Back to Journals » International Journal of Nanomedicine » Volume 15
Advanced Mg, Zn, Sr, Si Multi-Substituted Hydroxyapatites for Bone Regeneration
Authors Garbo C, Locs J, D'Este M, Demazeau G, Mocanu A ![]() , Roman C, Horovitz O
, Roman C, Horovitz O ![]() , Tomoaia-Cotisel M
, Tomoaia-Cotisel M ![]()
Received 24 August 2019
Accepted for publication 3 December 2019
Published 13 February 2020 Volume 2020:15 Pages 1037—1058
DOI https://doi.org/10.2147/IJN.S226630
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Corina Garbo,1 Janis Locs,2 Matteo D’Este,3 Gerard Demazeau4,†, Aurora Mocanu,1 Cecilia Roman,5 Ossi Horovitz,1 Maria Tomoaia-Cotisel1,6
1Babes-Bolyai University of Cluj-Napoca, Faculty of Chemistry and Chemical Engineering, Physical Chemistry Centre, Chemical Engineering Department, Cluj-Napoca 400028, Romania; 2Rudolfs Cimdins Riga Biomaterials Innovations and Development Centre of RTU, Institute of General Chemical Engineering, Faculty of Materials Science and Applied Chemistry, Riga Technical University, Riga LV-1007, Latvia; 3AO Research Institute Davos, Davos Platz 7270, Switzerland; 4HPBioTECH, Leognan 33850, France; 5INCDO INOE 2000, Research Institute for Analytical Instrumentation, Cluj-Napoca 400293, Romania; 6Academy of Romanian Scientists, Bucharest 050094, Romania
†Dr. Gerard Demazeau passed away on November 3, 2017
Correspondence: Maria Tomoaia-Cotisel
Babes-Bolyai University of Cluj-Napoca, Faculty of Chemistry and Chemical Engineering, Physical Chemistry Centre, Chemical Engineering Department, 11 Arany Janos Street, Cluj-Napoca 400028, Romania
Tel +40 264 593833
Email [email protected]
Purpose: Compositional tailoring is gaining more attention in the development of advanced biomimetic nanomaterials. In this study, we aimed to prepare advanced multi-substituted hydroxyapatites (ms-HAPs), which show similarity with the inorganic phase of bones and might have therapeutic potential for bone regeneration.
Materials: Novel nano hydroxyapatites substituted simultaneously with divalent cations: Mg2+ (1.5%), Zn2+ (0.2%), Sr2+ (5% and 10%), and Si (0.2%) as orthosilicate (SiO44-) were designed and successfully synthesized for the first time.
Methods: The ms-HAPs were obtained via a wet-chemistry precipitation route without the use of surfactants, which is a safe and ecologically friendly method. The composition of synthesized materials was determined by inductively coupled plasma optical emission spectrometry (ICP-OES). The materials were characterized by X-ray powder diffraction (XRD), FT-IR and FT-Raman spectroscopy, BET measurements and by imaging techniques using high-resolution TEM (HR-TEM), FE-SEM coupled with EDX, and atomic force microscopy (AFM). The ion release was measured in water and in simulated body fluid (SBF).
Results: Characterization methods confirmed the presence of the unique phase of pure stoichiometric HAP structure and high compositional purity of all synthesized nanomaterials. The doping elements influenced the crystallite size, the crystallinity, lattice parameters, morphology, particle size and shape, specific surface area, and porosity. Results showed a decrease in both nanoparticle size and crystallinity degree, coupled with an increase in specific surface area of these advanced ms-HAP materials, in comparison with pure stoichiometric HAP. The release of biologically important ions was confirmed in different liquid media, both in static and simulated dynamic conditions.
Conclusion: The incorporation of the four substituting elements into the HAP structure is demonstrated. Synthesized nanostructured ms-HAP materials might inherit the in vivo effects of substituting functional elements and properties of hydroxyapatite for bone healing and regeneration. Results revealed a rational tailoring approach for the design of a next generation of bioactive ms-HAPs as promising candidates for bone regeneration.
Keywords: nanomaterials, hydroxyapatite, multi-substituted hydroxyapatites, bioceramics, synthesis, characterization, ions release
Introduction
Multi-element substituted hydroxyapatites (ms-HAPs) are receiving increasing attention, as in these nanomaterials the typical properties of hydroxyapatite can be modified toward specific biological responses.1–12 In hydroxyapatite, even a small amount of doping can create a significant alteration of critical properties such as solubility, particle size, morphology, specific surface area, porosity, and aspect ratio.3–6,9 Moreover, the doping elements will eventually be released in vivo, making these materials as multi-functional systems for the release of biologically active ions.1,2 Given this complexity, it is to be understood that the mechanisms leading to the putative modification of biological properties are still debated.11
It is known that osteoporosis affects bone homeostasis, protein synthesis as well as cell reactivity to both signaling ions and signaling biomolecules, weakening bone remodeling and healing.13,14 Innovative strategies are currently in progress for bone regeneration, especially based on bone grafting using biophysical and biochemical stimulation of osteoblasts and simultaneously decreasing the osteoclast activities. Moreover, for a substantial supply of graft biomaterials avoiding donor site injury, hydroxyapatite (HAP) and ms-HAPs,15–17 mimicking the composition of the main inorganic phase of bone,18–20 are still under development.
It is already recognized that biological hydroxyapatite comprises diverse foreign ions, such as stable nonradioactive strontium, Sr2+, ions.21 Accordingly, the development of strontium-containing biomaterials has received an important attention.20,22–36 Sr is reported as a vector for bone regeneration.7 Sr is recognized to adjust bone turnover in favor of bone formation by stimulating osteoblast activity and their proliferation,23,34 as well as by reducing bone resorption and mediating inhibition of osteoclast activity.25 It was suggested that Sr concentration is greater in the region of high metabolic turnover and in new bone than in old bone.22,27
Mg is associated with mineral phase in hard tissue and makes about 0.44 wt% of human enamel, 1.23 wt% of dentin, and 0.72 wt% of bone.17 The effect of Mg, as a substituting element in HAP-Mg, on HAP characteristics is a subject of numerous different studies.16,17,37–43 Clearly, Mg is involved in skeletal metabolism and bone growth by increasing osteoblast cell activity and avoiding osteopenia and bone fragility, and thus playing an essential role in bone remodeling.16,17,37 It was also suggested that Mg deficiency is linked to osteoporosis.
Zinc was found to trigger an inhibitory effect, similar to Sr, on osteoclasts in vitro,44,45 increasing bone formation. Zn stimulates alkaline phosphatase activity and DNA synthesis in rats,46 enhancing bone mineralization. Due to Zn beneficial effects on bone formation, the synthesis of substituted HAP with Zn (HAP-Zn) has been carried out by using chemical precipitation methods for different Zn substitution levels into the HAP lattice.16,47–50
Silicon, Si, was found to be essential in bone growth and development.51 This discovery has inspired the development of bioceramics based on Si-substituted hydroxyapatite (HAP-Si),52–57 primarily by using precipitation routes.52–54 The role of HAP-Si biomaterial on bone formation, mineralization, and remodeling by stimulating the bioactivity of human osteoblasts and promoting bone regeneration was tackled both in vitro and in vivo studies.53–57 It was suggested that the higher dissolution rates of the HAP-Si compared to HAP might explain some of the biological effects of HAP-Si biomaterials.57
As shown above, there are many experimental studies on mono-substituted HAP with Sr, Mg, Zn, or Si, whereas there are only a few about double-substituted HAPs with these elements, for instance HAP-Mg-Zn.50 Co-substitution of Mg and Sr within HAP lattice (HAP-Mg-Sr) has been carried out41,58 and ensured an improved biological response. The Sr and Zn co-substituted HAP was shown to present excellent osteoinductivity and antibacterial properties.26 Another double-substituted HAP, with Mg and Si, was recently obtained.56 It is important to mention that a compensation effect was also determined in the structural modifications, which prevents both a destabilization of the HAP structure and a potential decomposition during the heat treatment through the calcination process.
In addition, a triple-substituted HAP with Ag (0.25 wt%), Au (0.025 wt%), and Zn (0.2 wt%), HAP-Ag-Au-Zn, was produced by wet chemical method and used as carrier for silver nanoparticles embedded together into a polymeric matrix,59 and a strong antimicrobial effect was evidenced against several pathogens.
Moreover, we first reported on substituting Mg2+, Zn2+, and SiO44- ions, in the amount of 0.6 wt%, 0.2 wt%, and 0.2 wt%, respectively, simultaneously into the HAP lattice, while retaining a HAP structure.60 The resulted HAP-0.6%Mg-0.2%Zn-0.2%Si biomaterial was found to enhance the adhesion, growth, and proliferation as well as the collagen synthesis of human osteoblasts in vitro. We also demonstrated that hydroxyapatite substituted with Si, HAP-Si, stimulated the behavior of human osteoblast cells in culture better than pure hydroxyapatite,54 even for a low level of substitution such as HAP-0.47 wt% Si. Recently, it was shown that HAP-Sr, particularly HAP-5%Sr and HAP-10%Sr, scaffolds revealed an excellent biocompatibility and high performance in osteoblast cell culture,34 promoting osteoblast adhesion and proliferation.
Undoubtedly, chemical doping and topographical features of these biomaterials are responsible for the in vitro response, namely their nanostructure, appropriate crystallinity, and rather large specific surface area. A further understanding of the specific compositional role of doping elements on the HAP structure and physical properties of ms-HAPs is of great interest particularly in designing synthetic hydroxyapatites for bone regeneration. In this respect, the ms-HAPs comprising different physiological elements, such as Mg, Zn, Sr, and Si (eg, HAP-Mg-Zn-Si and HAP-Mg-Zn-Sr-Si), might have a great potential in bone regeneration,2 due to the biological role of these elements,1 and to the excellent biocompatibility of native HAP ceramic.
For the goal of the present work, a rational strategy was used to design ms-HAPs on the basis of our in vitro previous studies,54,60 using Mg, Zn, and Si as doping elements. In addition, Sr was also selected for HAP doping due to its excellent role on osteoblasts in bone regeneration.1,34 The strategy consisted in establishing a starting tailored composition as a new complex hydroxyapatite, noted HAPc, namely HAP-1.5%Mg-0.2%Zn-0.2%Si, where the doping elements are in total under 2 wt%. This choice is based on our previous results,19,60 and is in accordance with the given data for co-substitution of Mg and Si in the HAP lattice,5 showing that HAP structure can retain both elements in the total amount of 2 wt%. Further, a working hypothesis has been generated, and it consisted in the addition of Sr within HAPc structure, based on the idea that the four-substituted HAP ceramics are expected to inherit the demonstrated beneficial effects due to HAP structure and to essential elements present in its structure. This was done by adjusting the compositions as follows: HAPc, HAPc-5%Sr, and HAPc-10%Sr. Up to now, no study investigated these multi-substituted HAPs with possible applications for bone regeneration.
Specifically, a wet chemistry precipitation approach was employed54,60,61 for the simultaneous multi-doping with Mg, Zn, Sr, and Si in HAP structure. The influence of this type of doping on the physical and chemical characteristics of ms-HAPs has not yet been reported. The new synthesized nanomaterials have been developed particularly for paste (precipitate) and nanopowder applications, like carriers for local drug delivery,62,63 injectable gels,64 or as green (non-sintered) nanoparticles for scaffolding in cell culture,54,60 as well as for chemical coating onto metallic implants for bone regeneration and healing.19
Materials and Methods
The Synthesis of HAP Powders
Stoichiometric hydroxyapatite (HAP) was synthesized by a wet chemical route, as previously reported,35,49,54,59,60 but avoiding the use of any surfactant or other template molecules. This wet chemical precipitation method is a simple one-pot synthesis and the reaction temperature is under 100°C. The low processing temperature makes the process cost-effective, resulting in products of very high purity. In addition, the wet chemical method is safe and ecologically friendly, directing to HAP or ms-HAP nanomaterials, and ammonium nitrate, which is nontoxic. The process parameters, such as the concentration and addition rate of reactants, reaction temperature, and heat treatment conditions are crucial factors and can be accurately controlled and stabilized. This novel procedure offers optimum working conditions to monitor and control the size and shape of particles at nanoscale, as well as the crystallinity degree, specific surface area, and porosity of the obtained ceramic products. The purpose is to realize the reproducibility of the HAP and ms-HAP powders related to the stoichiometry, particle size and shape, and morphology of the synthesized powders having the same properties, such as specific surface areas, crystallinity degree, and porosity.
For the preparation of pure HAP, two aqueous solutions were used, namely a 0.25 M calcium nitrate solution obtained from Ca(NO3)2·4H2O (purity >99%, Sigma-Aldrich, Germany) and a 0.15 M diammonium hydrogen phosphate, (NH4)2HPO4 (purity 99.4%, Chempur, Poland), solution. Each compound was dissolved in ultrapure water. The pH was brought to 11.5 for both solutions by adding a 25% ammonia solution (pure p.a. Chempur, Poland).
Equal volumes of these two solutions were mixed at 22°C, assuring a Ca/P mole ratio of 5/3 (1.67), characteristic for stoichiometric HAP. Fast and thorough mixing was assured using a peristaltic pump Masterflex L/S Digital Drive, 600 RPM, 115/230 VAC, EW-07523-80 and an impact reactor type “Y” for the two liquid flows, with a flow rate of the reagents of 1000 mL/min.
The maturation of the obtained suspension was performed in two stages, first by maintaining it for 24 h at 22ºC, and then for other 24 h at 70ºC, in the reactor, under stirring at 60 rpm. Next, the suspension was filtered (Filter Disks Munktell, grade: 389), washed up repeatedly with double-distilled water to nitrate free, and the HAP precipitate (crude paste) was dried by lyophilization resulting in lyophilized HAP (sample 1) or ms-HAP nanopowders, named also green nanopowders.
These lyophilized powders were further calcined at 300°C for a dwelling time of 1 h, to make sure that possible traces of ammonium nitrate were eliminated from the synthesized HAP materials. The thermal decomposition of ammonium nitrate takes place in a temperature range of 140°C to 200°C65 and the resulting calcined HAP powder is named sample 2.
The Synthesis of ms-HAP Powders
The multi-substituted hydroxyapatites (ms-HAPs), namely: complex hydroxyapatite (HAPc) with 1.5 wt% Mg, 0.2 wt% Zn, and 0.2 wt% Si, and hydroxyapatites with the same Mg, Zn, and Si content and with 5 wt% Sr (HAPc-5% Sr) as well as with 10 wt% Sr (HAPc-10% Sr) were synthesized in an analog manner, the only difference relates to the composition of the two initial solutions. Solution 1 contained the cations (0.25 M in Ca2++Mg2++Zn2++Sr2+) and was obtained from the corresponding nitrates Ca(NO3)2·4H2O, Mg(NO3)2·6H2O (purity 99%), Zn(NO3)2·6H2O (purity >98%), and Sr(NO3)2 (purity 99.995%) (all from Sigma-Aldrich, Germany), dissolved in ultrapure water, in the ratios required by the composition of the synthesized product.
The anions containing solution 2 (0.15 M in PO43-+SiO44-) was obtained from (NH4)2HPO4, and tetraethyl orthosilicate, TEOS, Si(OC2H5)4 (98%, Alfa Aesar, Germany) as source for silicate ions, both dissolved in ultrapure water, in the ratios required by the composition of the synthesized product. All reagents were of the highest purity and were used as received.
The pH was adjusted to 11.5 by adding a 25% ammonia solution. Solutions 1 and 2 (equal volumes) were mixed at 22°C, so the mole ratio (Ca+Mg+Zn+Sr)/(P+Si) was maintained at the theoretical value 5/3 (1.67). The subsequent maturation and the processing of obtained precipitate were the same as in the case of pure HAP. Further on in this work, the lyophilized (noncalcined) powders are indicated as samples 3 (HAPc), 5 (HAPc-5%Sr), and 7 (HAPc-10%Sr), while the calcined lyophilized powders as samples 4 (HAPc), 6 (HAPc-5%Sr), and 8 (HAPc-10%Sr).
Digestion of Powdered Samples for Elemental Analysis
For the determination of the chemical composition, the digestion of the hydroxyapatite samples (powders) was performed in a Teflon beaker. Each sample (200 mg) was processed by microwave digestion system (Berghof speed wave MWS-3+, Eningen, Germany); for 5 min at 140°C (power 800 W), then 25 min at 220°C (1000 W), and 15 min at 100°C (1000 W), followed by ventilation for 10 min. The composition of the solution used for digestion was: 40% hydrofluoric acid, HF (2.5 mL), boric acid, H3BO3 (2.5 g), and 65% nitric acid HNO3 (5 mL). After cooling down at room temperature, the sample was diluted to 25 mL with ultrapure water, prepared by using Milli-Q, Millipore, Bedford, USA, and filtered through cellulose filter paper. The solutions for establishing the calibration curves were prepared from multi-element IV storage solutions (for Ca, Mg, Zn, and Sr), and mono-element storage solutions for P and Si (all from Merck, Germany).
Characterization Methods and Instrumentation
Elemental Analysis
The elemental analysis, namely the determination of the Ca, Mg, Zn, Sr, P, and Si amount, of the obtained hydroxyapatites was performed by using an inductively coupled plasma optical emission spectrometer (ICP-OES) OPTIMA 5300DV (Perkin-Elmer, USA), and complied with ISO 11885:2009.66 The limit of quantification (LQ) is the lowest concentration of an analyte that can be determined at an acceptable level of repeatability and reliability.66 For the present measurements, the limits of quantification for the determined elements are: Ca (LQ=0.008 mg/L), Mg (LQ=0.005 mg/L), Zn (LQ=0.005 mg/L), Sr (LQ=0.010 mg/L), P (LQ = 0.05 mg/L), and Si (LQ=0.005 mg/L). These values are lower or at least equal to those requested in ISO 11885:2009.
Ions Release from HAP and ms-HAP
The behavior of HAP and HAPc-5%Sr samples, both noncalcined and calcined, in the presence of water and simulated body fluid (SBF) was investigated. Therefore, the Ca, Mg, and Sr amounts were measured by immersion of 0.15 g of each sample in 15 mL ultrapure water, respectively, in SBF. Incubation of the samples was done at 37°C in separated closed flasks for each sample/day. The sample content in the mixtures was 10 g/L.
The SBF was prepared according to Kokubo’s SBF solution, containing the following ions (mmol/dm3): Na+ (142.0); K+ (5.0); Mg2+ (1.5); Ca2+ (2.5); Cl− (147.8); HCO3− (4.2); HPO42- (1.0); SO42- (0.5), and buffered at the physiologic pH 7.40 at 37°C, with tris(hydroxymethyl)amino methane and hydrochloric acid.
After 1, 3, 7, 14, 21, 30, 60, and 90 days, the supernatant was filtered through 0.45 µm paper filter and Ca, Mg, and Sr amount was determined using ICP-OES. Consequently, the release of Ca, Mg, and Sr from HAP and HAPc-5%Sr was comparatively evaluated in water and in SBF, in static conditions without the replacement of immersion fluid.
In order to study the Ca, Mg, and Sr released from HAP and HAPc-5%Sr in water, as well as the Sr released from HAPc-5%Sr in SBF, in simulated dynamic conditions, the experimental samples were prepared as above, but the immersion liquid was changed every day, for 7 days. Each day the supernatant (after centrifugation) was filtered, and a new volume of water, respectively SBF, was added to the individual sample.
The regular replacement technique of different immersion fluids represents an in vitro appropriate simulation of in vivo dynamic conditions, which involve a continuous flow of fluid in real system. This technique has advantages of good reliability of data without interference with the fluid rate in the system, which can cause an issue in actual conditions, while still remaining a simulation of in vivo dynamic conditions.
Mechanism Evaluation of Ions Release from HAP and ms-HAP
For the description of ion release mechanism, the Korsmeyer-Peppas model67 can be used. This model proposes a simple equation:
(1)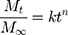
where Mt/M∞ is the fraction of species (type of ions) released at time t, Mt is the amount of chosen ions (eg, Ca2+, Mg2+, or Sr2+) released at time t, M∞ is the total amount of selected ions, k is the kinetic constant, and n is the diffusional exponent (also named the release constant), which is characteristic of the release mechanism. Usually, equation (1) is used to evaluate the release of ions68 when more than one release mechanism could be involved. For spherical (or cylindrical) particles, the value n = 0.43 (or 0.45) is characteristic for Fickian diffusion,67 and higher n values up to 1 describe an anomalous (non-Fickian) transport, while n =1 means a zero-order process (linear release).
The n value can be obtained from the linear form of equation (1):
(2)
as the slope of the straight line: lg(Mt/M∞) = f(lg t).
Crystal Structure
The crystal structure, specifically the calculation of unit cell constants (a = b and c) and crystal lattice planar spacing, d, was achieved by using XRD patterns and the following formula:4,43
(3)
where h, k and l represent the Miller indices and d is the interplanar spacing
(4)
for each diffraction plane, at Bragg’s angle (θ) and λ is the wavelength of the incident X-rays. Accordingly, the lattice parameters are calculated for every as-synthesized sample by using the peak maxima (ie, 2θ) of XRD pattern, for different hkl sets, which characterize the crystal structure. The diffracted X-rays exhibit constructive interferences, leading to the lattice fringes.
Instrumentation
X-Ray diffraction (XRD) investigations were carried out using a DRON-3 diffractometer, in Bragg–Brentano geometry, equipped with an X-ray tube with cobalt Kα radiation, wavelength 1.79026 Å, 25 kV/20 mA. The XRD powder patterns were collected from 2θ angle scale (10° −80°) with a step size of 0.02° and a normalized count time of 1s/step to 2s/step. The diffractometer settings allow a resolution of 0.02° and a signal/noise ratio greater than 20, and in majority of cases greater than 50, for the peak 211 of the HAP.
FT-IR spectra were measured on KBr pellets, containing the sample powders (0.5 wt%), with an FT-IR spectrometer JASCO 6100 in the 4000–400 cm−1 range of wavenumbers, with a 4 cm−1 resolution.
FT-Raman spectra on the solid samples were obtained with an FRA 106/S FT-Raman Module attached to Bruker EQUINOX 55, using the Nd:YAG laser (wavelength 1064 nm) and a liquid nitrogen-cooled germanium detector (D418-T). The FT-Raman spectra were recorded for wavenumbers below 3600 cm−1, with 400 scans and a resolution of 4 cm−1.
An FEI Tecnai F20 field emission, high-resolution transmission electron microscope (HR-TEM) operating at an accelerating voltage of 200 kV was used to obtain the HR-TEM images. Electron micrographs were recorded on Eagle 4k CCD camera. The hydroxyapatite samples were dispersed in pure water and then deposited on Cu grids previously covered with carbon film. The fast Fourier transform (FFT) was performed by using Gatan Digital Micrograph software for the evaluation of lattice spacing in HR-TEM images.
Field emission scanning electron microscope (FE-SEM or SEM), Hitachi SU-8230, operated at 30 kV, was used to explore the nanostructure of HAP samples. FE-SEM was equipped with Oxford energy-dispersive X-ray spectrometer (EDS) for elemental analysis (energy-dispersive X-ray, EDX, spectra). SEM grids were of Cu, covered by a carbon layer of 10 to 20 nm thickness. SEM samples were prepared by deposition of HAP samples, as powder, in thin layers on SEM grids. FE-SEM&EDS equipment was also used on ms-HAPs for elemental analysis.
Atomic force microscopy (AFM) images were obtained using an AFM JEOL 4210 equipment, operated in tapping mode, using standard cantilevers with silicon nitride tips (resonant frequency in the range of 200–300 kHz, spring constant 17.5 N/m). The particles were adsorbed from their aqueous dispersion on optically polished glass support. The HAPs aqueous dispersions needed for AFM imaging were homogenized using a high-intensity ultrasonic processor Sonics Vibra-Cell, model VCX 750, for 5 min, at room temperature, 22°C.
Brunauer, Emmett, and Teller (BET) analysis was achieved with an automated Sorptomatic 1990 instrument, with nitrogen adsorption at 77 K. The calculation of the surface area was made in the P/Po range between 0.03 and 0.3, and the total pore volume was determined at P/Po = 0.95. Before the BET analysis, the samples were first dried at 120°C in oven until constant weight, and then, they were outgassed for 6 h, at 70°C.
Statistical Analysis
All quantitative assessments were taken in triplicate (n = 3), unless otherwise stated, and results are expressed as mean ± standard deviation (SD). Statistical comparison of data for different compositions was carried out via a one-way analysis of variance (ANOVA) using the Origin 8.5 program from OriginLab. Differences were considered statistically significant at p < 0.05.
Results
Chemical Composition of Synthesized Nano Materials
The chemical composition of synthesized products, using rational design strategy, is summarized in Table 1 for the calcined lyophilized powders, noted as samples: 2 (HAP), 4 (HAPc), 6 (HAPc-5%Sr), and 8 (HAPc-10%Sr). The cations Mg2+, Zn2+, and Sr2+ partially substitute Ca2+ ions in the hydroxyapatite lattice. Si is assumed to form silicate (SiO44-) anions, partially substituting phosphate (PO43-) anions, while the number of hydroxide (OH−) anions is correspondingly decreased to assure the electro-neutrality of the lattice. With these assumptions, we calculated the theoretical formulas given in Table 1 for the multi-substituted hydroxyapatites.
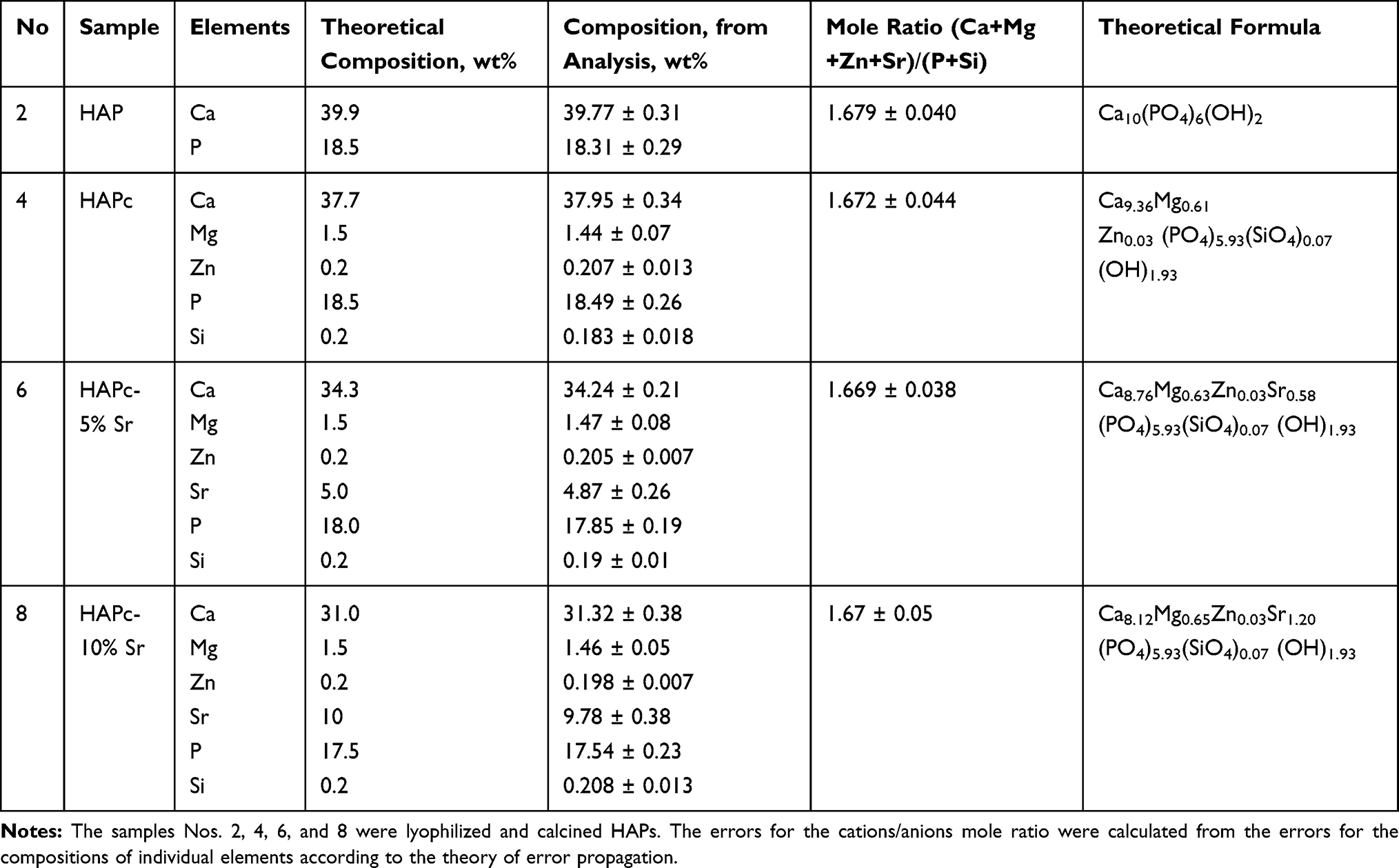 |
Table 1 Theoretical and Experimental Composition of Synthesized Hydroxyapatites, and Their Theoretical Formulas |
Theoretical compositions based on the synthesis and the experimental values from ICP-OES chemical analysis are also compared in Table 1. For the experimental amounts, the standard deviations, calculated from six measurements (n = 6) on six different samples (calcined at 300°C) prepared for each synthesized nanomaterial of a given theoretical composition, are indicated in Table 1.
X-Ray Powder Diffraction: XRD
XRD data were collected for 2θ between 10º and 80º (Figure 1). The XRD patterns for the noncalcined HAPs are given in Figure 1A and for the calcined HAPs are shown in Figure 1B. Phases were identified by comparing the peak positions of the experimental X-ray powder diffraction patterns with those of the ICDD (International Centre for Diffraction Data) powder diffraction files (PDF), such as PDF 74–0566 and PDF 09–0432 of HAP used for stoichiometric hydroxyapatite, and PDF 34–0484 of Ca9Sr(PO4)6(OH)2 for calcined HAPc-Sr10%, as exemplified in Figure S1. No matches could be found for diffraction peaks of β-tricalcium phosphate, β-TCP (PDF 09–0169), α-TCP (PDF 09–0348), CaO (PDF 82–1690), or tetracalcium phosphate, TTCP (PDF 25–1137).
The average crystallite size for these samples was evaluated by the Scherrer method, from the width of the most intense diffraction peaks, measured at half-maximum, as shown previously in the case of HAP mono substituted with various Si amounts.54 The degree of crystallinity was estimated using Reflex computer program part of Material Studio software (Accelrys Software Inc. Materials Studio, Release 5.5, 2010) as the ratio of the area of all crystalline peaks to the total area (crystalline peaks and amorphous halo).
 |
Figure 1 Experimental XRD powder patterns for noncalcined (A) and calcined (B) samples. |
The crystal lattice parameters, a=b (for hexagonal lattice) and c, in the HAP and ms-HAP ceramics were calculated from XRD data (Figure 1), using the equations (3) and (4), as given in the characterization methods.4,43 The results are given in Table 2.
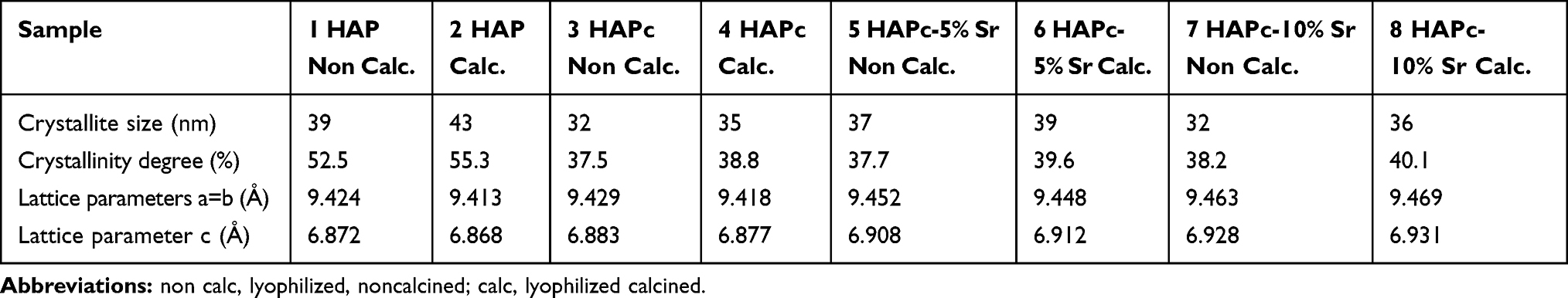 |
Table 2 Crystallites Size, Crystallinity Degree, and Lattice Parameters, Estimated from XRD for Multi-Substituted Hydroxyapatites and for Pure Hydroxyapatite |
FT-IR and FT-Raman Spectroscopy
FT-IR spectra are compared in Figure 2A for the noncalcined samples and in Figure 2B for the calcined ones. All the spectra are normalized to 1 for the highest absorption maximum. The positions of the main absorption maxima are given in Table S1.
 |
Figure 2 FT-IR spectra of noncalcined samples: 1, 3, 5, and 7 (A), and calcined samples: 2, 4, 6, and 8 (B); FT-Raman spectra of noncalcined samples: 1, 3, 5, and 7 (C), and calcined samples: 2, 4, 6, and 8 (D); all the spectra are normalized. |
FT-Raman spectra in the wavenumbers range from 1500 to 400 cm−1 are given in Figure 2C and D, for noncalcined and calcined samples, respectively.
The FT-IR spectra for all samples are similar, presenting all absorption peaks for the PO4 group in hydroxyapatites: asymmetric stretching ν3 (the most intense band, doublet), symmetric stretching ν1 (low intensity, since it is forbidden in IR), and the low intensity bending modes, such as asymmetric ν4 (doublet) and symmetric ν2. The characteristic peak for the structural OH groups in HAP69–71 is also present at about 3570 cm−1, while the libration peak at about 630 cm−1 is visible only as a shoulder in some samples. Some vibrations assigned to carbonate ions71 appear as broad and very weak shoulders denoting the presence of carbonate ions but only in traces within HAP structure. However, their presence may somewhat improve the bioactivity of HAPs rather than being a cause of concern, since the bone mineral phase is carbonated.
In FT-IR spectra the bands assigned to vibrations of non-apatitic HPO42- ions, at 530–540 cm−1 or at 1125 and 1145 cm−1,72 as well as maxima characteristic for non-stoichiometric apatites (1018 cm−1) are missing.72
All characteristic vibrations for the PO4 group also appear in the FT-Raman spectra (Figure 2C and D). The most intense is the symmetric stretching vibration ν1, at 960–962 cm−1, which is forbidden in IR.
Nanostructure and Morphology: HR-TEM, FE-SEM, and AFM
High-resolution TEM (HR-TEM) images are given in Figure 3, for noncalcined lyophilized powder of pure HAP (A, B, C), and calcined lyophilized powder of HAPc-5% Sr (D, E, F) and for HAPc-10% Sr (G, H, I).
HR-TEM images reveal the morphology, the shape, and size of HAP (Figure 3A–C), HAPc-5%Sr (Figure 3D–F) and HAPc-10% Sr (Figure 3G–I) nanoparticles. In these images, synthesized HAP nanoparticles are well illustrated and display nanorods and elongated shapes, like needles.
Therefore, these powders are well dispersed in water and their dispersions have good stability as proved by HR-TEM images. The size of particles decreases with doping of HAP lattice (Figure 3) in substantial accord with XRD patterns (Figure 1) and Table 2.
Figure 3A, D, and G reveal that nanoneedles/nanorods are almost transparent. This feature suggests that those nanoparticles are very thin, whereas some black rods (or plates) might result from loose associated particles in agreement with the FE-SEM observations (Figure 4), or from the standing projection of self-assembled nanoparticles as reported on related materials.73
 |
Figure 3 HR-TEM images for noncalcined HAP sample (A–C), and for the calcined samples HAPc-5%Sr (D–F) and HAPc-10%Sr (G–I); the bars are 50 nm (A, D, G) and 100 nm (B, E), 20 nm (H), 10 nm (F, I), 5 nm (C). In figures (C, F, and I), crystal structure is selected in (black or white) square with nearby amorphous (non-crystalline) zone. |
Further, Figure 3 evidences that the nanoneedles (or nanorods) are built from small spherical nanoparticles that are interconnected with each other to form predominantly not aggregated needle-like structure. The length of needles or rods is between 20 and 50 nm, and their thickness is from about 5 to 20 nm, considering more than 200 particles from images similar to those given in Figure 3. Furthermore, the Sr, Mg, Zn, and Si doping of HAP appears to play a role in the formation of loose self-assembled nanoparticles with a porous structure, illustrated as black arrangements in Figure 3D and G. Accordingly, it can be assumed that the ion doping can change the crystallinity and solubility of HAP. Consequently, ms-HAPs might have a superior biological performance against pure stoichiometric HAP.
The detailed structural analysis is further illustrated in Figure 3C for HAP, Figure 3F for HAPc-5%Sr, and in Figure 3I for HAPc-10%Sr samples. These HR-TEM images show a mixture of crystalline nanostructures and amorphous ones. The continuous lattice fringes, for instance, those in the black or white squares, indicate individual crystalline nature for these samples. The clearly resolved fringes by fast Fourier transform, FFT, correspond to FFT patterns, shown as inset in those Figure 3C, F and I. The calculated lattice spacing of 0.34 nm corresponds to the interplanar distance of the 002 planes for the hexagonally structured HAP and is given in FFT patterns, demonstrating the unique crystal structure of HAP, found also in HAPc-5%Sr and HAPc-10%Sr.
FE-SEM coupled with EDS is used for chemical analysis of HAP samples. Further, FE-SEM&EDS gives the EDX spectrum and distribution maps of elements in all as-synthesized HAP samples. In the following example, the EDX spectrum is given in Figure 4A and the multi-color distribution map of Ca, P, Mg, Zn, Sr, Si, and O is presented in Figure 4B in the case of calcined lyophilized HAPc-5%Sr powder, sample 6 in Table 1. Moreover, the uniform distribution of all elements is evident both in multi-color distribution map (Figure 4B) and in the individual maps for each element (Figure S2). Similar results were obtained for the other as-synthesized nanomaterials in substantial agreement with chemical analysis by ICP-OES, indicating a homogeneous distribution of all elements in these powders.
 |
Figure 4 Calcined lyophilized HAPc-5%Sr (sample 6, in Table 1); EDX spectrum (A), multi-color distribution map showing jointly all elements on FE-SEM image (B). Gold appears in the EDX spectrum due to the imaging technique that uses coating of biomaterial with a gold layer. |
Atomic force microscopy, AFM images were obtained for all synthesized nanomaterials. Different areas from 10 μm x 10 μm to 0.5 μm x 0.5 μm were scanned on a particular nanomaterial, adsorbed from its aqueous dispersion on optically polished glass plates.74–76 As an example, the AFM images, ie, 2D-topography (A) and phase image (B), as well as cross-section profiles (C) for noncalcined, lyophilized powders of hydroxyapatites: HAP, HAPc, HAPc-5% Sr and HAPc-10% Sr are presented in Figure 5.
AFM images on needles, rods or spindle-shaped particles of HAP and ms-HAPs gave spherical or oval shapes of nanoparticles, as exemplified in Figure 5. The average size of particles is 41 ± 3 nm for HAP, 38 ± 2 nm for HAPc, 36 ± 2 nm for HAPc-5%Sr, and 35 ± 3 nm for HAPc-10%Sr, as determined from cross-section profiles of each noncalcined sample. These values are comparable with those given by XRD (Table 2). A similar trend is also found for the average size of particles in calcined, lyophilized HAP and ms-HAPs.
 |
Figure 5 AFM images of nanoparticles from noncalcined HAPs adsorbed on glass support; 2D topography (A) and phase (B) images; cross-section profiles (C) along the arrow in panel (A); scanned area of 1 µm x 1 µm. |
Specific Surface Area (SSA) and Porosity
The Brunauer-Emmett-Teller (BET) theory explains the physical adsorption-desorption of gas molecules on a solid surface and represents the basis for the analysis of measured data. For exemplification, some representative adsorption-desorption isotherms and pores size distributions are given in Figure 6.
 |
Figure 6 Adsorption-desorption isotherms (A, C), and pore size distributions (B, D) for calcined HAP (A, B) sample 2 in Table 1, and calcined HAPc-10%Sr (C, D), sample 8 in Table 1; the relative pressure p/p0 is the ratio of equilibrium pressure to saturation pressure of the adsorbate gas (N2). |
Specifically, the nitrogen adsorption-desorption isotherm and pores radius distribution for HAP and HAPc-10%Sr, calcined samples, are shown in Figure 6A–D, respectively. Similar results were obtained for the other six samples as-synthesized in this study. The isotherms exhibited type IV isotherm behavior with a distinct hysteresis loop, indicating that the as-synthesized HAPs contained mainly mesopores in the size (radius) range of about 2 nm to 20 nm.
The pores of about 20 nm in diameter were also evidenced in HR-TEM (Figure 3) and AFM images (Figure 5). These pores were probably placed at about 20 nm as voids among the needle/spindle-shaped HAP and ms-HAP particles. Therefore, BET measurements and HR-TEM and AFM images demonstrated that the as-prepared HAP and ms-HAP nanoparticles were well-dispersed in aqueous systems from mesoporous powders.
BET analysis in terms of specific surface area and pores specific volume is presented in Table 3, for eight nanomaterials, eg, calcined lyophilized and noncalcined lyophilized powders of HAP, HAPc, HAPc-5%Sr, and HAPc-10%Sr. The determinations were made in triplicate, and the standard deviations for specific surface areas are given as errors in Table 3, and in Figure S3.
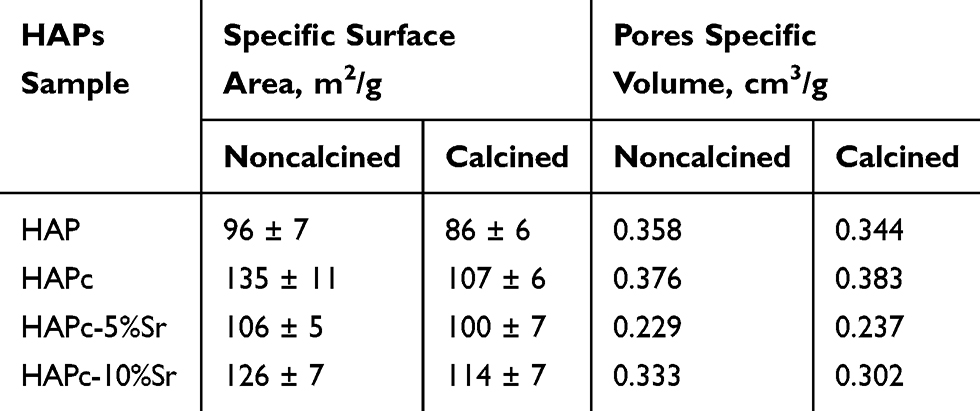 |
Table 3 Some Characteristics from BET Analysis on Eight Samples of Hydroxyapatites (HAPs), as Calcined Lyophilized and Noncalcined Lyophilized Powders |
A statistical analysis by one-way ANOVA revealed that for the noncalcined samples, at the 0.05 significance level, the means are significantly different, except for the pairs HAP compared to HAPc-5%Sr and HAPc compared to HAPc-10%Sr. For the calcined samples, the means are not significantly different at the 0.05 level, except for the pairs HAP&HAPc and HAP&HAPc-10%Sr. The mean specific surface area for the same type of sample, noncalcined and calcined, differs significantly only for HAPc, where a significant SSA decrease was produced by calcination process.
The SSA values are larger for HAPc, HAPc-5%Sr, and HAPc-10%Sr than those of mesoporous HAP nanoparticles, both in noncalcined and calcined powders. Generally, the as-prepared ms-HAPs have higher surface area than HAP, indicating that these ms-HAPs might have superior potential applications in drug delivery, taking into account the higher absorption capacity besides the possibility of ions released from ms-HAP in vivo conditions.
The crystalline structure of mesoporous HAP and ms-HAP nanomaterials was confirmed by XRD (Figure 1, Table 2) and by HR-TEM, Figure 3C, F and I. Actually, a unique HAP lattice structure was identified with lattice fringes and interplanar spacing of 0.34 nm, corresponding to (002) planes.
Ions Release from HAP and ms-HAP in Water and in SBF
The results for ions release in static conditions (without changing the medium) for 90 days are presented graphically in Figure 7 for Ca, Mg, and Sr release in water (A, B, and C) and in SBF (D, E, and F). The Zn and Si released amount could not be monitored in this study, because as tracers they were under the detection limit of ICP-OES.
For the ions release in simulated dynamic conditions, with changed liquid each day, the results are given in Figure 8 for Ca (A) and Mg (B) release in water, and for Sr release both in water and in SBF (C and D).
The kinetics of the Sr2+ release was investigated by using the Korsmeyer–Peppas model on the values of cumulative ion release (Figure 8D), both in water and in SBF, measured for a daily change of the medium (simulated dynamic method). The M∞ value for Sr (500 mg/L) was calculated from the formula of the HAPc-5%Sr, sample 6 in Table 1, for a sample weight of 10,000 mg (10 g), corresponding to 1 L solution, as the released amount of Sr2+ ions is given in mg/L, as in Figure 8D. The Mt values were the cumulated release values for the 1 to 7 days.
The regression lines corresponding to equation (2) are represented in Figure S4, for strontium release from HAPc-5%Sr noncalcined (A) and calcined sample (B), in water, and for HAPc-5%Sr, noncalcined (C) and calcined sample (D), in SBF. The k parameter is obtained from the y-intercept, and the release parameter n is obtained from the slope of line. These values are given in Table 4, along with their standard errors and the coefficients of determination, denoted r2.
 |
Figure 7 Variation of Ca (A), Mg (B), and Sr (C) amount in water, and Ca (D), Mg (E), and Sr (F) amount in SBF, in the presence of HAP and HAPc-5%Sr, for calcined and noncalcined samples. The vertical bars represent the standard deviations of the measured values. |
 |
Figure 8 Variation of Ca (A) and Mg (B) amount in water and Sr (C) amount in water and in SBF, in the presence of HAP and HAPc-5%Sr; cumulative release of Sr from HAPc-5%Sr in water and in SBF (D). The vertical bars represent the standard deviations of the measured values. The immersion liquid was changed every day to simulate dynamic conditions. |
 |
Table 4 Korsmeyer–Peppas Parameters of Sr2+ Release from HAPc-5%Sr in Water and in SBF |
The k value is the ratio of Mt/M∞ for the time, t, corresponding to the first day of the Sr2+ ions release calculated from Figure 8D, by using equation (2). The k values are higher for the Sr2+ ions released from the calcined samples against noncalcined ones, both in water and in SBF, in substantial agreement with the release rate in the first day, which is higher for the calcined HAPc-5%Sr in immersion fluids, water and SBF (Figure 8C).
Discussion
There is a very good correspondence between calculated and measured content, for each constituent element (Table 1). The mole ratio of the total amount of cations (Ca2+ + Mg2+ + Zn2+ + Sr2+) to the amount of anions (PO43- + SiO44-) for all samples is very close to that of theoretical value (1.67) characteristic for the stoichiometric hydroxyapatite structure. No impurities (such as Mg or Sr traces) could be found by the ICP-OES analysis either in the HAP samples, or in the raw materials used in their synthesis.
The diffraction patterns indicate that each of these powders is synthesized at the nanoscale level. Only the HAP phase was found by XRD; hence, the calcination at 300°C for 1 h could not lead to the decomposition of obtained ms-HAP structures. The highest diffraction maxima (Figure 1) positioned at a 2θ value of about 32° correspond to the (211) diffraction maximum of HAP structure.77 Thus, although the substitution ions carry different charges, or have various sizes, they can facilitate the formation of a stable isostructural lattice for this range of composition (Table 1). Additionally, the diffraction peaks for ms-HAPs were apparently broader than those of pure stoichiometric HAP, suggesting that the multi-ionic substitution within HAP lattice resulted in a decrease of crystallinity degree for the ms-HAP materials, in agreement with similar data for co-substituted HAPs with Si and Sr (HAP-Si-Sr).4
The average size of crystallites is at the nanoscale level, with the highest value for pure HAP and the lowest value for lyophilized noncalcined HAPc-10%Sr. The degree of crystallinity (Table 2) is higher for pure HAP (52–55%) compared to all multi-substituted HAPs (37–40%). This is expected as the presence of the dopants and the heterogeneity of the lattice composition hinders the formation of periodical structures on a wider length scale. A reduction of both the crystallite size and degree of crystallinity after Ca substitution by Mg was also reported.42
Consequently, almost the same reduction in the crystallinity degree of pure HAP was observed for all multi-substituted HAPs, rather independent of the increasing amount of Sr in HAPc lattice. Therefore, it clearly appears that the Sr amount has a minor impact on the crystallinity of triple-substituted HAPc, in this range of concentration.
The calcination at 300°C for 1 h caused only a slight increase of crystallite size for all obtained nanopowders: HAP, HAPc, and for HAPc containing Sr. The crystallinity degree was slightly increased by calcination and is in agreement with other similar observations.78 Due to multi-elements doping, a stabilizing effect is clearly induced in structural modifications, which prevents both a destabilization of the HAP structure and a potential decomposition during the heat treatment through the calcination process.
The calcination at 300°C slightly modified the HAP lattice parameters (Table 2) compared to values obtained for samples, as-synthesized and only lyophilized. The substitutions in the HAP lattice resulting in HAPc (for doping element amount <2%) brought about only small increases of the lattice parameters. Further, the substitution of Sr2+ for Ca2+ in the HAPc led to HAPc-5%Sr and HAPc-10%Sr, where the presence of the larger Sr2+ ions caused a gradual increase in the lattice parameters (Table 2) with increasing Sr content, similar to that observed for strontium-substituted HAP, HAP-Sr.35 For example, HAP-5%Sr, the estimated parameters were a = 9.443 Å, and c = 6.903 Å, while for HAP-10%Sr they were: a = 9.463, and c = 6.926 Å.35
These findings (Table 2) obtained with increasing content of doping elements/ions can be considered as an important proof of the integration of foreign ions into the HAP structure causing a slight gradual increase in the a and c values, while the HAP lattice was not disrupted, and is in very good agreement with related data.35,36,61
FT-IR and FT-Raman spectra confirmed the high phase purity of all synthesized HAP samples, as it was evident also from the XRD patterns.
The HR-TEM images give an insight into the details of particle morphology. They show that the oblong particles consist of arrays of nanodomains with mostly spherical symmetry. The average size of particles is 20 to 50 nm. HR-TEM images at the highest resolution (Figure 3C, F, and I) allowed us to observe in all samples both crystalline areas (evidenced in these images by white and black squares) and predominantly amorphous zones. These results are in substantial agreement with XRD data related to the crystallinity degree of tailored hydroxyapatites: HAP, HAPc, HAPc-5%Sr, and HAPc-10%Sr.
The crystalline structure of all HAP and ms-HAP nanoparticles was also confirmed by HR-TEM (Figure 3). For instance, Figure 3A–C showed the rods and needle-like morphology of synthesized HAP nanoparticles. Figure 3C displays the lattice fringes (marked zone in the black square) of HAP nanoparticles, which were further analyzed by fast Fourier transform. The FFT pattern (inset in Figure 3C) revealed that the lattice fringes correspond to the plane (002), with an interplanar spacing of 0.34 nm, which is in very good agreement with the calculated value of 0.343 nm for the (002) peak in XRD (Figure 1). The HR-TEM images of HAPc-5%Sr are shown in Figure 3D-F and revealed needle-like shapes. The data in Figure 3F revealed the lattice fringes (shown in white square) with an interplanar spacing of 0.34 nm, as illustrated in FFT pattern (inset in Figure 3F) with respect to plane (002) in substantial agreement with the calculated value of 0.344 by using XRD (Figure 1). This d value coincides with the distance (d) between successive parallel planes of atoms in a HAP crystal, with an interplanar spacing of 0.344 nm, which was assigned to the (002) planes of the HAP crystal.73 The morphology of HAPc-10%Sr nanoparticles is also illustrated in Figure 3G–I, as rods and needles. The FFT pattern (inset in Figure 3I) of the HR-TEM fringes (included in black square) indicated the interplanar spacing of 0.34 nm corresponding to plane (002), which is in substantial agreement with a calculated value of 0.345 nm by using XRD (Figure 1). These results confirm that a unique HAP structure is formed in all as-synthesized nanoparticles, either in lyophilized noncalcined or lyophilized calcined samples, in excellent agreement with XRD patterns.
According to FE-SEM analysis (Figure 4) of calcinated HAPc-5%Sr powders at 300°C for 1 h, this thermal treatment does not bring about significant changes in the aspect of particles; however, a loose self-assembly of particles was observed.
The EDX results are reasonably close to the expected values from synthesis and to those determined by ICP-OES chemical analysis (Table 1).
The EDX spectrum (Figure 4A) and the multi-color map (Figure 4B) on elemental composition indicate a fairly homogeneous distribution of all elements for calcined lyophilized HAPc-5%Sr nanopowder, in total agreement with the individual elemental maps (Figure S2). Similarly, the same results were found for each of the eight synthesized powders, in this work. These results are consistent with the presence of a unique phase of hydroxyapatite structure, both in pure HAP, and in HAP substituted simultaneously with Mg, Zn, and Si or with Mg, Zn, Sr, and Si.
AFM images and cross-section profiles (Figure 5) indicated that the nanosuspensions of HAP powders are formed of nanoparticles of size, in the range of 35 to 41 nm, in good agreement with crystalline domains found in XRD data (32–39 nm, Table 2). These particles seem somewhat bigger than values given by XRD probably due to particle clustering caused by the adsorption and drying process. They had a near-spherical shape with slight deformation to oval shape.
All eight HAP samples present a rather high specific surface area (SSA), as determined from BET analysis (Table 3, Figure S3), due to the high degree of dispersion of powders, as well as due to the synthesis by an aqueous precipitation method that is recognized to lead to nanomaterials with high SSA.58
In the case of HAPc-5%Sr, SSA values are higher in comparison with those of pure HAP but are smaller with respect to those of HAPc powders. This finding suggests that a dual effect might be responsible for SSA values, mainly the presence of Mg might cause an increase in SSA with respect to HAP, but Sr might be responsible for a slight decrease in SSA, as found in related compounds.58 For HAPc-10%Sr sample with Sr (10 wt%) content that specific trend is not further observed.
By calcination, the specific surface area is diminished as an effect of changes that occurred in the structure of the nanoparticles. The specific surface areas of the samples are comparable with reported data on similar nanomaterials. For instance, crystalline HAP was reported to possess a surface area 90 m2/g after oven-drying and 113 m2/g after freeze-drying.79.
The adsorption–desorption isotherms are of type IV. These samples can be classified as mesoporous materials, according to the IUPAC notation,80 with pore diameters between 2 and 50 nm. Since the nanoparticles themselves (according to HR-TEM images) are apparently not porous, this porosity results mainly from the self-assemblies of nanoparticles (interparticle porosity), as seen in FE-SEM images. However, the presence of intraparticle nano-porosity of HAPs cannot be completely excluded.
The non-substituted pure HAP, as calcined powder, presents a narrower distribution of pores sizes (about 8 nm diameter, Figure 6B) in comparison with the multi-substituted HAPs, namely calcined HAPc-10%Sr powder (Figure 6D), where most pore diameters between 10 and 20 nm (Figure 6D). The same trend is generally determined for noncalcined ms-HAPs. This is a consequence of foreign ions being present in the HAP lattice.
The SSA and porosity might be considered as critical factors for the behavior of the ms-HAP bioceramics at the interface with the surrounding medium, because they can affect the dissolved species in that medium.81 The released elements can interact with cells and tissue leading to an improved in vivo behavior of the ms-HAP bioceramics.19 Furthermore, these findings also suggest that the synthesized ms-HAP bioceramics can be employed as appropriate orthopedic implants and as coatings on metallic implants for bone repair and regeneration.
The ions release in water (Figure 7A–C) for cations is increasing in time, slowing down at a longer time, but without attaining a saturation value. It is always higher for calcined samples than for noncalcined ones. The substituted hydroxyapatites release a higher amount of each element than pure HAP. Thus, the presence of the substituting elements clearly increases the solubility of doped hydroxyapatite. The dissolution of HAPs is incongruent,82,83 and the equilibrium state has not been reached. The amount of released Mg (Figure 7B) and Sr (Figure 7C) is higher than the quantities expected from their content in the solid phase, ie, the Mg/Ca and the Sr/Ca ratios are higher in solution than in the solid. Moreover, magnesium seems to be released the easiest.5,42
Although the Zn amount and Si amount in ms-HAPs were very low, their release even as tracers in various liquid media can be important for a collective synergistic effect on cells and tissues, due to the simultaneous release of the other ions from ms-HAP bioceramics.
In SBF (Figure 7D), the Ca2+ concentration is decreasing in time in the presence of HAP, from its initial value in SBF (100 mg/L). The diminution is rapid in the first 20 days, and then it continues, but much slower, and in a similar way for all the samples. This behavior can be explained by the formation of HAP from the ions in SBF and/or the adsorption of Ca2+ ions on the HAP lattice. The Sr release in SBF is somewhat higher (Figure 7F) than in water (Figure 7C), as signaled also in literature.84 However, the ionic exchange with Ca2+ and perhaps Mg2+ from SBF could contribute to this. The Mg amount (Figure 7E) in SBF (36 mg/L) strongly decreases in presence of HAP, while with HAPc-5%Sr the decrease is limited probably by the higher release of Mg2+ from the dispersed solid, and possibly by an ionic exchange with the Ca2+ ions from SBF.85
The observed behavior of HAPs can be described in terms of competing processes, ion release, dissolution, and ion adsorption as well as precipitation of hydroxyapatite. For example, the decrease in Ca2+ ion concentration can be attributed to the precipitation process, namely calcium and phosphate precipitate to form the HAP layer. The other ions might act as nucleation sites for the formation of hydroxyapatite layer. Thus, multi-substituted HAPs could act as a source of retarded release of valuable ions for biological processes.
The daily release rate of Ca2+ ions and of substituting elements (Sr2+ and Mg2+ ions) from HAP and HAPc-5%Sr (noncalcined or calcined) in water and in SBF is displayed in Figure 8A–C. The release rates of Ca2+ in water were higher from HAPc-5%Sr than from HAP, and particularly from calcined than from noncalcined samples (Figure 8A). A similar trend was obtained for the release rate of Mg2+ ions from HAPc-5%Sr in water (Figure 8B). Specifically, the Sr2+ ions amount released in SBF was around twice as much as that in water and more evidenced for calcined HAPc-5%Sr particles than for noncalcined ones (Figure 8C).
Further, a two-step release was observed in these simulated dynamic conditions. The graphs show that the release of Ca2+, Mg2+, and Sr2+ ions progressed rapidly in the first 2 days. The initial fast release of ions could be explained by an accelerated process, which is probably due to the release of ions from the surface of HAP nanoparticles. This behavior is in agreement with data reported on related materials, but the understanding of release is still limited.86,87 Therefore, in the initial stage, the ions are released mainly from the surface of nanoparticles by diffusion into the surrounding (immersion) medium. Then, a diffusion of ions from the interior of particles to the immersion fluid could take place. Accordingly, the ions release process involves dissolution and diffusion, resulting in a sustained release behavior.
Furthermore, the cumulative profile for the release of Sr2+ ions from HAPc-5%Sr (noncalcined or calcined) in water and in SBF is illustrated in Figure 8D, in simulated dynamic conditions. It is observed that the Sr2+ ions are released in larger amounts in SBF than in water. At 7 days, the released Sr amount in SBF is at least twice than in water.
To study the influence of dynamic and static conditions on Sr2+ ion release, the data given in Figure 8D, Figure 7C and F were used. A comparative analysis of Sr release profiles revealed that the Sr amount released in water (at 7 days) increased at least 3 times in dynamic conditions (Figure 8D) than in static conditions (Figure 7C), being higher from calcined HAPc-5%Sr against noncalcined one. A similar trend was observed between release profiles of Sr in SBF (Figures 8D and 7F), but at 7 days, the cumulative released amount of Sr in simulated dynamic conditions is much (about 5 times) higher than its value in static conditions. Therefore, cumulative release of Sr2+ ions was much faster in SBF than in water, and the release was more efficient in dynamic conditions than in static conditions. Evidently, the Sr profiles have a similar trend, but the sustained release is higher in SBF, Figure 8D (simulated dynamic conditions) than in Figure 7F (static conditions). For biological applications, ms-HAPs can be considered as a source of trace elements and might have promising applications for bone regeneration.
To study the mechanism of Sr2+ ions release from HAPc-5%Sr, particularly in simulated dynamic conditions (Figure 8D), the Korsmeyer–Peppas model was applied. The linearity of the regression lines (Figure S4) according to equation (2) is good, as shown by the coefficients of determination (r2 about 0.98–0.99, in Table 4). Thus, the Korsmeyer–Peppas model is suitable for describing the Sr release. The values of the release parameter (n) in water are between 0.7 and 0.9 (Table 4), indicating a non-Fickian transport. It could be understood as a combination of diffusion and dissolution of the material, in substantial agreement with similar reported data.68 For the release in water, the dissolution seems to have a higher contribution for the noncalcined sample (higher n value). In SBF this difference is attenuated, both samples presenting an n-value of about 0.83 (Table 4), which could be explained by the combination of diffusion, dissolution, and ion-exchange processes. The tendency of n values toward 1 is correlated with the near-linear release (zero-order process) in the last days of the experiment.
Moreover, as synthesized ion-doped hydroxyapatites, HAP-Mg-Sr-Zn-Si, could have promising applications as bone/tooth materials, primarily due to trace release of therapeutically active ions. For instance, it is already known that the low dose administration of silicon and strontium could increase bone mass and strength by inhibiting bone resorption and enhancing bone formation.88,89 Certainly, a local release of these trace elements, Sr, Mg, Zn, and Si, in a low dosage from coatings on metallic implants might improve bone regeneration and have a strong positive impact on clinical applications.
The present work provides evidence for triple- and four-substituted HAPs as powders, particularly about the effect of co-doping, with different elements, such as Mg, Zn, Sr, and Si within HAP lattice, on physical and chemical properties, such as nanostructure, crystallite size, crystallinity degree, morphology, nanoparticle size, lattice parameters, specific surface area, porosity and ion release.
A correlation between lattice parameters (Table 2) and ion release (Figures 7 and 8) can be used to confirm this aspect in accord with recent published data.90 The lattice parameters revealed an increase with the increased substituting ions within HAP lattice, which can explain the rather high release of ions in water and in SBF.
Apart from ion release, the unique pure nano HAP structure of these ms-HAPs bioceramics plays an important role in regulating cell viability and functionality, in substantial agreement with similar data.91 As mentioned already, HAP has an excellent biocompatibility and these ms-HAPs might inherit this quality.
The combined effect of ions released from ms-HAPs might promote an increased performance of these ms-HAPs scaffolds compared to pure HAP ones in cell culture. Further in vitro and in vivo studies using these nanomaterials are in progress in our laboratories.
In this work, we demonstrated how the wet chemical synthesis route is suitable for producing complex multi-substituted hydroxyapatites preserving the main crystallographic hydroxyapatite features. The absence of surfactants makes the synthesized materials suitable for direct use or in combination with other biomaterials to prepare composites for applications in tissue engineering and regenerative medicine.
Our synthesis approach has several advantages over other preparation techniques used for pure HAP and/or substituted HAP with various ions, eg, simplicity, low processing temperature, and production of highly pure products. In addition, this chemical method is safe and ecologically friendly and offers the possibility to monitor and control the size and shape of nanoparticles as well as the crystallinity, surface area, and porosity of the resulted products.
The physical characterization revealed that the average size of crystallites is at the nanoscale level, with the highest value for pure HAP and the lowest value for lyophilized noncalcined HAPc-10%Sr. The degree of crystallinity is higher for pure HAP (52–55%) compared to all multi-substituted HAPs (37–40%). This finding shows that almost the same reduction in the crystallinity degree of pure HAP was observed for all multi-substituted HAPs, rather independent of the increasing amount of Sr in HAPc lattice. Therefore, it clearly appears that the Sr amount does not essentially modify the crystallinity of triple-substituted HAPc, in this range of concentration. Additionally, this evidence represents an important proof that these functional elements are incorporated into the HAP structure and a stabilizing effect is prompted due to this multi-elements doping, in total agreement with slight modifications in lattice parameters.
The control over complex multi-substituted hydroxyapatite synthesis and physicochemical properties is an important step toward the preparation of bio-minerals embracing the complexity of biological hydroxyapatite, in understanding the uniqueness of the hard tissues and in the design of biomaterials with improved biological response.
Conclusion
The synthesized nanomaterials represent a meaningful addition to the field, and their optimal composition was tailored to obtain a unique multi-doped hydroxyapatite phase of very high purity for all ms-HAP biomaterials developed in this study. Moreover, these novel biomaterials might possess the therapeutic effects of substituting functional elements and the properties of hydroxyapatite for bone formation, and thus, they are well suited for biomedical applications in bone regeneration.
Our present study clearly revealed a rational tailoring approach for the design of a next generation of multi-substituted bioactive hydroxyapatites as promising candidates for bone regeneration and bone replacement applications as well as implants or coatings on metallic implants.
Acknowledgments
This work was supported by the European Joint Research EURONANOMED II project “Multifunctional injectable nano HAP composites for the treatment of osteoporotic bone fractures,” NanoForOsteo, namely from the Swiss National Science Foundation through the ERANet under the frame of EuroNanoMed-II (Project number: 31NM30-152035), the Romanian Executive Agency for Higher Education, Research, Development and Innovation Funding, UEFISCDI (grant 4-005, grant 241 and grant 83), the Academy of Science, Latvia, Participation in European Union programs for research and technology development, grant Z/14/1187. Authors gratefully thank Dr. L. Barbu-Tudoran and Dr. A. Vulpoi for electron microscopy assistance and Dr. G. Borodi for XRD assistance. Dr. Gerard Demazeau passed away on Nov. 3, 2017. He was an active member of the international research team of the NanoForOsteo project. He participated in the structural investigation of pure stoichiometric hydroxyapatite and multi-substituted hydroxyapatites using the HR-TEM images and XRD spectra.
Disclosure
The authors report no conflicts of interest in this work.
References
1. O’Neill E, Awale G, Daneshmandi L, Umerah O, Lo KWH. The roles of ions on bone regeneration. Drug Discov Today. 2018;23(4):879–890. doi:10.1016/j.drudis.2018.01.049
2. Ratnayake JTB, Mucalo M, Dias GJ. Substituted hydroxyapatites for bone regeneration: a review of current trends. J Biomed Mater Res. 2017;105(5):1285–1299. doi:10.1002/jbm.b.33651
3. Lowry N, Han Y, Meenan BJ, Boyd AR. Strontium and zinc co-substituted nanophase hydroxyapatite. Ceram Int. 2017;43(15):12070–12078. doi:10.1016/j.ceramint.2017.06.062
4. Zhang N, Zhai D, Chen L, Zou Z, Lin K, Chang J. Hydrothermal synthesis and characterization of Si and Sr co-substituted hydroxyapatite nanowires using strontium containing calcium silicate as precursors. Mater Sci Eng C Mater Biol Appl. 2014;37:286–291. doi:10.1016/j.msec.2014.01.011
5. Sprio S, Tampieri A, Landi E, et al. Physico-chemical properties and solubility behaviour of multi-substituted hydroxyapatite powders containing silicon. Mater Sci Eng C Mater Biol Appl. 2008;28(1):179–187. doi:10.1016/j.msec.2006.11.009
6. Baba IYM, Wimpenny I, Bretcanu O, Dalgarno K, El Haj AJ. Development of multisubstituted hydroxyapatite nanopowders as biomedical materials for bone tissue engineering applications. J Biomed Mater Res A. 2017;105(6):1775–1785. doi:10.1002/jbm.a.v105.6
7. Frasnelli M, Cristofaro F, Sglavo VM, et al. Synthesis and characterization of strontium-substituted hydroxyapatite nanoparticles for bone regeneration. Mater Sci Eng C Mater Biol Appl. 2017;71:653–662. doi:10.1016/j.msec.2016.10.047
8. Begam H, Kundu B, Chanda A, Nandi SK. MG63 osteoblast cell response on Zn doped hydroxyapatite (HAp) with various surface features. Ceram Int. 2017;43(4):3752–3760. doi:10.1016/j.ceramint.2016.12.010
9. Goga F, Forizs E, Avram A, et al. Synthesis and thermal treatment of hydroxyapatite doped with magnesium, zinc and silicon. Rev Chim (Bucharest). 2017;68(6):1193–1200.
10. Ran J, Jiang P, Sun G, et al. Comparisons among Mg, Zn, Sr, and Si doped nano-hydroxyapatite/chitosan composites for load-bearing bone tissue engineering applications. Mater Chem Front. 2017;1(5):900–910. doi:10.1039/C6QM00192K
11. Bohner M. Silicon-substituted calcium phosphates – A critical view. Biomaterials. 2009;30(32):6403–6406. doi:10.1016/j.biomaterials.2009.08.007
12. Boanini E, Gazzano M, Bigi A. Ionic substitutions in calcium phosphates synthesized at low temperature. Acta Biomater. 2010;6(6):1882–1894. doi:10.1016/j.actbio.2009.12.041
13. Larsson S, Fazzalari NL. Anti-osteoporosis therapy and fracture healing. Arch Orthop Trauma Surg. 2014;134(2):291–297. doi:10.1007/s00402-012-1558-8
14. Boanini E, Torricelli P, Fini M, Bigi A. Osteopenic bone cell response to strontium-substituted hydroxyapatite. J Mater Sci Mater Med. 2011;22(9):2079–2088. doi:10.1007/s10856-011-4379-3
15. Kumta PN, Sfeir C, Lee DH, Olton D, Choi D. Nanostructured calcium phosphates for biomedical applications: novel synthesis and characterization. Acta Biomater. 2005;1(1):65–83. doi:10.1016/j.actbio.2004.09.008
16. Cox SC, Jamshidi P, Grover LM, Mallick KK. Preparation and characterisation of nanophase Sr, Mg, and Zn substituted hydroxyapatite by aqueous precipitation. Mater Sci Eng C Mater Biol Appl. 2014;35:106–114. doi:10.1016/j.msec.2013.10.015
17. Shepherd JH, Shepherd DV, Best SM. Substituted hydroxyapatites for bone repair. J Mater Sci Mater Med. 2012;23(10):2335–2347. doi:10.1007/s10856-012-4598-2
18. Bigi A, Cojazzi G, Panzavolta S, et al. Chemical and structural characterization of the mineral phase from cortical and trabecular bone. J Inorg Biochem. 1997;68(1):45–51. doi:10.1016/S0162-0134(97)00007-X
19. Oltean-Dan D, Dogaru GB, Tomoaia-Cotisel M, et al. Enhancement of bone consolidation using high frequency pulsed electromagnetic short-waves and titanium implants coated with biomimetic composite embedded into PLA matrix: in vivo evaluation. Int J Nanomed. 2019;14:5799–5816. doi:10.2147/IJN.S205880
20. Ge M, Ge K, Gao F, et al. Biomimetic mineralized strontium-doped hydroxyapatite on porous poly(L-lactic acid) scaffolds for bone defect repair. Int J Nanomed. 2018;13:1707–1721. doi:10.2147/IJN.S154605
21. Younesi M, Javadpour S, Bahrololoom ME. Effect of heat treatment temperature on chemical compositions of extracted hydroxyapatite from bovine bone ash. J Mater Eng Perform. 2011;20(8):1484–1490. doi:10.1007/s11665-010-9785-z
22. Marie PJ, Ammann P, Boivin G, Rey C. Mechanisms of action and therapeutic potential of strontium in bone. Calcif Tissue Int. 2001;69(3):121–129. doi:10.1007/s002230010055
23. Caverzasio J. Strontium ranelate promotes osteoblastic cell replication through at least two different mechanisms. Bone. 2008;42(6):1131–1136. doi:10.1016/j.bone.2008.02.010
24. Peng SL, Zhou GQ, Luk KDK, et al. Strontium promotes osteogenic differentiation of mesenchymal stem cells through the ras/MAPK signalling pathway. Cell Physiol Biochem. 2009;23(1–3):165–174. doi:10.1159/000204105
25. Hurtel-Lemaire AS, Mentaverri R, Caudrillier A, et al. The calcium-sensing receptor is involved in strontium ranelate induced osteoclast apoptosis: new insights into the associated signaling pathways. J Biol Chem. 2009;284(1):575–584. doi:10.1074/jbc.M801668200
26. Wang Q, Tang P, Ge X, et al. Experimental and simulation studies of strontium/zinc-codoped hydroxyapatite porous scaffolds with excellent osteoinductivity and antibacterial activity. Appl Surf Sci. 2018;462:118–126. doi:10.1016/j.apsusc.2018.08.068
27. Pilmane M, Salma-Ancane K, Loca D, Locs J, Berzina-Cimdina L. Strontium and strontium ranelate: historical review of some of their functions. Mater Sci Eng C Mater Biol Appl. 2017;78:1222–1230. doi:10.1016/j.msec.2017.05.042
28. Bigi A, Boanini E, Capuccini C, Gazzano M. Strontium substituted hydroxyapatite nanocrystals. Inorg Chim Acta. 2007;360(3):1009–1016. doi:10.1016/j.ica.2006.07.074
29. O’Donnell MD, Fredholm Y, de Rouffignac A, Hill RG. Structural analysis of a series of strontium-substituted apatites. Acta Biomater. 2008;4(5):1455–1464. doi:10.1016/j.actbio.2008.04.018
30. Zhang W, Shen Y, Pan H, et al. Effects of strontium in modified biomaterials. Acta Biomater. 2011;7(2):800–808. doi:10.1016/j.actbio.2010.08.031
31. Landi E, Tampieri A, Celotti G, Sprio S, Sandri M, Logroscino G. Sr-substituted hydroxyapatites for osteoporotic bone replacement. Acta Biomater. 2007;3(6):961–969. doi:10.1016/j.actbio.2007.05.006
32. Ni GX, Chiu KY, Lu WW, et al. Strontium-containing hydroxyapatite bioactive bone cement in revision hip arthroplasty. Biomaterials. 2006;27(24):4348–4355. doi:10.1016/j.biomaterials.2006.03.048
33. Ni GX, Yao ZP, Huang GT, Liu WG, Lu WW. The effect of strontium incorporation in hydroxyapatite on osteoblasts in vitro. J Mater Sci Mater Med. 2011;22(4):961–967. doi:10.1007/s10856-011-4264-0
34. Rapuntean S, Frangopol PT, Hodisan I, et al. In vitro response of human osteoblasts cultured on strontium substituted hydroxyapatites. Rev Chim (Bucharest). 2018;69(12):3537–3544.
35. Frangopol PT, Mocanu A, Almasan V, et al. Synthesis and structural characterization of strontium substituted hydroxyapatites. Rev Roum Chim. 2016;61(4–5):337–344.
36. Cheng G, Zhang Y, Yin H, Ruan Y, Sun Y, Lin K. Effects of strontium substitution on the structural distortion of hydroxyapatite by Rietveld refinement and Raman spectroscopy. Ceram Int. 2019;45(8):11073–11078. doi:10.1016/j.ceramint.2019.02.194
37. Bigi A, Foresti E, Gregorini R, Ripamonti A, Roveri N, Shah JS. The role of magnesium on the structure of biological apatites. Calcif Tissue Int. 1992;50(5):439–444. doi:10.1007/BF00296775
38. Landi E, Logroscino G, Proietti L, Tampieri A, Sandri M, Sprio S. Biomimetic Mg-substituted hydroxyapatite: from synthesis to in vivo behavior. J Mater Sci Mater Med. 2008;19(1):239–247. doi:10.1007/s10856-006-0032-y
39. Laurencin D, Almora-Barrios N, de Leeuw NH, et al. Magnesium incorporation into hydroxyapatite. Biomaterials. 2011;32(7):1826–1837. doi:10.1016/j.biomaterials.2010.11.017
40. Stipniece L, Salma-Ancane K, Borodajenko N, Sokolova M, Jakovlevs D, Berzina-Cimdina L. Characterization of Mg-substituted hydroxyapatite synthesized by wet chemical method. Ceram Int. 2014;40(2):3261–3267. doi:10.1016/j.ceramint.2013.09.110
41. Stipniece L, Narkevica I, Salma-Ancane K. Low-temperature synthesis of nanocrystalline hydroxyapatite: effect of Mg and Sr content. J Am Ceram Soc. 2017;100(4):1697–1706. doi:10.1111/jace.2017.100.issue-4
42. Andres NC, D’Elía NL, Ruso JM, Campelo AE, Massheimer VL, Messina PV. Manipulation of Mg2+- Ca2+ switch on the development of bone mimetic hydroxyapatite. ACS Appl Mater Interfaces. 2017;9(18):15698–15710. doi:10.1021/acsami.7b02241
43. Ince T, Kaygili O, Tatar C, Bulut N, Koytepe S, Ates T. The effects of Ni-addition on the crystal structure, thermal properties and morphology of Mg-based hydroxyapatites synthesized by a wet chemical method. Ceram Int. 2018;44(12):14036–14043. doi:10.1016/j.ceramint.2018.04.257
44. Moonga BS, Dempster DW. Zinc is a potent inhibitor of osteoclastic bone-resorption in vitro. J Bone Miner Res. 1995;10(3):453–457. doi:10.1002/jbmr.5650100317
45. Yamaguchi M, Goto M, Uchiyama S, Nakagawa T. Effect of zinc on gene expression in osteoblastic MC3T3-E1 cells: enhancement of Runx2, OPG, and regucalcin mRNA expressions. Mol Cell Biochem. 2008;312(1–2):157–166. doi:10.1007/s11010-008-9731-7
46. Yamaguchi M, Yamaguchi R. Action of zinc on bone metabolism in rats. Increases in alkaline-phosphatase activity and DNA content. Biochem Pharmacol. 1986;35(5):773–777. doi:10.1016/0006-2952(86)90245-5
47. Ofudje EA, Adeogun AI, Idowu MA, Kareem SO. Synthesis and characterization of Zn-doped hydroxyapatite: scaffold application, antibacterial and bioactivity studies. Heliyon. 2019;5(5):e01716. doi:10.1016/j.heliyon.2019.e01716
48. Ren FZ, Xin RL, Ge X, Leng Y. Characterization and structural analysis of zinc-substituted hydroxyapatites. Acta Biomater. 2009;5(8):3141–3149. doi:10.1016/j.actbio.2009.04.014
49. Garbo C, Sindilaru M, Carlea A, et al. Synthesis and structural characterization of novel porous zinc substituted nanohydroxyapatite powders. Part Sci Technol. 2017;35(1):29–37. doi:10.1080/02726351.2015.1121180
50. Alioui H, Bouras O, Bollinger JC. Toward an efficient antibacterial agent: Zn- and Mg- doped hydroxyapatite nanopowders. J Environ Sci Health a Toxic/Hazard Subst Environ Eng. 2019;54(4):315–327. doi:10.1080/10934529.2018.1550292
51. Carlisle EM. Silicon: a requirement in bone formation independent of vitamin D1. Calcif Tissue Int. 1981;33(1):27–34. doi:10.1007/BF02409409
52. Gibson IR, Best SM, Bonfield W. Effect of silicon substitution on the sintering and microstructure of hydroxyapatite. J Am Ceram Soc. 2002;85(11):2771–2777. doi:10.1111/j.1151-2916.2002.tb00527.x
53. Qiu ZY, Noh IS, Zhang SM. Silicate-doped hydroxyapatite and its promotive effect on bone mineralization. Front Mater Sci. 2013;7(1):40–50. doi:10.1007/s11706-013-0193-9
54. Tomoaia G, Mocanu A, Vida-Simiti I, et al. Silicon effect on the composition and structure of nano calcium phosphates. In vitro biocompatibility to human osteoblasts. Mater Sci Eng C Mater Biol Appl. 2014;37:37–47. doi:10.1016/j.msec.2013.12.027
55. Khan AF, Saleem M, Afzal A, Ali A, Khan A, Khan AR. Bioactive behavior of silicon substituted calcium phosphate based bioceramics for bone regeneration. Mater Sci Eng C Mater Biol Appl. 2014;35:245–252. doi:10.1016/j.msec.2013.11.013
56. Szurkowska K, Zgadzaj A, Kuras M, Kolmas J. Novel hybrid material based on Mg2+ and SiO44- co-substituted nano-hydroxyapatite, alginate and chondroitin sulphate for potential use in biomaterials engineering. Ceram Int. 2018;44(15):18551–18559. doi:10.1016/j.ceramint.2018.07.077
57. Porter AE, Botelho CM, Lopes MA, Santos JD, Best SM, Bonfield W. Ultrastructural comparison of dissolution and apatite precipitation on hydroxyapatite and silicon-substituted hydroxyapatite in vitro and in vivo. J Biomed Mater Res A. 2004;69(4):670–679. doi:10.1002/jbm.a.30035
58. Aina V, Lusvardi G, Annaz B, et al. Magnesium- and strontium-co-substituted hydroxyapatite: the effects of doped-ions on the structure and chemico-physical properties. J Mater Sci Mater Med. 2012;23(12):2867–2879. doi:10.1007/s10856-012-4767-3
59. Mocanu A, Furtos G, Rapuntean S, et al. Synthesis; characterization and antimicrobial effects of composites based on multi-substituted hydroxyapatite and silver nanoparticles. Appl Surf Sci. 2014;298:225–235. doi:10.1016/j.apsusc.2014.01.166
60. Tomoaia G, Soritau O, Tomoaia-Cotisel M, et al. Scaffolds made of nanostructured phosphates, collagen and chitosan for cell culture. Powder Technol. 2013;238:99–107. doi:10.1016/j.powtec.2012.05.023
61. Safarzadeh M, Ramesh S, Tan CY, et al. Effect of multi-ions doping on the properties of carbonated hydroxyapatite bioceramic. Ceram Int. 2019;45(3):3473–3477. doi:10.1016/j.ceramint.2018.11.003
62. Loca D, Sokolova M, Locs J, Smirnova A, Irbe Z. Calcium phosphate bone cements for local vancomycin delivery. Mater Sci Eng C Mater Biol Appl. 2015;49:106–113. doi:10.1016/j.msec.2014.12.075
63. Fulgione A, Ianniello F, Papaianni M, et al. Biomimetic hydroxyapatite nanocrystals are an active carrier for Salmonella bacteriophages. Int J Nanomed. 2019;14:2219–2232. doi:10.2147/IJN.S190188
64. Mishra D, Bhunia B, Banerjee I, Datta P, Dhara S, Maiti TK. Enzymatically crosslinked carboxymethyl–chitosan/gelatin/nano-hydroxyapatite injectable gels for in situ bone tissue engineering application. Mater Sci Eng C Mater Biol Appl. 2011;31(7):1295–1304. doi:10.1016/j.msec.2011.04.007
65. Chaturvedi S, Dave PN. Review on thermal decomposition of ammonium nitrate. J Energy Mater. 2013;31(1):1–26. doi:10.1080/07370652.2011.573523
66. ISO 11885. Water quality - Determination of selected elements by inductively coupled plasma optical emission spectrometry (ICP-OES); 2009.
67. Ritger PL, Peppas NA. A simple equation for description of solute release I. Fickian and non-Fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release. 1987;5(1):23–36. doi:10.1016/0168-3659(87)90034-4
68. Lindahl C, Xia W, Lausmaa J, Engqvist H. Incorporation of active ions into calcium phosphate coatings, their release behavior and mechanism. Biomed Mater. 2012;7(4):045018. doi:10.1088/1748-6041/7/4/045018
69. Bakan F, Lacin O, Sarac H. A novel low temperature sol–gel synthesis process for thermally stable nanocrystalline hydroxyapatite. Powder Technol. 2013;233:295–302. doi:10.1016/j.powtec.2012.08.030
70. Suchanek WL, Shuk P, Byrappa K, Riman RE, Ten Huisen KS, Janes VF. Mechanochemical–hydrothermal synthesis of carbonated apatite powders at room temperature. Biomaterials. 2002;23(3):699–710. doi:10.1016/S0142-9612(01)00158-2
71. Gopi D, Nithiya S, Shinyjoy E, Kavitha L. Spectroscopic investigation on formation and growth of mineralized nanohydroxyapatite for bone tissue engineering applications. Spectrochim Acta a Molec Biomolec Spectr. 2012;92:194–200. doi:10.1016/j.saa.2012.02.069
72. Ou-Yang H, Paschalis EP, Boskey AL, Mendelsohn R. Two-dimensional vibrational correlation spectroscopy of in vitro hydroxyapatite maturation. Biopolymers. 2000;57(3):129–139. doi:10.1002/(ISSN)1097-0282
73. Jiang SD, Yao QZ, Ma YF, Zhou GT, Fu SQ. Phosphate-dependent morphological evolution of hydroxyapatite and implication for biomineralisation. Gondwana Res. 2015;28(2):858–868. doi:10.1016/j.gr.2014.04.005
74. Horovitz O, Tomoaia G, Mocanu A, Yupsanis T, Tomoaia-Cotisel M. Protein binding to gold auto-assembled films. Gold Bull. 2007;40(4):295–304. doi:10.1007/BF03215603
75. Tomoaia G, Tomoaia-Cotisel M, Pop LB, et al. Synthesis and characterization of some composites based on nanostructured phosphates, collagen and chitosan. Rev Roum Chim. 2011;56(10–11):1039–1046.
76. Tomoaia G, Horovitz O, Mocanu A, et al. Effects of doxorubicin mediated by gold nanoparticles and resveratrol in two human cervical tumor cell lines. Colloids Surf B Biointerfaces. 2015;135:726–734. doi:10.1016/j.colsurfb.2015.08.036
77. Manocha S, Joshi P, Patel B, Manocha LM. Synthesis and characterization of hydroxyapatite nanoparticles using sol-gel method. Eurasian Chem Techn J. 2011;13(1–2):85–88. doi:10.18321/ectj71
78. Pang YX, Bao X. Influence of temperature, ripening time and calcination on the morphology and crystallinity of hydroxyapatite nanoparticles. J Eur Ceram Soc. 2003;23(10):1697–1704. doi:10.1016/S0955-2219(02)00413-2
79. Bouyer E, Gitzhofer F, Boulos MI. Morphological study of hydroxyapatite nanocrystal suspension. J Mater Sci Mater Med. 2000;11(8):523–531. doi:10.1023/A:1008918110156
80. Rouquerol J, Avnir D, Fairbridge CW, et al. Recommendations for the characterization of porous solids (Technical report). Pure Appl Chem. 1994;66(8):1739–1758. doi:10.1351/pac199466081739
81. Barba A, Diez-Escudero A, Maazouz Y, et al. Osteoinduction by foamed and 3D-printed calcium phosphate scaffolds: effect of nanostructure and pore architecture. ACS Appl Mater Interfaces. 2017;9(48):41722–41736. doi:10.1021/acsami.7b14175
82. Dorozhkin SV. Surface reactions of apatite dissolution. J Colloid Interf Sci. 1997;191(2):489–497. doi:10.1006/jcis.1997.4942
83. Kaufman HW, Kleinberg I. Studies on the incongruent solubility of hydroxyapatite. Calcif Tissue Intern. 1979;27(12):143–151. doi:10.1007/BF02441177
84. Beuvelot J, Mauras Y, Mabilleau G, Marchand-Libouban H, Chappard D. Adsorption and release of strontium from hydroxyapatite crystals developed in simulated body fluid (SBF) on poly (2-hydroxyethyl) methacrylate substrates. Dig J Nanomater Biostruct. 2013;8(1):207–217.
85. Stötzel C, Müller FA, Reinert F, Niederdraenk F, Barralet JE, Gbureck U. Ion adsorption behaviour of hydroxyapatite with different crystallinities. Colloids Surf B Biointerfaces. 2009;74(1):91–95. doi:10.1016/j.colsurfb.2009.06.031
86. Xia W, Grandfield K, Schwenke A, Engqvist H. Synthesis and release of trace elements from hollow and porous hydroxyapatite spheres. Nanotechnology. 2011;22(30):305610. doi:10.1088/0957-4484/22/30/305610
87. Huang X, Brazel CS. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J Control Release. 2001;73(2–3):121–136. doi:10.1016/S0168-3659(01)00248-6
88. Grynpas MD, Marie PJ. Effects of low doses of strontium on bone quality and quantity in rats. Bone. 1990;11(5):313–319. doi:10.1016/8756-3282(90)90086-E
89. Pietak AM, Reid JW, Stott MJ, Sayer M. Silicon substitution in the calcium phosphate bioceramics. Biomaterials. 2007;28(28):4023–4032. doi:10.1016/j.biomaterials.2007.05.003
90. Cacciotti I. Multisubstituted hydroxyapatite powders and coatings: the influence of the codoping on the hydroxyapatite performances. Int J Appl Ceram Technol. 2019;16(5):1864–1884. doi:10.1111/ijac.v16.5
91. Webster TJ, Ergun C, Doremus RH, Siegel RW, Bizios R. Enhanced functions of osteoblasts on nanophase ceramics. Biomaterials. 2000;21(17):1803–1810. doi:10.1016/S0142-9612(00)00075-2
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.