcinoma, HCC and lung cancer.26,27 To this end, we speculated that OX40L-expressing OV would show promising results and hold potential for the treatment of cancer.
In recent years, several studies have focused on targeted therapy and immunotherapy of HCC.28,29 We previously reported the generation of recombinant influenza A viruses (IAV) encoding PD-1 or GM-CSF and showed that these recombinant viruses exhibit selective cytotoxicity in vitro without damaging normal tissues and inhibit tumor growth in vivo.30,31 Herein, we engineered a recombinant oncolytic influenza virus strain, IAV-OX40L, using reverse genetics (RG) technology. The novel virus contained the OX40L gene in the background of the nonstructural (NS) fragment of influenza virus A/PuertoRico/8/34 (PR8). The oncolytic effect of IAV-OX40L was explored on HCC cells in vitro and in vivo.
Materials and Methods
Cells, Plasmids, Viruses, Medium
African green monkey kidney cells (COS-I) and canine kidney cells (MDCK) were purchased from Shanghai Institutes of Biological Sciences, Chinese Academy of Sciences (Shanghai, China). Normal human liver cells (MIHA), and human hepatoma cells (HepG2, Huh7 and Hep3B) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The mouse hepatoma cell line H22 was provided by the Fifth Medical Center of PLA General Hospital (Beijing, China). The use of the cell lines was approved by the Ethics Committee of the Fifth Medical Center of Chinese PLA General Hospital. Influenza virus A/Puerto Rico/8/34 (PR8) was maintained in our laboratory.
Eight plasmid backbones of the influenza virus PR8, namely pHW191-PB2, pHW192-PB1, pHW193-PA, pHW194-HA, pHW195-NP, pHW196-NA, pHW197-M, and pHW198-NS were stored in our laboratory. Escherichia coli DH5α was purchased from TianGen (Beijing, China). The medium were bought from Beijing EallBio Biomedical Technology Co.,Ltd.
Experimental Animals
Specific pathogen-free (SPF) chicken embryos and female BALB/c mice aged 4–6 weeks were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All animal experiments were conducted in accordance with the guidelines of the Animal Care and Utilization Committee and the Ethics Committee of the Fifth Medical Center of PLA General Hospital.
Construction of Recombinant Plasmid pFlu-OX40L-NS
The OX40L gene sequences were downloaded from the National Center for Biotechnology Information (NCBI, Bethesda, MD, USA). After the OX40L gene sequence was optimized and bioinformatically analyzed, the open reading frame sequence of this gene was inserted into site 406 of the NS sequence in one of the PR8 influenza virus backbone plasmids. The recombinant sequence was synthesized by Sangon Bioengineering (Shanghai) Co. Ltd. (Shanghai, China), cloned into the pHW2000 vector, and validated by DNA sequencing.
Rescue of the Recombinant Virus IAV-OX40L
COS-I and MDCK cells were plated in 6-well plates in a 2:1 ratio (4 × 105 cells/well) and placed in an incubator under 5% CO2 at 37°C. When the cells reached 70% confluence, the medium was replaced with serum-free DMEM (EallBio, Beijing, China). The remaining seven plasmid fragments, pHW191-PB2, pHW192-PB1, pHW193-PA, pHW194-HA, pHW195-NP, pHW196-NA, and pHW197-M, from the recombinant plasmid pFlu-OX40L-NS and PR8, were diluted to 200 ng/μL in a volume of 1 μL each. The plasmids were transfected into the COS-I/MDCK co-cultured cells using an Effectene Transfection Kit (Qiagen, Germantown, MD, USA) according to the manufacturer’s instructions. The cells were grown in an incubator under 5% CO2 at 37°C for 72 h, and then stored at –80°C.
Hemagglutination Test
The transfected cells were freeze-thawed once and injected into 9-day-old SPF chicken embryos (200 μL of cells per embryo), with the eggs then placed in an incubator at 37°C after sealing with a wax block. After incubation for 72h, hemagglutination by 1% chicken red blood cells was determined with 100 µL allantoic fluid that was diluted with PBS.
Median Tissue Culture Infectious Dose (TCID50)
MDCK cells were plated in 96-well plates (2 × 104 cells/well) and placed in an incubator under 5% CO2 at 37°C. When the cells reached 70% confluence, they were inoculated with 200 µL of IAV-OX40L serially diluted from 10–1 to 10–12 in serum-free DMEM and placed in an incubator under 5% CO2 at 37°C. The hemagglutination titer of the allantoic fluid was determined with 50 μL/well of supernatant with 1% chicken red blood cells after 5 days.
Morphology of IAV-OX40L Virus Under Electron Microscopy
The recombinant virus was diluted to 1:5000 and injected into SPF chicken embryos. The allantoic fluid was collected after incubation for 72 h and centrifuged at 4000 rpm for 25 min. The resulting supernatant was centrifuged at 32,000 rpm for 4 h through a 30% and 60% sucrose concentration gradient to absorb virus bands. After dilution with PBS, the resulting solution was centrifuged at 32,000 rpm for 2 h to harvest the purified virus. The morphology and size distribution were examined by negative staining under transmission electron microscopy (TEM).
RT-PCR Identification
RNA was extracted from 500 µL of purified recombinant IAV-OX40L virus using Trizol (Invitrogen, Carlsbad, CA, USA). The RNA was reverse transcribed into cDNA and amplified by PCR with NS universal primers for the influenza A virus (Figure S1). The amplified products were subjected to agarose gel electrophoresis and sequenced. pHW198-NS was used as a control.
Western Blot (WB)
MDCK and HepG2 cells were seeded in 6-well plates. When the cell density reached 70%, the cells were inoculated with IAV-OX40L. After 24 h, cell precipitates were collected and proteins extraction was performed using RIPA buffer (Solarbio R0010, Beijing, China). Protein concentrations were then determined by the BCA Protein Assay kit according to the manufacturer’s instructions. Next, 10 µg of protein was resolved by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membranes. Nonspecific binding was blocked with 5% bovine serum albumin (BSA) blocking buffer (SolarbioSW3015). NP polyclonal antibody (1:3000 dilution, GeneTex, GTX636247) and OX40L monoclonal antibody (1:1500 dilution, CST, 14,991) were applied overnight at 4°C. The secondary antibody was HRR-conjugated goat anti-rabbit (1:1000 dilution, ZSGB-bio, ZB2306) followed by HRR-conjugated goat anti-mouse (1:10,000 diluent, Protentech, SA00001-1). An enhanced chemiluminescence (ECL) detection system was used for observation.
Immunofluorescence Assay (IFA)
Glass microscope slides were set inside a 12-well plate on which to culture MDCK cells (5 × 104 cells/well). When the cells reached 70% confluence, they were inoculated with recombinant influenza virus IAV-OX40L at a multiplicity of infection (MOI) of 1 in serum-free DMEM. PR8 was used as a control. After incubation for 24 h, the cells were fixed with 4% paraformaldehyde at room temperature for 30 min and then permeabilized using 0.5% Triton-100. The slides were washed three times with PBS and 1% normal goat serum (NGS) for 30 min, then 50 µL of anti-nucleoprotein (NP) antibody (GeneTex, Los Angeles, CA, USA) and recombinant anti-OX40L/TNFSF4 antibody (EPR23155–317, Abcam, Cambridge, UK) diluted 1:50 in PBS were added. Slides were placed at 4°C for 16 h, followed by the addition of 100 μL/well goat anti-mouse IgG (DyLight 594, GeneTex) and goat anti-rabbit IgG H&L (Alexa Fluor® 488, Abcam) diluted 1:200. After incubation in the dark for 1 h, the slides were washed three times with PBS and 1% NGS for 30 min. Then, 200 µL DAPI was added to stain the nuclei for 5 min, after which a fluorescence-quenching agent was added. The slides were covered with slide glasses and images were acquired by confocal laser scanning microscopy (Zeiss). HepG2 were seeded at 5×104 cells/well in the 12-well plates. Following the same method, IAV-OX40L was detected by IFA to determine whether the recombinant virus could produce OX40L protein in HepG2.
Growth Curve of IAV-OX40L Virus
MDCK cells were plated in 96-well plates (2 × 104 cells/well) and placed in an incubator under 5% CO2 at 37°C. When the cells reached 70% confluence, they were inoculated with 200 µL of IAV-OX40L serially diluted from 10−1 to 10−12 in serum-free DMEM. The hemagglutination titer of the culture supernatant was determined with 50 μL/well at 24, 48, 72, 96 h, respectively.
Cell Viability
MIHA, HepG2, and HuH7 cells were plated in 96-well plates (2 × 104 cells/well) and placed in an incubator under 5% CO2 at 37°C. When the cells reached 70% confluence, they were washed three times with PBS, and the medium was replaced with serum-free DMEM. The cells were inoculated with IAV-OX40L at an MOI of 0.1, 1, and 3, and incubated at 37°C in an atmosphere of 5% CO2. The MTS assay was performed using a commercial kit (Promega) according to the manufacturer’s instructions. Cytotoxicity was monitored and analyzed by measuring the optical density at 450 nm (OD450) at 24, 48, and 72 h.
Cell Death Analysis by Flow Cytometry
MIHA and HepG2 cells were plated in 6-well plates (4 × 104 cells/well) and placed in an incubator under 5% CO2 at 37°C. When the cells reached 70% confluence, they were washed three times with PBS, and the medium was replaced with serum-free DMEM. The cells were inoculated with IAV-OX40L at an MOI of 1 and 3 and incubated at 37°C in an atmosphere of 5% CO2 for 48 h. The cells were examined for apoptosis by using flow cytometry and an Annexin V-FITC Apoptosis Detection Kit (Biyantian Biotechnology, Shanghai, China) according to the manufacturer’s instructions. The data were analyzed with FlowJo 10 software.
Antitumor Efficacy of IAV-OX40L Virus in vivo
Murine hepatoma H22 cells were inoculated into the abdominal cavity of 4-6-week-old female BALB/C mice. Seven days later, the ascites that formed were centrifuged at 3000 rpm for 5 min. The precipitate was suspended in PBS and used to inoculate mice with 5 × 106 H22 cells. When the tumors reached 80–120 mm3, mice were divided randomly into three group and inoculated with PBS, PR8 influenza virus, or recombinant virus IAV-OX40L (100 μL every other day for seven times). The vital signs of mice were monitored and tumor volume was measured every day. After the last inoculation on day 7, the mice were killed and their tumor tissues were collected to measure their volume and weight.
Viral Loads
The heart, liver, spleen, lung, kidney, brain, and tumors of mice were separated, cut into pieces, ground, and centrifuged at 4000 rpm for 10 min. The resulting supernatants were harvested and the median tissue culture infectious dose (TCID50) was determined.
Pathological Changes in Mice
The tumor, liver, and lung tissues of the mice were embedded with paraffin and cut into sections that were 4 μm thick. The sections were stained with hematoxylin and eosin (H&E). Briefly, the sections were deparaffinized by incubation in xylene I and II for 10 min, rehydrated in 100%, 90%, 80%, and 70% alcohol for 5 min each, and covered with water. Next, the sections were incubated with hematoxylin for 6 min and rinsed under running water then they were destained with 1% hydrochloric acid, stained for 6 min with eosin, and rinsed with running water. Finally, the sections were dehydrated with 70%, 80%, 90%, and 100% alcohol for 10s each and then washed with xylene for 1 min. Fourteen days after the last inoculation, the remaining mice were killed to isolate the tumor tissues, heart, liver, spleen, lung, kidney, and brain tissues of the mice. The liver and lung tissues were obtained for H&E staining.
Lymphocyte Activation
Mouse spleens were dissected out, ground, and filtered, and the filtrate was slowly added into 6 mL of mouse lymphocyte separation solution (Beijing Dakewe Biotechnology Co. Ltd., Beijing, China) along the wall of the centrifuge tube to isolate the mouse lymphocytes. The antibody system was configured with 100 μL of FACE solution (saline containing 2% fetal bovine serum) and 1 μL each of Brilliant Violet 510™-conjugated anti-mouse CD3, FITC-conjugated anti-mouse CD4, PerCP/Cyanine5.5-conjugated anti-mouse CD8a, APC-conjugated anti-mouse CD45 and PE-conjugated anti-mouse CD69 antibodies (BioLegend, San Diego, CA, USA) to every sample tube. After mixing, the cells were incubated at 4°C for 30 min in the dark. The excess antibodies were washed off with FACE solution, and the supernatant was discarded after centrifugation at 8000 rpm for 1 min. The sediment was dissolved with 200 μL of 4% paraformaldehyde in the dark for 20 min and the cells were detected by flow cytometry.
Real-Time Quantitative Polymerase Chain Reaction (qPCR)
CD4 T cells were isolated from human peripheral blood mononuclear cells using Miltenyi Biotec magnetic beads. The blood was provided by the Fifth Medical Center of PLA General Hospital (Beijing, China) and approved by the Ethics Committee of the Fifth Medical Center of Chinese PLA General Hospital. IAV-OX40L was infected the Hep3B. After 24h, the supernatant was aspirated and discarded. CD4 T cells were co-cultured at 6×104/well with 2mL of 10% 1640 medium, 2.5 µg/mL, CD3, and 5 µg/mL CD28. After 6 h, cells were collected for RNA extraction as the instruction of the R1200RNA Isolation Kit (Solarbio, Beijing, China. cDNA was synthesized using a cDNA Synthesis Mix Kit (Tiangen, KR118 Beijing, China). Then using the SYBR Green qPCR premix (Tiangen, FP207, Beijing, China) for Quantitative real-time polymerase chain reaction. The primer sequences of the OX40L were: 5′-GCGTGATCATCAACTGCGAC-3′.The reverse primer sequences of the OX40L were: 5′-TGTTCACGCTCCTCACCTTC-3′.
Statistical Analysis
Statistical analyses were performed using a single-factor t-test using GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA). The animal experiment was repeated twice. In all analyses, P<0.05 was taken to indicate statistical significance.
Results
Design and Rescue of Chimeric IAV-OX40L Virus
To establish a chimeric IAV that expresses OX40L during the virus life cycle, we explored fragments of oncolytic influenza virus NS protein. First, the OX40L gene sequence was obtained from the NCBI. After sequence optimization, the open reading frame of OX40L with a length of 552 bp was cloned into the NS sequence in one of the PR8 backbone plasmids. A schematic diagram of the cloning strategy is shown in Figure 1A. The recombinant plasmid which was 1477 bp was cloned into the pHW2000 vector and synthesized by Sangong Bioengineering (Shanghai) Co., Ltd. The plasmids pFlu-OX40L-NS and pHW198-NS were identified by agarose gel electrophoresis. The fragment size of pFlu-OX40L-NS (Figure 1B) was as expected, indicating that the plasmid had been constructed successfully. The recombinant plasmid pFlu-OX40L-NS was mixed with the remaining seven plasmids of the PR8 virus pHW191-PB2, pHW192-PB1, pHW193-PA, pHW194-HA, pHW195-NP, pHW196-NA, and pHW197-M which were co-transfected into COS-I/MDCK cells and identified via agarose gel electrophoresis (Figure 1C).
 |
Figure 1 Design and construction of the recombinant IAV-OX40L virus. (A) Schematic diagram of the recombinant plasmid. (B) Agarose gel electrophoresis confirmed that the size of the recombinant plasmid was consistent with expectations. (C) Fragment sizes of the eight plasmids were confirmed by agarose gel electrophoresis. |
Characterization of the IAV-OX40L Virus
To further characterize of the recombinant influenza IAV-OX40L virus, Hemagglutination (HA) titers and stability of IAV-OX40L virus were evaluated from passage 1 to 5. The HA titers of the first generation were 25–26 as measured with 1% chicken blood and were stable at 27–28 after five generations (Figure 2A), and the virus titer was 7–8 logTCID50/mL (Figure 2B).
 |
Figure 2 Characterization of the IAV-OX40L virus. (A) The recombinant virus was repeatedly subcultured and the hemagglutination titer of P5 was stabilized at 27–8. (B) The virulence of IAV-OX40L increased gradually during continuous culture and ultimately reached 7–8 LogTCID50/mL. (C and D) The morphology and size distribution of the virus were examined with transmission electron microscopy. (E) The NS fragment was amplified by RT-PCR and identified via agarose gel electrophoresis. (F) Expression of the OX40L protein was confirmed in MDCK and HepG2 cells by Western blot. (G) Immunofluorescence staining of IAV-OX40L virus-infected MDCK and HepG2 cells for the influenza NP protein (red), OX40L protein (green), and DAPI (blue). |
The recombinant influenza virus IAV-OX40L was purified via ultracentrifugation through a sucrose concentration gradient. In negative staining TEM, the virus was spherical, with cilia on the surface of the capsule at the surface of the nuclear capsid (Figure 2C). The size of the virus particles ranged from 80 to 120 nm (Figure 2D), consistent with the morphological characteristics of wild-type influenza virus.
RNA from IAV-OX40L virus was extracted and universal primers were used to amplify NS gene fragments. The pHW198-NS plasmid was used as positive control. The NS fragment size of the recombinant plasmid was 4437 bp (Figure 2E). After identification and DNA sequencing, the sequence of the PCR product was consistent with pFlu-OX40L-NS.
The presence of OX40L protein was examined by WB after infection of MDCK and HepG2 cells with PBS, PR8, and IAV-OX40L, respectively. The results showed that IAV-OX40L could express OX40L protein in MDCK and HepG2 cells, but PR8 could not express OX40L protein in any cells. This confirmed that IAV-OX40L could express the OX40L protein (Figure 2F).
The production of OX40L by IAV-OX40L in MDCK and HepG2 cells was subsequently validated by indirect fluorescent antibody test (IFA). As expected, IFA showed that NP and OX40L were strongly positive in MDCK after IAV-OX40L infection, whereas the cells inoculated with PR8 was only positive for NP. Consistent results were observed in HepG2 cells (Figure 2G). These findings proved that IAV-OX40L and PR8 were NP-centered influenza viruses, and IAV-OX40L could express OX40L in cells, but PR8 could not.
Cytotoxic Effect of IAV-OX40L Virus in vitro
The cytotoxic effect of IAV-OX40L virus in vitro was detected by cell viability and apoptosis assays. MDCK cells were inoculated with IAV-OX40L diluted in serum-free DMEM and the HA titers were measured at 24, 48, 72, and 96 h. As shown in Figure 3A, viral titers of the recombinant IAV-OX40L reached a peak of 27 at 72 h, and then decreased to 26 at 96 h post infection. MIHA, HepG2, and HuH7 cells were inoculated with IAV-OX40L at various doses. An MTS assay revealed that MIHA cell viability was unchanged, while HepG2 and HuH7 cell viability decreased gradually with time and increasing viral dose (Figure 3B). Subsequently, apoptosis was determined by flow cytometry using an Annexin V-FITC Apoptosis Detection Kit according to the manufacturer’s instructions at 48 h. The apoptosis rates of HepG2 cells in the IAV-OX40L group were 10.91% and 19.08% with MOI 1 and 3 respectively. IAV-OX40L virus induced apoptosis of HepG2 cells in a dose-dependent manner, and the apoptosis-inducing effect was greater in HepG2 cells than in MIHA cells when an MOI of 3 was used (Figure 3C). The finding indicated that IAV-OX40L had a selective oncolytic efficacy in vitro.
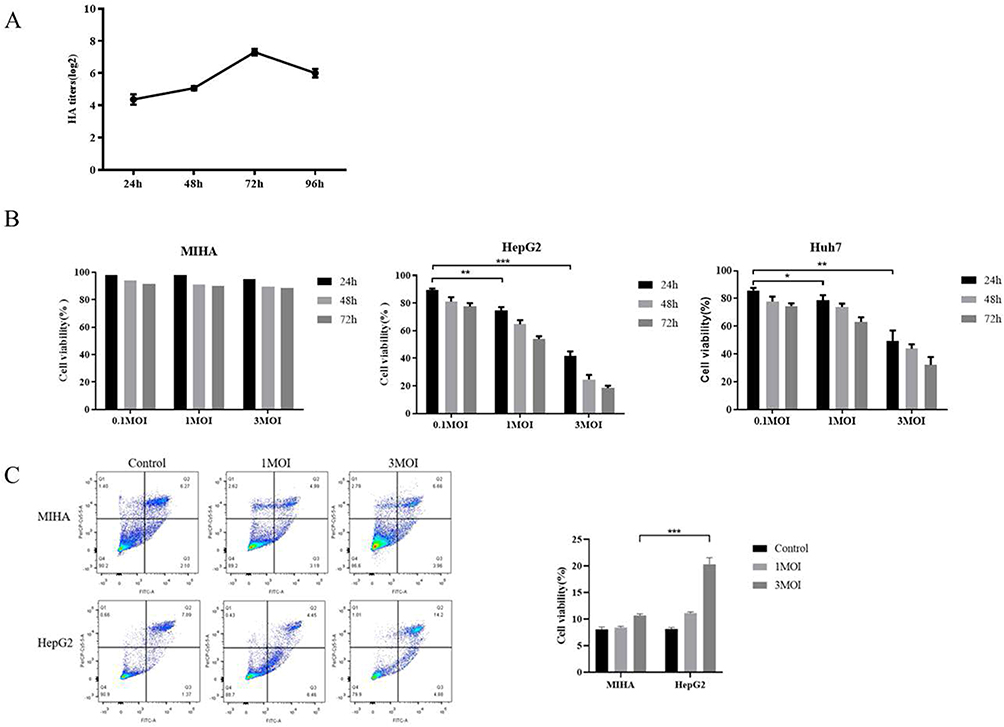 |
Figure 3 Cytotoxic effects of IAV-OX40L virus in vitro. (A) The growth curve of IAV-OX40L virus was consistent with that of the influenza virus and peaked at 27–8 at 72 h. (B) MTS assays showed that the virus inhibited the growth of tumor cells in vitro in a time- and dose-dependent manner but had no effect on normal cells. (C) Flow cytometry showed that IAV-OX40L significantly promoted the apoptosis of tumor cells, with the most obvious effect at an MOI of 3 but no effect on normal cells. (*P< 0.05, **P< 0.01, ***P< 0.001). |
IAV-OX40L Virus Inhibits Tumor Growth in vivo
To further investigate the oncolytic efficacy of IAV-OX40L virus in vivo, murine liver cancer H22 cells were used to establish the subcutaneous tumor in BALB/c mice. Tumor-bearing mice were randomly divided to inoculate with PBS, PR8, or IAV-OX40L virus every other day for seven times, respectively. Changes in tumor volume were monitored every two days (Figure 4A) until the last inoculation and then the mice were killed and the tumor tissue was isolated to measure volume (Figure 4B) and weight (Figure 4C). The tumor volume was smallest in the IAV-OX40L group, followed by the PR8 group, and was largest in the PBS group and the differences in tumor volume between the IAV-OX40L group and the other two groups were significant (Figure 4D). In addition, viral titers were determined in the supernatants of ground organs (heart, liver, spleen, lung, kidney and brain) and tumors from each group of mice. The viral titers of tumor tissues of mice in the IAV-OX40L and PR8 groups were 7–8 LogTCID50/mL, whereas there was no measurable viruses in other tissues (heart, liver, spleen, lung, kidney, brain) of the IAV-OX40L virus-treated mice (Figure 4E). These data revealed that IAV-OX40L significantly inhibited tumor growth in the HCC mouse model.
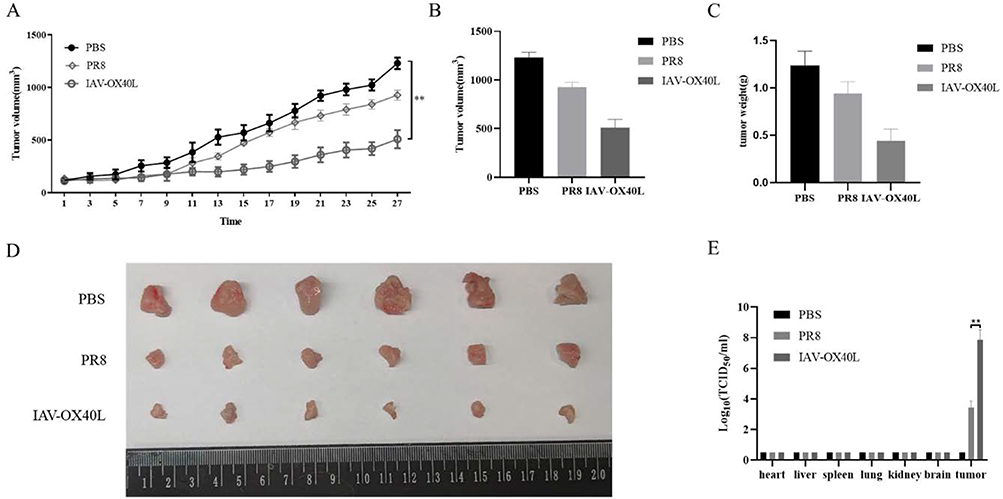 |
Figure 4 Antitumor efficacy of IAV-OX40L virus in a murine HCC model. (A) The change in tumor volume in the group inoculated with IAV-OX40L virus was significantly slower than in the PR8 and PBS groups. (B–D) Tumor volume and weight in mice in the IAV-OX40L group were significantly smaller than those in the PR8 and PBS groups (6 mice per group). The animal experiment was repeated twice. (E) The viral load in tumor tissues of the IAV-OX40L group was significantly higher than that of the PR8 group, while no viral titer was measurable in other tissues (heart, liver, spleen, lung, kidney, brain) (**P< 0.01). |
Biological Activity of IAV-OX40L Virus in vivo
To evaluate the toxicity of the IAV-OX40L virus in mice, the liver, lung, and tumor tissues of mice in each group were cut into sections and examined histologically with H&E staining. The liver and lung tissues showed exhibited no significant changes, whereas obvious tumor lysis and necrosis were observed in the IAV-OX40L group. In contrast, there were no obvious pathological changes in the PR8 and PBS groups (Figure 5A). Pathological examination of liver and lung tissues isolated from mice at 14 days after the last inoculation showed no obvious alterations, implying that the IAV-OX40L virus was relatively safe within the 14-day observation period (Figure S2).
 |
Figure 5 Toxicity and immunological activity induced by IAV-OX40L virus in vivo (A) H&E staining of pathological alterations of liver, lung, and tumor tissues in IAV-OX40L virus-treated mice showed that the virus significantly destroyed tumor tissue without damaging liver and lung tissues. (B) CD4+CD69+ and CD8+CD69+ T cell numbers in the spleen of mice inoculated with IAV-OX40L virus were significantly higher than in the PR8 and PBS groups (***P< 0.001). (C) IAV-OX40L expressed OX40L protein and stimulated CD4 T cells to produce IL-2 and IFN-γ in the presence of CD3/CD28 agonist. CD4 T cells also produces a small amount of T bet and GATA3. Therefore, IAV-OX40L mainly stimulates Th1 immune cells. However, the activation of T cells was also demonstrated by the increase of CD25. |
Subsequently, we explored the activation of the immune responses induced by IAV-OX40L virus by determining the frequency of CD4+CD69+ and CD8+CD69+ T cells in the spleen by flow cytometry, the frequencies were 34.4% and 33.5% in the IAV-OX40L group, 21.2% and 11.7% in the PR8 group, and 13.7% and 8.55% in the PBS group, respectively. These results suggest that IAV-OX40L significantly activated T cells to stimulate the immune system and had a cytotoxic effect on tumor cells (Figure 5B).
To further clarify the underlying mechanisms of the IAV-OX40L virus, Hep3B cells were co-cultured with CD4+ T cells and CD3/CD28 and were then infected with IAV-OX40L. CD3/CD28, IAV-OX40L and NC were set as controls. RNA was collected 6h later for qPCR. The group of IAV-OX40L CD4T cells significantly produced interleukin-2 (IL-2) and interferon-γ (IFN-γ), and the expression of T-bet was also significantly increased under the stimulation of CD3/CD28. OX40L expressed by IAV-OX40L mainly stimulated Th1 immune cells. IAV-OX40L stimulated IL4 expression, and to a lesser extent GATA3 expression, indicating that OX40L also stimulated Th2 cells to a lesser extent. The activation of T cells was demonstrated by an increase in CD25. The increase of OX40 proved that OX40L expression increases the expression of the interacting target OX40 (Figure 5C).
Discussion
Virotherapy is a promising novel strategy for the treatment of advanced tumors, achieving encouraging results in several preclinical studies and clinical trials against various malignant tumors.32 OVs can dynamically modulate the immune system to attack cancer cells.33,34 The immune system is enhanced by regulating the immune microenvironment, enabling immune cells to attack and eliminate tumor cells at several important nodes.35 Several recombinant OV products or immunotherapeutic agents are currently available, such as RIGVIR, Oncorine, and T-VEC. The safety and efficacy of OV products have been widely recognized.36–38 However, in contrast to the DNA virus vectored-OVs, influenza virus as an RNA virus avoids the potential risk of its genome being incorporated into the human genome, thereby further improving safety. In addition, influenza virus has several advantages such as its genome being easily cooperated using RG technology, the availability of sensitive mice and ferret models, and the ability to easily express foreign genes. However, further studies are needed to determine the appropriate combination therapy according to different targets to improve the antitumor effects of OVs.39,40 In addition, the selectively targeted therapy of OVs must be improved to reduce toxicity and side effects.
In recent years, accumulating studies have focused on the underlying mechanism of action of co-stimulation with OX40—a member of the TNF receptor family (TNF) receptor family—and its ligand OX40L. Ligands of OX40 (OX40L, CD252, and TNFSF4) belong to the TNF superfamily and can be induced by APCs such as B cells, DCs, and macrophages. Previous studies have shown that OX40–OX40L interactions affect T cell expansion mediated by a potent costimulatory signal to activated CD4+ and CD8+ T cells.41,42 Thus, OX40L has become an attractive target for cancer therapy.
In this study, the OX40L gene was successfully inserted into the NS1 segment of PR8 virus. The recombinant virus, IAV-OX40L, was then successfully rescued by RG technology. The virus titer was stable at 7–8LogTCID50/mL as confirmed by virus subculture. The morphology and size distribution of purified IAV-OX40L (80–120 nm) were the same as those of PR8 by TEM. It was confirmed that the recombinant virus could significantly reduce the viability and promote apoptosis of HCC cells at a MOI of 3 by MTS and flow cytometry, respectively. We demonstrated that IAV-OX40L could express OX40L protein in both HCC cells and MDCK cells by WB and immunofluorescence. In vivo experiments showed that IAV-OX40L significantly inhibited tumor growth by activating CD4+CD69+ and CD8+CD69+ T cells. In terms of T cell activation, IAV-OX40L is more advantageous than OV chimeric antibodies of CTLA4 and PD-1.30,43 Finally, the main immune pathway of IAV-OX40L was verified. After infection of CD4 T and Hep3B co-cultured cells by IAV-OX40L, the cytokines IL-2 and IFN-γ were significantly increased with the addition of CD3/CD28 stimulator, which confirmed that IAV-OX40L predominantly stimulated Th1 immune cells. However, the underlying mechanisms of OX40L action warrant further investigation in our future studies. A limitation of our study is that we only evaluated the cytotoxicity and apoptosis-inducing effect of IAV-OX40L on two HCC cell lines, which may not represent the heterogeneity of tumors in vivo. Consequently, additional investigations should be conducted to validate the oncolytic effect of IAV-OX40L not only on HCC cells, but also on other solid tumor cells. Nevertheless, this report represents major progress toward the development of influenza virus as a novel virotherapeutic candidate for the treatment of liver cancer. Further evaluation of the safety of IAV-OX40L in vivo is required and the potential for viral shedding and transmission following IAV-OX40L administration needs to be investigated in our subsequent studies.
Conclusion
In summary, we engineered a chimeric novel influenza virus, IAV-OX40L, and demonstrated that the recombinant virus exhibits selective oncolytic efficacy on HCC cells in vitro and in vivo. However, there are several limitations that should be explored in follow-up studies, including the pharmacokinetics of the IAV-OX40L in vivo, the optimal dose and method of administration, the neutralizing antibodies against IAV-OX40L after repeated treatments, and whether a combination of other agents with the virus have synergistic effects. Furthermore, it will be necessary to examine whether a recombinant virus carrying multiple target genes could be engineered to achieve better oncolytic effects in the future.
Data Sharing Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethical Approval
This study was approved by the Ethics Committee of Fifth Medical Center of Chinese PLA General Hospital, and carried out according to the Declaration of Helsinki.
Disclosure
The authors declare no competing of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3
3. Anwanwan D, Singh SK, Singh S, Saikam V, Singh R. Challenges in liver cancer and possible treatment approaches. Biochim Biophys Acta Rev Cancer. 2020;1873(1):188314.
4. Llovet JM, De Baere T, Kulik L, et al. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2021;18(5):293–313. doi:10.1038/s41575-020-00395-0
5. Nault JC, Cheng AL, Sangro B, Llovet JM. Milestones in the pathogenesis and management of primary liver cancer. J Hepatol. 2020;72(2):209–214. doi:10.1016/j.jhep.2019.11.006
6. Sangro B, Sarobe P, Hervas-Stubbs S, Melero I. Advances in immunotherapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2021;18(8):525–543. doi:10.1038/s41575-021-00438-0
7. Brown ZJ, Greten TF, Heinrich B. Adjuvant treatment of hepatocellular carcinoma: prospect of immunotherapy. Hepatology. 2019;70(4):1437–1442. doi:10.1002/hep.30633
8. Pinter M, Scheiner B, Peck-Radosavljevic M. Immunotherapy for advanced hepatocellular carcinoma: a focus on special subgroups. Gut. 2021;70(1):204–214. doi:10.1136/gutjnl-2020-321702
9. Feng GS, Hanley KL, Liang Y, Lin X. Improving the efficacy of liver cancer immunotherapy: the power of combined preclinical and clinical studies. Hepatology. 2021;73(1):104–114. doi:10.1002/hep.31479
10. Hemminki O, Dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol. 2020;13(1):84. doi:10.1186/s13045-020-00922-1
11. Abd-Aziz N, Poh CL. Development of oncolytic viruses for cancer therapy. Transl Res. 2021;237:98–123. doi:10.1016/j.trsl.2021.04.008
12. Zhang B, Cheng P. Improving antitumor efficacy via combinatorial regimens of oncolytic virotherapy. Mol Cancer. 2020;19(1):158. doi:10.1186/s12943-020-01275-6
13. Shin DH, Nguyen T, Ozpolat B, et al. Current strategies to circumvent the antiviral immunity to optimize cancer virotherapy. J Immunother Cancer. 2021;9(4):e002086. doi:10.1136/jitc-2020-002086
14. Wan PK, Ryan AJ, Seymour LW. Beyond cancer cells: targeting the tumor microenvironment with gene therapy and armed oncolytic virus. Mol Ther. 2021;29(5):1668–1682. doi:10.1016/j.ymthe.2021.04.015
15. Duan Q, Zhang H, Zheng J, Zhang L. Turning cold into hot: firing up the tumor microenvironment. Trends Cancer. 2020;6(7):605–618. doi:10.1016/j.trecan.2020.02.022
16. Watanabe N, McKenna MK, Rosewell Shaw A, Suzuki M. Clinical CAR-T cell and oncolytic virotherapy for cancer treatment. Mol Ther. 2021;29(2):505–520. doi:10.1016/j.ymthe.2020.10.023
17. Guedan S, Alemany R. CAR-T cells and oncolytic viruses: joining forces to overcome the solid tumor challenge. Front Immunol. 2018;9:2460. doi:10.3389/fimmu.2018.02460
18. Feola S, Russo S, Ylosmaki E, Cerullo V. Oncolytic ImmunoViroTherapy: a long history of crosstalk between viruses and immune system for cancer treatment. Pharmacol Ther. 2021;236:108103. doi:10.1016/j.pharmthera.2021.108103
19. Webb GJ, Hirschfield GM, Lane PJ. OX40, OX40L and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol. 2016;50(3):312–332. doi:10.1007/s12016-015-8498-3
20. Hill GR, Koyama M. Cytokines and costimulation in acute graft-versus-host disease. Blood. 2020;136(4):418–428. doi:10.1182/blood.2019000952
21. Fu N, Xie F, Sun Z, Wang Q. The OX40/OX40L axis regulates T follicular helper cell differentiation: implications for autoimmune diseases. Front Immunol. 2021;12:670637. doi:10.3389/fimmu.2021.670637
22. Lu X. OX40 and OX40L Interaction in Cancer. Curr Med Chem. 2021;28(28):5659–5673. doi:10.2174/0929867328666201229123151
23. Deng J, Zhao S, Zhang X, et al. OX40 (CD134) and OX40 ligand, important immune checkpoints in cancer. Onco Targets Ther. 2019;12:7347–7353. doi:10.2147/OTT.S214211
24. Fu Y, Lin Q, Zhang Z, Zhang L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm Sin B. 2020;10(3):414–433. doi:10.1016/j.apsb.2019.08.010
25. Gajdasik DW, Gaspal F, Halford EE, et al. Th1 responses in vivo require cell-specific provision of OX40L dictated by environmental cues. Nat Commun. 2020;11(1):3421. doi:10.1038/s41467-020-17293-3
26. Hewitt SL, Bai A, Bailey D, et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36gamma, and OX40L mRNAs. Sci Transl Med. 2019;11:477.
27. Porciuncula A, Morgado M, Gupta R, et al. Spatial mapping and immunomodulatory role of the OX40/OX40L pathway in human non-small cell lung cancer. Clin Cancer Res. 2021;27(22):6174–6183. doi:10.1158/1078-0432.CCR-21-0987
28. Xiong Z, Chan SL, Zhou J, et al. Targeting PPAR-gamma counteracts tumour adaptation to immune-checkpoint blockade in hepatocellular carcinoma. Gut. 2023;72(9):1758–1773. doi:10.1136/gutjnl-2022-328364
29. Conche C, Finkelmeier F, Pesic M, et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023;72(9):1774–1782. doi:10.1136/gutjnl-2022-327909
30. Lei G, Li B, Hao Y, et al. Therapeutic efficacy of an oncolytic influenza virus carrying an antibody against programmed cell death 1 in hepatocellular carcinoma. Hum Gene Ther. 2022;33:309–317. doi:10.1089/hum.2021.167
31. Penghui Y, Fang S, Ruilan W, et al. Oncolytic activity of a novel influenza a virus carrying granulocyte-macrophage colony-stimulating factor in hepatocellular carcinoma. Hum Gene Ther. 2019;30(3):330–338. doi:10.1089/hum.2018.095
32. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. 2019;18(3):175–196. doi:10.1038/s41573-018-0006-z
33. Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 2020;70(2):86–104. doi:10.3322/caac.21596
34. Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018;175(2):313–326. doi:10.1016/j.cell.2018.09.035
35. Kruger S, Ilmer M, Kobold S, et al. Advances in cancer immunotherapy 2019 - latest trends. J Exp Clin Cancer Res. 2019;38(1):268.
36. Hastie E, Cataldi M, Moerdyk-Schauwecker MJ, Felt SA, Steuerwald N, Grdzelishvili VZ. Novel biomarkers of resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus. Oncotarget. 2016;7(38):61601–61618. doi:10.18632/oncotarget.11202
37. Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. 2006;98(5):298–300. doi:10.1093/jnci/djj111
38. Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene Laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am J Clin Dermatol. 2017;18(1):1–15. doi:10.1007/s40257-016-0238-9
39. Burugu S, Dancsok AR, Nielsen TO. Emerging targets in cancer immunotherapy. Semin Cancer Biol. 2018;52(Pt 2):39–52. doi:10.1016/j.semcancer.2017.10.001
40. Yarchoan M, Johnson BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17(4):209–222. doi:10.1038/nrc.2016.154
41. Chen P, Wang H, Zhao L, et al. Immune Checkpoints OX40 and OX40L in small-cell lung cancer: predict prognosis and modulate immune microenvironment. Front Oncol. 2021;11:713853. doi:10.3389/fonc.2021.713853
42. Tian L, Liu T, Jiang S, et al. Oncolytic Newcastle disease virus expressing the co-stimulator OX40L as immunopotentiator for colorectal cancer therapy. Gene Ther. 2021;30:64–74. doi:10.1038/s41434-021-00256-8
43. Yang H, Lei G, Sun F, et al. Oncolytic activity of a chimeric influenza A virus carrying a human CTLA4 antibody in hepatocellular carcinoma. Front Oncol. 2022;12:875525. doi:10.3389/fonc.2022.875525
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.