Back to Journals » OncoTargets and Therapy » Volume 13
Activated Hepatic Stellate Cells (HSCs) Exert Immunosuppressive Effects in Hepatocellular Carcinoma by Producing Complement C3
Authors Xu Y ![]() , Huang Y, Xu W, Zheng X, Yi X, Huang L, Wang Y, Wu K
, Huang Y, Xu W, Zheng X, Yi X, Huang L, Wang Y, Wu K
Received 16 October 2019
Accepted for publication 1 February 2020
Published 18 February 2020 Volume 2020:13 Pages 1497—1505
DOI https://doi.org/10.2147/OTT.S234920
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Gaetano Romano
Yaping Xu,1,2 Yihao Huang,1 Wanqiong Xu,1 Xiaohui Zheng,1 Xue Yi,1,2 Liyue Huang,1 Yuxiao Wang,1 Kangni Wu3
1Key Laboratory of Functional and Clinical Translational Medicine, Fujian Province University, Department of Physiology, Xiamen Medical College, Xiamen 361023, People’s Republic of China; 2Xiamen Key Laboratory of Respiratory Diseases, Xiamen 361023, People’s Republic of China; 3Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, Medical College of Xiamen University, Xiamen 361003, People’s Republic of China
Correspondence: Yaping Xu; Kangni Wu Tel +86-159 5923 5683
Email [email protected]; [email protected]
Objective: Hepatic stellate cells (HSCs) are the important players in liver cirrhosis and liver cancer. They also act as critical mediators of immunosuppression in hepatocellular carcinoma (HCC). In this study, we hypothesized that HSCs promote HCC progression via C3.
Methods: C3 in HSCs was knocked down using a shRNA retroviral plasmid. The conditioned medium from HSCs or shC3 HSCs (knockdown of C3 by shRNA in HSCs) was collected to detect their effects on bone marrow (BM) and T cells (including expansion and apoptosis) in vitro, and in an HCC in situ model in mice.
Results: We found that HSCs promoted T-cell apoptosis and decreased their proliferation, inhibited dendritic cell (DC) maturation, and induced myeloid-derived suppressor cell (MDSC) expansion through the C3 pathway in vitro. In addition, the knockdown of C3 suppressed HSC-promoted HCC development in the orthotopic transplantation tumor model of HCC in mice.
Conclusion: These findings provide more insights into the immunomodulatory roles of HSCs in HCC progression and indicate that modulation of the C3 pathway might be a novel therapeutic approach for liver cancer.
Keywords: hepatocellular carcinoma, hepatic stellate cells, complement C3, myeloid-derived suppressor cells, T cells
Introduction
Hepatocellular carcinoma (HCC) is the second leading cause of cancer-related death worldwide.1,2 Due to drug resistance by immune evasion in HCC, there is currently no comprehensive treatment for this disease. The tumor microenvironment, which consists of hepatic stellate cells (HSCs), myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and tumor-associated macrophages (TAMs), plays an important role in immune evasion in HCC.3,4 Reciprocal communication between tumor cells and stromal cells in the tumor microenvironment is fundamental to the initiation and progression of HCC.5,6
HSCs are the important non-parenchymal cells in the liver, and the co-transplantation of HSCs with allogeneic islets exerts immunomodulatory effects.4 HSCs are also the critical stromal cells that accelerate hepatocarcinogenesis and potentiate the metastasis of HCC by increasing the expression of N-methyltransferase.7 Moreover, HSCs secrete high levels of interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), the key regulators of Th17 cell differentiation, leading to a poor prognosis.8,9 MDSCs are another key regulator of immune responses.10 MDSCs are a heterogeneous population of immature myeloid cells (IMCs), including myeloid progenitors and precursors of macrophages, granulocytes, and dendritic cells (DCs). In pathological conditions such as infections and cancer, the differentiation of IMCs into mature myeloid cells is partially blocked, resulting in the expansion of MDSCs.11 Active HSCs play a key role in HCC by producing abundant cytokines, such as activated third component of complement (C3), prostaglandin E2 (PGE2), stem cell factor (SCF), macrophage colony-stimulating factor (M-CSF), IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), and vascular endothelial growth factor (VEGF), which induce MDSC expansion.11–16 We previously found that HSCs increased Tregs and MDSC levels in the HCC microenvironment, which lead to the growth of HCC.17 Although HSCs play an important role in immunosuppression in HCC, it is still unclear how HSCs modulate the MDSCs and form a negative network that leads to HCC immune escape.
The complement system is the major component of both innate immunity and the acquired immune system. C3 is the central component of the complement system and promotes growth, proliferation, migration, and stemness of cutaneous squamous cell carcinoma.18,19 Activation of C3 triggers resistance of tumor cells to programmed death-ligand 1 (PD-L1) antibody by modulating TAMs.20 In C3-deficient mice, the growth and metastasis of primary tumors were strongly inhibited in lung cancer, which was ascribed to the increased numbers of CD4+ and CD8+ T cells.21 C3 has varied functions in HCC progression, and the role of C3 in HSC-mediated HCC immune escape is not fully known. HSCs promote the development of MDSCs in the DC culture through C3.12 Moreover, in a mouse model of HCC, HSCs induced expansion of MDSCs and granulocytic-MDSCs (G-MDSCs) via a COX2/PEG2-dependent pathway, but affect monocytic-MDSCs (Mo-MDSCs) expansion through COX2/PEG2-independent signaling. The mechanisms that HSCs trigger in Mo-MDSC expansion are still unclear.22,23
We hypothesized that HSCs promote HCC progression via C3. Thus, the conditioned medium from HSCs or shC3 HSCs (knockdown of C3 by shRNA in HSCs) was collected to detect their effects on bone marrow (BM) and T cells, and both in vitro and in vivo results demonstrated that HSCs promoted T-cell apoptosis and inhibited their proliferation through a C3 pathway. Current studies provide more insights into the immunomodulatory roles of HSCs in HCC progression, indicating that the modulation of the C3 pathway might be a novel therapeutic approach for liver cancer.
Materials and Methods
Animals
Eight-week-old male BALB/c (H-2d, haplotype) mice were purchased from the National Rodent Laboratory Animal Resources (Shanghai, China). They were housed under pathogen-free conditions in the animal center of Xiamen University. The mice were randomly assigned to three groups as follows: (1) the control group received an intrahepatic injection of 1 × 106 H22 cells; (2) the HSC group received an intrahepatic injection of 1 × 106 H22 cells plus 2 × 105 activated HSCs; and (3) the shC3 HSC group received an intrahepatic injection of 1 × 106 H22 cells plus 2 × 105 shC3 HSC. Each group had at least six mice. All animal experimental protocols were performed in compliance with the Guidelines for the Institutional Animal Care and Use Committee of Xiamen University.
Cell Culture
The H22 cells were purchased from Shanghai Cell Bank (Chinese Academy of Sciences) and grown in Roswell Park Memorial Institute (RPMI) 1640 medium (HyClone, Logan, UT, USA), supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 U/mL streptomycin at 37°C and 5% CO2. The HSCs were isolated from the livers of BALB/c mice as previously described.22 The conditional medium from HSCs (HSC-CM) or shC3 HSCs (shC3-CM) was obtained as follows: The cells were grown in 10-cm plates, whereas the HSCs or shC3 HSCs were grown to 100% confluence, and then the media were replaced with 10 mL fresh FBS-free medium. After 24 hrs of culture, the media were collected to culture with bone marrow or T cells.
Knockdown of C3 in HSCs
Knockdown of C3 in HSCs was performed using a shRNA retroviral plasmid containing a puromycin resistance gene, which was purchased from Clontech (Mountain View, CA, USA). Lentiviral pLKO.1 vector was used to express short hairpin RNA (shRNA) directed against C3. Turbofect transfection reagent (Thermo Scientific, Waltham, MA, USA) was used for the transfection. The stable cells were selected by 1 μL/mL puromycin for 1 week. The transfection efficiency was assessed by Western blot assay and real-time quantitative polymerase chain reaction (RT-qPCR). The primers for C3 were 5ʹ-GTGGTCACTCAGGGATCTAATG-3ʹ (forward) and 5ʹ- TCTGGGAGAGTGTCCTTCTT-3ʹ (reverse). Western blot assay and RT-qPCR were performed as descriptions.24,25
Cell Apoptosis Assay
T cells were isolated from BALB/c mice spleens by nylon wool-elution, as described previously.23 T cells were cultured with RPMI 1640 medium, HSC-CM, or shC3 HSC-CM in 12-well plates, respectively, and then stimulated with 1 μg/mL of anti-CD3 mAb and CD28 (R&D Systems, Minneapolis, MN, USA). After 24 hrs, cells were harvested and subjected to flow cytometry (CytoFLEX LX, Beckman Coulter, Boulevard Brea, CA, USA) for cell apoptosis assay by incubation with Annexin-V-FITC and PI (BD PharMingen, San Diego, CA, USA).
T-Cell Proliferation Assay
Proliferation of T cells was analyzed by carboxyfluorescein succinimidyl ester (CFSE) dilution assay. T-cell proliferation assay was performed in 12-well plates (Corning, Corning, NY, USA). The T cells were stimulated with 1 μg/mL of anti-CD3 mAb and CD28. After 72 hrs of culture, cells were harvested and divided into two groups to be stained with APC-CD4 and APC-CD8 antibodies, respectively. Flow cytometry was performed to test T-cell proliferation.
Flow Cytometry Analysis
APC-CD11b, PE Gr-1, FITC-Ly-6G, PE-Ly-6C, APC-CD4, and APC-CD8 were purchased from Biosciences (San Jose, CA, USA). CD40, CD86, CD80, CD274, and MHCII were purchased from eBioscience (San Diego, CA, USA). Tumor cells were prepared for flow cytometry analysis as previously described.23 Fluorescent-labeled cells were analyzed with CytoFLEX LX.
Statistical Analysis
Randomization was used to assign mice to different experimental groups and to collect and process data. All of the histology samples were scored blindly and independently by at least two investigators. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology. Data were analyzed using Prism6.0 (GraphPad Software, USA), and they are presented as the mean ± standard deviation (SD). Each experiment was performed at least three times. Group comparisons were performed using one-way analysis of variance (ANOVA). For all ANOVA, post hoc tests were run only if F achieved p < 0.05 and there was no significant variance inhomogeneity. P < 0.05 was considered statistically significant.
Results
Downregulation of C3 Expression by shRNA in HSCs
In our previous study, we reported that the conditioned medium from HSCs (HSC-CM) induced MDSC expansion (especially G-MDSCs) through COX2-PEG2.23 However, the mechanism by which HSCs induced Mo-MDSC expansion was unclear. To explore the putative mechanism, we tested three shRNA (sh1, sh2, sh3) in the context of a lentiviral vector for their activity to downregulate C3 expression in HSCs. The results showed that all three shRNA but not their vector significantly decreased the protein levels and mRNA expression of C3 (Figure 1A and B). The sh3 was chosen for further studies.
 |
Figure 1 Expression of C3 was down-regulated in HSCs via shC3 transfection. (A) Representative western blot bands and quantification of C3a abundances in HSCs (HSC) or treatment with vehicle (shCon) or shRNA after cells generated with puromycin for 1 week. The relative intensity was normalized to actin and relative to the HSC. (B) The C3 mRNA level of HSCs, shCon HSCs, or shC3 HSCs was measured by real-time qPCR cells generated with puromycin for 1 week. The statistical significance was labeled as ***p < 0.001, ns, p > 0.05, n = 3. |
HSCs Inhibit Bone Marrow (BM)-Derived DC Differentiation Through the C3 Pathway
Encouraged by the knockdown efficiency of sh3 on C3 in HSCs, we further tested the role of C3 in BM-derived DC differentiation in HSCs. The conditional media from HSCs and shC3 HSCs (shC3-CM) were collected to culture with BM, and the biomarker of BM-derived DC differentiation was measured by flow cytometry analysis. Consistent with previous findings,23 the expressions of CD11c, CD80, CD86, and MHC II were decreased and CD274 expression was slightly increased in BM co-cultured with HSC-CM (Figure 2A). However, shC3-CM increased the expression of CD11c, CD86, and MHC II in BM when compared with the HSC-CM group (Figure 2A), suggesting that HSCs inhibited BM cell differentiation into immature DCs and macrophages through C3.
 |
Figure 2 HSCs inhibit bone marrow (BM)-derived DC differentiation through the C3 pathway. (A) Flow cytometry analysis of cell surface marker expression in DC differentiation in Ctrl medium (1640 medium), HSC-CM (conditional medium from HSCs), or shC3-CM (conditional medium from shC3 HSCs). (B) The gating strategy of MDSC subsets. (C) The MDSC expression in Ctrl, HSC-CM, or shC3-CM media. Number is percent of the cell population represented by MDSCs. (D) The G-MDSC and Mo-MDSC expression in Ctrl, HSC-CM, or shC3-CM media. Percent G-MDSCs was calculated as follows: corrected G-MDSC percent = 100% × CD11b+ percent × Ly6G+Ly6Clow percent. Corrected Mo-MDSC percent = 100% × CD11b+ percent × Ly6G−Ly6Chigh percent. The statistical significance was labeled as ***p < 0.001; **p < 0.01; ns, p > 0.05, n = 3. |
MDSCs are critical mediators of tumor-induced immune dysfunction and cancer progression.26 To detect the effect of HSCs on MDSC accumulation in the liver cancer microenvironment, the expression of MDSCs and their subsets were then detected. Murine MDSCs co-express CD11b and Gr-1, and their subsets consist of granulocytic-MDSCs (G-MDSCs, CD11b+Ly6G+Ly6Cint/low) and monocytic-MDSCs (Mo-MDSCs, CD11b+ Ly6G−Ly6Chigh).11 Thus, in our study, the gating strategy of these cells was performed as shown in Figure 2B. MDSC expression was significantly increased in BM co-cultured with HSC-CM, whereas C3 knockdown significantly suppressed the increment of MDSC expression. We also investigated the influence of C3 on the subsets of MDSCs, G-MDSCs, and Mo-MDSCs. HSC-CM induced both G-MDSC and Mo-MDSC expansion, whereas C3 knockdown effectively decreased the Mo-MDSCs expansion but not the increased G-MDSCs expression (Figure 2D). When combined, these results suggest that HSCs induce MDSC and Mo-MDSC expansion via the C3 pathway.
HSCs Induce T-Cell Apoptosis and Suppress T-Cell Proliferation Through the C3 Pathway
HSCs also exhibit immunomodulatory activities;4 therefore, we further explored the role of C3 in the immunosuppressive effects of HSCs on T cells. HSC-CM and shC3-CM were collected to culture with T cells for 24 hrs and 72 hrs, and the apoptosis and cell proliferation of T cells were analyzed by flow cytometry, respectively. Data showed that HSC-CM induced T-cell apoptosis, whereas the knockdown of C3 in HSCs markedly decreased the increment of the apoptosis ratio of T cells (Figure 3A). In addition, the proliferation of CD4+ and CD8+ T cells was measured by CFSE label. As shown in Figure 3B and C, cell proliferation of CD4+ and CD8+ T cells was inhibited by HSCs, whereas this inhibitory trend was weakened by C3 knockdown. In addition, we had added the recombinant C3a into the proliferation and apoptosis of T cells. The data showed that HSC knockdown C3 lost the ability to inhibit T-cell proliferation and promoted T-cell apoptosis, and when supplemented with C3a, the T cell ability was partly recovered (Supplement Figure 1).
 |
Figure 3 HSCs induce T-cell apoptosis and suppress T-cell proliferation through the C3 pathway. T cells were co-cultured with Ctrl (1640 medium), HSC-CM (conditional medium from HSCs), or shC3-CM (conditional medium from shC3 HSCs), followed by measurement of (A) apoptosis using Annexin V/PI assay after co-culturing for 24 h and (B, C) proliferation of CD4+ and CD8+ T cells after co-culturing for 72 h using CFSE staining assay. T cells were simulated with CD3 and CD28 antibody. The statistical significance was labeled as ***p < 0.001; **p < 0.01; n = 3. |
Knockdown of C3 Suppressed HSC-Promoted HCC Development
HSCs have been reported to enhance HCC development,27–29 and C3 contributed to the growth of HCC (Supplement Figure 2); thus, we further studied the role of C3 in HSCs in the mouse model of HCC. The intra-hepatic injection of H22 cells into male BALB/c mice induced significant HCC symptoms on day 10 after the operation, whereas the administration of H22 cells together with HSCs further enhanced tumor growth (Figure 4A). However, co-injection of H22 cells with C3 knockdown shC3 HSCs effectively suppressed the HSC-promoted HCC development (Figure 4A). We also analyzed the MDSCs and the subsets of the cell population in the tumor. The expressions of MDSCs and their subsets, G-MDSCs and Mo-MDSCs, were increased in tumor tissues from HSC-treated mice, but were decreased in the shC3 HSC group (Figure 4B and C). These results showed that C3 contributed to the immunosuppressive effects of HSCs in vivo.
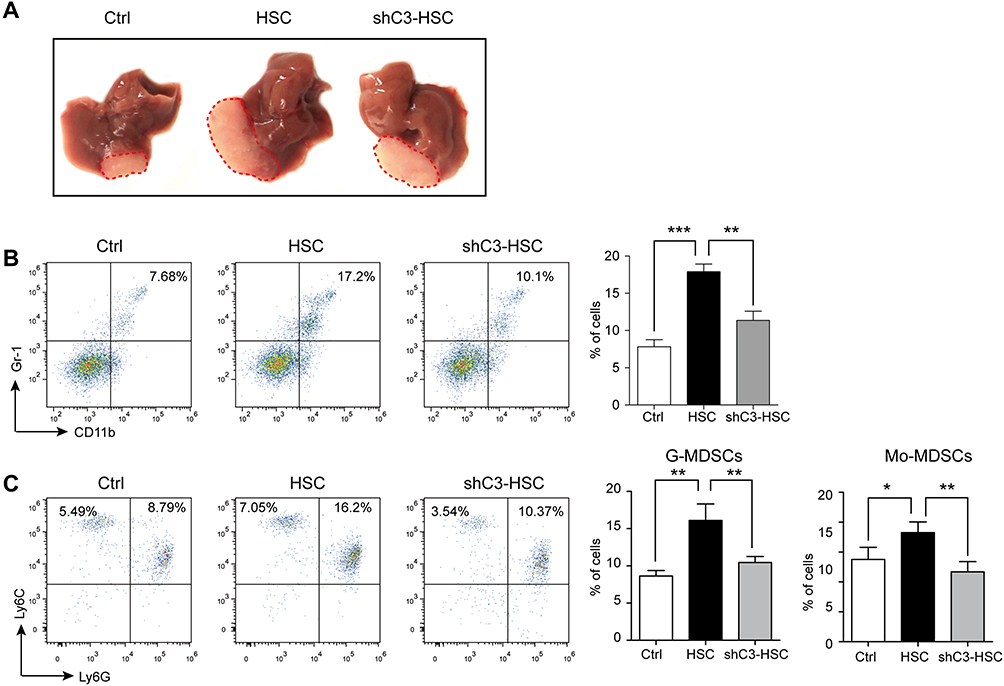 |
Figure 4 Knockdown of C3 suppressed HSCs promoted HCC development. (A) Representative images of typical tumors 10 days after intra-hepatic injection of H22 cells (Ctrl), together with vehicle HSC or shC3 HSC into male BALB/c mice. (B) MDSC accumulation in tumors was counted. Number is percent of the cell population represented by MDSCs (right panels). (C) G-MDSCs and Mo-MDSCs in tumors were measured with flow cytometry. Number is percent of the cell population represented by G-MDSCs and Mo-MDSCs. Percent G-MDSCs was calculated as follows: corrected G-MDSC percent = 100% × CD11b+ percent × Ly6G+Ly6Clow percent. Corrected Mo-MDSC percent = 100% × CD11b+ percent × Ly6G−Ly6Chigh percent. The statistical significance was labeled as ***p < 0.001, **p < 0.01, and *p < 0.05. |
Discussion
The tumor environment plays a critical role in tumor development. In the HCC environment, different players, such as HSCs, MDSCs, and Tregs, contribute to HCC progression by building a negative network that leads to immune escape.3 In the last decade, many researchers have focused on understanding the modulation of the tumor microenvironment to reveal the mechanism of liver cancer.30,31 The tumor microenvironment that consists of liver-resident cells (e.g., liver sinusoidal endothelial cells and HSCs) and infiltrating immune cells (e.g., DCs, T cells, MDSCs, NK cells, Treg, and macrophages) is known to play an important role in HCC development in the liver. These cells promote HCC growth by the formation of negative immune responses.3 T cells (namely cytotoxic T cells), NK cells, and macrophages have the ability to kill tumor cells and eradicate established tumors.32–34 DCs as antigen-presenting cells stimulated T-cell activation. However, in the tumor microenvironment, their abilities were generally suppressed by MDSCs and Tregs. HSCs exist in the space of Disse for the storage of vitamin A and play a central role in liver fibrosis and HCC development. In the liver cancer microenvironment, activated HSCs are capable of shaping an immunosuppressive environment through inducing an accumulation of MDSCs and inhibiting T-cell proliferation and function. However, the underlying mechanism was not entirely clear. In this study, we found that C3 plays an important role in HSC-mediated HCC progression. We demonstrated the capability of C3 to suppress BM-derived DC differentiation, enhance MDSCs and their subset expansion, promote T-cell apoptosis, and inhibit the proliferation of CD4+ and CD8+ T cells in vitro. We also investigated the role of C3 in HSCs in the mouse model of HCC (Figure 5). In a previous study, HSCs were found to induce G-MDSC expansion but not Mo-MDSC expansion through the COX2-PGE2 pathway.23 However, there was no study on the effects of C3 in HSC-mediated HCC progression before our study, and for the first time, we reported the immunomodulatory roles of C3 produced by HSCs in liver cancer.
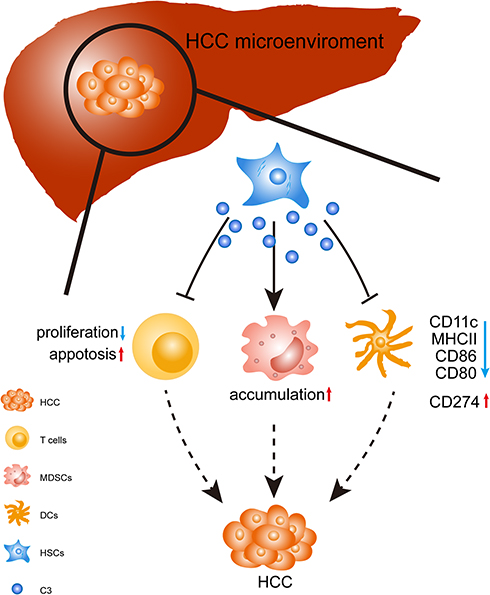 |
Figure 5 Schematic diagram of the stimulatory effect of C3 produced by HSCs in HCC. In the HCC microenvironment, HSCs produced C3 to induce T-cell apoptosis and reduced the inhibition of CD4+ and CD8+ T-cell proliferation. In addition, HSCs induced MDSCs expansion via C3; HSCs attenuated DC activity through C3 by the decreased expressions of CD11c, CD80, CD86, and MHCII, and slightly increased CD274 expression. HSCs disrupted the immunological balance in HCC, resulting in dominant immunosuppression and tumor progression. |
MDSCs are one of the most important regulators of immune responses in liver cancer.10 MDSCs expand in pathological conditions, such as cancer, infections, sepsis, trauma, transplantation, and some autoimmune disorders.11 MDSCs could be induced from mature human monocytes through HSCs dose-dependently.35 Furthermore, activated human hepatic stellate cells induce myeloid-derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion.36 Bone marrow cells cultured with HSC-CM express lower levels of CD11c, CD80, CD86, and MHCII (Figure 2B), suggesting that HSCs inhibit the differentiation of IMCs into mature myeloid cells and increase the expansion of MDSCs. These results were consistent with our previous results and those of other researchers.23,37 However, knockdown of C3 in HSCs reverses this effect. Notably, knockdown of C3 in HSCs decreased the expansion of Mo-MDSCs but had no effect on G-MDSCs. Previous results showed that HSCs induced MDSCs and G-MDSCs via a COX2/PEG2-dependent pathway,23 which seemed to suggest that HSCs induced expansion of G-MDSCs via a COX2/PEG2-dependent pathway, but increased the expression of Mo-MDSCs through C3 signaling.
HSCs also influence immune suppression by promoting T-cell apoptosis via the C3 pathway.15 The effect of C3 on the immune response is dual. Local C3 secretion by macrophages leads to IL-17A-mediated inflammatory cell Th17 cell infiltration into the kidney.38 However, macrophages from C3−/- mice showed a reduced capability to stimulate an alloreactive T-cell response.39 Human plasma C3 is not essential for the development of T cells.40 However, our results demonstrated that C3 derived from HSCs had a suppressive effect on T-cell proliferation. The inconsistent effect of C3 on immune response may be due to other cell populations or coexisting factors in the local inflammatory environment, which could directly or indirectly modulate C3 signaling on immune cells. In this study, the role of C3 was in accordance with that reported in islet transplantation.12 In addition, C3 is produced by hepatocytes and is abundant in the liver;41 thus, why C3-deficient HSCs lose their immunosuppressive abilities in vivo is of special interest. In theory, the C3 expression of HCC cells would affect the DCs and T cells. Interestingly, the local environment theory showed, in many circumstances, such as the regulation of T-cell response42,43 and osteoclast differentiation,44 that the effector complement components are locally produced by related cells, not by systemic complement components produced by hepatocytes. Another possibility is that HSCs produce a C3 isoform that is different from that produced by hepatocytes in structure and function. A more thorough investigation of the isoform of C3 is needed to support this hypothesis.
Conclusion
Our data showed that HSCs promoted HCC progression, which suggests that the cellular and molecular mechanisms mediating the immunomodulatory activity of HSCs are required to provide more insight into the progression of HCC and boost the development of therapeutic strategies for HCC.
Ethical Statement
All animal experimental protocols were performed in compliance with the Guidelines for the Institutional Animal Care and Use Committee of Xiamen University. (The date of animal research approval: 2016-03-14).
Acknowledgments
We thank our colleagues for their valuable efforts and comments on this article. This work was supported by grants from the National Natural Science Foundation of China (81602500); Outstanding Youth Science Research Personnel Training Plan of Fujian Province Colleges and Universities (2017); Doctoral Start-up Foundation of Xiamen Medical College (K2016-07); and Xiamen Key Laboratory of Respiratory Diseases Major projects (HXJB-02).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Bruix J, Reig M, Sherman M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology. 2016;150(4):835–853. doi:10.1053/j.gastro.2015.12.041
2. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi:10.1002/ijc.29210
3. Eggert T, Greten TF. Tumor regulation of the tissue environment in the liver. Pharmacol Ther. 2017;173:47–57. doi:10.1016/j.pharmthera.2017.02.005
4. Hsieh CC, Hung CH, Lu L, Qian S. Hepatic immune tolerance induced by hepatic stellate cells. World J Gastroenterol. 2015;21(42):11887–11892. doi:10.3748/wjg.v21.i42.11887
5. Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144(3):512–527. doi:10.1053/j.gastro.2013.01.002
6. Zhou BY, Gong JH, Cai XY, et al. An imbalance between stellate cells and gammadeltaT cells contributes to hepatocellular carcinoma aggressiveness and recurrence. Hepatol Int. 2019;13:631–640. doi:10.1007/s12072-019-09969-w
7. Li J, You S, Zhang S, et al. Elevated N-methyltransferase expression induced by hepatic stellate cells contributes to the metastasis of hepatocellular carcinoma via regulation of the CD44v3 isoform. Mol Oncol. 2019. doi:10.1002/1878-0261.12544
8. Liao R, Sun TW, Yi Y, et al. Expression of TREM-1 in hepatic stellate cells and prognostic value in hepatitis B-related hepatocellular carcinoma. Cancer Sci. 2012;103(6):984–992. doi:10.1111/j.1349-7006.2012.02273.x
9. Liao R, Sun J, Wu H, et al. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. J Exp Clin Cancer Res. 2013;32:3. doi:10.1186/1756-9966-32-3
10. Yang WC, Ma G, Chen SH, Pan PY. Polarization and reprogramming of myeloid-derived suppressor cells. J Mol Cell Biol. 2013;5(3):207–209.
11. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi:10.1038/nri2506
12. Hsieh CC, Chou HS, Yang HR, et al. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood. 2013;121(10):1760–1768. doi:10.1182/blood-2012-06-440214
13. Pan PY, Wang GX, Yin B, et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood. 2008;111(1):219–228. doi:10.1182/blood-2007-04-086835
14. Hui AY, Dannenberg AJ, Sung JJ, et al. Prostaglandin E2 inhibits transforming growth factor beta 1-mediated induction of collagen alpha 1(I) in hepatic stellate cells. J Hepatol. 2004;41(2):251–258. doi:10.1016/j.jhep.2004.04.033
15. Yu MC, Chen CH, Liang X, et al. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology. 2004;40(6):1312–1321. doi:10.1002/hep.20488
16. Maher JJ. Interactions between hepatic stellate cells and the immune system. Semin Liver Dis. 2001;21(3):417–426. doi:10.1055/s-2001-17555
17. Zhao W, Zhang L, Xu Y, et al. Hepatic stellate cells promote tumor progression by enhancement of immunosuppressive cells in an orthotopic liver tumor mouse model. Lab Invest. 2014;94(2):182–191. doi:10.1038/labinvest.2013.139
18. Riihila P, Nissinen L, Farshchian M, et al. Complement component C3 and complement factor B promote growth of cutaneous squamous cell carcinoma. Am J Pathol. 2017;187(5):1186–1197. doi:10.1016/j.ajpath.2017.01.006
19. Fan Z, Qin J, Wang D, Geng S. Complement C3a promotes proliferation, migration and stemness in cutaneous squamous cell carcinoma. J Cell Mol Med. 2019;23(5):3097–3107. doi:10.1111/jcmm.2019.23.issue-5
20. Zha H, Wang X, Zhu Y, et al. Intracellular activation of complement C3 leads to PD-L1 antibody treatment resistance by modulating tumor-associated macrophages. Cancer Immunol Res. 2019;7(2):193–207. doi:10.1158/2326-6066.CIR-18-0272
21. Kwak JW, Laskowski J, Li HY, et al. Complement activation via a C3a receptor pathway alters CD4(+) T lymphocytes and mediates lung cancer progression. Cancer Res. 2018;78(1):143–156. doi:10.1158/0008-5472.CAN-17-0240
22. Zhao W, Zhang L, Yin Z, et al. Activated hepatic stellate cells promote hepatocellular carcinoma development in immunocompetent mice. Int J Cancer. 2011;129(11):2651–2661. doi:10.1002/ijc.25920
23. Xu Y, Zhao W, Xu J, et al. Activated hepatic stellate cells promote liver cancer by induction of myeloid-derived suppressor cells through cyclooxygenase-2. Oncotarget. 2016;7(8):8866–8878. doi:10.18632/oncotarget.6839
24. Zhou P, Xiang L, Yang Y, et al. N-Acylethanolamine acid amidase (NAAA) inhibitor F215 as a novel therapeutic agent for osteoarthritis. Pharmacol Res. 2019;145(07):104264. doi:10.1016/j.phrs.2019.104264
25. Wu K, Xiu Y, Zhou P, Qiu Y, Li Y. A new use for an old drug: carmofur attenuates Lipopolysaccharide (LPS)-induced acute lung injury via inhibition of FAAH and NAAA activities. Front Pharmacol. 2019;10:818. doi:10.3389/fphar.2019.00818
26. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi:10.1146/annurev.immunol.25.022106.141609
27. Shao S, Duan W, Xu Q, et al. Curcumin suppresses hepatic stellate cell-induced hepatocarcinoma angiogenesis and invasion through downregulating CTGF. Oxid Med Cell Longev. 2019;2019:8148510. doi:10.1155/2019/8148510
28. Imai Y, Yoshida O, Watanabe T, et al. Stimulated hepatic stellate cell promotes progression of hepatocellular carcinoma due to protein kinase R activation. PLoS One. 2019;14(2):e0212589. doi:10.1371/journal.pone.0212589
29. Yan C, Yang Q, Gong Z. Activation of hepatic stellate cells during liver carcinogenesis requires fibrinogen/integrin alphavbeta5 in zebrafish. Neoplasia. 2018;20(5):533–542. doi:10.1016/j.neo.2018.02.002
30. Ringelhan M, Pfister D, O’Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol. 2018;19(3):222–232. doi:10.1038/s41590-018-0044-z
31. Roth GS, Decaens T. Liver immunotolerance and hepatocellular carcinoma: patho-physiological mechanisms and therapeutic perspectives. Eur J Cancer. 2017;87:101–112. doi:10.1016/j.ejca.2017.10.010
32. Chao MP, Weissman IL, Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24(2):225–232. doi:10.1016/j.coi.2012.01.010
33. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi:10.1038/nrc3239
34. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12(4):239–252. doi:10.1038/nri3174
35. Heine A, Schilling J, Grunwald B, et al. The induction of human myeloid derived suppressor cells through hepatic stellate cells is dose-dependently inhibited by the tyrosine kinase inhibitors nilotinib, dasatinib and sorafenib, but not sunitinib. Cancer Immunol Immunother. 2016;65(3):273–282. doi:10.1007/s00262-015-1790-5
36. Hochst B, Schildberg FA, Sauerborn P, et al. Activated human hepatic stellate cells induce myeloid derived suppressor cells from peripheral blood monocytes in a CD44-dependent fashion. J Hepatol. 2013;59(3):528–535. doi:10.1016/j.jhep.2013.04.033
37. Charles R, Chou HS, Wang L, Fung JJ, Lu L, Qian S. Human hepatic stellate cells inhibit T-cell response through B7-H1 pathway. Transplantation. 2013;96(1):17–24. doi:10.1097/TP.0b013e318294caae
38. Liu Y, Wang K, Liang X, et al. Complement C3 produced by macrophages promotes renal fibrosis via IL-17A secretion. Front Immunol. 2018;9:2385. doi:10.3389/fimmu.2018.02385
39. Zhou W, Patel H, Li K, Peng Q, Villiers MB, Sacks SH. Macrophages from C3-deficient mice have impaired potency to stimulate alloreactive T cells. Blood. 2006;107(6):2461–2469. doi:10.1182/blood-2005-08-3144
40. Jimenez-Reinoso A, Marin AV, Subias M, et al. Human plasma C3 is essential for the development of memory B, but not T, lymphocytes. J Allergy Clin Immunol. 2018;141(3):1151–1154 e1114. doi:10.1016/j.jaci.2017.09.037
41. Peng Q, Li K, Anderson K, et al. Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a-C3aR interaction. Blood. 2008;111(4):2452–2461. doi:10.1182/blood-2007-06-095018
42. An F, Li Q, Tu Z, et al. Role of DAF in protecting against T-cell autoreactivity that leads to experimental autoimmune uveitis. Invest Ophthalmol Vis Sci. 2009;50(8):3778–3782. doi:10.1167/iovs.08-3264
43. Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112(5):1759–1766. doi:10.1182/blood-2008-04-151068
44. Tu Z, Bu H, Dennis JE, Lin F. Efficient osteoclast differentiation requires local complement activation. Blood. 2010;116(22):4456–4463. doi:10.1182/blood-2010-01-263590
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.