Back to Journals » International Journal of Nanomedicine » Volume 14
Acid-responsive nanoparticles as a novel oxidative stress-inducing anticancer therapeutic agent for colon cancer
Authors Zhao C, Cao W, Zheng H, Xiao Z, Hu J, Yang L, Chen M, Liang G, Zheng S, Zhao C ![]()
Received 6 October 2018
Accepted for publication 7 January 2019
Published 28 February 2019 Volume 2019:14 Pages 1597—1618
DOI https://doi.org/10.2147/IJN.S189923
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Chengwei Zhao,1,* Weilan Cao,2,* Hailun Zheng,1,3,* Zhongxiang Xiao,1 Jie Hu,1,4 Lehe Yang,1 Min Chen,1 Guang Liang,1 Suqing Zheng,1 Chengguang Zhao1
1Chemical Biology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, Zhejiang 325035, China; 2Department of Coloproctology, The Second Affiliated Hospital and Yuying Children’s Hospital, Wenzhou Medical University, Wenzhou, Zhejiang 325035, China; 3Department of Pharmacy, The Second Affiliated Hospital and Yuying Children’s Hospital, Wenzhou Medical University, Wenzhou, Zhejiang 325035, China; 4State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Taipa, Macau 999078, China
*These authors contributed equally to this work
Objective: Nanoparticles can efficiently carry and deliver anticancer agents to tumor sites. Mounting evidence indicates that many types of cancer cells, including colon cancer, have a weakly acidic microenvironment and increased levels of reactive oxygen species. The construction of nano drug delivery vehicles “activatable” in response to the tumor microenvironment is a new antitumor therapeutic strategy.
Methods: Cinnamaldehyde (CA) was designed to link directly with dextran to form a polymer through an acid cleavable acetal bond. Herein, a novel pH-sensitive drug delivery system was constructed with co-encapsulated 10-hydroxy camptothecin (HCPT). Dynamic light scattering (DLS) analysis, transmission electron microscopy (TEM) analysis, and release kinetics analysis of HCPT-CA-loaded nanoparticles (PCH) were conducted to investigate the physical and chemical properties. The cellular uptake signatures of the nanoparticles were observed by confocal microscopy and flow cytometry. Cell viability, cell scratch assay, apoptosis assay, and colony formation assay were performed to examine the potent antiproliferative and apoptotic effects of the PCH. The antitumor mechanism of the treatment with PCH was evaluated by Western blotting, flow cytometry, and TEM analysis. The pharmacokinetics of PCH were examined in healthy Sprague Dawley rats within 6 hours after sublingual vein injection. We lastly examined the biodistribution and the in vivo anticancer activity of PCH using the xenograft mouse models of HCT116 cells.
Results: Both HCPT and CA were quickly released by PCH in an acidic microenvironment. PCH not only induced cancer cell death through the generation of intracellular reactive oxygen species in vitro but also facilitated the drug uptake, effectively prolonged drug circulation, and increased accumulation of drug in tumor sites. More attractively, PCH exhibited excellent therapeutic performance and better in vivo systemic safety.
Conclusion: Overall, PCH not only utilized the tumor microenvironment to control drug release, improve drug pharmacokinetics, and passively target the drug to the tumor tissue, but also exerted a synergistic anticancer effect. The acid-responsive PCH has enormous potential as a novel anticancer therapeutic strategy.
Keywords: cinnamaldehyde, hydroxy camptothecin, ROS, pH-responsive nanoparticles, colon cancer
Introduction
In the last 2 decades, the use of stimulus-responsive nanoparticles has emerged as a significant approach to selectively deliver antitumor drugs to cancerous sites within the human body. External or physiological stimuli, such as light,1–3 temperature,4,5 ultrasound,6–8 magnetic force,9–11 enzymes,12,13 pH,14–17 reductive18 or oxidative stress,19,20 have been used for triggering drug delivery and controlled release. Stimulus-responsive nanoparticles could provide a platform to reduce the side effects of free drugs, high toxicity, nonspecific biodistribution, and even multidrug resistance.21–23 Due to the wide variation of the pH value in the human body, the pH value has been extensively exploited for stimuli-responsive drug delivery. As the pH value has been found to be decreased in most solid tumors, drug delivery systems that respond to the slightly acidic extracellular environment of solid tumors have been developed as a powerful strategy for tumor targeting.24
Reactive oxygen species (ROS), such as H2O2, oxygen radicals (O2.−) and hydroxyl radicals, are normal byproducts of a normal cellular metabolism in all aerobic organisms.25,26 Moderate increase of ROS can promote cell proliferation and differentiation,27,28 while excessive ROS production can lead to oxidative damage of lipids, proteins, and DNA.29 Therefore, the maintenance of ROS homeostasis is essential for cell growth and survival. Compared to normal cells, cancer cells are usually in an uncontrolled state and have higher ROS levels and antioxidant activity. However, cancer cells cannot tolerate additional oxidative stress and are susceptible to excessive ROS.30,31 Therefore, the regulation of ROS levels is a strategy that selectively kills cancer cells without causing serious toxicity to surrounding normal cells.
Cinnamaldehyde (CA) is a major component in the bark of cinnamon trees, which has been widely used as dietary factor and food additive. Notably, CA and its analogs have been reported to inhibit the growth of various human cancer cells and induce cancer cell death through ROS generation, ROS-mediated mitochondrial permeability transition, and caspase activation.32–34
Although CA shows potent anticancer activity, its clinical application is still limited by the ease with which aldehyde is oxidized in vivo and its lack of specificity for pathological tissues.35,36 In this study, CA was directly linked with dextran, biocompatible polymer of glucose, through acetal linkage. The polymer can spontaneously form acid-responsive nanoparticles in water. Camptothecin (CPT) is a kind of quinoline alkaloid with cancer cell cytotoxicity, which can inhibit the activity of the DNA enzyme topo I.37 In clinical trials, CPT showed significant anticancer activity, but its solubility is low and adverse drug reactions occur. Additionally, CPT has been reported to induce apoptosis by promoting ROS production.37 Moreover, its derivative, 10-hydroxy camptothecin (HCPT), has been shown to be more potent and less toxic than CPT. In this study, HCPT was chosen as model drug to play a synergistic role with CA in promoting ROS production.
HCPT-CA-loaded nanoparticles (PCH) can release HCPT and CA in an acidic microenvironment quickly. In addition, PCH not only induced cancer cell death through the enhancement of ROS level in vitro but also facilitated the drug uptake, effectively prolonged drug circulation, and increased accumulation drug in tumor sites. Notably, the PCH showed excellent therapeutic performance and better in vivo systemic safety. It is anticipated that the acid-responsive PCH have enormous potential as a novel anticancer therapeutic strategy.
Materials and methods
Materials
Dextran (Mw =20,000) was obtained from Leuconostoc spp.; HCPT, CA, CPT, pyrene, and trimethoxymethane were purchased from Energy Chemical (Shanghai, China). Pyridinium p-toluenesulfonate, indocyanine green (ICG), DMSO, methanol, and other organic solvents, unless otherwise specified, were purchased from Aladdin (Shanghai, China).
Cell lines and antibodies
Human colon cancer cell lines SW620 and HCT116 (3–4 generations) were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). HCT116 cells were routinely cultured in McCoy’s 5A medium containing 10% heat-inactivated FBS. SW620 cells were routinely cultured in DMEM containing 10% heat-inactivated FBS. The cells were cultured in a humidified cell incubator with an atmosphere of 5% CO2 at 37°C. Antibodies including anti-cleaved PARP and anti-P53 were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Antibodies including anti-mouse IgG-HRP (7076S), anti-rabbit IgG-HRP (7074S), anti-BCL-2, anti-BAX, anti-CHOP, anti-ATF4, anti-p-elf2α, and anti-GAPDH were purchased from Cell Signaling Technology (Danvers, MA, USA). The annexin fluorescein isothiocyanate-labeled annexin V (Annexin V-FITC) apoptosis detection kit I and propidium iodide (PI) were purchased from BD (Franklin Lakes, NJ, USA).
Synthesis of pH-responsive dextran-CA acetal conjugates polymers (PC)
CA (1 g, 6.17 mmol), trimethoxymethane (2.94 g, 27.75 mmol), and pyridinium p-toluenesulfonate (0.307 g, 1.23 mmol) were dissolved in a suitable amount of methanol. Then the mixture was stirred for 3 hours at 60°C and monitored by thin layer chromatography, with the mobile phase: petroleum ether/ethyl acetate =20:1. After completion, the mixture was quenched with NaHCO3 saturated solution, the reaction mixture was extracted with ethyl acetate twice and the organic layers were collected, dried over Na2SO4, and then evaporated using a rotary evaporator. The reaction residue was used in the next step without further purification.
To synthesize dextran-CA acetal conjugates polymers (PC), dextran (100 mg) was dissolved in anhydrous DMSO (2 mL), then CA acetal (0.75 g, 2eq per hydroxyl group), molecular sieves (5 Å, 130 mg), and pyridinium p-toluene sulfonate (25 mg, 5% mol per hydroxyl group) were added to the mixture. The mixture was stirred for 72 hours at 60°C under N2 atmosphere. After completion, the molecular sieves were removed by filter under vacuum. The product obtained (PC) was precipitated in cold ethanol several times. Finally, the dextran derivative was dissolved in deionized (DI) water and dialyzed for 48 hours in DI water (MWCO: 3.5 kDa) upon stirring to remove DMSO and other little molecular compound. Pure PC were obtained after freeze-drying and verified by proton nuclear magnetic resonance (1H-NMR) spectra (AVANCE 500 spectrometer, Bruker Corporation, Billerica, MA, USA).
Preparation and characterization of PC nanoparticles
To 20 mL of PBS (pH 7.4) was added 1.0 mL of polymer PC solution in DMSO (5 mg/mL) dropwise under stirring. The resulting mixture was stirred for 5 minutes. The size and zeta potential of nanoparticles (resuspended in PBS, 0.25 mg/mL) were measured by dynamic light scattering (DLS) (Malvern Zetasizer Nano ZS, Malvern Instruments, Malvern, UK) with 2-minute data acquisition time. Transmission electron microscopy (TEM) samples were prepared by resuspension of nanoparticles in DI water with a concentration of 0.125 mg/mL. The samples were dropped onto the copper grid, dried in the air, and then observed with TEM (H-7500, Hitachi Ltd., Tokyo, Japan).
Determination of critical micelle concentration (CMC) of PC nanoparticles
First, 1 mL of nanoparticles solution with different concentrations was added to a 1.5 mL tube which contained the same concentration and excessive pyrene (6×10−7 M), kept at room temperature for 24 hours. The fluorescence emission spectra of pyrene-loaded nanoparticles were recorded using a fluorospectrometer with emission at 390 nm. The excitation intensity ratio (I339/I336) was plotted against the logarithm of the polymer concentration.
The stability and responsiveness of PC micelles
An amount of 1.0 mL of polymer PC solution (DMSO, 5 mg/mL) was added dropwise to 20 mL of PBS (pH 7.4 and pH 6.5) under stirring. At appropriate time intervals, the size and zeta potential of nanoparticles (resuspended in PBS, 0.25 mg/mL) were measured by DLS (Malvern Zetasizer Nano ZS) with 2-minute data acquisition time.
Preparation of PCH and determination of the encapsulation efficiency (EE) of nanoparticles
First, we mixed the 0.5 mL PC polymer solution (DMSO, 10 mg/mL) with 0.5 mL HCPT solution (DMSO, 6 mg/mL), vortexed it for 2 minutes, then added the solution to the 20 mL PBS (pH 7.4) under stirring and stirred the mixture for 0.5 hour. The mixture was dialyzed in DI water (MWCO: 3.5 kDa) upon stirring to remove DMSO and unencapsulated HCPT for 24 hours. The PCH were lyophilized from water. After measuring the absorption intensity, the amount of HCPT loaded in the nanoparticles was calculated against the calibration curve. The EE is calculated with the following equation: EE (%) = (weight of loaded drug)/(weight of initially added drug)×100.
Release kinetics of PCH
In order to determine the release characteristics of HCPT and CA in vitro environment, we transferred 3 mL of PCH solution (0.25 mg/mL) into a dialysis bag (MWCO =1,000) and soaked in 50 mL of PBS (pH 5.0 or 7.4) at 37°C. The medium was collected at different intervals and the same amount of fresh medium was added. The release amount of HCPT and CA was calculated respectively according to the calibration curve.
Cellular uptake assay
Cells were treated with PCH (including ICG) for 1 hour, 2 hours, and 4 hours in McCoy’s 5A medium (pH 7.4 or 6.5), washed twice with PBS, and stained with Hoechst 33258 staining solution and then we captured images by using a confocal laser scanning microscope (Nikon Corporation, Tokyo, Japan) with 40× amplification. Three microscopic fields were selected randomly in each group. Quantitative data of ICG uptake were measured by flow cytometry (FACSCalibur flow cytometer, BD Biosciences, San Jose, CA, USA).
MTT cell viability assay
Cells were plated at a density of 8×103 cells per well into 96-well plates and then allowed to attach overnight in McCoy’s 5A medium containing 10% heat-inactivated FBS. Cells were incubated with various concentrations of CA (3.1 μg/mL), HCPT (10 μg/mL), CA + HCPT (3.1+10 μg/mL), PC (36.2 μg/mL), PC + HCPT (36.2+10 μg/mL), and PCH (46.2 μg/mL) (10 μg/mL equivalent HCPT). The cell viability was determined by MTT assay after 48 hours.
Colony formation assay
We plated 1,000 cells on six-well plates overnight and then incubated with drugs (equivalent to 10 μg/mL HCPT) or DMSO for 24 hours. Then cells were washed twice with PBS and allowed to grow in McCoy’s 5A medium for 1 week. Cells were washed twice with PBS and fixed with paraformaldehyde for 10 minutes. The cells were washed twice with PBS and stained with 1% crystal violet (25% methanol) at room temperature for 15 minutes, washed, and dried.
Cell migration assay (in vitro wound-healing assay)
HCT116 cells were plated on six-well plates overnight, and the white tip was used to scratch the cells with the same width, then washed twice with PBS. After washing, cells were incubated with CA (3.1 μg/mL), HCPT (10 μg/mL), CA + HCPT (3.1+10 μg/mL), PC (36.2 μg/mL), PC + HCPT (36.2+10 μg/mL), PCH (46.2 μg/mL) or DMSO for 12 hours. It was then washed twice with PBS and allowed to grow in McCoy’s 5A medium for 48 hours. The microscopic camera system (Nikon Corporation) was used to capture images. The wound healing rate was calculated with the formula: (percent wound healing) = average of [(gap area: 0 hour)−(gap area: 48 hours)]/[gap area (DMSO): 0 hours].
Cell apoptosis analysis
HCT116 cells were plated on six-well plates for 24 hours, and then incubated with different concentrations of PCH (23.1, 46.2 or 92.4 μg/mL), N-acetyl-cysteine (NAC) (5 mM), NAC + 2PCH (5 mM +92.4 μg/mL) or DMSO for 12 hours. Cells were collected, washed twice with ice-cold PBS, and stained with FITC conjugated Annexin V and PI for 30 minutes in the binding buffer. Apoptosis was analyzed by flow cytometry (FACSCalibur flow cytometer; BD Biosciences).
Western blot analysis
The cells were harvested within 24–36 hours at drug or DMSO concentration of approximately 80%, and then lysed in a cold cell lysis buffer containing phosphatase inhibitors and protease inhibitors. Then the sample was disposed by centrifugation (12,000 rpm, 4°C) for 10 minutes. Protein concentrations in all samples were detected by the Bradford protein assay kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). After adding the sample to loading buffer, the protein sample was transferred to PVDF transfer membrane by electrophoresis and then blocked for 1.5 hours with fresh 5% non-fat milk in tris-buffered saline with Tween 20 buffer at room temperature. Membranes were incubated with a 1:1,000 dilution of specific primary antibody and 1:10,000 HRP conjugated secondary antibody. Membranes were investigated using ImageJ computer software (National Institute of Health, Bethesda, MD, USA).
Electron microscopy
HCT116 cells were incubated with DMSO, NAC (5 mM), PCH (46.2 μg/mL) or NAC + PCH (5 mM +46.2 μg/mL) for 6 hours. Then the cells were harvested and soaked in 2.5% glutaraldehyde overnight at 4°C. The cells were post-fixed in 1% glutaraldehyde at room temperature for 60 minutes, stained with 1% OsO4, dehydrated through graded acetone solutions, and embedded in Epon. The areas containing cells were collected and cut into 70 nm slices, and detected by electron microscopy (H-7500, Hitachi Ltd.).
Measurement of ROS generation
Flow cytometry (FACSCalibur flow cytometer) was used to measure the generation of ROS in cells. HCT116 cells were plated on six-well plates, incubated overnight, and treated with PCH for 4 hours, HCPT for 1 hour, and CA for 6 hours, then, ROS probe 2, 7,-dichlorofluorescein diacetate (DCFH-DA) (10 μM) was added to fresh medium and incubated in darkness for 30 minutes. Cells were harvested and analyzed for fluorescence intensity using a flow cytometer (BD Biosciences). Before the cells were exposed to the compound, they were pretreated with 5 mm NAC for 0.5 hour and then the amount of ROS was analyzed. The statistics were analyzed using FlowJo VX.
In vivo imaging and biodistribution
All animal experiments complied with the Wenzhou Medical University’s Policy on the Care and Use of Laboratory Animals. Protocols for animal studies were approved by the Wenzhou Medical University Animal Policy and Welfare Committee. BALB/cA nu/nu female mice (18–22 g) bearing HCT116 tumors with volume approximately 150 mm3 were intravenously (iv) injected with free ICG (2 mg/kg) or ICG-loaded PCH nanoparticles (ICG/PCH) (2 mg/kg equivalent ICG) (n=6). At 6 hours, 24 hours, and 48 hours after injection, the intensity of fluorescence signal in animals was observed using an in vivo imaging system (CRi maestro, Cambridge Research & Instrumentation, Inc., Hopkinton, MA, USA) with the excitation wavelength of 704 nm and a filter of 735 nm. Some mice in each group were sacrificed at 24 hours after injection, and the organs such as heart, liver, spleen, lung, kidneys, and tumor were excised for fluorescent imaging.
Pharmacokinetics study of PCH nanoparticles in vivo
All animal experiments complied with the Wenzhou Medical University’s Policy on the Care and Use of Laboratory Animals. Protocols for animal studies were approved by the Wenzhou Medical University Animal Policy and Welfare Committee. PCH (equivalent to 5 mg/kg HCPT) or free HCPT (5 mg/kg) were iv injected into female Sprague Dawley rats weighing 200–220 g to analyze pharmacokinetic characteristics of PCH. Blood samples (200 μL) were collected from the tail vein into heparinized tubes at appropriate intervals (5, 15, 30, 45, 60, 90, 120, 180, 240, 360 minutes) after administration (n=5 at each time point), centrifuged (10,000× rpm for 10 minutes at 4°C) to harvest the plasma, and stored at −25°C until required for analysis. The samples were pre-incubated with acetic acid (10%) for 24 hours, HCPT in plasma was extracted with methanol. The HCPT concentrations in plasma were detected using high performance liquid chromatography (1260 VWD, Agilent Technologies, Santa Clara, CA, USA) with CPT as internal standard.
In vivo antitumor study
All animal experiments complied with the Wenzhou Medical University’s Policy on the Care and Use of Laboratory Animals. Protocols for animal studies were approved by the Wenzhou Medical University Animal Policy and Welfare Committee. For in vivo experiments, 5-week old athymic BALB/cA nu/nu female mice (18–22 g) were purchased from Vital River Laboratories (Beijing, China). Standard rodent diet and water were fed to laboratory animals under standard experimental animal conditions. The mice were randomized to four groups of six per group. A subcutaneous tumor was established by injecting HCT116 cells (5×106 cells in 0.1 mL PBS) into the right flank of nude mice. According to the dosage of 5 mg/kg HCPT, HCPT + CA and PCH water-soluble preparations were intraperitoneally injected into the nude mice when the tumor volume reached 100–200 mm3. For the control group, liposome carrier in PBS was injected. The volume of the tumor (V=0.5×l×w2) was calculated by measuring the length (l) and width (w) of the tumor at the specified time point. At the end of the experiments, animals were sacrificed, and the tumors were excised and weighed for use in the histology and Western blot analysis.
H&E staining
The harvested heart, kidney, and liver tissues were soaked in 4% formaldehyde, dehydrated with an ethanol gradient, embedded in paraffin, and the paraffin tissue sections (5 μm) were stained with H&E. Images of each slice were taken using an optical microscope, (kidney: 40× amplification, heart and liver: 20× amplification, Nikon Corporation).
Statistical analysis
Data were presented as mean ± standard error (SE). The Student’s t-test or one-way ANOVA for multiple comparisons in GraphPad Prism 7 (GraphPad Software, Inc., La Jolla, CA, USA) were used to determine the statistical differences between groups. P<0.05 was considered statistically significant.
Results
Synthesis and characterization of PC nanoparticles
PC can be easily synthesized from CA through a two-step reaction. First, CA is reacted with trimethyl orthoformate in methanol in the presence of pyridinium p-toluenesulfonate to produce CA acetal, which reacts with dextran to generate the corresponding product dextran CA acetal copolymers (Scheme S1). The successful connection between CA and dextran was confirmed by nuclear magnetic resonance. Based on the 1H-NMR spectrum of the copolymer, the calculated conjugation ratio was 10%–11%. To further confirm the successful modification and clearly demonstrate the degradation behaviors of the polymer in an acidic environment, acid-induced polymer decomposition was investigated in an acid solution by 1H-NMR spectroscopy (Figure 1). When the pH value was adjusted from 7.4 to 6.5, acetal protons (~6.2 ppm) disappeared and aldehyde protons appeared at 9.5 ppm. These changes demonstrated that CA was degraded from dextran CA acetal conjugates under acidic conditions.
 | Figure 1 1H-NMR of dextran-cinnamaldehyde acetal conjugates. |
Amphiphilic dextran CA acetal copolymers have the ability to self-assemble in aqueous solution to form stable nanoparticles with a hydrophilic dextran shell and CA segments in the hydrophobic core. The average diameter is approximately 182.3 nm (polydispersity index [PDI] ~2.25×10−2), as determined by DLS and TEM analysis (Figure 2A). Studies have shown that when the nanoparticle size is <200 nm, they preferentially accumulate in tumor sites due to enhanced permeability and retention effects. The zeta potential of the nanoparticles was determined to be ~−3.86 mV at pH 7.4.
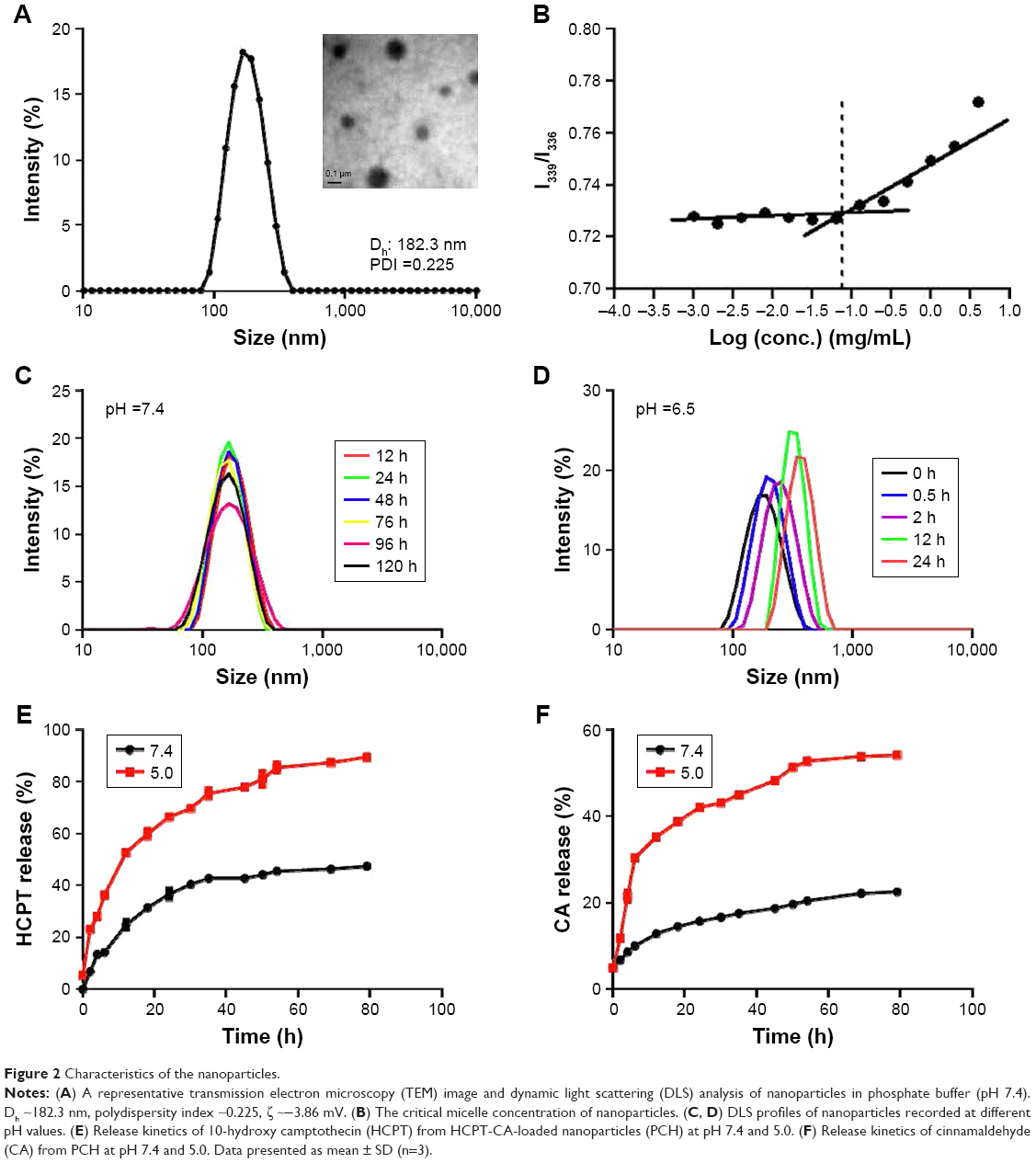 | Figure 2 Characteristics of the nanoparticles. |
The CMC of dextran CA acetal copolymers
Pyrene was selected as a hydrophobic fluorescent probe to determine the CMC of the nanoparticles. The pyrene was combined with the different concentrations of polymer and the fluorescence intensity of the pyrene-loaded micelles was monitored at an excitation wavelength of 339 and 336 nm and at an emission wavelength of 390 nm. Their ratio (I339/I336) was measured as an indicator of micelle hydrophobicity. The plot of I339/I336 vs the logarithm of the polymers’ concentrations demonstrated that the concentration of fluorescent probe encapsulated in the hydrophobic core forming thermodynamically stable micelles was higher than 70.7 g/mL (Figure 2B).
Acid-responsive degradation of PC nanoparticles in vitro
The acetal linkage is usually stable at physiological neutral pH and is rapidly hydrolyzed at acidic pH. Along with the hydrolysis of the CA acetal from dextran, the nanoparticles are broken into water-soluble fragments, resulting in the rapid dissociation of nanoparticles. The pH-dependent stability of PC at pH 7.4 and 6.5 was examined by DLS analysis. As shown in Figure 2C and D, the average particle size increased from 182 nm to 600 nm over 24 hours at pH 6.5 (Figure 2C), whereas the average size remained stable at pH 7.4 (Figure 2D) for 5 days.
Characterization of PCH and acid-responsive drug release in vitro
The hydrophobic anticancer drug HCPT was chosen for encapsulation in PC nanoparticles to determine whether PC nanoparticles can effectively release the encapsulated drugs in a weakly acidic environment. The drug loading content and EE were calculated to be 0.216 mg/mg and 57.6%, respectively. The average nanoparticle size decreased from ~182.3 nm to ~166.4 nm (PDI ~0.15) as a result of increased hydrophobic core. The zeta potential of the nanoparticles decreased to ~16.4 mV, which indicated that the drug was effectively encapsulated. The release kinetics of HCPT and CA from PCH were investigated at different pH values. As shown in Figure 2E, at pH 5.0, most of the HCPT was released within 10 hours and 90% of the HCPT was released within 80 hours, while at pH 7.4 only a very small amount (45%) of HCPT was released after 80 hours. The amount of CA released at pH 5.0 within 80 hours was more than twice that at physiological pH 7.4 (Figure 2F). These findings validated our hypothesis that PCH nanoparticles have good pH-responsiveness and the ability to act as a pH-responsive drug delivery system.
Uptake of nanoparticles by cancer cells in vitro at different pH values
Since free drugs are taken up into cells through a rapid diffusion process, and drug carrier particles are internalized into cells by endocytosis relying on energy, drug carriers are usually taken up slower, and more easily affected by the microenvironment conditions. We chose ICG as a model molecule to examine the cellular uptake of nanoparticles into the colon cancer HCT116 cells at different pH values. The ICG uptake by HCT116 cells was examined by confocal microscopy at different pH values. As shown in Figure 3A, after 2 hours’ incubation, a sharp near-infrared (NIR) fluorescence signal could be detected at a slightly more acidic condition (pH 6.5) than at a normal pH condition, indicating that ICG is more effectively distributed into cells under acidic conditions. More significantly, ICG encapsulated by PC can be more effectively translocated into nucleus after internalization. To further confirm the differences in the subcellular localization of ICG, quantitative data were collected by flow cytometry (Figure 3B and C). Compared to the impact on uptake at different pH values, the uptake increased as the pH value decreased. The results revealed that more ICG was present in HCT116 cells incubated at pH 6.5, than in cells incubated at pH7.4 (95.74% fluorescence intensity of pH 6.5 group), which is consistent with the previously mentioned observation. These results indicate that the weakly acidic tumor microenvironment can accelerate the delivery of drugs carried by nanoparticles into HCT116 cells, particularly into the nuclei.
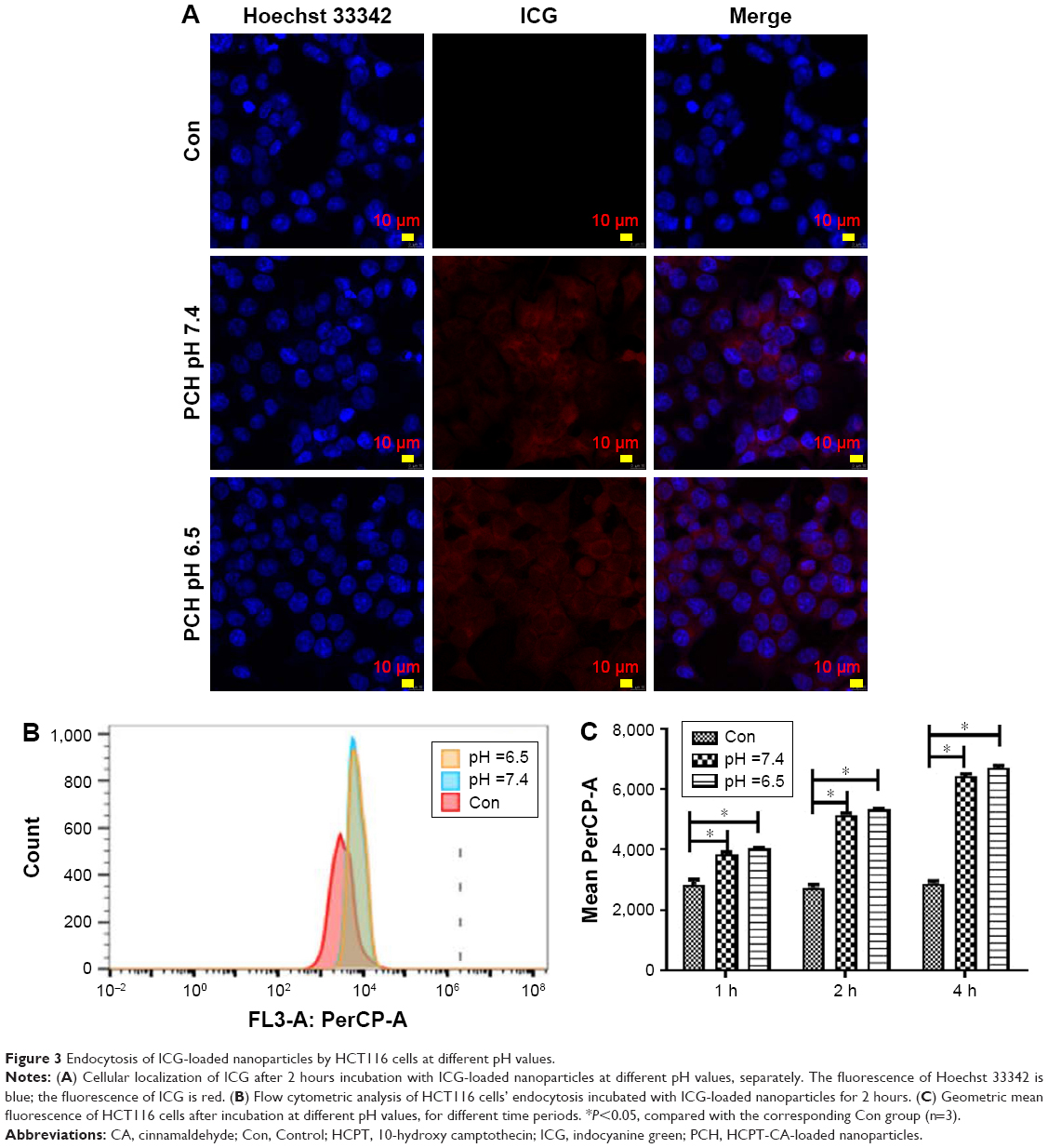 | Figure 3 Endocytosis of ICG-loaded nanoparticles by HCT116 cells at different pH values. |
The anti-proliferation and anti-migratory effects of PCH
Mounting evidence suggests that normal cells have lower ROS levels and an appropriate increase of ROS levels can promote cell proliferation and differentiation without causing cell damage. In comparison, ROS levels in various types of cancer cells have higher ROS levels than normal cells.38–40 Consequently, cancer cells are more susceptible to ROS damage induced by exogenous substances that either weaken antioxidant defenses or increase ROS production to levels or thresholds at which cells cannot survive. It has been reported that both CA and HCPT can induce intracellular ROS production.32–34,37 Accordingly, we wondered whether PCH could selectively kill malignant tumor cells by increasing ROS levels without causing significant toxicity to normal cells. The effect of PCH nanoparticles on cell viability was assessed by the MTT assay in non-malignant human lung epithelial BEAS-2B cells, human vascular endothelial HUVEC cells, human cardiomyocytes 4910, and human colon cancer HCT116 cells at 48 hours. As shown in Figure 4A, PC and PCH showed negligible cytotoxicity against normal BEAS-2B cells, these results suggested that PC or PCH nanoparticles had good cell biocompatibility compared with other free non-selective therapeutic drugs, with significant cytotoxicity against normal cells. HUVEC cells and 4910 cells showed similar results (Figure 4B and C). In addition, cell viability of colon cancer HCT116 cells was also investigated. CA and PC showed minimal cytotoxicity, due to the lower drug efficacy of CA, which is consistent with previous studies.41 However, HCPT and HCPT combined with CA or PC plus HCPT all exhibited significantly cytotoxic effects. Notably, PCH also showed significant cytotoxicity against colon cancer HCT116 cells (Figure 4D). These data confirmed that PCH, once absorbed by cancer cells, can effectively release the HCPT and CA, thereby making the encapsulated drug as effective as the free drugs.
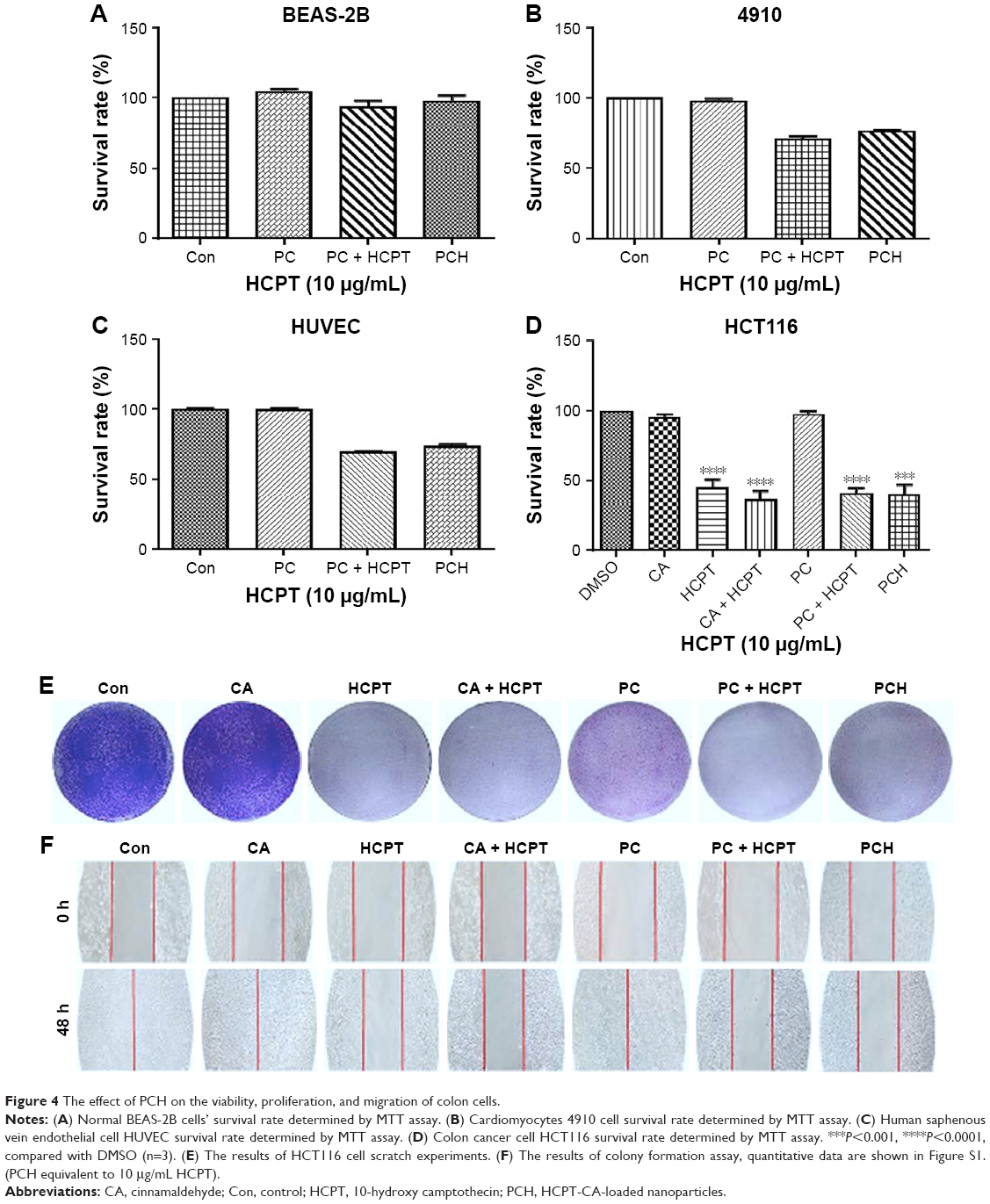 | Figure 4 The effect of PCH on the viability, proliferation, and migration of colon cells. |
To further evaluate the effects of PCH on cancer cell growth, colony formation assay was performed with SW620 colon cancer cells (Figure 4E). CA alone had no impact on the growth and proliferation of colon cancer cells. PC alone exhibited moderate anti-proliferation activity against SW620 colon cancer cells. Similar to the free drugs, PCH significantly inhibited the growth and proliferation of human colon cancer cells, leading to apoptosis.
As is widely known, malignant cancer cells have invasive and migratory abilities. To determine whether PCH has any effect on the migratory capacity of cancer cells, cell scratch assays were conducted with HCT116 cells. As shown in Figure 4F, CA and PC showed a negligible inhibitory effect on the migratory ability of HCT116 cells and even promoted it. In comparison, PCH exhibited a similar effect to that of the combination of HCPT with CA, leading to a potent inhibition of cell migration, which is one of the keys to cancer treatment. Quantitative data are shown in Figure S1. However, the mechanism remains unknown, thus further studies are required to confirm our hypothesis.
PCH induced apoptosis in colon cancer cells
Apoptosis induction in colon cancer cells was studied by flow cytometric analysis using Annexin V-FITC and PI staining to assess the level of apoptosis and cell viability following treatment with PCH. After PCH treatment for 24 hours, the colon cancer cells underwent apoptosis in a dose-dependent apoptotic manner (Figure 5A). Noteworthy, NAC, a specific inhibitor of ROS production, significantly alleviated PCH-induced apoptosis in colon cancer cells. Quantitative data are presented in Figure 5B. To further confirm PCH-induced ROS-mediated apoptosis in HCT116 colon cancer cells, the expression of apoptosis-related proteins was analyzed by Western blot analysis. As shown in Figures 5C and S2, PCH dose-dependently increased the expression of pro-apoptotic proteins PARP, P53 and BAX and decreased the expression of anti-apoptotic protein BCL-2. However, these changes were partially mitigated by pre-incubation with NAC. Besides being involved in apoptosis, studies have shown that BAX and BCL-2 play important roles in the mitochondrial pathways. Mitochondria are known to be important in the regulation of apoptosis. To further confirm whether PCH has an effect on mitochondria morphology, we performed electron microscopy analysis to examine the mitochondrial morphology in HCT116 colon cancer cells exposed to PCH for 6 hours in the presence or absence of NAC. As shown in Figure 5D, the mitochondria in cells treated with PCH were abnormally enlarged or swollen, showing shredded cristae and impaired outer membrane integrity. In addition, the NAC pre-treatment reversed the PCH-induced mitochondrial swelling. Noteworthy, the NAC alone had no effect on mitochondrial morphology. These results demonstrated that PCH may induce ROS production and cause cancer cells’ mitochondrial dysfunction and apoptosis.
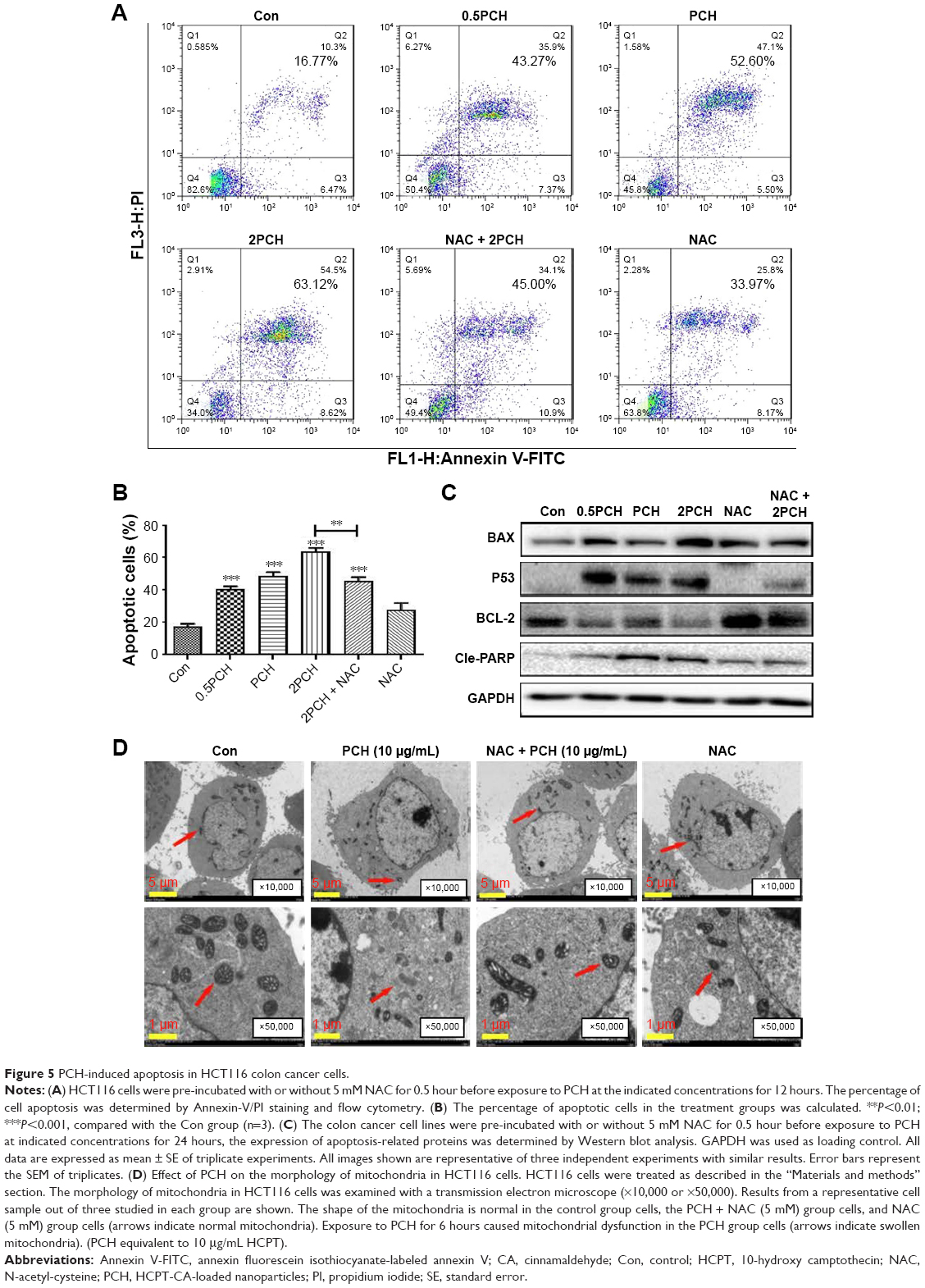 | Figure 5 PCH-induced apoptosis in HCT116 colon cancer cells. |
PCH-activated endoplasmic reticulum (ER) stress signaling by promoting the accumulation of ROS
To further confirm our hypothesis, ROS levels in HCT116 colon cancer cells were determined by flow cytometry using the redox-sensitive fluorescent probe DCFH-DA. As shown in Figure 6A, both CA and HCPT showed a moderate ability to increase the ROS level, while PCH caused a sharp increase in ROS levels. NAC significantly inhibited PCH-induced increase of ROS production. These results indicated that PCH can remarkably enhance intracellular ROS accumulation in HCT116 cells.
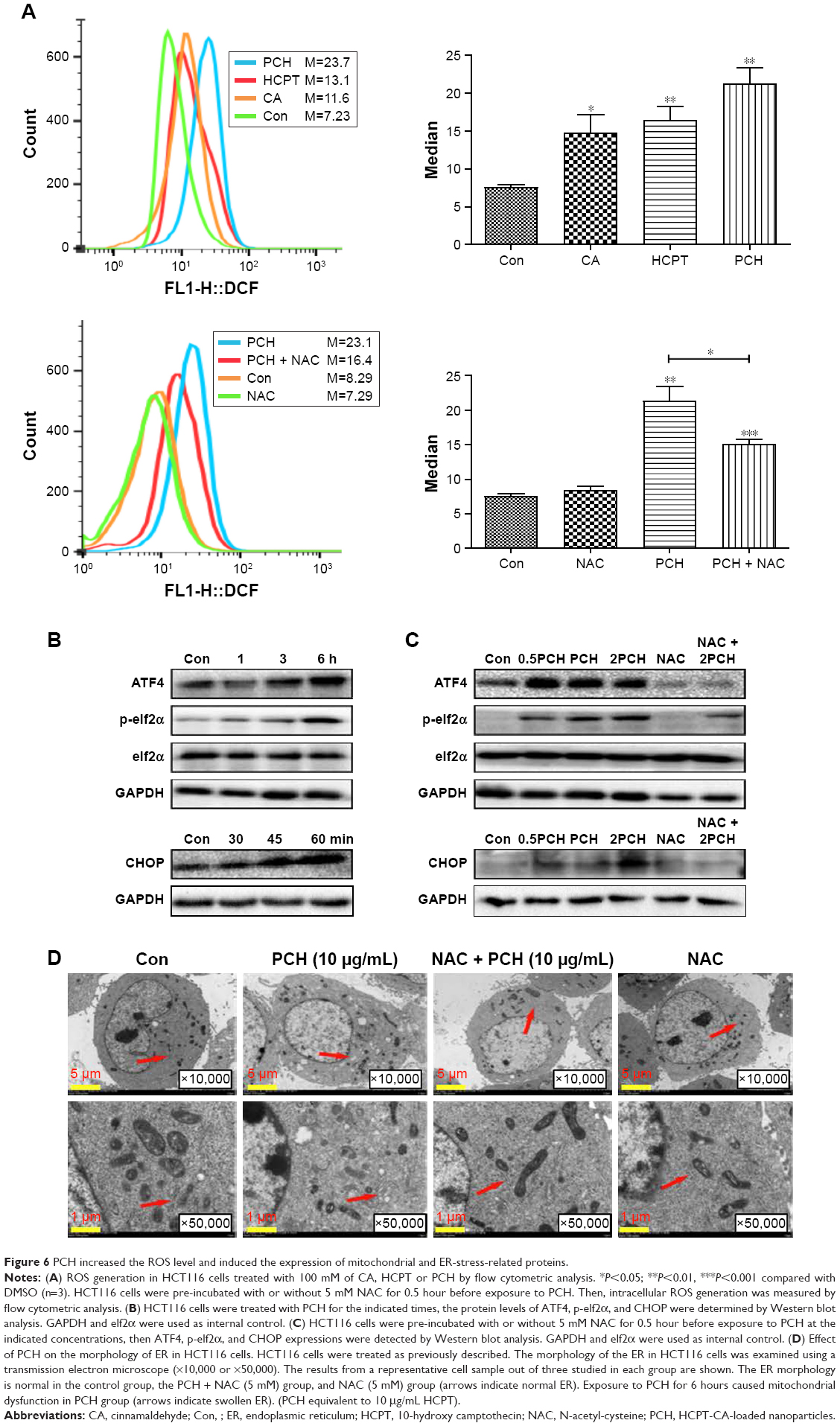 | Figure 6 PCH increased the ROS level and induced the expression of mitochondrial and ER-stress-related proteins. |
Research has shown that oxidative stress can induce tumor cell apoptosis by activating ER stress.42 Accordingly, we hypothesized that PCH might activate ER stress by increasing ROS production. We confirmed our hypothesis by Western blot analysis. PCH regulated the expression of ER-stress-related proteins (such as ATF4, p-elf2α, and CHOP) in a time-dependent and dose-dependent manner (Figures 6B and C, S3, S4). However, pre-treatment with NAC clearly reversed these changes. Electronic microscopy was used to examine the effect of PCH on the morphology of the ER in HCT116 cells. As shown in Figure 6D, the ER in HCT116 cells treated with DMSO (arrow) had a smooth appearance. Instead, the ER in HCT116 cells treated with PCH for 6 hours appeared swollen (arrow), suggesting the accumulation of misfolded protein in the ER. However, in cells pretreated with NAC, the ER showed the familiar smooth morphology, which is also shown in cells treated with NAC alone. In summary, PCH activated the ER stress pathway by increasing ROS level.
PCH effectively enhanced the accumulation of drug in tumor tissues
Prolonging the drug circulation time and enhancing drug accumulation in cancerous sites are the main advantages of nanoparticle drug delivery systems. In this study, the pharmacokinetics of HCPT and PCH were evaluated within 6 hours after sublingual vein injection in healthy Sprague Dawley rats (Figure 7A and B). In contrast to free HCPT, PCH showed a significantly extended half-life (t1/2=7.042 h) and decreased clearance rate (0.437 mL/h). These data showed that PCH nanoparticles prolonged the circulation time, which would potentially enhance the efficacy of chemotherapy.
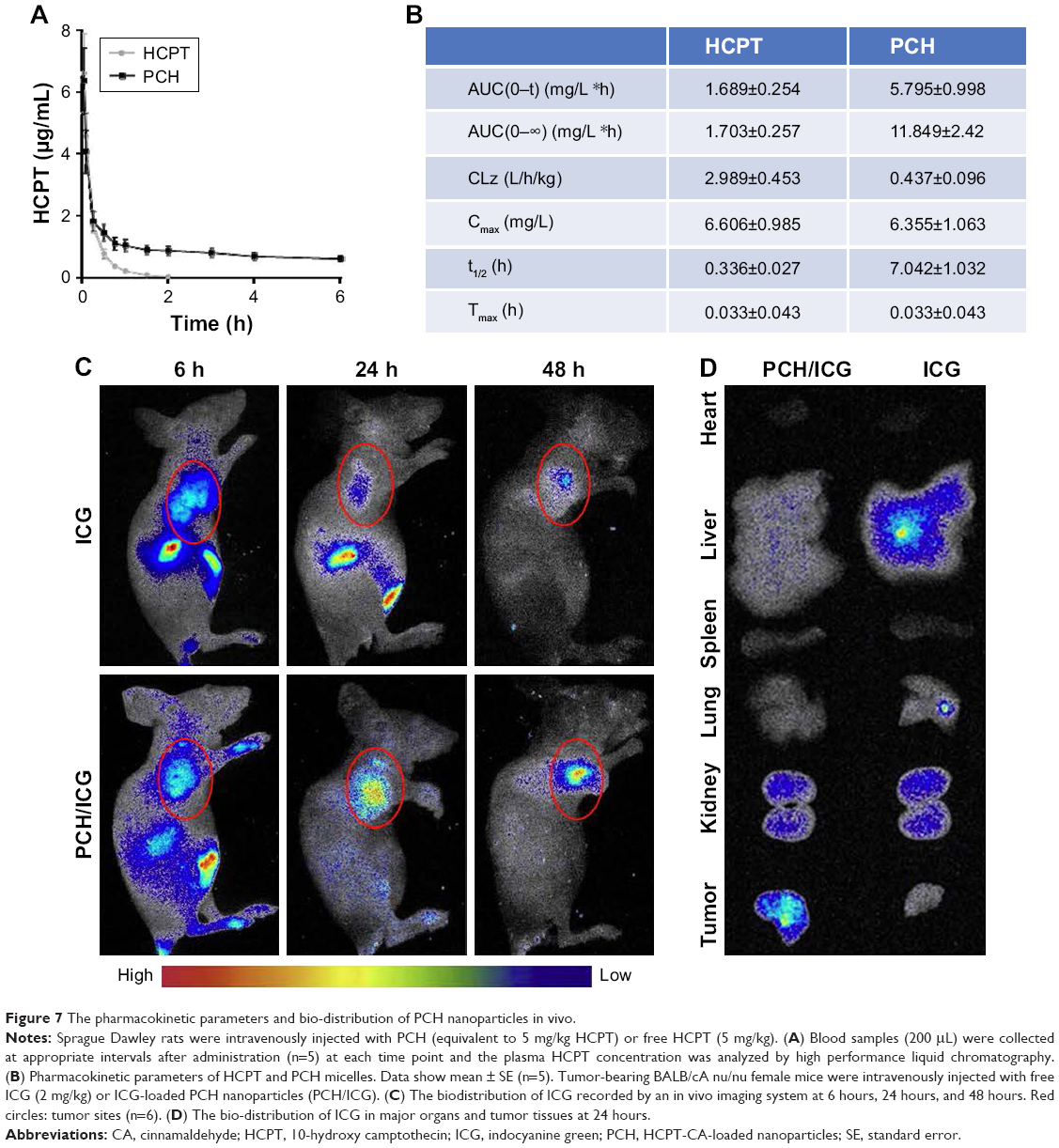 | Figure 7 The pharmacokinetic parameters and bio-distribution of PCH nanoparticles in vivo. |
The in vivo biodistribution of PCH nanoparticles was further investigated. ICG, an NIR fluorescent dye, was simultaneously encapsulated in PCH to produce ICG/PCH. ICG/PCH or free ICG was injected into tumor-bearing nude mice, and an in vivo imaging system was used to monitor the biodistribution of the ICG (Figure 7C). At 24 hours after injection, free ICG only resulted in a weak fluorescent signal, and rapidly declined afterward. In contrast, ICG/PCH generated an intense fluorescent signal at 6 hours after injection at the tumor site, which peaked at 24 hours and remained detectable until 48 hours after injection. These results clearly demonstrated the increased accumulation of PCH at tumor sites, indicating its passive tumor-targeting ability. They also clearly showed that the PCH has a strong ability to accumulate at tumor sites, passively targeting the tumor.
In order to further study the biodistribution of PCH nanoparticles, the in vivo imaging system was used to examine the major organs and tumors excised from the tumor-bearing nude mice (Figure 7D). The fluorescent images confirmed that PCH mostly accumulated in tumors, weakly accumulated in kidneys, and was hardly detectable in other organs. In comparison, the free ICG dye mostly accumulated in liver and partially in kidneys. In general, the systemic administration of PCH nanoparticles improved the efficacy of chemotherapy by effectively promoting drug accumulation in tumors.
The in vivo anticancer effect and biosafety of PCH
We further investigated the in vivo anticancer effects of PCH using xenograft HCT116 cells tumor models. Female nude mice bearing HCT116 cell-induced tumors (tumor volume ~100 mm3) were randomly divided into several groups and administered various formulations at the equivalent amounts of HCPT (5 mg/kg) every 3 days. As shown in Figure 8A–C, the blank control group administered PBS showed gradual and continuous tumor growth. The tumor volume was successfully suppressed by treatment with HCPT or HCPT combined with CA. Notably, PCH showed the strongest ability to suppress tumor growth and the tumor volume remained steady, which was better than mechanically mixed drugs. Overall, PCH exhibited effective in vivo inhibitory effects on human colon cancer cells.
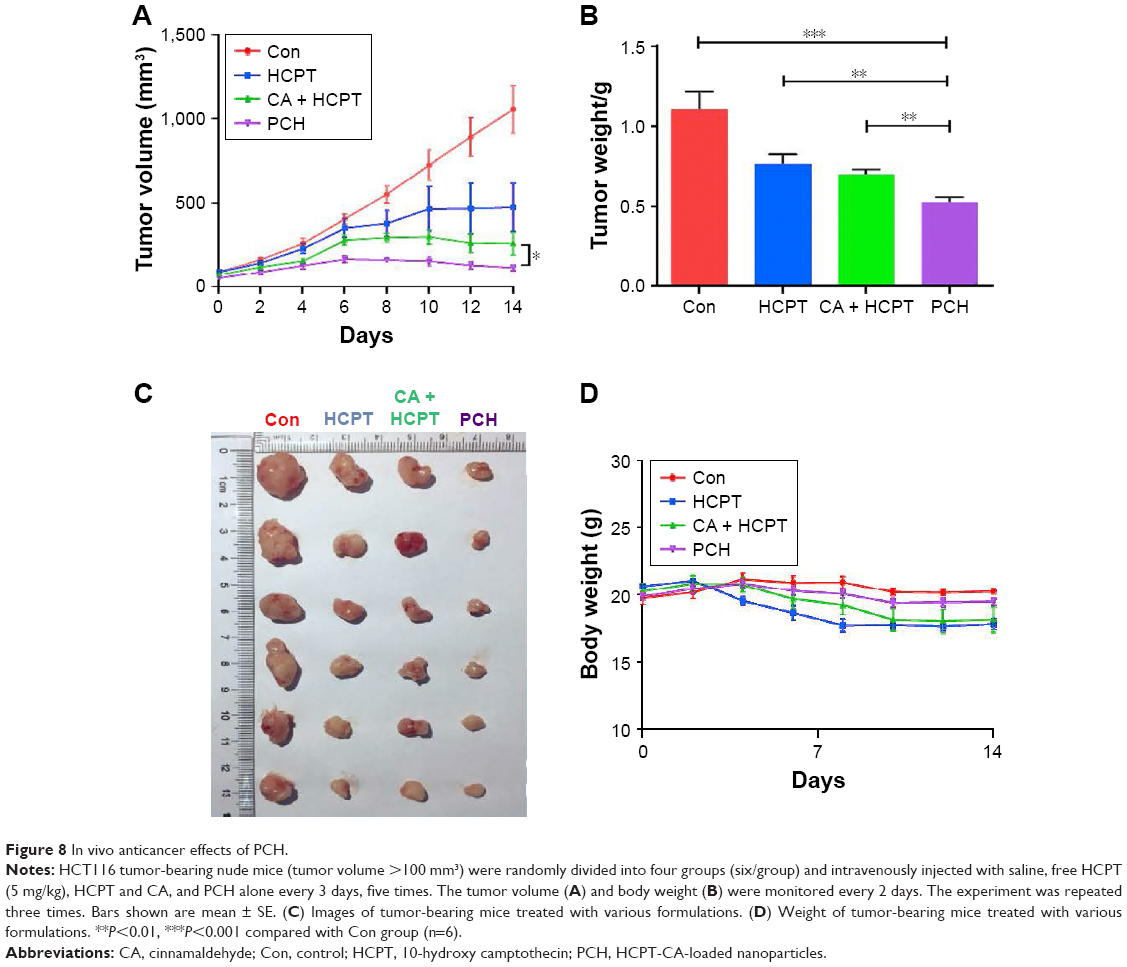 | Figure 8 In vivo anticancer effects of PCH. |
The change in body weight was simultaneously measured after administration of the drugs, which serves as an indicator of general toxicity. The results shown in Figure 8D reveal that the body weight of the group treated with free drugs was significantly decreased compared with the negligible changes detected for the PCH-treated group, indicating the remarkable in vivo systemic safety of the PCH. In addition, to further confirm the potential toxicity of PCH, histologic sections of the major organs including kidneys, liver, and heart were stained with H&E and examined by microscopy. As shown in Figure 9A, livers and hearts in all groups showed no noticeable abnormality. However, both HCPT alone and HCPT with CA caused kidney damage, including structural disturbance in areas of the kidney. In comparison, PCH treatment only caused negligible tissue damage or lesions in the kidney. These results clearly indicated the safety of PCH as cancer treatment.
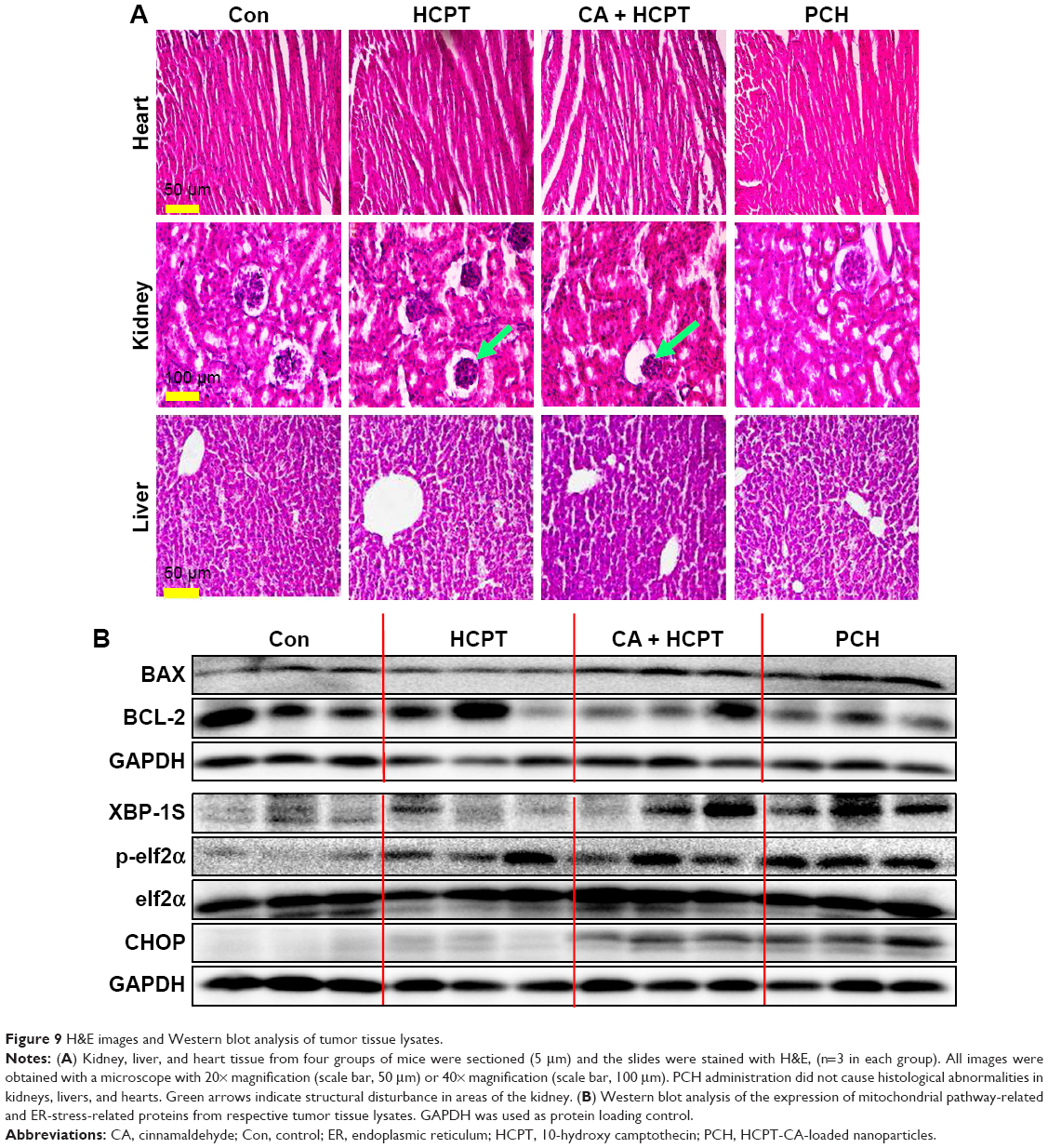 | Figure 9 H&E images and Western blot analysis of tumor tissue lysates. |
To further confirm our hypothesis that PCH can activate the ER-stress pathway and cause mitochondrial dysfunction by increasing ROS level, the mouse tumors were analyzed by Western blot analysis. The results revealed that PCH treatment regulated the expression of proteins associated with the ER-stress pathway, including XBP-1S, p-elf2α, and CHOP (Figure 9B), and mitochondrial dysfunction, including BAX and BCL-2 (Figure 9B), which increased apoptosis in colon cancer cells. In conclusion, the PCH-induced apoptosis in HCT116 cells is closely related to the activation of ER-stress and resulting mitochondrial dysfunction in vivo.
Discussion
It has been well-established that ROS has double-edged effects on living cells, depending on their concentration and duration, which can determine the fate of cancer cells.31 Based on these properties, many efforts have been made to induce oxidative stress and thus preferentially kill cancer cells. CA is the main ingredient of cinnamon, and it turns out that it can increase the ROS levels in tumor cells and cause cancer cell death.32–34 Although CPT has been considered as a drug with a unique antitumor activity spectrum which is mediated through the selective inhibition of eukaryotic DNA topo-I, its analog HCPT has also been reported to induce ROS.37 There are a few studies which found that CA combined with HCPT induces ROS production, especially when delivered as an acidic microenvironment-responsive nanoparticle.
The Fenton reaction-performing polymer (PolyCAFe) micelle is a new ROS manipulation anticancer therapeutic agent. Amphiphilic PolyCAFe conjugates benzoyloxy CA and iron-containing compounds in its skeleton and self-assembles to form micelles which generate cytotoxic hydroxyl radicals that preferentially kill cancer cells.43 In addition, nanoparticles designed to incorporate CA in its pH-sensitive hydrophobic backbone via acid-cleavable acetal linkages and encapsulate CPT, which induced apoptotic cell death through the generation of intracellular ROS and their apoptotic activities were significantly enhanced with a payload of CPT and have been used in in vitro and in vivo model of SW 620 colon tumor-bearing mice.44 However, their uptake by cells, biodistribution, safety and toxicity pharmacokinetics in normal tissues and organs were not thoroughly investigated. Also, although the nanoparticles induced ROS production to cause cancer cell apoptosis, the related downstream mechanism is still unknown. In particular, these are important potential targets that need to be evaluated for the development of new therapeutics for cancer, and thus have great significance for further research.
In this work, we developed pH-responsive nanoparticles by linking CA with biocompatible dextran, which was then allowed to self-assemble to form the nanoparticles (PC), which were further used to encapsulate HCPT (PCH) for the purpose of developing a synergistic therapeutic for use against colon cancer. PCH nanoparticles dissociated rapidly and showed a significantly faster release kinetics in vitro in acidic environments. The results showed that PCH nanoparticles have good pH-responsiveness and have the ability to serve as a drug delivery system in response to a decrease in the pH value in tumor environments. Cellular uptake experiment verified that PCH can be internalized into cells more easily under acidic conditions (Figure 3), and further accumulated in the nuclei of cancer cells. As the structure of PCH nanoparticles was designed to target the tumor sites, the in vivo imaging system also revealed that different from free ICG, the PCH nanoparticles preferentially accumulated in the tumor sites. In summary, PCH nanoparticles have been shown to be a smart drug delivery system, which is responsive to a weakly acidic microenvironment.
In addition, PCH nanoparticles significantly inhibited the growth and proliferation of human colon cancer cells in vitro. PCH-treated cells showed large proportion of early apoptotic cells (in the lower right quadrant; Figure 5A) and late apoptotic cells (in the upper right quadrant; Figure 5A). Notably, NAC, a specific ROS inhibitor, attenuated the apoptotic effect exerted by PCH, which indicated that the apoptotic activity of PCH is closely related to ROS. The results of flow cytometry, Western blot analysis, and electron microscopy analysis revealed that PCH induced the expression of proteins associated with mitochondrial apoptosis pathway and ER stress, which led to mitochondrial dysfunction and ER damage, as shown in Figure 10. Consistent with the in vitro studies’ results, in vivo studies in animals have shown that PCH particles have significant advantages over free HCPT and CA, as they show more effective tumor growth inhibition and better systemic safety. In this context, the kidney toxicity was notably decreased by PCH treatment compared with other free therapeutic drugs, in terms of tissue damage or lesions in kidneys. We anticipate that novel pH-responsive PCH nanoparticles have enormous potential for efficient cancer treatment.
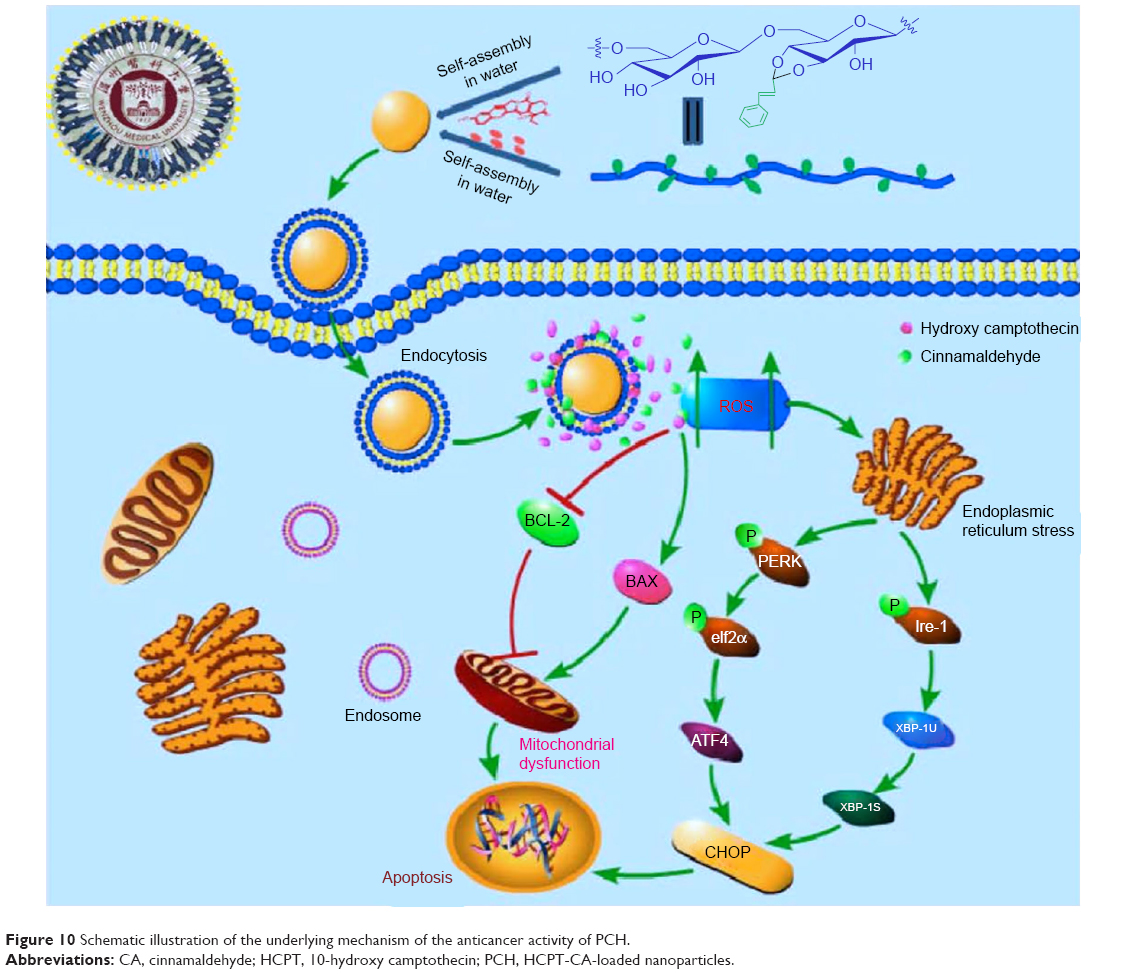 | Figure 10 Schematic illustration of the underlying mechanism of the anticancer activity of PCH. |
Conclusion
PCH not only exerted synergistic anticancer activity through the increase of ROS production in vitro but also facilitated the uptake of drug, effectively prolonged drug circulation, and increased accumulation of the drug in the tumor sites. More importantly, PCH showed excellent therapeutic performance and better systemic safety in vivo, which significantly alleviated the renal damage caused by HCPT. It is anticipated that acid-responsive PCH holds great promise for the treatment of cancer.
Acknowledgments
This work was financially supported by the National Key R&D Program of China (2017YFA0506000), National Natural Science Funding of China (21502144, 81502912, and 81622043), Zhejiang Provincial Natural Science Foundation of China (LR16H310001), and Medical Scientific Research Fund of Zhejiang Province (2019322308).
Disclosure
The authors report no conflicts of interest in this work.
References
Jiang J, Tong X, Morris D, Zhao Y. Toward photocontrolled release using light-dissociable block copolymer micelles. Macromolecules. 2006;39(13):4633–4640. | ||
Chang YT, Liao PY, Sheu HS, Tseng YJ, Cheng FY, Yeh CS. Near-infrared light-responsive intracellular drug and siRNA release using Au nanoensembles with oligonucleotide-capped silica shell. Adv Mater. 2012;24(25):3309–3314. | ||
Kang H, Trondoli AC, Zhu G, et al. Near-infrared light-responsive core-shell nanogels for targeted drug delivery. ACS Nano. 2011;5(6):5094–5099. | ||
Langereis S, Keupp J, van Velthoven JL, et al. A temperature-sensitive liposomal 1H CEST and 19F contrast agent for Mr image-guided drug delivery. J Am Chem Soc. 2009;131(4):1380–1381. | ||
Bikram M, West JL. Thermo-responsive systems for controlled drug delivery. Expert Opin Drug Deliv. 2008;5(10):1077–1091. | ||
Huang SL. Liposomes in ultrasonic drug and gene delivery. Adv Drug Deliv Rev. 2008;60(10):1167–1176. | ||
Ferrara KW. Driving delivery vehicles with ultrasound. Adv Drug Deliv Rev. 2008;60(10):1097–1102. | ||
Hernot S, Klibanov AL. Microbubbles in ultrasound-triggered drug and gene delivery. Adv Drug Deliv Rev. 2008;60(10):1153–1166. | ||
Zhao X, Kim J, Cezar CA, et al. Active scaffolds for on-demand drug and cell delivery. Proc Natl Acad Sci. 2011;108(1):67–72. | ||
Hoare T, Santamaria J, Goya GF, et al. A magnetically triggered composite membrane for on-demand drug delivery. Nano Lett. 2009;9(10):3651–3657. | ||
Hoare T, Timko BP, Santamaria J, et al. Magnetically triggered nanocomposite membranes: a versatile platform for triggered drug release. Nano Lett. 2011;11(3):1395–1400. | ||
de La Rica R, Aili D, Stevens MM. Enzyme-responsive nanoparticles for drug release and diagnostics. Adv Drug Deliv Rev. 2012;64(11):967–978. | ||
Lee M-R, Baek K-H, Jin HJ, Jung Y-G, Shin I. Targeted enzyme-responsive drug carriers: studies on the delivery of a combination of drugs. Angew Chem Int Ed. 2004;43(13):1675–1678. | ||
Gao W, Chan JM, Farokhzad OC. pH-responsive nanoparticles for drug delivery. Mol Pharm. 2010;7(6):1913–1920. | ||
Felber AE, Dufresne MH, Leroux JC. pH-sensitive vesicles, polymeric micelles, and nanospheres prepared with polycarboxylates. Adv Drug Deliv Rev. 2012;64(11):979–992. | ||
Ulbrich K, Subr V. Polymeric anticancer drugs with pH-controlled activation. Adv Drug Deliv Rev. 2004;56(7):1023–1050. | ||
Fattal E, Couvreur P, Dubernet C. “Smart” delivery of antisense oligonucleotides by anionic pH-sensitive liposomes. Adv Drug Deliv Rev. 2004;56(7):931–946. | ||
Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Control Release. 2011;152(1):2–12. | ||
Mahmoud EA, Sankaranarayanan J, Morachis JM, Kim G, Almutairi A. Inflammation responsive logic gate nanoparticles for the delivery of proteins. Bioconjug Chem. 2011;22(7):1416–1421. | ||
Broaders KE, Grandhe S, Fréchet JM. A biocompatible oxidation-triggered carrier polymer with potential in therapeutics. J Am Chem Soc. 2011;133(4):756–758. | ||
Jhaveri AM, Torchilin VP. Multifunctional polymeric micelles for delivery of drugs and siRNA. Front Pharmacol. 2014;77:1–26. | ||
Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–1822. | ||
Sawant RR, Torchilin VP. Multifunctionality of lipid-core micelles for drug delivery and tumour targeting. Mol Membr Biol. 2010;27(7):232–246. | ||
Lx L, Bai Z, Levkin PA. Boronateedextran: an acid-responsive biodegradable polymer for drug delivery. Biom. 2013;34:8504–8510. | ||
Fang J, Seki T, Maeda H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv Drug Deliv Rev. 2009;61(4):290–302. | ||
Fruehauf JP, Meyskens FL. Reactive oxygen species: a breath of life or death? Clin Cancer Res. 2007;13(3):789–794. | ||
Boonstra J, Post JA. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene. 2004;337:1–13. | ||
Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30(11):1191–1212. | ||
Perry G, Raina AK, Nunomura A, Wataya T, Sayre LM, Smith MA. How important is oxidative damage? Lessons from Alzheimer’s disease. Free Radic Biol Med. 2000;28(5):831–834. | ||
Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. | ||
Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. | ||
Gan FF, Chua YS, Scarmagnani S, et al. Structure-activity analysis of 2′-modified cinnamaldehyde analogues as potential anticancer agents. Biochem Biophys Res Commun. 2009;387(4):741–747. | ||
Huang T-C, Fu H-Y, Ho C-T, Tan D, Huang Y-T, Pan M-H. Induction of apoptosis by cinnamaldehyde from indigenous cinnamon Cinnamomum osmophloeum Kaneh through reactive oxygen species production, glutathione depletion, and caspase activation in human leukemia K562 cells. Food Chem. 2007;103(2):434–443. | ||
Ka H, Park HJ, Jung HJ, et al. Cinnamaldehyde induces apoptosis by ROS-mediated mitochondrial permeability transition in human promyelocytic leukemia HL-60 cells. Cancer Lett. 2003;196(2):143–152. | ||
Hong SH, Kim J, Kim JM, et al. Apoptosis induction of 2′-hydroxycinnamaldehyde as a proteasome inhibitor is associated with ER stress and mitochondrial perturbation in cancer cells. Biochem Pharmacol. 2007;74(4):557–565. | ||
Lee K, Kwon BM, Kim K, et al. Plasma pharmacokinetics and metabolism of the antitumour drug candidate 2′-benzoyloxycinnamaldehyde in rats. Xenobiotica. 2009;39(3):255–265. | ||
Sen N, das BB, Ganguly A, et al. Camptothecin induced mitochondrial dysfunction leading to programmed cell death in unicellular hemoflagellate Leishmania donovani. Cell Death Differ. 2004;11(8):924–936. | ||
Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51(3):794–798. | ||
Kawanishi S, Hiraku Y, Pinlaor S, Ma N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol Chem. 2006;387(4):365–372. | ||
Toyokuni S, Okamoto K, Yodoi J, Hiai H. Persistent oxidative stress in cancer. FEBS Lett. 1995;358(1):1–3. | ||
Singh R, Koppikar SJ, Paul P, Gilda S, Paradkar AR, Kaul-Ghanekar R. Comparative analysis of cytotoxic effect of aqueous cinnamon extract from Cinnamomum zeylanicum bark with commercial cinnamaldehyde on various cell lines. Pharm Biol. 2009;47(12):1174–1179. | ||
Moserova I, Kralova J. Role of ER stress response in photodynamic therapy: ROS generated in different subcellular compartments trigger diverse cell death pathways. PLoS One. 2012;7(3):e32972. | ||
Kwon B, Han E, Yang W, et al. Nano-Fenton reactors as a new class of oxidative stress amplifying anticancer therapeutic agents. ACS Appl Mater Interfaces. 2016;8(9):5887–5897. | ||
Kim B, Lee E, Kim Y, Park S, Khang G, Lee D. Dual acid-responsive micelle-forming anticancer polymers as new anticancer therapeutics. Adv Funct Mater. 2013;23(40):5091–5097. |
Supplementary materials
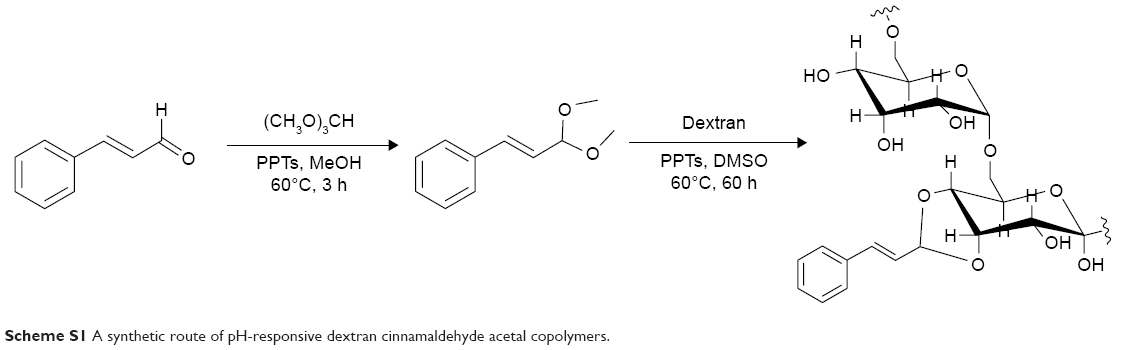 | Scheme S1 A synthetic route of pH-responsive dextran cinnamaldehyde acetal copolymers. |
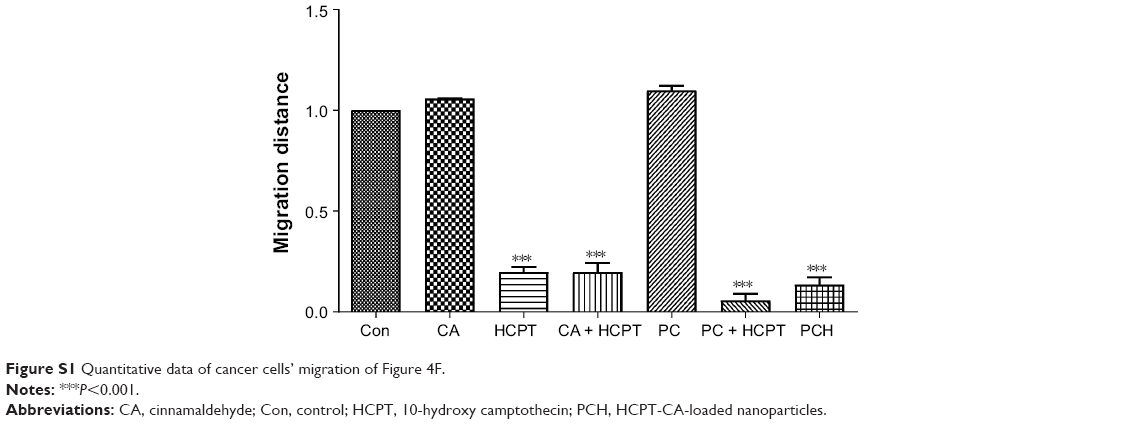 | Figure S1 Quantitative data of cancer cells’ migration of Figure 4F. |
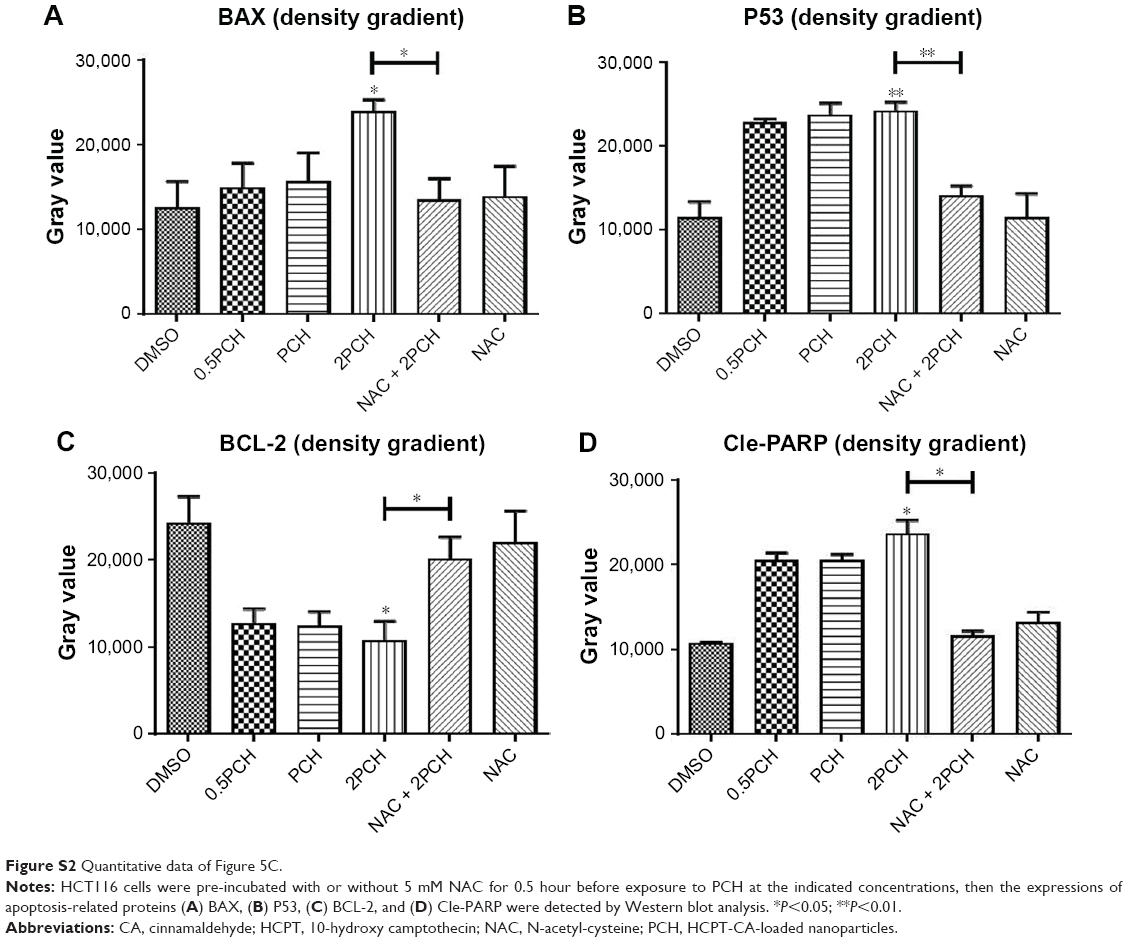 | Figure S2 Quantitative data of Figure 5C. |
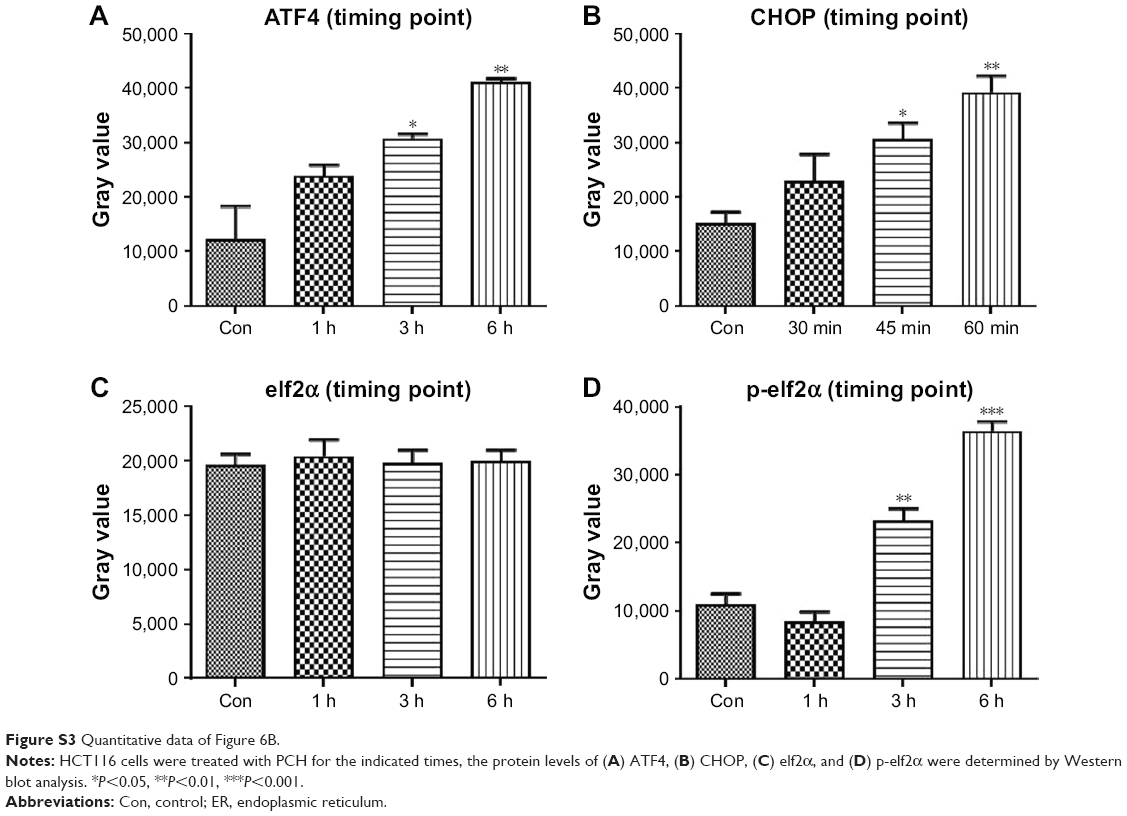 | Figure S3 Quantitative data of Figure 6B. |
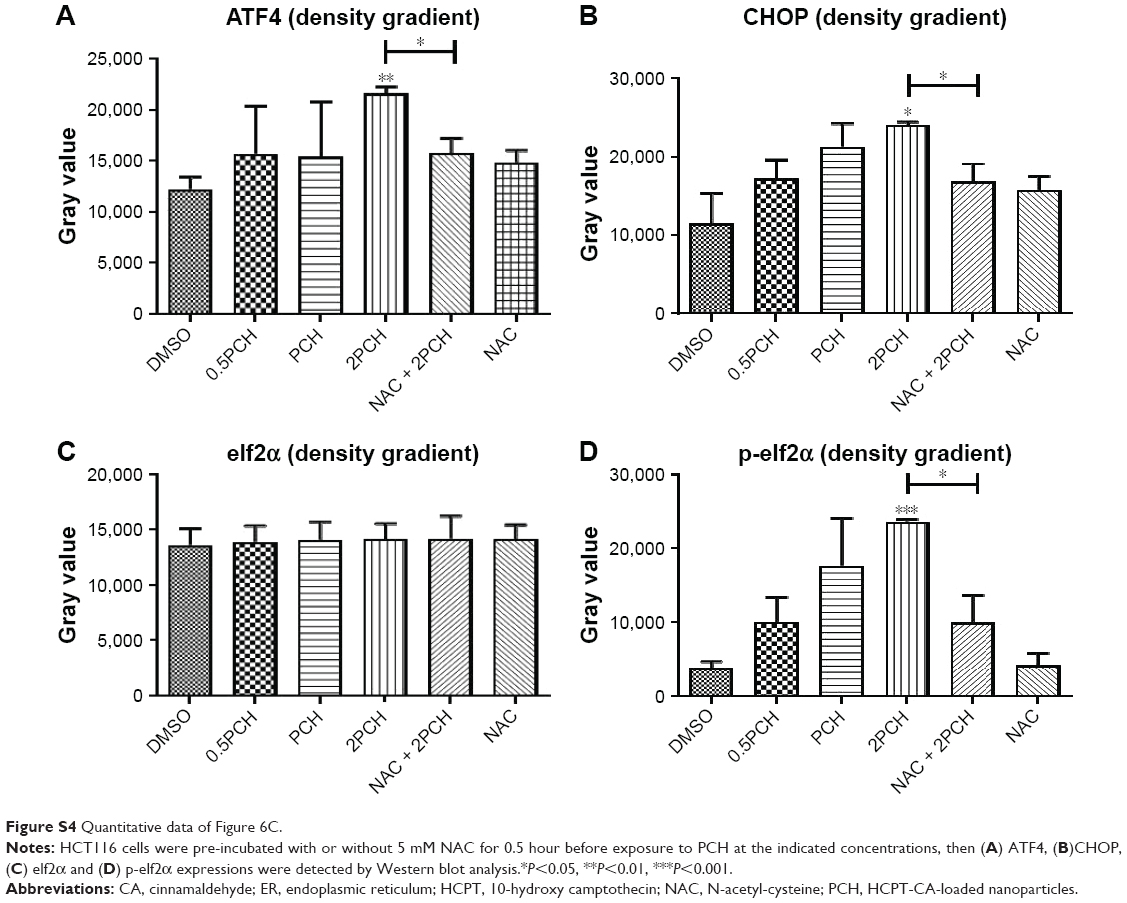 | Figure S4 Quantitative data of Figure 6C. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.