Back to Journals » Journal of Blood Medicine » Volume 16
Advances in Complement Inhibition Therapies for Paroxysmal Nocturnal Hemoglobinuria and Autoimmune Hemolytic Disorders
Authors Tamdin T ![]() , Rodgers GM
, Rodgers GM ![]()
Received 28 May 2025
Accepted for publication 3 November 2025
Published 12 November 2025 Volume 2025:16 Pages 559—572
DOI https://doi.org/10.2147/JBM.S543272
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Tenzin Tamdin,1 George M Rodgers2
1Department of Internal Medicine, Danbury Hospital/Nuvance Health, Danbury, CT, 06810, USA; 2Division of Hematology and Hematologic Malignancies, Department of Internal Medicine, University of Utah Health Sciences Center, Salt Lake City, UT, 84132, USA
Correspondence: George M Rodgers, Division of Hematology and Hematologic Malignancies, Department of Internal Medicine, University of Utah Health Sciences Center, Salt Lake City, UT, 84132, USA, Email [email protected]
Abstract: Paroxysmal Nocturnal Hemoglobinuria (PNH) is a rare hematologic disorder characterized by intravascular hemolysis through complement activation, bone marrow failure, and thrombosis. The advancement of complement biology has enabled better therapeutic approaches that lead to better clinical outcomes in patients with PNH and other complement-driven hemolytic disorders. The terminal complement inhibitor, eculizumab, was the initial drug available which significantly reduced hemolysis and thrombotic events but failed to resolve residual extravascular hemolysis and transfusion requirements. New therapeutic agents which target proximal complement factors C3, factor B and factor D demonstrate better control of intra- and extravascular hemolysis while decreasing transfusion requirements and improving patient quality of life. This review highlights the evolving therapeutic landscape in complement inhibition by summarizing clinical evidence for the terminal complement inhibitors, as well as pegcetacoplan, iptacopan, and danicopan as emerging agents for treatment of PNH and autoimmune hemolytic anemias—warm AIHA and cold agglutinin disease (CAD). The review also examines ongoing clinical trials and proposes future directions to optimize therapeutic outcomes to address remaining clinical challenges.
Keywords: paroxysmal nocturnal hemoglobinuria, complement inhibitors, eculizumab, ravulizumab, iptacopan, danicopan, crovalimab
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare acquired disorder of hematopoiesis with an incidence of 12–13 cases per 1 million population in the United States.1 PNH was associated with high mortality before the advent of eculizumab. The disease results from somatic mutations in the X-linked PIGA gene. A key study by Ware et al in 1994 provided strong evidence that PIGA mutations are the cause of PNH, resulting in a deficiency of glycosylphosphatidylinositol (GPI)-anchored proteins,2 rendering red blood cells more susceptible to complement lysis. This leads to intravascular hemolysis, thrombosis and bone marrow failure (Figure 1). The approval of C5 inhibitors such as eculizumab3 and ravulizumab4 has greatly improved clinical outcomes in PNH by decreasing hemolysis and preventing thrombotic complications. More recently, therapies that target other components of the complement pathway such as C3 (pegcetacoplan), factor B (iptacopan), and factor D (danicopan) have shown promise for a more comprehensive and effective disease management, including patients with persistent extravascular hemolysis. However, breakthrough hemolysis, infection risk, and the high cost of therapy remain persistent challenges in treating PNH (Table 1). The use of complement inhibitors in PNH is well established, but their use in other hemolytic and immune-mediated diseases is still under investigation.
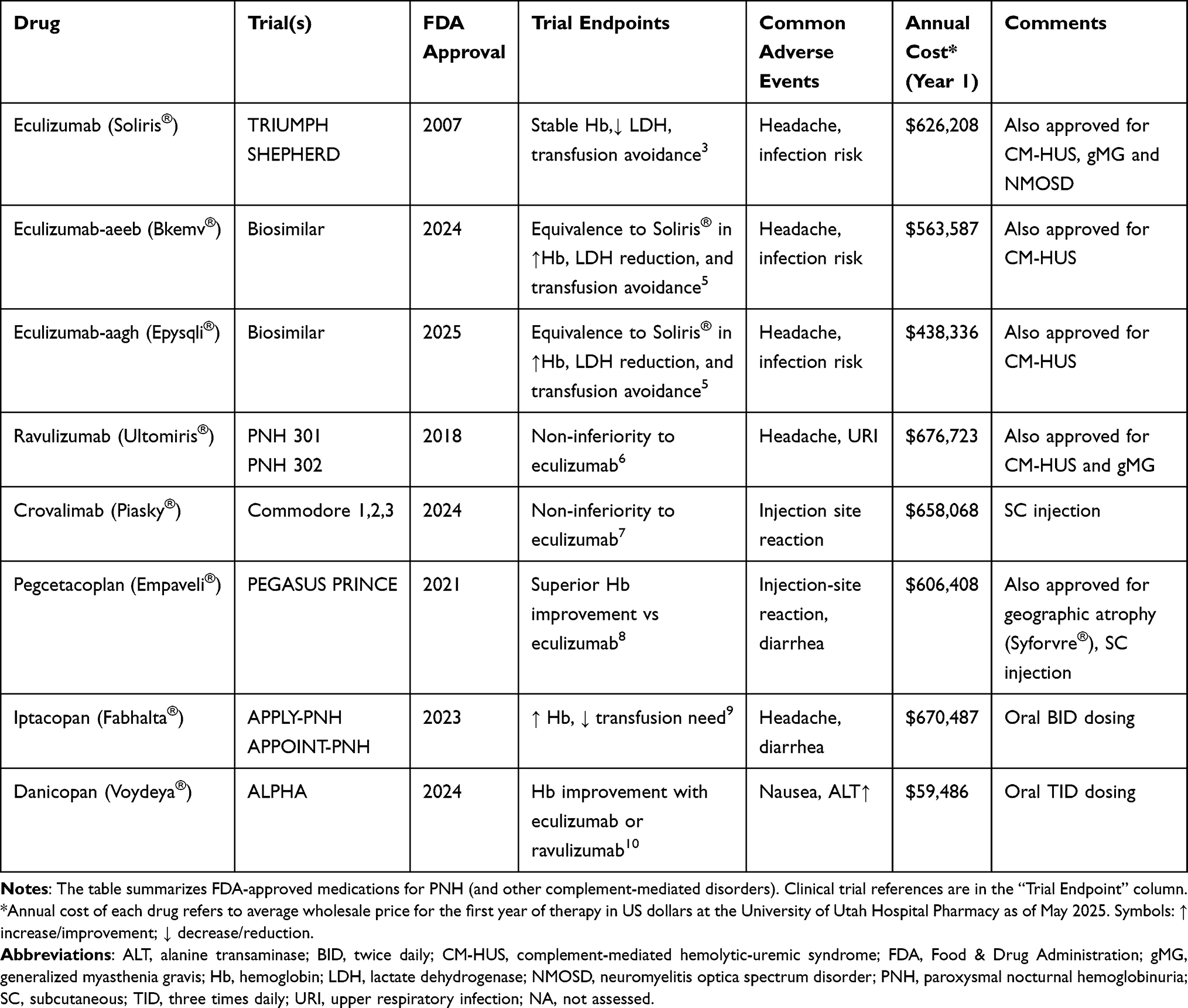 |
Table 1 FDA-Approved Medications for PNH |
 |
Figure 1 Role of CD55 and CD59 in preventing hemolysis: Normal vs PNH. The top panel shows normal red blood cells (RBCs) while the bottom panel shows PNH RBCs during complement activation. Normal RBCs maintain protection of their surface through CD55 (also referred to as decay-accelerating factor (DAF)) and CD59 (also referred to as membrane inhibitor of reactive lysis (MIRL)) proteins which are anchored by GPI and function to block C3 convertase formation and membrane attack complex (MAC) assembly, respectively, thus preventing hemolysis. The PIGA gene mutation in PNH cells results in loss of CD55 and CD59 from the RBC membrane surface. This absence of CD55 and CD59 on RBC membranes allows uncontrolled complement activation to generate MAC resulting in intravascular hemolysis. The ongoing destruction of red blood cells through hemolysis leads to thrombosis, a major reason for both morbidity and mortality in PNH patients. Created in BioRender. tamdin, t. (2025) https://BioRender.com/02bto4h. |
In this narrative review, we first group the terminal C5 inhibitors (eculizumab, ravulizumab, and crovalimab) as a class, then explain why C3-opsonization under C5 blockade sustains extravascular hemolysis. We next summarize the evidence for proximal inhibition (C3, factor B, factor D), integrate a practical framework for patient-tailored and combination therapy, and briefly extend the discussion to other complement-mediated hemolytic anemias. We close with real-world monitoring, safety considerations, and future directions.
Materials and Methods
A narrative review was conducted to analyze recent and upcoming therapeutic approaches for the complement system in PNH while briefly exploring their use in other hemolytic anemias. PubMed database was searched for articles from January 1, 2005 through April 1, 2025 that included the following keywords: “paroxysmal nocturnal hemoglobinuria”, “complement inhibitors”, “eculizumab”, “pegcetacoplan”, “iptacopan”, “danicopan”, “crovalimab”, and “autoimmune hemolytic anemia.”
The research focused on Phase 2 and Phase 3 clinical trials together with systematic reviews and meta-analyses and key studies about PNH pathophysiology and complement biology. The major trials included TRIUMPH, PEGASUS, APPLY-PNH, PRINCE, ALPHA and COMMODORE. The review included both recent discoveries alongside essential foundational research from previous decades especially from the 1980s and 1990s to establish the historical background.
Our analysis combined data about drug mechanism of action together with drug clinical effectiveness and safety aspects, as well as cost-related information. This review aims to provide a current overview to assist clinicians in better understanding the treatment options for PNH and complement-related hemolytic disorders.
Understanding the Complement System and PNH Pathophysiology
The complement system functions as a vital component of the innate immune response to identify pathogens while eliminating them through a controlled sequence of protein interactions. The protective complement system functions as a threat to patients with PNH because excessive complement activation hemolyzes red blood cells (RBCs). The PIGA gene mutation leads to this condition because this gene controls glycosylphosphatidylinositol (GPI) anchor production. The surface attachment of important complement regulatory proteins CD55 and CD59 to blood cells requires these GPI anchors. RBCs remain vulnerable to complement-mediated damage when they lack these essential protective proteins (Figure 1). Research conducted by Nicholson-Weller et al11 revealed that red cells from PNH patients lacked CD55, while Holguin et al12 discovered CD59 deficiency, a protein which blocks terminal complement cascade activation. Bessler et al13 discovered frameshift mutations in the PIGA gene which created premature stop codons that resulted in loss of GPI-anchored proteins from hematopoietic cells.
The absence of CD55 and CD59 creates unregulated complement system activation primarily affecting alternative and terminal pathways, thus exposing RBCs to continuous complement activation damage. Uncontrolled activation results in the formation of the membrane attack complex (MAC) and intravascular hemolysis (IVH) which causes the primary PNH symptoms of dark urine (hemoglobinuria), severe fatigue and increased thrombotic risk. Bordet’s foundational research into complement pathways (classical, lectin, and alternative) established the mechanism by which these pathways merge at C3 activation to produce MAC.14
Clinical Manifestations of PNH
PNH patients typically show diverse clinical symptoms, including complement-driven intravascular hemolysis together with bone marrow failure and an elevated risk of thrombosis.15 The clinical manifestations are largely determined by the size of the PIGA-mutant clone and the degree of associated bone marrow failure.15 A review by Parker emphasized that the peripheral blood in PNH patients is typically a mosaic of normal and GPI-anchored protein (GPI-AP) deficient cells, with clone size directly impacting symptom severity and risk of complications.15 Parker also classified PNH into three clinical subtypes: classic PNH (overt intravascular hemolysis, large clone), PNH associated with another bone marrow failure syndrome (moderate clone, variable symptoms), and subclinical PNH (small clone, no evident hemolysis). Patients with large PNH clones typically develop severe hemolytic anemia in addition to marrow failure that may cause pancytopenia or aplastic anemia.
PNH patients experience thrombotic events that occur in unusual locations. Classic hepatic vein thrombosis leads to Budd-Chiari syndrome,16 yet clots can also form in portal and mesenteric venous sinuses, as well as cerebral venous sinuses, dermal veins, and pulmonary vasculature.17,18 The exact mechanism underlying the thrombophilia in PNH is not fully understood, but it is believed to be related to the exposure of procoagulant particles from hemolyzed red cells, and the activation of platelets and the coagulation cascade. Intravascular hemolysis plays a major role in the thrombophilia of PNH.15 Intravascular hemolysis releases free hemoglobin that scavenges nitric oxide, leading to nitric-oxide depletion and smooth muscle dystonia, a proposed contributor to PNH-associated thrombophilia.19
The development of PNH may occur in patients with bone marrow failure, particularly those with aplastic anemia (AA). Studies have shown that patients with PNH often have prior AA or marrow hypoplasia in 50–70% of cases. The majority of patients with severe AA contain small PNH clones according to Sun et al.20 Patients who present with pancytopenia face increased risks of infection and bleeding complications, and have a higher chance of developing myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) due to clonal evolution. The risk of developing secondary MDS or AML stands at 2–6% among PNH patients during the first ten years following diagnosis.20 Patients with PNH also face an additional major life-threatening complication: progressive renal failure from chronic hemoglobinuria.21
Eculizumab as a C5-complement inhibitor brought substantial changes to treatment approaches resulting in better survival rates and enhanced quality of life for patients who have PNH.22 Table 1 summarizes the available complement inhibitors with information on their pivotal clinical trials, current approved indications, and cost.
Terminal Complement Inhibitors: Eculizumab, Ravulizumab and Crovalimab
Eculizumab
The approval of eculizumab which is a humanized monoclonal antibody against complement protein C5 marked a significant advancement in the treatment of PNH (Figure 2).3 Patients experienced a major disease burden before the availability of this treatment because chronic intravascular hemolysis required frequent blood transfusions for severe anemia. The main reasons for death in PNH patients during the pre-complement inhibition period were thromboembolic events which caused 40–67% of all fatalities.18 Thrombosis occurred most frequently in the hepatic veins (Budd-Chiari syndrome) and cerebral sinuses which resulted in early patient fatalities. The median survival duration of patients who received only supportive care was 10–20 years according to Sørensen et al.23
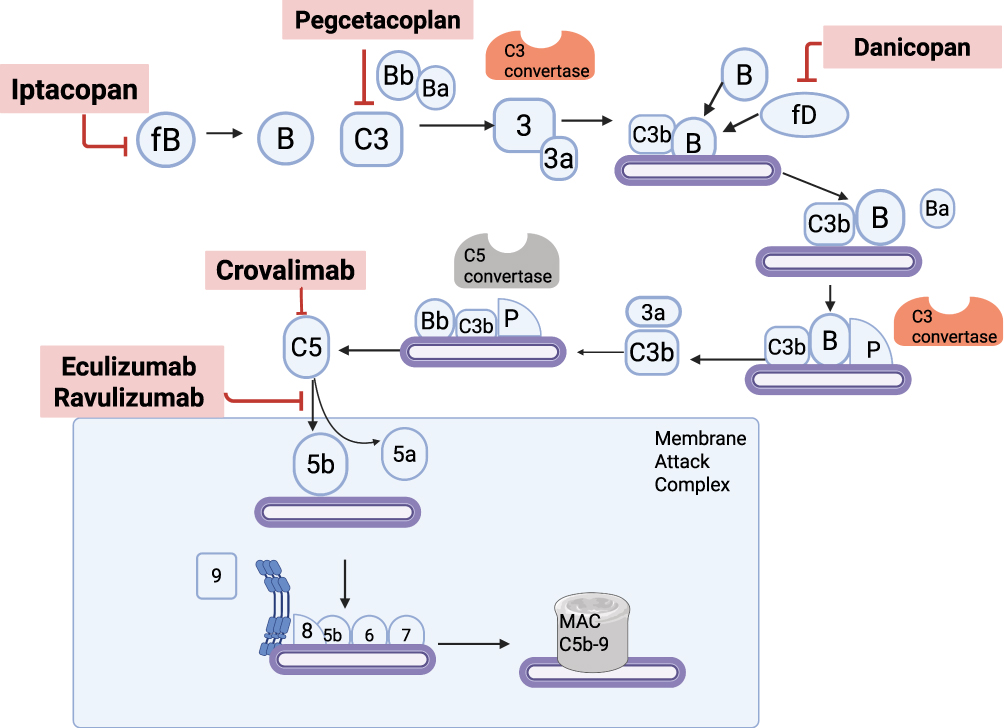 |
Figure 2 The alternative complement pathway and sites of therapeutic inhibition. This schematic illustrates key steps in the activation and amplification of the alternative complement pathway, culminating in formation of the membrane attack complex (MAC, C5b-9). Multiple complement inhibitors target distinct components of this cascade to prevent overactivation and downstream hemolysis. Iptacopan inhibits Factor B, blocking formation of the C3 convertase (C3bBb). Danicopan inhibits Factor D, preventing the cleavage of Factor B. Pegcetacoplan inhibits C3, halting the upstream cascade. Eculizumab and ravulizumab bind to C5, preventing its cleavage into C5a and C5b. Crovalimab also targets C5, but binds to a distinct epitope. Created in BioRender. tamdin, t. (2025) https://BioRender.com/4o0gznd. Abbreviations: fB, Factor B; fD, Factor D; P, Properdin; Ba/Bb, Inactive/active fragments of Factor B; C3, C5, Complement components 3 and 5; C3a, C5a, Anaphylatoxins released during cleavage; C3b, C5b, Active fragments contributing to convertase or MAC; C3 convertase, C3bBb; C5 convertase, C3bBbC3b or C3bBbP; MAC, Membrane Attack Complex (C5b-9). |
Eculizumab established a new treatment paradigm through its mechanism of blocking terminal complement activation to prevent MAC formation. The TRIUMPH trial3 demonstrated the clinical benefit of eculizumab, with intravascular hemolysis decreasing by 85% based on lower LDH levels; transfusion requirements decreased dramatically. 50% of patients receiving eculizumab achieved stable hemoglobin levels without transfusions; the placebo group showed no such response. Patients treated with eculizumab experienced both reduced hemolysis rates and major improvements in quality of life along with a significant decrease in thrombotic events from 33% to less than 5%.24 Two eculizumab biosimilars are now available (Table 1). Their safety and efficacy are comparable to the original drug, but the biosimilars offer cost savings of ~10-30%.
Although the occurrence of thrombotic events has decreased, thrombosis remains a major reason for complications leading to death in PNH patients.18 These events tend to occur in unusual venous sites, so healthcare providers need to consider occult thrombosis in patients with suspicious symptoms. Patients who have aplastic anemia–PNH as an overlapping bone marrow failure syndrome fail to achieve complete treatment benefits from eculizumab. The medication eliminates hemolysis, but fails to normalize hematopoiesis so these patients may need ongoing blood transfusions even when receiving eculizumab.22 The limited response to complement inhibition in such cases indicates that marrow failure plays a dominant role in causing anemia in these patients.
More recent research suggests that additional mutations, such as those in DNMT3A or BCOR, may occur within the same clone harboring the PIGA mutation, or in separate hematopoietic clones, reflecting the clonal complexity often seen in PNH. Patients with PNH may have genetic mutations in DNMT3A and BCOR genes which are typical for clonal hematopoiesis and myeloid malignancies.6,25 These mutations appear either before or alongside PIGA mutations and help drive clonal evolution which may affect both short-term disease progression and long-term patient outcomes, including the development of MDS or AML.
The understanding of PNH pathophysiology has led to the development of additional complement inhibitors. For example, ravulizumab as a long-acting C5 inhibitor delivers comparable effectiveness through its 8-week dosing schedule (Figure 3). The oral factor B inhibitor iptacopan shows promise as a treatment for patients who have persistent anemia after receiving eculizumab. Iptacopan demonstrates the ability to manage both intravascular and extravascular hemolysis, as well as enhance hemoglobin values in this specific group of patients.9
 |
Figure 3 Illustrative trough-level suppression: eculizumab vs ravulizumab. Changes in serum C5 inhibitor concentrations with eculizumab dosing every two weeks and ravulizumab dosing every eight weeks. The graph displays pharmacokinetic trends through arbitrary units that do not represent actual serum values. The dashed red line shows the lowest concentration required to block terminal complement activity effectively. The drug ravulizumab maintains its trough-level suppression throughout its dosing interval, but eculizumab may drop below therapeutic levels before the next dose which could lead to breakthrough hemolysis in specific patients. |
Ravulizumab
Ravulizumab functions as a long-acting C5 inhibitor (Figure 2) which was developed to overcome two major eculizumab limitations in PNH treatment: the requirement for biweekly infusions and intermittent hemolytic events due to suboptimal trough levels26 (Figure 3). The phase 3 clinical trials (PNH 301 and 302) demonstrated that ravulizumab provided equivalent efficacy to eculizumab for both new patients and those previously receiving eculizumab. The medication achieved equivalent results for all main study endpoints which included transfusion prevention, LDH normalization and hemoglobin control, and fatigue symptom reduction.9,26 Breakthrough hemolysis occurred less frequently among patients treated with ravulizumab, with 4.0% of treatment-naïve patients experiencing this compared to 10.7% of patients on eculizumab.26 The switch trial demonstrated that no breakthrough hemolysis occurred in ravulizumab patients, but eculizumab patients developed 5.1% of such events.27
The safety profile of ravulizumab matched that of eculizumab with no difference in serious adverse events occurring during treatment. There were no meningococcal infections, and headache side effects appeared at equivalent frequencies between both treatment groups. Ravulizumab provides an easier and potentially more enduring solution for PNH management because it only requires dosing every 8 weeks while maintaining consistent C5 inhibition. The residual anemia or ongoing transfusion needs in some patients may be due to the fact that ravulizumab does not address C3-mediated extravascular hemolysis, similar to eculizumab. Ravalizumab still requires intravenous administration and carries the typical infection risks seen with terminal complement blockade. These remaining challenges have led to growing interest in targeting earlier steps of the complement cascade—particularly at the level of C3 or factor B—as a strategy to provide more complete control of hemolysis.
Crovalimab
Crovalimab functions as a next-generation anti-C5 monoclonal antibody which delivers long-acting effects through subcutaneous self-injection at four-week intervals using low-volume doses (Figure 2). The COMMODORE phase 3 trials demonstrated that crovalimab provided equivalent treatment outcomes as eculizumab among patients who received complement inhibitors for the first time and those transitioning from eculizumab treatment.7 Patients achieved equivalent control of hemolysis, transfusion avoidance, and hemoglobin stabilization. A small number of patients developed mild immune-complex reactions following therapy changes, but all patients recovered without medical intervention.7 This clinical trial showed that crovalimab had the same safety profile as eculizumab without introducing any new safety concerns.
The monthly dosing and self-administration features of crovalimab may be attractive to patients who want more convenient treatment that does not require frequent hospital visits. The trial results showed that crovalimab was preferred by more than 80% of participants compared to IV eculizumab. Crovalimab shares the same limitation as other terminal complement inhibitors because it fails to stop C3-mediated extravascular hemolysis, which means that it may not completely resolve anemia in specific patients. Nevertheless, the combination of strong efficacy and safety and user-friendly design makes crovalimab an attractive option for PNH patients.
Under C5 blockade, upstream activation persists, depositing C3b/C3d fragments on PNH erythrocytes. These opsonized cells are then cleared by hepatic and splenic macrophages, producing extravascular hemolysis (EVH) despite adequate terminal-pathway inhibition.28 This biology provides the rationale for proximal inhibition: blocking C3 (Pegcetacoplan)8 prevents opsonization, while inhibiting the alternative pathway (factor B with iptacopan, factor D with danicopan) reduces amplification and downstream C3 fragment deposition, aiming for comprehensive control of both IVH and EVH.9,10 The newer drugs aim to manage both intravascular and extravascular hemolysis more effectively, while also offering alternative routes of administration and greater flexibility in individualizing therapy.
Proximal Complement Inhibitors
C5 inhibition was the first therapeutic breakthrough in PNH and remains foundational. Eculizumab, ravulizumab, and the newer subcutaneous agent crovalimab share a terminal-pathway mechanism, preventing C5 cleavage and membrane attack complex formation, to suppress intravascular hemolysis and markedly reduce thrombosis. Key differences are dosing interval, route, and epitope binding (with crovalimab retaining activity in certain rare C5 variants unresponsive to eculizumab). Class limitations include ongoing C3 deposition on erythrocytes and extravascular hemolysis (EVH), which motivate proximal-pathway strategies.
Pegcetacoplan
Pegcetacoplan is a C3 inhibitor that acts upstream of the C5 inhibitors: eculizumab, ravulizumab, and crovalimab, to block both intravascular and extravascular hemolysis (Figure 2) in contrast to the terminal complement inhibitors that do not block extravascular hemolysis.8 The subcutaneous administration of pegcetacoplan twice per week during the phase 3 PEGASUS trial produced better outcomes than intravenous eculizumab in patients who needed anemia treatment beyond C5 inhibitor monotherapy. Treatment with pegcetacoplan resulted in major improvements of hemoglobin levels and achieved higher transfusion independence rates since 85% of patients needed no transfusions; however, 15% of eculizumab patients required transfusions.8 The PRINCE trial showed that pegcetacoplan therapy achieved transfusion independence in 91% of complement inhibitor–naïve patients, whereas this endpoint was only achieved by 6% of patients receiving best supportive care (without complement inhibition).29 Patients can self-administer pegcetacoplan subcutaneously at home which removes the burden for hospital visits. This therapy poses infection risks, but Hillmen et al30 reported no meningococcal infections in trial participants who received proper vaccinations. The most frequent adverse events with pegcetacoplan included gastrointestinal problems such as diarrhea and injection site reactions. The drug demonstrated comparable safety aspects as C5 inhibitors. Interest in oral complement inhibitors (iptacopan, danicopan), and long-acting subcutaneous complement inhibitors like crovalimab has emerged because healthcare providers and PNH patients want more flexible and individualized treatment approaches.
Iptacopan
Iptacopan functions as an oral first-in-class complement factor B inhibitor which targets the alternative pathway to manage both intravascular and extravascular hemolysis in PNH patients (Figure 2).9 The APPLY-PNH trial showed that iptacopan monotherapy was more effective than standard C5 inhibitors (eculizumab or ravulizumab) in patients with persistent anemia. Iptacopan treatment resulted in hemoglobin increases of at least 2 g/dL for 85% of patients, while all patients who received C5 inhibitors required transfusions.31 Iptacopan therapy led to significant improvements in hemolysis markers and fatigue scores, and 95% of patients did not require transfusions throughout the 24-week study period.
The oral delivery method of iptacopan provides patients with an easy-to-use treatment option that differs from standard IV and SC-based therapies. Headache appeared as the most frequent adverse event, and complement inhibitors continue to pose infection risks, but no new safety signal emerged. These results demonstrate that iptacopan therapy provides both effective hemolysis management and user-friendly treatment for patients. The complement-inhibitor–naïve patient population in the APPOINT-PNH study experienced rapid and sustained hemoglobin improvement along with high transfusion independence rates, as demonstrated by Peffault de Latour et al.31 These results support the use of iptacopan as an initial oral monotherapy for PNH.
Danicopan
The oral drug danicopan functions as an inhibitor of complement factor D which plays a central role in the alternative pathway before C3 (Figure 2). The phase 3 ALPHA trial evaluated danicopan as an addition to ongoing C5 therapy for PNH patients who had persistent anemia. The combined treatment led to major improvements in hemoglobin levels together with reduced transfusion needs without creating additional safety issues.10 Danicopan serves as an effective dual inhibition treatment because it targets both intravascular and extravascular hemolysis. The oral dosing requirement of danicopan as a three times daily drug along with IV C5 therapy administration presents potential limitations in convenience compared to single-agent treatment options. Additionally, this dual therapy comes at a substantially higher cost (Table 1).
Complement Inhibition in Other Hemolytic Disorders
Autoimmune Hemolytic Anemia (AIHA) and Cold Agglutinin Disease (CAD)
AIHA comprises warm AIHA (wAIHA) and cold agglutinin disease (CAD), two biologically distinct entities that require different treatment; CAD is predominantly complement-mediated and therefore the clearest use-case for complement inhibitors. Red blood cells are targets of autoantibodies in autoimmune hemolytic anemia (AIHA). Warm antibody AIHA (wAIHA) features RBC antibodies which bind at room temperature leading to spleen-based extravascular hemolysis. Intravascular red cell destruction results from complement activation when patients have complement-fixing antibodies. Cold agglutinin disease (CAD) is characterized by the presence of IgM antibodies which attach to RBCs at low temperatures thereby activating the complement cascade and resulting primarily in complement-mediated intravascular hemolysis, though some C3b-opsonized red blood cells may undergo extravascular clearance by hepatic macrophages.
The drug class of complement-targeted treatments has shown effectiveness in AIHA patients, particularly in CAD patients because their hemolysis is mainly driven by uncontrolled complement activation.32 Although patients with PNH have benefited greatly from C5 inhibitors such as eculizumab and ravulizumab which decrease terminal complement-mediated intravascular hemolysis, effectiveness of eculizumab in AIHA treatment, especially CAD is limited because it requires IV infusion every two weeks and does not inhibit complement activation at C3 which results in only minor improvements in hemoglobin levels.33
Sutimlimab is a monoclonal antibody that inhibits C1s which stops classical complement pathway activation at its starting point. Through this mechanism, the drug inhibits C3b deposition and MAC formation in CAD. Sutimlimab therapy resulted in long-lasting increases in hemoglobin levels together with reduced transfusion needs in two major phase 3 clinical trials: CARDINAL (studying transfusion-dependent CAD patients) and CADENZA (investigating CAD patients without recent transfusions).34,35 A total of 54–73% of patients treated with sutimlimab reached their primary composite endpoints, whereas only ~15% of placebo recipients met this endpoint.34,35 As a result of these trials, sutimlimab became the first FDA-approved treatment for CAD.
The complement-inhibiting drug, pegcetacoplan which was initially developed for PNH treatment has shown effectiveness in treating AIHA patients. An open-label Phase II trial with CAD and wAIHA patients showed that pegcetacoplan increased hemoglobin values by 2.4 g/dL in CAD patients and 1.7 g/dL in wAIHA patients during a 48-week study period.32 This treatment requires a subcutaneous injection or infusion pump administration which gives patients an easier treatment experience compared to eculizumab. No serious complement-related adverse events occurred during the study, but injection site reactions were a primary side effect reported by patients.32
Safety remains an important consideration with all complement inhibitors. All of these medications elevate infection risks, especially for encapsulated pathogens, yet sutimlimab’s targeted classical pathway block may preserve alternative and lectin pathway activity, thus minimizing the risk of infections.35
The use of complement inhibitors is being studied in AIHA for applications beyond CAD treatment. C5 inhibitors have been used successfully in case reports for treating severe refractory wAIHA when patients did not respond to standard immunosuppressive treatments.36 Investigational research is also focused on applying complement-targeted treatments in hereditary spherocytosis and sickle cell disease because complement activation may worsen the hemolytic burden in these disorders, but clinical evidence of drug benefit in these disorders remains limited.
Challenges
Breakthrough Hemolysis (BTH)
The most well-known side effect of complement inhibition therapy is breakthrough hemolysis (BTH), which is hemolysis that persists while the patient is receiving complement inhibitors. Complement activation can be initiated by infections, trauma, or surgery. Although eculizumab can control hemolysis effectively in most patients, approximately 5–15% of patients have hemolysis recurrence before their next dose. These episodes are generally characterized by elevated LDH values and the return of typical PNH symptoms. Common management approaches include changing the dosing frequency or increasing the dose to maintain drug concentrations within the therapeutic range.37 “Pharmacodynamic breakthrough” is another cause of sudden and unpredictable increase in complement activation that occurs during periods of severe complement activation, which may be triggered by infections or surgery that exceeds the inhibitory capacity of C5 inhibitors38 (Figure 3). Even though eculizumab levels are appropriate, these episodes mimic traditional PNH hemolytic crises.39 The occurrence of BTH has been reduced with the use of newer agents. Because of its weight-based dosing and extended 8-week dosing interval, ravulizumab provides a more stable and sustained inhibition of C5, significantly reducing end-of-dose hemolysis as compared to eculizumab’s 2-week dosing interval.40 Also, crovalimab, a subcutaneous anti-C5 monoclonal antibody, was found to be noninferior to eculizumab in Phase III trials, with a relatively low (~10%) rate of BTH.41 In fact, crovalimab binds to a different epitope on C5, allowing it to remain effective in the rare cases of eculizumab resistance due to C5 mutations like p.Arg885His.42
Two patterns of breakthrough hemolysis are recognized. Pharmacokinetic BTH occurs when drug concentrations fall near end-of-dose; mitigation involves dose/interval optimization or agents with longer half-life (eg, ravulizumab). Pharmacodynamic BTH occurs when acute triggers (infection, surgery, trauma) generate complement activation that exceeds inhibitory capacity despite therapeutic levels; management focuses on trigger control and, when recurrent, escalation to upstream therapy (C3 or alternative-pathway blockade).
Research in this area is presently centered on optimizing dosing schedules, enhancing drug delivery systems, and designing the next generation of C5 inhibitors that will provide more consistent and prolonged complement inhibition. The goal is to abolish both PK- and PD-related BTH, thus guaranteeing better disease control and quality of life for PNH patients. A practical caveat of proximal complement inhibition is that, by suppressing both intravascular and C3-mediated extravascular hemolysis, the survival of circulating PNH erythrocytes increases, and clinical studies of C3 blockade show expansion of the circulating PNH RBC fraction—a shift that can modify baseline hemolysis and the phenotype of breakthrough events. Breakthrough hemolysis—whether pharmacokinetic or precipitated by complement-amplifying triggers—may itself promote thrombosis, underscoring the need for rapid trigger control and optimized complement blockade. Given heterogeneity in thrombotic risk, antithrombotic strategies should be individualized.43,44
Extravascular Hemolysis
While eculizumab and ravulizumab provide robust control of intravascular hemolysis through terminal complement inhibition at C5, they fail to prevent upstream complement activity that results in C3b fragment deposition on PNH erythrocytes. This results in persistent extravascular hemolysis, as opsonized red cells are cleared by macrophages in the reticuloendothelial system. As a result, many patients continue to experience anemia despite adequate C5 blockade. The opsonization of red cells by C3b fragments results in their continued destruction by macrophages in the reticuloendothelial system which causes persistent extravascular hemolysis.45 The clinical data indicate that eculizumab treatment leads to hemoglobin normalization in 10–33% of patients while many patients still need blood transfusions.38
Extravascular hemolysis in PNH can be identified by laboratory markers such as elevated indirect bilirubin, increased reticulocyte count and a positive direct antiglobulin test for C3d.45 These findings indicate ongoing opsonization despite terminal complement blockade.10 New therapeutic approaches aim to address this limitation by blocking proximal components of the complement cascade to prevent C3 activation and extravascular hemolysis. The C3 inhibitor pegcetacoplan in the PEGASUS trial outperformed eculizumab by increasing mean hemoglobin levels by 3.8 g/dL and decreasing transfusion requirements.8,10 Similarly, the addition of danicopan, a factor D inhibitor, to standard C5 therapy resulted in a 2.4 g/dL increase in hemoglobin levels among patients with residual anemia.10 These findings demonstrate that proximal complement inhibition controls hemolysis better and improves hematologic outcomes in PNH.
Infection Risk
Complement inhibitors in patients trigger major infection risks because the complement system functions as a primary defense mechanism against bacteria through both opsonization and bacterial lysis. C5 inhibitors, eculizumab and ravulizumab, block the production of MAC which is essential for preventing Neisseria species infection.46 These patients develop invasive meningococcal disease at significantly higher rates than the general population while taking these therapies, even after receiving vaccination.47
A 10-year global pharmacovigilance study of PNH patients treated with eculizumab revealed meningococcal infection rates were 0.25 cases per 100 patient-years together with a fatal outcome occurring in 10% of cases.48 The data indicate that patients must absolutely adhere to infection prevention protocols that include mandatory vaccination before beginning therapy. Patients undergoing C5 inhibitor treatment should also receive antibiotic prophylaxis with penicillin daily as part of their standard protocol.47,48 Proximal complement inhibitors which target C3 and the alternative pathway create a higher complexity of risk since they also block C3b opsonization and increase patient susceptibility to encapsulated pathogens. Patients must receive complete vaccination against Streptococcus pneumoniae, Haemophilus influenzae type B, and Neisseria according to the black-box label warning of pegcetacoplan before beginning therapy.46 The 48-week APPLY-PNH trial of iptacopan which is a factor B inhibitor did not report any serious infections because the trial used strict vaccination procedures.9 The need for real-world surveillance persists. Patients who undergo complement blockade treatment have also experienced rare bacterial infections from Neisseria gonorrhoeae, Capnocytophaga, and other encapsulated bacteria.48
Comprehensive infection prevention practices form an essential part of the medical care provided to PNH patients who receive complement inhibitors. The safe long-term use of these life-changing therapies depends on a complete approach which combines vaccination with patient education, antibiotic use, and registry-based safety monitoring.48
Monitoring Complement Inhibition
The cornerstone of PNH management relies on laboratory monitoring which evaluates complement inhibition and clinical effectiveness. The monitoring of PNH requires regular laboratory tests of LDH levels together with bilirubin because these tests reflect disease activity. Breakthrough hemolysis or insufficient complement suppression becomes apparent when any of these markers increase, especially when LDH levels rise.38 CH50 has limited contemporary utility and is largely superseded in many laboratories; when used, it should be interpreted cautiously. Patients who have elevated complement turnover require trough drug level measurements to enable personalized dosing.38 Flow-cytometric monitoring of PNH clone size at baseline and periodically is recommended and considered standard practice, informing disease burden, response, and clonal evolution.44,49
The assessment of upstream complement activity requires specific tools such as the Alternative Pathway Hemolytic 50% (AH50) assay which measures the functional activity of the alternative complement pathway, along with C3/C3d ratios, and emerging biomarkers including C5a and sC5b-9 when using proximal complement inhibitors like C3 or factor B blockers.50 In the context of factor D inhibition, it is indirectly assessed through biomarkers such as Bb fragment levels, LDH, and C3 deposition.50 These parameters have been validated as reliable indicators of alternative pathway inhibition in preclinical studies and the phase II trial of danicopan. While direct measurement of factor D levels is not routinely performed, its functional blockade is inferred from these biomarkers.51
The evaluation of bone marrow failure becomes necessary whenever cytopenias persist despite controlled hemolysis, thus requiring a complete monitoring system that extends beyond hemolysis assessment.
Personalised Medicine
As the treatment landscape for PNH continues to expand, a personalized approach is becoming increasingly important. Tailoring therapy begins with identifying the dominant type of hemolysis and evaluating the patient’s response to initial C5 inhibition. For example, a patient who has good LDH control and stable hemoglobin levels with eculizumab may do well with continued C5 monotherapy—perhaps transitioning to ravulizumab for its extended 8-week dosing schedule. On the other hand, a patient with ongoing anemia and elevated bilirubin—despite C5 inhibition—likely has significant C3-mediated extravascular hemolysis. In such cases, switching to a proximal complement inhibitor may offer better control. The choice can be individualized based on preferences and lifestyle: pegcetacoplan (a C3 inhibitor) offers robust control via twice-weekly subcutaneous administration, while iptacopan (a factor B inhibitor) is a convenient oral alternative.31
In the APPLY-PNH trial, nearly 94% of patients who transitioned from C5 inhibitors to iptacopan monotherapy remained transfusion-free and achieved hemoglobin levels near 12 g/dL,31 reinforcing its potential in patients with suboptimal responses to terminal pathway blockade. Pharmacogenomics also plays a role in personalizing therapy. For instance, patients with the rare C5 variant p.Arg885His do not respond to eculizumab, but may benefit from agents such as crovalimab, which targets a different C5 epitope, or from upstream inhibitors that bypass C5 altogether,41,42
Importantly, underlying bone marrow function must be considered. Many patients with PNH have overlapping aplastic anemia or myelodysplastic syndromes, limiting erythropoietic capacity. These patients may require concurrent therapies such as immunosuppressants, erythropoiesis-stimulating agents, or even hematopoietic stem cell transplantation alongside complement inhibition.38 Looking ahead, integrating biomarkers and genetic profiles into clinical decision-making will help stratify patients more effectively. Baseline C3 deposition, complement protein variants, or clonal mutation profiles may soon help predict which patients will do well with C5 inhibition alone, and those who may need upfront combination or proximal inhibition strategies. With multiple therapeutic drug classes now available—targeting C5, C3, factor D, and factor B—PNH management can move away from a “one-size-fits-all” approach toward truly individualized care, tailored to each patient’s biology, clinical course, and preferences.
Conclusion and Future Directions
Current PNH management is anchored by C5 inhibition, with proximal complement strategies (C3, factor B, factor D) expanding disease control; supportive measures - vaccination/infection mitigation and periodic flow-cytometric clone monitoring remain essential, and the same therapeutic logic extends to complement-mediated AIHA, particularly CAD and, more selectively, wAIHA. Future research in PNH is directed toward enhancing complement blockade, reducing residual thrombotic risk, and pursuing curative gene-based approaches, with current trials evaluating C5–proximal combinations as a leading strategy.
In addition to the agents reviewed above, as of September 2025, several investigational complement inhibitors for PNH are under study; tesidolumab (anti-C5 antibody),52 zilucoplan (subcutaneous C5 inhibitor),53 vemircopan/ALXN2050 (oral factor D inhibitor),54 and nomacopan/Coversin (dual C5 and leukotriene B4 inhibitor)55 have completed their pivotal studies and remain investigational, with no current approval for PNH. These programs are noted briefly for completeness, as their development has transitioned to follow-up analyses rather than active enrollment. In contrast, pozelimab in combination with cemdisiran (anti-C5 antibody plus siRNA targeting hepatic C5 synthesis)56 remains in active clinical development and represents the principal ongoing program of novel PNH treatments.
Emerging therapeutic strategies in PNH are also exploring upstream complement targets such as mannan-binding lectin-associated serine protease-2 (MASP-2)57 and mannan-binding lectin-associated serine protease-3 (MASP-3). MASP-2 drives lectin pathway activation, while MASP-3 links the lectin and alternative pathways by activating pro–Factor D.5 Inhibition of MASP-3, currently under investigation with agents like OMS906, offers a promising approach to attenuate alternative pathway activity and reduce hemolysis.58
Research findings indicate that dual therapy provides enhanced benefits to refractory patients through its ability to decrease hemolysis and minimize thrombotic risks.10 Research continues to evaluate the sustainability of these treatment regimens as well as their potential to be reduced to monotherapy. The development of crovalimab (subcutaneous monthly administration) and iptacopan (oral administration) has decreased the treatment burden for patients,59 while biosimilars have the potential to reduce treatment costs (Table 1).
The management of thrombosis persists as a significant issue even when hemolysis is successfully controlled. Research efforts are focused on investigating complement inhibition effects on coagulation pathways while testing anticoagulant and anti-inflammatory medications as potential thrombotic risk reduction agents. Research on long-term outcomes of PNH treatment has become essential because the condition now exists as a chronic illness instead of a fatal disease. Patients treated with ravulizumab exhibit improved quality of life and greater treatment satisfaction compared to those receiving eculizumab, as evidenced by patient preferences favoring ravulizumab’s less frequent dosing schedule and associated lifestyle benefits.60
Gene therapy and CRISPR-mediated correction of PIGA mutations represent potential curative approaches for stem cell-level cures, but remain in preliminary development. C5 inhibition remains the backbone of PNH care, while recognition of C3-opsonization and EVH has shifted goals toward comprehensive hemolysis control. Proximal strategies (C3, factor B, factor D) improve hemoglobin and transfusion independence and can be used alone or in combination when EVH persists or breakthrough hemolysis recurs. Care should include vaccination/infection-risk mitigation and periodic flow-cytometric clone monitoring, with simplified oral or infrequent SC regimens emerging. These principles also inform treatment of autoimmune hemolytic anemias—especially CAD, where complement dependence is strongest.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lopez MA, Rare Disease Advisor. Paroxysmal nocturnal hemoglobinuria epidemiology. 2022 [cited April 26, 2025]. Available from: https://www.rarediseaseadvisor.com/disease-info-pages/paroxysmal-nocturnal-hemoglobinuria-epidemiology/.
2. Ware RE, Rosse WF, Howard TA. Mutations within the Piga gene in patients with paroxysmal nocturnal hemoglobinuria. Blood. 1994;83(9):2418–2422. doi:10.1182/blood.V83.9.2418.2418
3. Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233–1243. doi:10.1056/NEJMoa061648
4. Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood. 2019;133(6):540–549. doi:10.1182/blood-2018-09-876805
5. Cohn-Emery D, Targeted Oncology. MASP-3 inhibition shows efficacy in PNH proof-of-concept study. 2023 [cited May 12, 2025]. Available from: https://www.targetedonc.com/view/masp-3-inhibition-shows-efficacy-in-pnh-proof-of-concept-study.
6. Awada H, Rahman S, Durrani J, et al. Leukemia evolving from paroxysmal nocturnal hemoglobinuria. Leukemia. 2019;34(1):327–329. doi:10.1038/s41375-019-0555-0
7. Scheinberg P, Clé DV, Kim JS, et al. Phase 3 randomized COMMODORE 1 trial: crovalimab versus eculizumab in complement inhibitor-experienced patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2024;99(9):1757–1767. doi:10.1002/ajh.27413
8. Hillmen P, Szer J, Weitz I, et al. Pegcetacoplan versus eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2021;384(11):1028–1037. doi:10.1056/NEJMoa2029073
9. Risitano AM, Kulasekararaj A, Roeth A, et al. Factor B inhibition with oral iptacopan monotherapy demonstrates sustained long-term efficacy and safety in anti-C5–treated patients with PNH and persistent anemia: final 48-week results from the Phase III APPLY-PNH trial. Blood. 2023;142(Suppl 1):571. doi:10.1182/blood-2023-180780
10. Lee JW, Griffin M, Kim JS, et al. Addition of danicopan to ravulizumab or eculizumab in patients with paroxysmal nocturnal haemoglobinuria and clinically significant extravascular haemolysis (ALPHA): a double-blind, randomised, phase 3 trial. Lancet Haematol. 2023;10(12):e955–65. doi:10.1016/S2352-3026(23)00315-0
11. Nicholson-Weller A, Spicer DB, Austen KF. Deficiency of the complement regulatory protein, decay-accelerating factor, on membranes of granulocytes, monocytes, and platelets in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1985;312(17):1091–1097. doi:10.1056/NEJM198504253121704
12. Holguin MH, Wilcox LA, Bernshaw NJ, Rosse WF, Parker CJ. Relationship between the membrane inhibitor of reactive lysis and the erythrocyte phenotypes of paroxysmal nocturnal hemoglobinuria. J Clin Invest. 1989;84(5):1387–1394. doi:10.1172/JCI114311
13. Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J. 1994;13(1):110–117. doi:10.1002/j.1460-2075.1994.tb06240.x
14. Heesterbeek DAC, Angelier ML, Harrison RA, Rooijakkers SHM. Complement and bacterial infections: from molecular mechanisms to therapeutic applications. J Innate Immun. 2018;10(5–6):455–464. doi:10.1159/000491439
15. Parker CJ. Paroxysmal nocturnal hemoglobinuria. Curr Opin Hematol. 2012;19(3):141–148. doi:10.1097/MOH.0b013e328351c348
16. Sonavane A, Saigal S, Sood N, Choudhary N. A rare case of Budd–Chiari syndrome secondary to paroxysmal nocturnal hemoglobinuria: salvaged with TIPSS. Trop Gastroenterol. 2020;41(3):190–193.
17. Chatzileontiadou S, Hatjiharissi E, Angelopoulou M, et al. Thromboembolic events in patients with paroxysmal nocturnal hemoglobinuria: real-world data of a Greek nationwide multicenter retrospective study. Front Oncol. 2023;13:1128994. doi:10.3389/fonc.2023.1128994
18. Höchsmann B, Peffault de Latour R, Hill A, et al. Risk factors for thromboembolic events in patients with paroxysmal nocturnal hemoglobinuria: a nested case–control study in the international PNH registry. Ann Hematol. 2023;102(11):2979–2988. doi:10.1007/s00277-023-05402-3
19. Brodsky RA. Advances in the diagnosis and therapy of paroxysmal nocturnal hemoglobinuria. Blood Rev. 2008;22(2):65–74. doi:10.1016/j.blre.2007.10.002
20. Sun L, Babushok DV. Secondary myelodysplastic syndrome and leukemia in acquired aplastic anemia and paroxysmal nocturnal hemoglobinuria. Blood. 2020;136(1):36–49. doi:10.1182/blood.2019000940
21. Naqvi E, Rare Disease Advisor. Paroxysmal nocturnal hemoglobinuria complications. Available from: https://www.rarediseaseadvisor.com/disease-info-pages/paroxysmal-nocturnal-hemoglobinuria-complications/.
22. Al-Ani F, Chin-Yee I, Lazo-Langner A. Eculizumab in the management of paroxysmal nocturnal hemoglobinuria: patient selection and special considerations. Ther Clin Risk Manag. 2016;12:1161–1170. doi:10.2147/TCRM.S96720
23. Sørensen AL, Hansen DL, Frederiksen H. Early mortality in paroxysmal nocturnal hemoglobinuria. Cureus. 2023;15(10):e47225. doi:10.7759/cureus.47225
24. Hillmen P, Muus P, Dührsen U, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110(12):4123–4128. doi:10.1182/blood-2007-06-095646
25. Lee SCW, Abdel-Wahab O. The mutational landscape of paroxysmal nocturnal hemoglobinuria revealed: new insights into clonal dominance. J Clin Invest. 2014;124(10):4227–4231. doi:10.1172/JCI77984
26. Lee JW, Sicre de Fontbrune F, Wong LL, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naïve to complement inhibitors: the 301 study. Blood. 2019;133(6):530–539. doi:10.1182/blood-2018-09-876136
27. Kulasekararaj AG, Griffin M, Langemeijer S, et al. Long-term safety and efficacy of ravulizumab in patients with paroxysmal nocturnal hemoglobinuria: 2-year results from two pivotal phase 3 studies. Eur J Haematol. 2022;109(3):205–214. doi:10.1111/ejh.13783
28. Risitano AM, Notaro R, Marando L, et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009;113(17):4094–4100. doi:10.1182/blood-2008-11-189944
29. Wong RSM, Navarro-Cabrera JR, Comia NS, et al. Pegcetacoplan controls hemolysis in complement inhibitor–naïve patients with paroxysmal nocturnal hemoglobinuria. Blood Adv. 2023;7(11):2468–2478. doi:10.1182/bloodadvances.2022009129
30. Hillmen P, Risitano AM, Peffault de Latour R. Pegcetacoplan versus eculizumab in PNH. N Engl J Med. 2021;385(18):1725–1726. doi:10.1056/NEJMc2106424
31. Peffault de Latour R, Röth A, Kulasekararaj AG, et al. Oral iptacopan monotherapy in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2024;390(11):994–1008. doi:10.1056/NEJMoa2308695
32. Roman E, Fattizzo B, Shum M, et al. Safety and efficacy of pegcetacoplan treatment for cold agglutinin disease and warm antibody autoimmune hemolytic anemia. Blood. 2025;145(4):397–408. doi:10.1182/blood.2023022549
33. Röth A, Bommer M, Hüttmann A, et al. Eculizumab in cold agglutinin disease (DECADE): an open-label, prospective, bicentric, nonrandomized phase 2 trial. Blood Adv. 2018;2(19):2543–2549. doi:10.1182/bloodadvances.2018024190
34. Röth A, Barcellini W, D’Sa S, et al. Sutimlimab in cold agglutinin disease. N Engl J Med. 2021;384(14):1323–1334. doi:10.1056/NEJMoa2027760
35. Röth A, Berentsen S, Barcellini W, et al. Sutimlimab in patients with cold agglutinin disease: results of the randomized placebo-controlled phase 3 CADENZA trial. Blood. 2022;140(9):980–991. doi:10.1182/blood.2021014955
36. Ma K, Caplan S. Refractory IgG warm autoimmune hemolytic anemia treated with eculizumab: a novel application of anticomplement therapy. Case Rep Hematol. 2016;2016:9181698. doi:10.1155/2016/9181698
37. Brodsky RA, de Latour RP, Rottinghaus ST, et al. Characterization of breakthrough hemolysis events observed in the phase 3 randomized studies of ravulizumab versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Haematologica. 2021;106(1):230–237. doi:10.3324/haematol.2019.236877
38. Risitano AM, Marotta S, Ricci P, et al. Anti-complement treatment for paroxysmal nocturnal hemoglobinuria: time for proximal complement inhibition? A position paper from the SAAWP of the EBMT. Front Immunol. 2019;10:1157. doi:10.3389/fimmu.2019.01157
39. Sica M, Rondelli T, Ricci P, De Angioletti M, Risitano AM, Notaro R. Eculizumab treatment: stochastic occurrence of C3 binding to individual PNH erythrocytes. J Hematol Oncol. 2017;10(1):1–10. doi:10.1186/s13045-017-0496-x
40. Griffin M, Gandhi S, Kelly RJ, et al. Terminal complement inhibition and control of hemolysis in patients with paroxysmal nocturnal hemoglobinuria who switched from high-dose eculizumab to ravulizumab: a Phase IV, single-arm clinical trial. Haematologica. 2025;110(4):967–971. doi:10.3324/haematol.2024.285553
41. Röth A, Kulasekararaj AG, Scheinberg P, Nishimura JI. Crovalimab in the paroxysmal nocturnal hemoglobinuria treatment landscape. Immunotherapy. 2024;16(20–22):1185–1196. doi:10.1080/1750743X.2024.2433410
42. Young DJ. A growing panoply of options for patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2024;99(9):1667–1669. doi:10.1002/ajh.27426
43. Notaro R, Luzzatto L. Breakthrough hemolysis in PNH with proximal or terminal complement inhibition. N Engl J Med. 2022;387(2):160–166. doi:10.1056/NEJMra2201664
44. Kulasekararaj AG, Kuter DJ, Griffin M, Weitz IC, Röth A. Biomarkers and laboratory assessments for monitoring the treatment of patients with paroxysmal nocturnal hemoglobinuria: differences between terminal and proximal complement inhibition. Blood Rev. 2023;59:101041. doi:10.1016/j.blre.2023.101041
45. Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124(18):2804–2811. doi:10.1182/blood-2014-02-522128
46. Kelly RJ, Nishimori H, Horneff R, et al. Thrombosis and meningococcal infection rates in pegcetacoplan-treated patients with paroxysmal nocturnal hemoglobinuria in clinical trial and postmarketing settings. Res Pract Thromb Haemost. 2024;8(4):102416. doi:10.1016/j.rpth.2024.102416
47. Schaap CCM, Grotens A, de Haan AFJ, Blijlevens NMA, Langemeijer SMC. Infections during eculizumab therapy in a dutch population of patients with paroxysmal nocturnal haemoglobinuria. Clin Microbiol Infect. 2021;27(10):1534–1536. doi:10.1016/j.cmi.2021.06.030
48. Socié G, Caby-Tosi MP, Marantz JL, et al. Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome: 10-year pharmacovigilance analysis. Br J Haematol. 2019;185(2):297–310. doi:10.1111/bjh.15790
49. Davis BH. CLSI H52-A2: Red Blood Cell Diagnostic Testing Using Flow Cytometry.
50. Wehling C, Amon O, Bommer M, et al. Monitoring of complement activation biomarkers and eculizumab in complement-mediated renal disorders. Clin Exp Immunol. 2017;187(2):304–315. doi:10.1111/cei.12890
51. Yuan X, Gavriilaki E, Thanassi JA, et al. Small-molecule factor D inhibitors selectively block the alternative pathway of complement in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Haematologica. 2017;102(3):466–475. doi:10.3324/haematol.2016.153312
52. Nishimura JI, Ando K, Masuko M, et al. Tesidolumab (LFG316) for treatment of C5-variant patients with paroxysmal nocturnal hemoglobinuria. Haematologica. 2022;107(6):1483–1488. doi:10.3324/haematol.2020.265868
53. Kulasekararaj AG, Lehtinen AE, Forsyth C, et al. Phase II trials of zilucoplan in paroxysmal nocturnal hemoglobinuria. Haematologica. 2024;109(3):929–935. doi:10.3324/haematol.2022.281780
54. ClinicalTrials.gov [Internet]. 2019 [cited September 14, 2025]. Identifier NCT04170023. Available from: https://www.clinicaltrials.gov/study/NCT04170023.
55. Kulasekararaj A, Weston-Davies W, Robak T, et al. Long-term nomacopan administration results in complete transfusion Independence in previously transfusion-dependent PNH patients. Blood. 2019;134(Suppl 1):4797. doi:10.1182/blood-2019-125263
56. ClinicalTrials.gov [Internet]. 2023 [cited September 14, 2025]. Identifier NCT05744921. Available from: https://www.clinicaltrials.gov/study/NCT05744921.
57. Jacobs S, Rare Disease Advisor. 2025 [cited May 12, 2025]. Ongoing phase 3 clinical trial evaluates IV Zaltenibart for patients with PNH. Available from: https://www.rarediseaseadvisor.com/news/ongoing-phase-3-clinical-trial-evaluates-iv-zaltenibart-for-patients-with-pnh/.
58. Jacobs S, Rare Disease Advisor. 2023 [cited May 12, 2025]. OMS906 has beneficial impact in paroxysmal nocturnal hemoglobinuria. Available from: https://www.rarediseaseadvisor.com/reports/paroxysmal-nocturnal-hemoglobinuria-oms906-hemoglobin-ldh-rbc-clone-size/.
59. Röth A, He G, Tong H, et al. Phase 3 randomized COMMODORE 2 trial: crovalimab versus eculizumab in patients with paroxysmal nocturnal hemoglobinuria naïve to complement inhibition. Am J Hematol. 2024;99(9):1768–1777. doi:10.1002/ajh.27412
60. Peipert JD, Kulasekararaj AG, Gaya A, et al. Patient preferences and quality of life implications of ravulizumab (every 8 weeks) and eculizumab (every 2 weeks) for the treatment of paroxysmal nocturnal hemoglobinuria. PLoS One. 2020;15(9):e0237497. doi:10.1371/journal.pone.0237497
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Managing Fatigue in Patients with Paroxysmal Nocturnal Hemoglobinuria: A Patient-Focused Perspective
Fattizzo B, Cavallaro F, Oliva EN, Barcellini W
Journal of Blood Medicine 2022, 13:327-335
Published Date: 17 June 2022
Impact of Ravulizumab on Patient Outcomes and Quality of Life in Generalized Myasthenia Gravis
Antozzi C, Mantegazza R
Patient Related Outcome Measures 2023, 14:305-312
Published Date: 18 October 2023
Cost-Utility Analysis Comparing Pegcetacoplan to Anti-C5 Monoclonal Antibodies in the Treatment of Paroxysmal Nocturnal Hemoglobinuria
Di Matteo S, Freilone R, Bruno GM, Notaro R, Moumene S, Martone N, Teruzzi C, Ciccarone A, Colombo GL
ClinicoEconomics and Outcomes Research 2024, 16:225-232
Published Date: 11 April 2024