Back to Journals » ImmunoTargets and Therapy » Volume 9
All are Equal, Some are More Equal: Targeting IL 12 and 23 in IBD – A Clinical Perspective
Authors Jefremow A, Neurath MF
Received 17 September 2020
Accepted for publication 10 November 2020
Published 26 November 2020 Volume 2020:9 Pages 289—297
DOI https://doi.org/10.2147/ITT.S282466
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
André Jefremow,1,2 Markus F Neurath1,2
1Department of Medicine 1, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany; 2Deutsches Zentrum Immuntherapie (DZI), Erlangen, Germany
Correspondence: André Jefremow
Department of Medicine 1, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
Email [email protected]
Abstract: Chronic inflammatory diseases like inflammatory bowel diseases (IBD) or psoriasis represents a worldwide health burden. Researchers provided great achievements in understanding the origin of these diseases leading to improved therapeutic options. The discovery of cytokines like tumor necrosis factor-α or transforming growth factor-β are examples for these efforts. Interleukin 12 (IL 12) and interleukin 23 (IL 23) represent different important cytokines in this regard. They both belong to the interleukin 12 family and are related by sharing the subunit p40. Ustekinumab is an antibody that blocks p40 and thereby interleukins 12 and 23. Trials showed promising results in treating IBD patients with this drug. Consequently, new questions arose about the distinct features of IL 12 and 23. This review focuses on these interleukins regarding their functions in the healthy and inflamed gut and provides an overview about the results from in vitro and in vivo studies as well as clinical trials.
Keywords: inflammatory bowel diseases, Crohn’s disease, ulcerative colitis, interleukin 12, interleukin 23
Introduction
Inflammatory bowel diseases (IBD) like Crohn’s disease (CD) or ulcerative colitis (UC) show chronic inflammation of the gut. Inflammation in Crohn’s disease can affect the whole intestine – from the mouth to the anus – and is transmural, UC is restricted to the large intestine and within here to the mucosa and submucosa. Both are often related to inflammation of other body sides like the joints or the eyes causing, e.g. arthritis and uveitis. Besides that, especially patients suffering from UC have a high risk to develop colon cancer, namely colitis-associated cancer (CAC). The risk increases depending on circumstances like the severity and duration of inflammation, the presence of primary sclerosing cholangitis, and the beginning of the disease in a younger age. The host’s immune system, genetic factors, microbiota, and a disturbed barrier function in the gut drive IBD in a complex interplay. There have been made great achievements regarding IBD therapy. The usage of anti-tumor necrosis factor-α (TNF- α) represents a milestone in IBD therapy.1–4 Still, not every patient has a benefit from this therapy and some patients become refractory to it. There, ustekinumab, an anti-p40 antibody blocking Interleukin 12 (IL 12) and Interleukin 23 (IL 23), has shown promising results in treating these patients. When ustekinumab entered the IBD therapy landmark, IL 23 was unknown, so people regarded it as an anti-IL 12 antibody. After the discovery of IL 23, which shares the p40 unit with IL 12, the role of IL 23 came into focus.
The Interleukins 12 and 23 are closely related and important players in the symphony orchestra of inflammation. So, there remain a lot of questions about the part each of them takes place in this orchestra regarding their pro- and anti-inflammatory features.5,6 This review provides an overview about IL 12 and IL 23 in vitro and in vivo studies regarding IBD, leads to their implication in clinical usage and gives, finally, an outlook about the steps, that could be taken next.
The Physiology of the Interleukins 12 and 23
Interleukin 12 (IL 12) and interleukin 23 (IL 23) belong to interleukin 12 family. Here, they are joined by interleukin 27 (IL 27) and 35 (IL 35). The family members differ in the composition of their subunits: IL 12 consists of the subunit p35 and shares the subunit p40 with IL 23. IL 23 has the additional subunit p19. IL 27 and IL 35 share EBI3. IL 27 consists additionally of p28, while IL 35 shares its subunit p35 with IL 12.6 They represent the only cytokine family with a heterodimeric structure, where p19, 28, and p35 are the α-subunits and p40 and EBI3 the β-subunits (Figure 1). Interestingly, beyond their similar structure, they show different biological features leading to proinflammatory and immune-modulating activities. IL 27 and IL 35 provide inhibitory characteristics, while IL 12 and IL 23 are supposed to act in a proinflammatory manner.7 But newer studies put the proinflammatory features of IL 23 in perspective attributing IL 12 a more balanced behavior regarding its pro- and anti-inflammatory role.8
 |
Figure 1 The interleukin 12 family: IL 12, IL 23, IL 27, and IL 35 are composed out of the α-subunits p19, p28, and p35 and the β-subunits p40 and EBI3. They interact with their different receptors and receptor chains. Created with Biorender.com. |
Like the cytokines themselves, the receptors for IL 12 and IL 23 share one receptor chain and differ in the other. They have IL12Rβ1 in common; the other chains are IL12Rβ2 for the IL 12 receptor and IL23αR for the IL 23 receptor, respectively.9–12 When IL 23 binds to its receptor, it activates the Janus kinases Janus kinase 2 (JAK2) and non-receptor tyrosin-protein kinase 2 (Tyk2). In response, signal transducer and activator of transcription 3 (STAT3) and 4 (STAT4) become active and shift to the nucleus.13 Activation of the IL 12 receptor leads rather to STAT4 than STAT 3 activation in naïve T cells.
The production of both, IL 12 and IL 23, takes place in different immune cells. Antigen-presenting cells (APCs) like dendritic cells (DCs) and macrophages represent the main source, mainly after Toll-like activation in tissues. Tissue-infiltrating neutrophils produce IL 23, too. On the other side, regulatory T cells (Tregs) can impair IL 23 production through CX3CR1+ macrophages using the immune checkpoint receptor Lymphocyte-activation gene 3 (LAG3) in the intestine. Besides that, vitamin D can decrease IL 23 activity by downregulating IL 23 receptor expression on innate lymphoid cells type 3 (ILC3).5,6,13–18
IL 12 and IL 23 affect different immune cells. Thereby, they show distinct differences regarding the affected cells and resulting consequences. IL 12 provides a T helper 1 (Th 1) response and activates natural killer cells (NK cells), innate lymphoid cells type 1 (ILC1), and type 1 cytotoxic T-cells (Tc1) leading to production of interferon-γ (IFN-γ), tumor necrosis factor alpha (TNF-α), granzymes, and perforin. Besides, IL 12 acts mainly on naïve T cells.19–22 On the other side, IL 23 interacts with memory T cells.9,12 These cells produce upon IL23R expression Th 17 cytokines like IL 17 A, IL 17 F, and IL 22 and promote T helper cells 17 (Th 17) activation.23,24 IL 23 expression upregulates IL23R expression on neutrophils and the transcription of factor retinoid acid receptor-related orphan receptor γ t (RORγt) and aryl-hydrocarbon receptor (Ahr).25 So, IL 23 activates neutrophils and mediates IL-17 and IL-22 production. It also leads to production of IL-17, IL-22, TNF-α and defensin by ILC3, γδ T cells, and type 17 cytotoxic T cells.19
Interleukin 23 in the Healthy Gut
A high amount of IL 23 is located in the small intestine and reaches a peak in the terminal ileum.26 CD8α and CD11b double-negative CD11c + lamina propria dendritic cells represent the main source for IL-12 p40 and are common immune cells in the lamina propria of the small intestine. At the same time, Th 17 cells are present in this area and receive immunosuppressive properties, so they become regulatory Th 17 cells.27 They produce IL 10 and TGF-β – immunosuppressive cytokines – to protect the mucosa from inflammation28,29 (Figure 2). On the other side, IL 23-deficient mice develop colitis, when their T cells cannot respond to TGF-β,30 so TGF-β can also show proinflammatory properties in the colon.31,32 Intestinal epithelial cells (IECs) express the IL23R under normal conditions and produce IL 22 – enhancing intestinal barrier - and antimicrobial peptides after IL 23 has bound to its receptor. Aden et al showed this protective function of IL 23 in mice, who lacked the IL23R in IECs (IL23RΔIEC3). These mice had lower levels of IL 22, showed more inflammation in a colitis model and presented a disturbed microbiota with a higher prevalence of flagellated bacterial groups.33
 |
Figure 2 Macrophages produce IL 23 leading to production of anti-inflammatory cytokines like IL 10 and TGF-β under healthy conditions in the small intestine. Created with BioRender.com. |
Bauché et al demonstrated how intestinal homeostasis based on an interplay between Foxp3+ regulatory T cells (Treg cells), CX3CR1+ tissue-resident macrophages and innate lymphoid cells (ILCs) type 3 in different colitis models. ILCs lack T cell receptor and produce cytokines like IL 22 and IL 17A. The explanation for this protective interaction bases on the immune checkpoint inhibitor Lymphocyte Activation Gene 3 (LAG-3) on Treg cells. It decreased CX3CR1+ tissue-resident macrophages activity and their production of IL 23 by interacting with their major histocompatibility complex (MHC) II. Additionally, Treg cells could decrease proinflammatory ILC 3 activity.16,17
Interleukin 12 and Interleukin 23 in Experimental Colitis Models
Much knowledge about the similar and distinct features of Interleukin 12 and Interleukin 23 raised from in vivo experiments using models mimicking IBD. Interestingly, there seems to be cross regulation of IL 12 and IL 23, and IL 23 can show pro- and anti-inflammatory features.6
An increased level of Interleukin 23 could be observed in different IBD models, such as dextran sodium sulfate (DSS) colitis, 2,4,6-trinitrobenzenesulfonic acid solution (TNBS) colitis, Helicobacter hepaticus colitis and, T cell transfer colitis.34–36
CD11c + monocytes and macrophages represent the main source of IL 23 after microbial stimulation like lipopolysaccharide (LPS) or heat-killed bacteria. IFN-γ reduced its production in macrophages. ILCs, T lymphocytes and, hematopoietic stem and progenitor cells (HPSC) drive colitis after IL 23 stimulation.34–39
Consequently, scientists tried to block IL-23 to ameliorate colitis. Before Oppmann et al discovered interleukin 23,9 researchers were successful in treating colitis by blocking p40 without knowing about the “double knockout” they achieved, blocking IL 12 and IL 23.40 This led to the question, if rather IL 12 is the driving colitis force or IL 23. Many approaches have been undertaken to distinct the features of both interleukins.5
Imamura et al compared an antibody against a specific IL 23 subunit with a p40 antibody in a transfer colitis model and could not discover relevant differences. This suggests the driving role for IL 23 in comparison with IL 12 in colitis.41 Further studies showed similar results: Kullberg and his team investigated Helicobacter hepaticus-triggered T cell-dependent colitis in mice deficient for p40, p19 or p35 and concluded also, that IL 23 is the main force in driving inflammation.42 While p19-deficiency ameliorated colitis in IL 10−/–mice, Yen et al could not observe a difference in p35 knock out mice regarding colitis.35 In comparison with wild type mice p19- and IL23R-deficient mice showed less colitis in the dextran sulfate sodium (DSS) model.43 Uhlig et al investigated IL 12 and IL 23 in an innate immune cell-mediated colitis induced by agonistic CD40 antibody treatment in T and B cell-deficient mice.44 While IL 23 showed responsibility for inflammation, IL 12 influenced cytokine production and wasting disease.
Other research groups tried different approaches to inhibit IL 23 or IL23R using bacteria or vaccination:
Chen and his team observed IL 23 and Th 17 cytokine suppressing after administration of Lactobacillus acidophilus in a DSS experiment.45 Bastaki et al created a recombinant Lactococcus lactis strain producing IL 23 blockers.46 Making a lactic acid bacteria (Lb. salivarius), that binds to IL 17, IL 23 and TNF, gave the opportunity to knock down three key inflammation drivers, as Kosler et al showed.47 A mouse p40 vaccine prevented TNBS-induced murine colitis reducing inflammation and fibrosis. A p19 vaccine decreased IL 23 production and ameliorated inflammation in a TNBS colitis model.48,49
So far, it seems to be clear, that IL 23 is the inflammation forcing protein in comparison with IL 12. But further studies revealed the complexity of their relationship.
Becker and his team demonstrated an increased colitis in C57Bl6 LacZ knock-in mice deficient for IL-23 p19 in comparison with wild type mice. A decreased production of IL 12 by DCs explained this finding and the fact, that blockade of IL 12 reduced inflammation supported it. Besides that, mice with a knock down of IL23R are susceptible to T cell transfer colitis, as Eken and his team showed.50 Above that, mice harboring a deficiency of the IL23R in IECs (IECΔILR23) present with a heavy inflammation in the colon.33,43 The colitis can be treated in these mice with administration of IL 22-Fc or Reg3b, which increased IL 22 production by neutrophils.
Interleukin 12 and Interleukin 23 in Human IBD
Lot of evidence from genetical studies let to the conclusion, that genetic variants of IL23R are associated with Crohn’s disease and ulcerative colitis but may also protect from these diseases. For example, the variant p.Arg381Gln protects against IBD, while G149R is associated with both, Crohn’s disease (CD) and ulcerative colitis (UC).51–55 Also, some single nucleotide polymorphisms (SNPs) show an increased risk for developing IBD,56 while another study could not show any association between genetical IL23R variants and UC development.57
Gheita et al discovered an almost twofold higher level of IL 23 in serum from IBD patients in comparison with healthy people; the levels were higher in patients with CD than with UC and especially high in patients suffering from IBD associated arthritis.58 Rafa et al presented another hint for the proinflammatory property of IL 23 by discovering a positive correlation between IL 23 and nitric oxide levels in serum from IBD patients.83 Nitric oxide is a reactive oxygen species (ROS) and severity of Crohn’s disease and ulcerative colitis correlates with the amount of reactive oxygen species (ROS). Furthermore, they act procarcinogen.60
Patients suffering from UC have high IL 23 serum levels as well and this correlates with disease severity and a reduced Treg/Th 17 cells ratio.6,60,61
Liu et al found a higher expression of IL 23 p19 mRNA and a greater amount of protein levels in the lamina propria from CD patients in comparison with UC patients and healthy volunteers.62 On the other side, Kobayashi et al detected higher amounts of IL 23 p19 mRNA in CD (with higher IFN-γ levels) and UC (with higher IL 17 levels).63 There seems to be also a crosstalk between IL 23 and TNF-α: Anti-TNF non-responders showed an upregulation of IL23 p19 and IL23R. This resulted in a pile of apoptosis-resistant mucosal TNFR2 +IL23R + T cells that produce Th1 and Th17 cytokines.64 So, it is reasonable to treat anti-TNF non-responders with anti-p40 antibodies.6
CD68 expressing macrophages and DCs represent the main source for IL 23. They also produce less IL 10 in comparison with healthy controls up on Toll-like receptor stimulation.65 In contrast, Kvedaraite et al showed tissue-infiltrating neutrophils as a main source for IL 23 in pediatric CD patients.84
The Functional Role of IL 23
IL 23 activates CD161 Th1 cells, that carry IL23R. Kobayashi et al showed the production of IL 17 of lamina propria T cells in UC after IL 23 stimulation.63 This could also be seen in CD patients.66 Above that, IL 23 was able to enhance the production of IFN- γ, TNF and IL 17 in IBD6,62 (Figure 3). Bloemendaal et al showed that the inhibition of TNF inhibited IL 12 and IL 23 secretion by macrophages.67 IFN- γ enhanced IL 23 production by T cells in CD patients revealing avirtuous circle.68 Additionally, IL 23 stimulates the expression of RAR-related orphan receptor C (RORC) and ensured a local TH 17 environment.6 Strikingly, Liu et al showed IL 23 mediated inhibition of IL 10.69 Geremia et al discovered CD127 + CD56 ILCs in CD patients. These cells produced proinflammatory cytokines after IL 23 stimulation.70
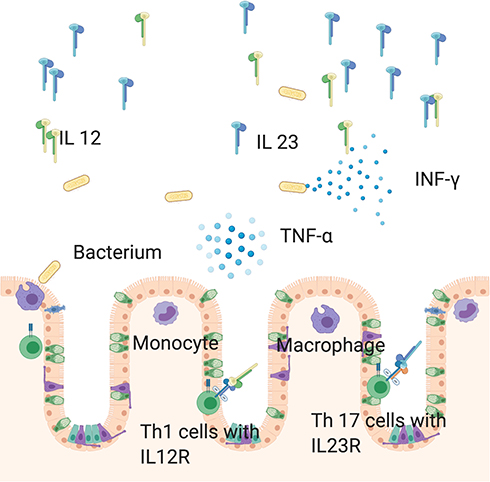 |
Figure 3 In the context of inflammation macrophages and monocytes stimulate production IL 12 and IL 23 in the large intestine. While IL 12 provides a Th 1 response, IL 23 is responsible for a Th17 response. Consequently, T cells produce proinflammatory cytokines like TNF-α and IFN-γ. Created with Biorender.com. |
Furthermore, IL 23 promoted activation of intraepithelial lymphocytes (IELs) and NK cell and cytotoxicity in IBD patients.62
Besides that, Stark et al found an IL 23 – IL 17A – colony-stimulating factors 3 (CFS3) axis leading to granulopoiesis with the result of neutrophils entering the inflammation side.15,71
Targeting P40
Ustekinumab
Ustekinumab is a monoclonal IgG1κ antibody blocking IL12/23 p40. Three studies – UNITI1, UNITI2, and IM-UNITI – showed its efficacy against CD. UNITI1 enrolled 741 patients, who were non-responders to TNFα-Inhibitors; UNITI2 enrolled 628 patients, who were refractory to conventional respond or had side effects. They received 130 mg or 6 mg/kg ustekinumab or placebo intravenously. IM-UNITI included 397 patients, who responded in UNITI1 and UNITI2 after 8 weeks; a Crohn’s Disease Activity score 150 or a decrease >100 defined response. After randomization, these patients received 90 mg ustekinumab every eight or twelve weeks. Response to ustekinumab was 34.3% and 33.7% vs. placebo 21.5% in UNITI1 and 55.5% and 48.8% against placebo (28.7%) after 44 weeks 53.1% and 48.8% were still in remission; 35.9 of the patients receiving placebo were in remission as well.72 In real life patients receive ustekinumab intravenously and subcutaneously eight weeks later. Good responder get into a 12-week schema and poor responder into an 8-week schema. Ustekinumab has to be stopped after 8 weight weeks, if there is no response and they receive an individual alternative, e.g. vedolizumab.
Notably, Biemans et al found, that ustekinumab showed superiority in comparison with vedolizumab regarding clinical and biochemical remission in CD.73 Alric et al supported these findings in a different trial with 239 patients.74
Sands et al investigated the efficacy of ustekinumab in ulcerative colitis. Here, 961 were enrolled patients and they also received 130 mg (or 6 mg/kg) ustekinumab or placebo intravenously and were assigned in responders and non-responders. Responders were administered ustekinumab or placebo every 8 or 12 weeks. After 8 weeks 15.5% of the patients, who had received 130 mg, and 15.6 of the patients, who received 6 mg/kg, showed clinical response. After 44 weeks 38.4% of the 12-week cohort and 43.8% of the 8-week cohort were in remission in comparison with placebo (24.0%).75 Ustekinumab is Food and Drug Administration (FDA) and European Medicines Agency (EMA) approved for moderate until severe CD and UC. Regarding the use of ustekinumab during pregnancy, it seems to be a safe drug, since the rate of spontaneous abortions and congenital anomalies is within the normal range. Ustekinumab crosses the placenta, but there is no evidence for an increased infection risk for the infants. Still, vaccinations with living vaccines are contraindicated during the first six month of life.76,77 But our knowledge about the use of ustekinumab during pregnancy is limited.
Briakinumab
Briakinumab is another monoclonal IgG1 anti-p40 antibody. Pannacioneet al investigated its efficacy in 246 CD patients.78 Therefore, Panaccione et al conducted a multicenter Phase II trial randomizing patients 1:1:1:3 (placebo, 200mg, 400mg, 700mg intravenously). The patients received it every four weeks. The primary endpoint – remission at week 6 – could not be reached. Even though 30% of the patients achieved remission in comparison with placebo (9%) the study was terminated early.8,78
Targeting P19
A different approach to inhibit IL 23 without directly affecting IL 12 is targeting p19. For example, risankizumab, brazikumab, guselkumab, and mirkizumab are anti-p19 antibodies, that are investigated in clinical trials.
Risankizumab
Risankizumab is a monoclonal IgG1 antibody targeting p19. Feagan et al investigated its efficacy in 121 CD patients in a phase II trial. They included patients with a (Crohn’s diseases activity index) CDAI from 220 to 450. Additionally, they needed to have ulcers in the terminal ileum or colon (or both) and a Crohn’s Disease Endoscopic Index of Severity (CDEIS) greater 7 (greater 4 in ileitis). They were randomised 1:1:1 to receive placebo, 200 mg or 600 mg risankizumab.
A CDAI <150 after 12 weeks defined the primary outcome. Secondary outcomes were endoscopic remission (CDEIS 4 or 2 for isolated ileitis), endoscopic response (CDEIS reduction >50% from baseline), mucosal healing (absence of ulcerations), and deep remission (combined endoscopic and clinical remission). Patients, who had received risankizumab showed more often clinical response (39% vs. 21%), endoscopic remission (17% vs. 3%), endoscopic response (32% vs. 13), and deep remission (7% vs. 0) compared with placebo.79 Interestingly, a sub-study revealed, that 1880 genes in the colon and 765 genes in the ileum associated with IL 23/IL 17 axis, Th 1 pathway, innate immunity, and tissue turnover were decreased.80
Brazikumab
Sands et al explored the effectiveness of the monoclonal IgG2 antibody targeting p19 in CD. They enrolled 119 CD patients with the following characteristics in a phase IIa study: previous primary non-response, secondary loss of response, or intolerance to at least one TNF antagonist, as well as objective demonstration of inflammation by biomarkers (CRP >/=5 mg/L, FCP >250 mg/g) or endoscopy (>/= 3 non-anastomotic ulcers, each >0.5 cm in diameter or </= 10 aphthous ulcers involving >/=10 cm of contiguous intestine). The patients were randomised 1:1 and received either placebo or 700 mg brazikumab intravenously at time point 0 and four weeks later. The primary outcome was a decrease of CDAI >100 or a total CDAI of <150. 49.2% of the verum group achieved clinical response, but only 26.7% of the placebo-treated patients. At week 24 clinical response maintained in 53.8% of the patients. Additionally, 57.7% of the patients, who switched from placebo to brazikumab after the double-blind period. Beyond that, IL 22 seemed to be a biomarker, for patients with IL 22 levels higher than 15.6 pg/mL showed a higher response rate.8,76,81
Guselkumab
The GALAXI trials investigate the efficacy of the monoclonal IgG1λ antibody against p19 (ClinicalTrials.gov identifiers: NCT03466411). GALAXI1 is a phase II trial over 48 weeks aiming to find the right dose. GALAXI 2 and 3 are supposed to be two Phase 3 studies. The investigators aim to enroll over 2000 probands. Beyond that, the control group receives no placebo but ustekinumab, making it a trial between two biologicals. Primary outcome is a clinical remission after 12 weeks with a CDAI <150.8,76
Mirikizumab
Mirikizumab is a monoclonal IgG4 antibody targeting p19. Currently, it is the only antibody against p19 that was investigated in respect to UC. Two hundred and forty-nine patients were enrolled for this multi-center, randomized, double-blind, placebo-controlled phase II trial. They received placebo, 50 mg mirikizumab, 200 mg mirikizumab, or 600 mg mirikizumab. Patients who got 50 or 200 mg were dose escalated, if the serum level were below 0.5 μL/mL respectively 2.0 μL/mL after four and eight weeks. The Mayo disease activity index score defined the primary endpoint after 12 weeks. 41.3–59.7% of all patients treated with mirikizumab compared with 20.6% of those treated with placebo archived this endpoint. The 200 mg arm showed a significant clinical remission in comparison with placebo; the other mirikizumab doses failed here. Above that, the investigators observed a higher amount of endoscopic healing in the 200 mg arm (22.6%) versus placebo (4.6%). A multicenter, randomized, parallel-arm, placebo-control phase II trial for patients with active CD is planned (ClinicalTrials.gov identifiers: NCT02891226)8,76,82 (Table 1).
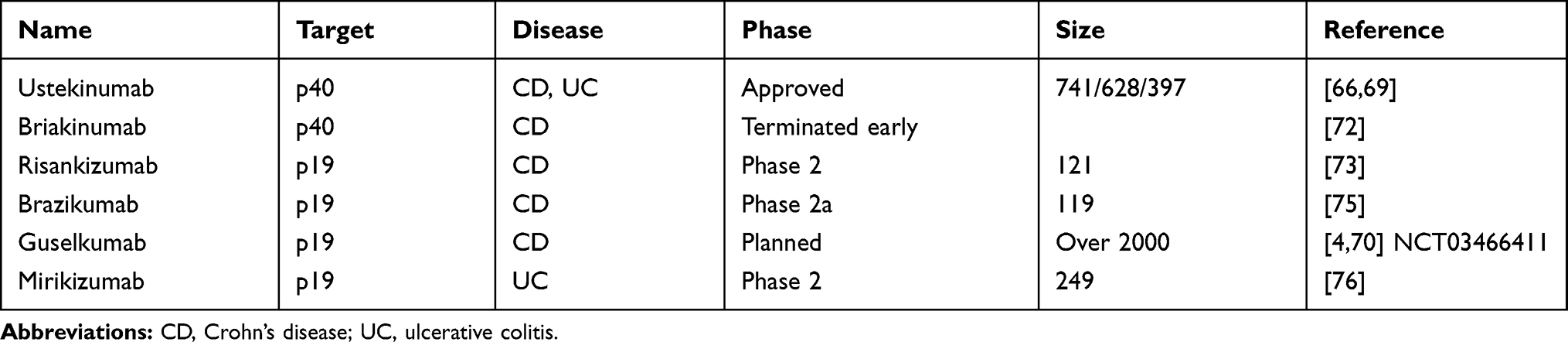 |
Table 1 Presents an Overview of the Different Anti-P40 and Anti-P19 Antibodies, That are Already Approved or are Tested in Clinical Trials |
Discussion and Conclusion
Crohn’s disease and ulcerative colitis are complex diseases regarding their multifactorial causes and features. Slowly, we begin to understand the complex pathogenesis, which includes genetic variants, microbiota, the host’s immune system and barrier dysfunctions in the gut. Still, many questions remain. Which microbiota composition favors development of IBD and which cytokine is important or more important than others? Also, why have some patients heavy diseases while others are only affected mildly?
It seems clear that IL 12 and IL 23 are major players in inflammation and – especially – in IBD. Still, the exact interplay of these interleukins and the role, they take in remains unclear. Both of them show pro- and anti-inflammatory features in experiments depending on the circumstances. Throughout the last years, the importance of IL 23 in inflammation became clearer. This led to the development of anti-IL 19 antibodies, that attack IL 23 in a targeted manner. Especially the GALAXI trials will give more insights into the IL 12/23 complex hopefully, for an anti-p40 antibody will be compared with an anti p19-antibody. These new antibodies must show their efficacy and safety. Currently, there is a lot of evidence to use anti-p40 antibodies after TNF-failure in IBD patients. In older patients and patients with a history of cancer anti-p40 antibodies may be preferred over anti-TNF-α-antibodies, since they seem to be less immunosuppressant.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
MFN was supported by the German Research Foundation (DFG) within the SPP1656 and the TRR241 (C04).
Disclosure
Markus Neurath declares personal fees from Boehringer & Abbvie, from Roche & Takeda, and from J&J, during the conduct of the study. André Jefremow declares speaker fees from AbbVie, MSD, and Eisai, traveller grants from Servier and Amgen, Sponsoring from Amgen, Cellgene, and Lilly, and personal fees from Servier und Amgen. The authors report no other potential conflicts of interest for this work.
References
1. Danese S, Fiocchi C. Ulcerative colitis. N Engl J Med. 2011;365(18):1713–1725.
2. Torres J, Mehandru S, Colombel JF, Peyrin-Biroulet L. Crohn’s disease. Lancet. 2017;389(10080):1741–1755. doi:10.1016/S0140-6736(16)31711-1
3. Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140(6):1807–1816.
4. Terzić J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138(6):2101–2114.e5. doi:10.1053/j.gastro.2010.01.058
5. Neurath MF. IL-23: a master regulator in Crohn disease. Nat Med. 2007;13(1):26–27.
6. Neurath MF. IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev. 2019;45(5):1–8.
7. Vignali DAA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13(8):722–728.
8. Ma C, Panaccione R, Khanna R, Feagan BG, Jairath V. IL12/23 or selective IL23 inhibition for the management of moderate-to-severe Crohn’s disease? Best Pract Res Clin Gastroenterol. 2019;38–39:101604. doi:10.1016/j.bpg.2019.02.006
9. Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13(5):715–725.
10. Bloch Y, Bouchareychas L, Merceron R, et al. Structural activation of pro-inflammatory human cytokine IL-23 by cognate IL-23 receptor enables recruitment of the shared receptor IL-12Rβ1. Immunity. 2018;48(1):45–58.e6.
11. Parham C, Chirica M, Timans J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168(11):5699–5708.
12. Croxford AL, Mair F, Becher B. IL-23: one cytokine in control of autoimmunity. Eur J Immunol. 2012;42(9):2263–2273. doi:10.1002/eji.201242598
13. Yang XO, Panopoulos AD, Nurieva R, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282(13):9358–9363.
14. Dennehy KM, Willment JA, Williams DL, Brown GD. Reciprocal regulation of IL-23 and IL-12 following co-activation of Dectin-1 and TLR signaling pathways. Eur J Immunol. 2009;39(5):1379–1386. doi:10.1002/eji.200838543
15. Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22(3):285–294.
16. Bauché D, Joyce-Shaikh B, Jain R, et al. LAG3+ regulatory T cells restrain interleukin-23-producing CX3CR1+ gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity. 2018;49(2):342–352.e5.
17. Rankin LC, Arpaia N. Treg cells: a lagging hand holds the double-edged sword of the IL-23 axis. Immunity. 2018;49(2):201–203. doi:10.1016/j.immuni.2018.08.008
18. Konya V, Czarnewski P, Forkel M, et al. Vitamin D downregulates the IL-23 receptor pathway in human mucosal group 3 innate lymphoid cells. J Allergy Clin Immunol. 2018;141(1):279–292.
19. Chyuan I-T, Lai J-H. New insights into the IL-12 and IL-23: from a molecular basis to clinical application in immune-mediated inflammation and cancers. Biochem Pharmacol. 2020;175:113928. doi:10.1016/j.bcp.2020.113928
20. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260(5107):547–549.
21. Cooper AM, Kipnis A, Turner J, Magram J, Ferrante J, Orme IM. Mice lacking bioactive IL-12 can generate protective, antigen-specific cellular responses to mycobacterial infection only if the IL-12 p40 subunit is present. J Immunol. 2002;168(3):1322–1327.
22. Lieberman LA, Cardillo F, Owyang AM, et al. IL-23 provides a limited mechanism of resistance to acute toxoplasmosis in the absence of IL-12. J Immunol. 2004;173(3):1887–1893.
23. Iwakura Y. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116(5):1218–1222. doi:10.1172/JCI28508
24. Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233–240.
25. Chen F, Cao A, Yao S, et al. mTOR mediates IL-23 induction of neutrophil IL-17 and IL-22 production. J Immunol. 2016;196(10):4390–4399.
26. Becker C, Wirtz S, Blessing M, et al. Constitutive p40 promoter activation and IL-23 production in the terminal ileum mediated by dendritic cells. J Clin Invest. 2003;112(5):693–706.
27. Esplugues E, Huber S, Gagliani N, et al. Control of TH17 cells occurs in the small intestine. Nature. 2011;475(7357):514–518.
28. Gagliani N, Vesely MCA, Iseppon A, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 2015;523(7559):221–225.
29. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14(5):329–342.
30. Izcue A, Hue S, Buonocore S, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28(4):559–570. doi:10.1016/j.immuni.2008.02.019
31. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–189. doi:10.1016/j.immuni.2006.01.001
32. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–434. doi:10.1038/nature06005
33. Aden K, Rehman A, Falk-Paulsen M, et al. Epithelial IL-23R signaling licenses protective IL-22 responses in intestinal inflammation. Cell Rep. 2016;16(8):2208–2218.
34. Karaboga İ, Demirtas S, Karaca T. Investigation of the relationship between the Th17/IL-23 pathway and innate-adaptive immune system in TNBS-induced colitis in rats. Iran J Basic Med Sci. 2017;20(8):870–879.
35. Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116(5):1310–1316. doi:10.1172/JCI21404
36. Becker C, Dornhoff H, Neufert C, et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J Immunol. 2006;177(5):2760–2764.
37. Arnold IC, Mathisen S, Schulthess J, Danne C, Hegazy AN, Powrie F. CD11c+ monocyte/macrophages promote chronic helicobacter hepaticus-induced intestinal inflammation through the production of IL-23. Mucosal Immunol. 2016;9(2):352–363.
38. Kamada N, Hisamatsu T, Okamoto S, et al. Abnormally differentiated subsets of intestinal macrophage play a key role in Th1-dominant chronic colitis through excess production of IL-12 and IL-23 in response to bacteria. J Immunol. 2005;175(10):6900–6908.
39. Sheikh SZ, Matsuoka K, Kobayashi T, Li F, Rubinas T, Plevy SE. Cutting edge: IFN-γ is a negative regulator of IL-23 in murine macrophages and experimental colitis. J Immunol. 2010;184(8):4069–4073.
40. Neurath MF, Fuss I, Kelsall BL, Stüber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182(5):1281–1290.
41. Imamura E, Taguchi K, Sasaki-Iwaoka H, Kubo S, Furukawa S, Morokata T. Anti-IL-23 receptor monoclonal antibody prevents CD4+ T cell-mediated colitis in association with decreased systemic Th1 and Th17 responses. Eur J Pharmacol. 2018;824:163–169. doi:10.1016/j.ejphar.2018.01.045
42. Kullberg MC, Jankovic D, Feng CG, et al. IL-23 plays a key role in helicobacter hepaticus–induced T cell–dependent colitis. J Exp Med. 2006;203(11):2485–2494.
43. Cox JH, Kljavin NM, Ota N, et al. Opposing consequences of IL-23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol. 2012;5(1):99–109.
44. Uhlig HH, McKenzie BS, Hue S, et al. Differential Activity of IL-12 and IL-23 in Mucosal and Systemic Innate Immune Pathology. Immunity. 2006;25(2):309–318
45. Chen L, Zou Y, Peng J, et al. Lactobacillus acidophilus suppresses colitis-associated activation of the IL-23/Th17 axis. J Immunol Res. 2015;2015:1–10. doi:10.1155/2015/206731
46. Bastaki SMA, Al Ahmed MM, Al Zaabi A, Amir N, Adeghate E. Effect of turmeric on colon histology, body weight, ulcer, IL-23, MPO and glutathione in acetic-acid-induced inflammatory bowel disease in rats. BMC Complement Altern Med. 2016;16(1):72. doi:10.1186/s12906-016-1057-5
47. Kosler S, Strukelj B, Berlec A. Lactic acid bacteria with concomitant IL-17, IL-23 and TNFα- binding ability for the treatment of inflammatory bowel disease. Curr Pharm Biotechnol. 2017;18(4):318–326.
48. Guan Q, Weiss CR, Wang S, et al. Reversing ongoing chronic intestinal inflammation and fibrosis by sustained block of IL-12 and IL-23 using a vaccine in mice. Inflamm Bowel Dis. 2018;24(9):1941–1952.
49. Guan Q, Burtnick HA, Qing G, et al. Employing an IL-23 p19 vaccine to block IL-23 ameliorates chronic murine colitis. Immunotherapy. 2013;5(12):1313–1322.
50. Eken A, Singh AK, Treuting PM, Oukka M. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol. 2014;7(1):143–154.
51. Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science (80-). 2006;314(5804):1461–1463.
52. Kim SW, Kim ES, Moon CM, et al. Genetic polymorphisms of IL-23R and IL-17A and novel insights into their associations with inflammatory bowel disease. Gut. 2011;60(11):1527–1536.
53. Turpin W, Goethel A, Bedrani L, Croitoru MDCMK. Determinants of IBD heritability: genes, bugs, and more. Inflamm Bowel Dis. 2018;24(6):1133–1148.
54. Newman WG, Zhang Q, Liu X, Amos CI, Siminovitch KA. Genetic variants in IL-23R and ATG16L1 independently predispose to increased susceptibility to crohnʼs disease in a Canadian Population. J Clin Gastroenterol. 2009;43(5):444–447.
55. Dubinsky MC, Wang D, Picornell Y, et al. IL-23 receptor (IL-23R) gene protects against pediatric Crohn’s disease. Inflamm Bowel Dis. 2007;13(5):511–515.
56. Bank S, Andersen PS, Burisch J, et al. Polymorphisms in the toll-like receptor and the IL-23/IL-17 pathways were associated with susceptibility to inflammatory bowel disease in a Danish Cohort. Chamaillard M, editor. PLoS One. 2015;10(12):e0145302.
57. Hayatbakhsh MM, Zahedi MJ, Shafiepour M, Nikpoor AR, Mohammadi M. IL-23 receptor gene rs7517847 and rs1004819 SNPs in ulcerative colitis. Iran J Immunol. 2012;2:128–135.
58. Gheita TA, El Gazzar II, El-Fishawy HS, Aboul-Ezz MA, Kenawy SA. Involvement of IL-23 in enteropathic arthritis patients with inflammatory bowel disease: preliminary results. Clin Rheumatol. 2014;33(5):713–717.
59. Roessner A, Kuester D, Malfertheiner P, Schneider-Stock R. Oxidative stress in ulcerative colitis-associated carcinogenesis. Pathol Res Pract. 2008;204(7):511–524.
60. Zhu X-M, Shi Y-Z, Cheng M, Wang D-F, Fan J-F. Serum IL-6, IL-23 profile and Treg/Th17 peripheral cell populations in pediatric patients with inflammatory bowel disease. Pharmazie. 2017;72(5):283–287.
61. Youssef T, Saleh SA, Rund A, Montasser I, Mohsen M, Hazem O. Evaluation of interleukin 23 (IL-23) as a non-invasive test of disease severity in patients with ulcerative colitis. Arab J Gastroenterol. 2018;19(3):116–120. doi:10.1016/j.ajg.2018.09.003
62. Liu Z, Yadav PK, Xu X, et al. The increased expression of IL-23 in inflammatory bowel disease promotes intraepithelial and lamina propria lymphocyte inflammatory responses and cytotoxicity. J Leukoc Biol. 2011;89(4):597–606. doi:10.1189/jlb.0810456
63. Kobayashi T, Okamoto S, Hisamatsu T, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut. 2008;57(12):1682–1689.
64. Schmitt H, Billmeier U, Dieterich W, et al. Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in Crohn’s disease. Gut. 2019;68(5):814–828.
65. Sakuraba A, Sato T, Kamada N, Kitazume M, Sugita A, Hibi T. Th1/Th17 immune response is induced by mesenteric lymph node dendritic cells in Crohn’s disease. Gastroenterology. 2009;137(5):1736–1745. doi:10.1053/j.gastro.2009.07.049
66. Kamada N, Hisamatsu T, Honda H, et al. TL1A produced by lamina propria macrophages induces Th1 and Th17 immune responses in cooperation with IL-23 in patients with Crohn’s disease. Inflamm Bowel Dis. 2010;16(4):568–575.
67. Bloemendaal FM, Koelink PJ, van Schie KA, et al. TNF-anti-TNF immune complexes inhibit IL-12/IL-23 secretion by inflammatory macrophages via an Fc-dependent mechanism. J Crohns Colitis. 2018;12(9):1122–1130.
68. Kamada N, Hisamatsu T, Okamoto S, et al. Unique CD14+ intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-γ axis. J Clin Invest. 2008;118(6):2269–2280.
69. Liu Z, Feng B-S, Yang S-B, Chen X, Su J, Yang P-C. Interleukin (IL)-23 suppresses IL-10 in inflammatory bowel disease. J Biol Chem. 2012;287(5):3591–3597.
70. Geremia A, Arancibia-Cárcamo CV, Fleming MPP, et al. IL-23–responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. 2011;208(6):1127–1133.
71. Leppkes M, Neurath MF. Cytokines in inflammatory bowel diseases – update 2020. Pharmacol Res. 2020;45:104835. doi:10.1016/j.phrs.2020.104835
72. Feagan BG, Sandborn WJ, Gasink C, et al. Ustekinumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2016;375(20):1946–1960.
73. Biemans VBC, van der Woude CJ, Dijkstra G, et al. Ustekinumab is associated with superior effectiveness outcomes compared to vedolizumab in Crohn’s disease patients with prior failure to anti-TNF treatment. Aliment Pharmacol Ther. 2020;52(1):123–134. doi:10.1111/apt.15745
74. Alric H, Amiot A, Kirchgesner J, et al. The effectiveness of either ustekinumab or vedolizumab in 239 patients with Crohn’s disease refractory to anti-tumour necrosis factor. Aliment Pharmacol Ther. 2020;51(10):948–957. doi:10.1111/apt.15706
75. Sands BE, Sandborn WJ, Panaccione R, et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2019;381(13):1201–1214.
76. Wong U, Cross RK. Expert opinion on interleukin-12/23 and interleukin-23 antagonists as potential therapeutic options for the treatment of inflammatory bowel disease. Expert Opin Investig Drugs. 2019;28(5):473–479. doi:10.1080/13543784.2019.1597053
77. Nguyen GC, Seow CH, Maxwell C, et al. The toronto consensus statements for the management of inflammatory bowel disease in pregnancy. Gastroenterology. 2016;150(3):734–757.e1. doi:10.1053/j.gastro.2015.12.003
78. Panaccione R, Sandborn WJ, Gordon GL, et al. Briakinumab for treatment of Crohn’s disease. Inflamm Bowel Dis. 2015;21(6):1.
79. Feagan BG, Panés J, Ferrante M, et al. Risankizumab in patients with moderate to severe Crohn’s disease: an open-label extension study. Lancet Gastroenterol Hepatol. 2018;10:671–680. doi:10.1016/S2468-1253(18)30233-4
80. Visvanathan S, Baum P, Salas A, et al. Selective IL-23 inhibition by risankizumab modulates the molecular profile in the colon and ileum of patients with active Crohn’s disease: results from a randomised Phase II Biopsy sub-study. J Crohns Colitis. 2018;12(10):1170–1179.
81. Sands BE, Chen J, Feagan BG, et al. Efficacy and safety of MEDI2070, an antibody against interleukin 23, in patients with moderate to severe Crohn’s disease: a Phase 2a Study. Gastroenterology. 2017;153(1):77–86.e6. doi:10.1053/j.gastro.2017.03.049
82. Sandborn WJ, Ferrante M, Bhandari BR, et al. 882 - efficacy and safety of anti-interleukin-23 therapy with mirikizumab (LY3074828) in patients with moderate-to-severe ulcerative colitis in a Phase 2 Study. Gastroenterology. 2018;154(6):
83. Rafa H, Saoula H, Belkhelfa M, et al. IL-23/IL-17A Axis Correlates with the Nitric Oxide Pathway in Inflammatory Bowel Disease: Immunomodulatory Effect of Retinoic Acid. J Interf Cytokine Res. 2013;33(7):355–368.
84. Kvedaraite E, Lourda M, Ideström M, et al. Tissue-infiltrating neutrophils represent the main source of IL-23 in the colon of patients with IBD. Gut. 2016;65(10):1632–1641.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.