Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 7
Advances in the treatment of relapsing–remitting multiple sclerosis: the role of pegylated interferon β-1a
Authors Furber KL, Van Agten M, Evans C ![]() , Haddadi A
, Haddadi A ![]() , Doucette JR, Nazarali AJ
, Doucette JR, Nazarali AJ
Received 23 September 2016
Accepted for publication 22 December 2016
Published 24 March 2017 Volume 2017:7 Pages 47—60
DOI https://doi.org/10.2147/DNND.S71986
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Müller
Kendra L Furber,1–3 Marina Van Agten,1–3 Charity Evans,2,4 Azita Haddadi,2 J Ronald Doucette,3–5,† Adil J Nazarali1–4
1Laboratory of Molecular Cell Biology, 2College of Pharmacy and Nutrition, 3Neuroscience Research Cluster, University of Saskatchewan, 4Cameco Multiple Sclerosis Neuroscience Research Center, City Hospital, 5Department of Anatomy and Cell Biology, College of Medicine, University of Saskatchewan, Saskatoon, SK, Canada
†Dr. J Ronald Doucette passed away on May 15, 2016
Abstract: Multiple sclerosis (MS) is a progressive, neurodegenerative disease with unpredictable phases of relapse and remission. The cause of MS is unknown, but the pathology is characterized by infiltration of auto-reactive immune cells into the central nervous system (CNS) resulting in widespread neuroinflammation and neurodegeneration. Immunomodulatory-based therapies emerged in the 1990s and have been a cornerstone of disease management ever since. Interferon β (IFNβ) was the first biologic approved after demonstrating decreased relapse rates, disease activity and progression of disability in clinical trials. However, frequent dosing schedules have limited patient acceptance for long-term therapy. Pegylation, the process by which molecules of polyethylene glycol are covalently linked to a compound, has been utilized to increase the half-life of IFNβ and decrease the frequency of administration required. To date, there has been one clinical trial evaluating the efficacy of pegylated IFN. The purpose of this article is to provide an overview of the role of IFN in the treatment of MS and evaluate the available evidence for pegylated IFN therapy in MS.
Keywords: interferon, pegylation, multiple sclerosis, relapsing–remitting, disease-modifying therapy
Multiple sclerosis: an overview
Multiple sclerosis (MS) is a progressive, neurodegenerative disease affecting roughly 2.3 million people worldwide.1 MS is characterized by infiltration of auto-reactive T cells, B cells and other immune mediators into the central nervous system (CNS), causing demyelinating lesions, axonal degeneration and formation of sclerotic plaques.2 Neural damage may manifest symptomatically as optic neuritis, numbness or tingling in the extremities, muscle weakness, slurred speech and bowel/bladder dysfunction.3–5 There is also an increasing appreciation of “soft” or “hidden” symptoms such as fatigue,6–8 cognitive impairment8–10 and comorbidity with other psychiatric disorders.11 Diagnosis typically occurs between 20 and 40 years of age but may occur at any stage in life, and women are affected two to three times more often than men.1 The etiology of MS remains unknown; however, it is likely an interplay between genetics and environmental factors.2,3,12–15
The first episode of neurological symptoms, characteristic of an inflammatory demyelinating event in the brain or spinal cord, is classified as a clinically isolated syndrome (CIS).16 This remains so until a definite diagnosis of MS, with “dissemination of demyelinating lesions in space and time”, is made based on clinical episodes, magnetic resonance imaging (MRI) and/or analysis of cerebrospinal fluid.17,18 There are three subtypes of MS based on the manifestation of clinical symptoms: relapsing–remitting MS (RRMS), secondary progressive MS (SPMS) and primary progressive MS (PPMS), which can be further classified as active- or non-active based on clinical or MRI criteria.16 The most common diagnosis is RRMS, which affects 80–85% of patients and is characterized by acute exacerbations followed by periods of remission.1,5,19 Exacerbations, referred to as attacks or relapses, are defined as new symptoms in the absence of fever reflecting decreased neurological function, lasting at least 24 h and separated from other new symptoms by at least 30 days.18 Remission from relapse may be partial or complete; neurologic recovery following a relapse tends to be better in early stages of the disease but becomes less complete with repeated relapses.3,5 Approximately 75% of patients presenting with RRMS will convert to SPMS within 35 years of the initial symptoms, which is marked by a decline in acute relapses with a steady increase in the progression of disability.5 PPMS is diagnosed in ~15% of cases and is characterized by a steady progression of disability from onset, which may occur with or without the presence of clinical relapses and/or MRI activity.1,5,19
Current therapies for disease management
There are currently 11 disease-modifying therapies (DMTs) approved for MS in Canada (Table 1), with several emerging therapies in Phase II and III clinical trials. All currently approved DMTs modulate immune functions and are indicated for treatment of CIS, RRMS and/or SPMS with relapses. To date, there are no approved DMTs to mitigate neurodegenerative disease mechanisms for progressive forms of MS, although some experimental therapies are targeted toward neural repair mechanisms. First-line therapies in Canada include interferon beta-1b (IFNβ-1b; Betaseron®, Extavia®), IFNβ-1a (Avonex®, Rebif®), pegylated IFNβ-1a (PEG-IFNβ-1a; Plegridy®), glatiramer acetate (Copaxone®), teriflunomide (Aubagio®) and dimethyl fumarate (Tecfidera®). Second-line therapies available are fingolimod (Gilenya®), natalizumab (Tysabri®) and alemtuzumab (Lemtrada®). Given that there is no cure for MS, the goals of current therapeutic interventions are to reduce the number and severity of relapses, minimize long-term disability and improve overall quality of life. In clinical trials, outcomes typically examined include annualized relapse rates (ARRs), MRI parameters such as disease burden (T2 lesion volume) or disease activity (number of new/newly enlarging T2 lesions or gadolinium-enhancing T1 lesions) and disability progression (Expanded Disability Status Scale [EDSS]20). The efficacy and safety profiles of currently available DMTs are briefly summarized in Table 1.
 | Table 1 Disease Modifying Therapies approved by Health Canada for the treatment of Multiple Sclerosisa Notes: aan additional DMT, daclizumab (humanized mAb that binds CD25) marketed as Zinbryta®, was approved by Health Canada for treatment of RRMS in December 2016 while manuscript was in print; befficacy reported as % reduction in relapse rate compared to placebo, except for alemtuzumab (compared to IFNβ-1a), in Phase III clinical trials; creported in Phase III clinical trials, except indicated as drecently reported by Health Canada (http://www.hc-sc.gc.ca; accessed January 14, 2017);42 eproposed mechanisms based on known pharmacology and/or current evidence from clinical trials and animal models.53 Abbreviations: BD, twice daily; CIS, clinically isolated syndrome; CNS, central nervous system; DMT, disease modifying therapy; GI, gastrointestinal; IFN, interferon; IM intramuscular; IV, intravenous; LFT, liver function tests; mAb; monoclonal antibody; MMP, matrix metalloproteinase; MS, multiple sclerosis; NF-κβ, nuclear factor-kappa β; Nrf2, nuclear factor-erythroid 2-related factor 2; PEG, polyethylene glycol; PO, per oral; PML, progressive multifocal leukoencephalopathy ; QAD, once every other day; QD, once daily; QW, once weekly; Q2W, once every 2 weeks; Q4W, once every 4 weeks; RRMS, relapsing-remitting MS; rxn, reaction; S1P, sphingosine-1-phosphate; SC, subcutaneous; SPMS, secondary progressive MS; TIW, three times weekly; VCAM-1, vascular cell adhesion molecule 1. |
Overall, treatment decisions are based on benefit/risk profile.21 First-line therapies have shown similar efficacy in placebo-controlled22–30 and head-to-head trials,27,31–35 which is supported by recent meta-analysis data.36–38 Second-line therapies show greater efficacy in reducing relapse rates and MRI activity but are also associated with more adverse side effects and potential toxicities.39–43 These second-line therapies are typically used only in patients who show disease activity while on first-line therapies, who cannot tolerate first-line therapies or who have extremely active RRMS from onset.21 Thus, the optimization of MS therapy ultimately depends on individual disease activity, response to treatment, tolerability and practitioner or patient preference. Proven efficacy and long-term safety information from numerous clinical trials make IFNβ an attractive first-line therapy; however, an important drawback is patient adherence due to frequent dosing schedules. Thus, the development of pegylated IFN44 (Plegridy®) has provided an alternative injectable agent that has similar efficacy and safety profiles as traditional IFNβ therapies but significantly reduces the frequency of administration.44–50 In this review, we focus our discussion on placebo-controlled trials of subcutaneous IFNβ-1a and head-to-head trials against other IFNβ formulations to evaluate the role of PEG-IFNβ-1a therapy in MS (Table 2).
 | Table 2 Summary of PRISMS, INCOMIN, EVIDENCE and ADVANCE trials Abbreviations: ARR, annualized relapse rate; EDSS, expanded disability status scale; IFNβ, interferon beta; IM, intramuscular; MS, multiple sclerosis; PEG, polyethylene glycol; QAD, once every other day; QW, once weekly; Q2W, once every 2 weeks; Q4W, once every 4 weeks; RRMS, relapsing–remitting MS; MRI, magnetic resonance imaging; SC, subcutaneous; TIW, three times weekly. |
Interferon β
IFNβ was the first biologic approved for MS therapy. IFNs are a family of cytokines involved in the regulation of innate and adapted immunity12,51–53 and are thus ideal candidates for immunomodulatory therapies in MS. Early attempts at IFN therapy in MS included the use of IFNγ, IFNα and IFNβ.54 Unexpectedly, administration of IFNγ resulted in activation of the immune system and increased the occurrence of relapses. Several different formulations of IFNα and IFNβ were explored with promising results, but IFNβ had a more acceptable patient safety profile. Both recombinant IFNβ-1b produced in Escherichia coli and recombinant IFNβ-1a produced in mammalian cells are approved for the treatment of MS (Table 1). Differences in post-translational modifications confer reduced biological activity of non-glycosylated IFNβ-1b compared to glycosylated IFNβ-1a,55 which is reflected in their dosages. Clinical trials with IFNβ have demonstrated a reduction in relapse rates and MRI activity.22–24 Delays in disease progression, as measured with EDSS, have also been reported over the 1- to 2-year clinical trial periods,23,24 and long-term benefits have been associated with continued IFNβ treatment in follow-up studies.56,57 However, a large retrospective study by Shirani et al58 reported no link between IFNβ treatment and the long-term disability progression, which has renewed debate around this issue.59–61
Dosage and administration
IFN treatment is currently indicated for the treatment of CIS, RRMS and SPMS with relapses. Commercially available formulations of IFNβ-1b include Betaseron® and Extavia®, which are administered subcutaneously (SC) every other day (QAD) at a dose of 250 µg. IFNβ-1a is marketed as Avonex® administered intramuscularly (IM) once weekly (QW) at a dose of 30 µg or Rebif® administered SC three times weekly (TIW) at a dose of 22 or 44 µg.
Mechanisms of action
The precise mechanisms of action of IFNβ in MS therapy are unknown but have been attributed to its antiproliferative, antiviral and immunomodulatory abilities.52 Systemic administration of IFNβ is believed to primarily modulate immune cell function in the periphery.62 In general, IFNβ appears to oppose the pathogenic processes associated with MS disease progression by shifting from a pro-inflammatory to an anti-inflammatory immune profile (Figure 1). A key player in MS pathology are auto-reactive T cells that migrate across the blood–brain barrier (BBB), initiating inflammatory cascades in the CNS inflicting damage on axons, neurons and myelin sheaths.2,63 IFNβ therapy is associated with a decrease in the expansion of pro-inflammatory Th1 and Th17 subtypes and promotes the expansion of anti-inflammatory Th2 subtype.64–68 IFNβ has also been shown to modulate the function of regulatory T cells,69 B cells,70,71 natural killer cells72 and dendritic cells.73 The complexity of immune cell signaling networks makes it difficult to delineate the direct and indirect effects of IFNβ on each cell type.52 Effects of IFNβ on T cell function are mostly likely mediated indirectly through up-regulation of anti-inflammatory mediators, such as interleukin (IL)-10,66,74–78 and down-regulation of pro-inflammatory cytokines, such as IL-17, osteopontin and tumor necrosis factor α (TNFα),64,73,74,78,79 secreted from other types of immune cells.
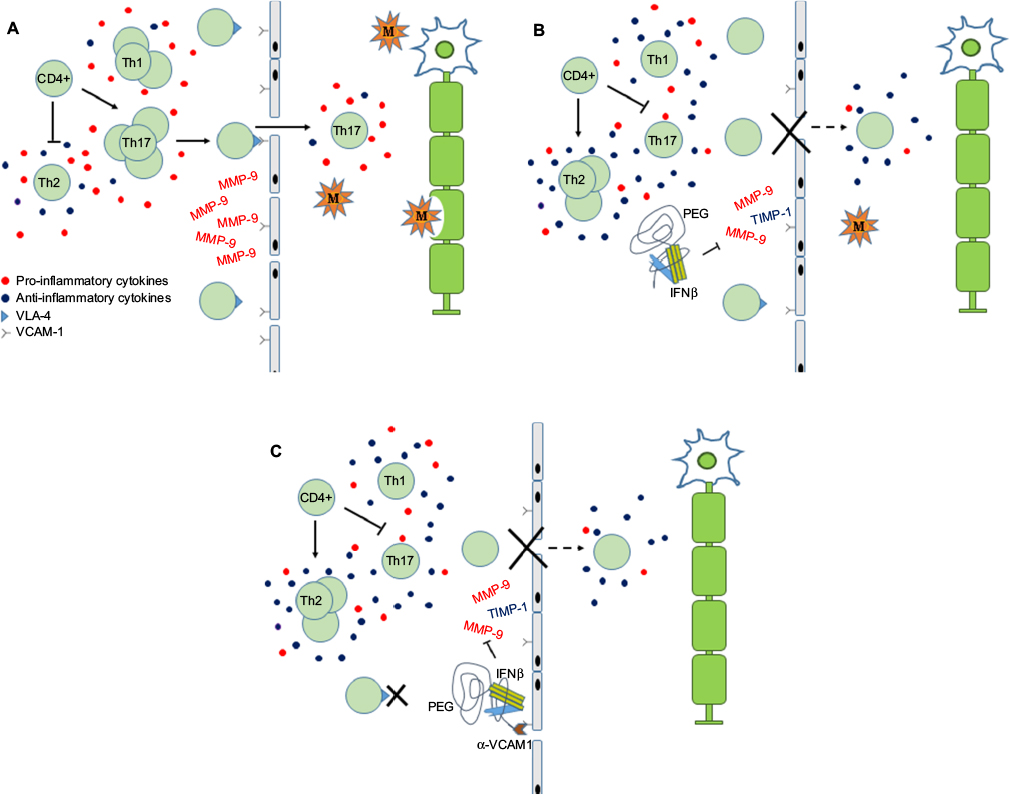 | Figure 1 Schematic depicting the role of PEG-IFNβ therapy in MS. Notes: (A) In MS, pro-inflammatory cytokines stimulate CD4+ cells to proliferate and differentiate into Th1 and Th17 effector cells. Activated T cells express VLA-4, which interacts with VCAM-1 on endothelial cells, to facilitate crossing the BBB. In the CNS, auto-reactive T cells and macrophages result in damage to the myelin sheath, axons and neurons. Inflammatory demyelinating lesions result in the clinical presentation of MS. (B) IFNβ is conjugated to PEG to increase the molecule’s serum concentration and half-life. Proposed actions of IFNβ include modulating cytokine milieu to favor anti-inflammatory pathways, which inhibits expansion of Th1/Th17 and promotes expansion of Th2 cells. Down-regulation of VLA-4 and inhibition of MMP-9 reduce migration of activated T cells into CNS. (C) Linking of anti-VCAM-1 antibodies to the PEG tail may enhance IFNβ anti-inflammatory actions by 1) blocking interaction of leukocytes expressing VLA-4 with VCAM-1 and 2) increasing local concentration of PEG-IFNβ at BBB. Abbreviations: BBB, blood–brain barrier; CNS, central nervous system; IFNβ, interferon beta; M, macrophage; MMP, matrix metalloproteinase; MS, multiple sclerosis; PEG, polyethylene glycol; PEG-IFNβ, pegylated interferon β; TIMP-1, tissue inhibitor of metalloproteinase-1; VCAM-1, vacular cell adhesion molecule 1; VLA-4, very late activation antigen-4. |
IFNβ has also been implicated in acting at the BBB, impeding migration of leukocytes into the CNS. Matrix metalloproteinases (MMPs), more specifically MMP-9 and tissue inhibitor of metalloproteinase-1 (TIMP-1), are involved in the degradation and remodeling of the extracellular matrix.80 IFNβ has been shown to decrease serum levels of MMP-9 and modulate MMP-9:TIMP-1 ratio, and this was associated with a decrease in the number of new and/or active MRI lesions.81,82 To facilitate crossing the BBB, activated T cells up-regulate adhesion molecules, such as very late activation antigen-4 (VLA-4), which interact with vascular cell adhesion molecule 1 (VCAM-1) receptor expressed by endothelial cells (Figure 1). Evidence suggests that administration of IFNβ down-regulates expression of VLA-4/α-4 integrin, thereby inhibiting lymphocyte migration into the CNS.83–85 Together, these mechanisms reduce the number of pro-inflammatory immune cells in circulation and suppress the ability of activated leukocytes to enter the CNS. In addition to immunomodulatory effects, IFNβ treatment has been associated with increased expression of neurotrophic factors,86–88 which may convey cytoprotective effects.
Clinical trials
The Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis (PRISMS) study23,89,90 was pivotal multi-centered, randomized, double-blind, placebo-controlled clinical trial investigating the benefit of SC IFNβ-1a administration TIW for RRMS (Table 2). Participants were randomized to either 22 or 44 μg or placebo groups with a primary outcome of number of relapses during the 2-year study period.23 The number of relapses was lower in both intervention groups compared to placebo: 27% (95% confidence interval [95% CI] 14–39) reduction for the 22 μg dose (1.82 vs 2.56) and 33% (95% CI 21–44) reduction for the 44 μg dose (1.73 vs 2.56) (Table 3). This included a delay in the time to relapse and a decreased number of severe relapses, steroid courses and hospitalizations with IFNβ-1a treatment. Monitoring of disease activity with MRI89 showed a corresponding decrease in the number of Gd-enhancing T1 lesions, number of active T2 lesions and reduction in burden of disease (T2-weighted). Participants receiving placebo showed a 10.9% increase in total lesion volume compared to a 1.2% decrease for the 22 μg intervention and 3.8% decrease for the 44 μg intervention. After 4 years, IFNβ-1a treatment continued to reduce ARR (0.90 in year 1 vs 0.44 in year 4 for high-dose group) and improve MRI outcomes.90 Furthermore, both 4-year treatment groups showed favorable outcomes compared to the delay-treatment, crossover groups. Follow-up analyses at both 891 and 15 years56 are indicative of long-term benefit of IFNβ-1a therapy on disease course, favoring early treatment intervention, higher dosages and longer exposure times.
 | Table 3 Comparison of currently available data on interferon β-1a and pegylated interferon β-1a Notes: aComparison of placebo-controlled studies; no head-to-head trials have directly compared IFNβ-1a to PEG-IFNβ-1a. bDefined as number of relapses over 2-year study period (PRISMS) or ARR at 48 weeks (ADVANCE). cDefined as change in EDSS over duration of study (PRISMS) or proportion of patients with 12-week sustained progression in EDSS ≥1.0 (ADVANCE). dDefined as change in total in T2 lesion volume from baseline (PRISMS, mm2; ADVANCE, cm3). eDefined as the sum of T1 Gd-enhancing and new or newly enlarging T2 lesions (PRISMS, clinically unique; ADVANCE new active). fReported at 2 years.122 gPRISMS reported adverse events in first 3 months of therapy and ADVANCE reported adverse events over the full 48 weeks. hMeasured over 8 h for 22 μg IFNβ-1a and measured over 168 h for 44 μg IFNβ-1a and 125 μg PEG-IFNβ-1a. Cmax, maximum serum concentration; Tmax, time to reach Cmax. Abbreviations: ARR, annualized relapse rate; AUC, area under curve; EDSS, Expanded Disability Status Scale; IFN, interferon; Q2W, once every 2 weeks; MRI, magnetic resonance imaging; n.a., not available; NAbs, neutralizing antibodies; PEG, polyethylene glycol; SC, subcutaneous; TIW, three times weekly. |
Following the PRISMS study, head-to-head trials further investigated the efficacy of different IFNβ formulations and dosage in the Independent Comparison of Interferon (INCOMIN) study31 and Evidence of Interferon Dose-response-European North American Comparative Efficacy (EVIDENCE) study.32 The INCOMIN trial31 compared 250 μg IFNβ-1b SC injections QAD with 30 μg IFNβ-1a IM injections QW over 2 years (Table 2). The primary outcome of the proportion of patients remaining relapse free favored IFNβ-1b therapy (49%) over IFNβ-1a therapy (33%); this was also reflected by a 29% reduction in the ARR (0.5 for IFNβ-1b vs 0.7 for IFNβ-1a). For MRI outcomes, IFNβ-1b SC therapy increased the proportion of patients who remained free from Gd-enhancing T1 lesions or new T2 lesions compared to IFNβ-1a. The burden of disease for participants receiving IFNβ-1b decreased 2.8% whereas IFNβ-1a increased 11.7%. However, due to multiple variables in treatment regimen, it is difficult to ascertain whether improved outcome relates to difference in formulation, dosage, frequency or route of administration.
The EVIDENCE trial32 compared IFNβ-1a treatment at a dosage of 44 μg administered by SC injections TIW (TIW) with a dosage of 30 μg administered by IM injections QW over 1 year (Table 2). The primary outcome was the proportion of patients remaining relapse free, which favored 44 μg SC TIW therapy (56%) over 30 μg IM QW therapy (48%). There was a 17% reduction in ARR in the 44 μg SC TIW group (0.54) compared to the 30 μg IM QW group (0.65), and the mean number of active T2 lesions was also reduced by 36%. Thus, both the IFNβ-1b SC (cumulative dose 28 MIU/week) in the INCOMIN study and the IFNβ-1a SC (cumulative dose 36 MIU/week) in the EVIDENCE trial showed greater efficacy than IFNβ-1a IM (cumulative dose 6 MIU/week). Pharmacokinetic studies indicate that the bioavailability of IFNβ does not appear to differ substantially between SC and IM administration.62 Taken together, data from the PRISMS, INCOMIN and EVIDENCE trials demonstrated that higher, more frequent dosing of IFNβ improve patient outcomes.
Therapeutic considerations
Safety profile
IFNs are naturally occurring cytokines and generally well tolerated. The PRISMS study23 reported common adverse events of IFNβ-1a treatment to include injection-site reactions (redness, swelling), flu-like symptoms, fever, headache and myalgia. However, flu-like symptoms were also commonly reported in placebo group (24% vs 25% and 27% in 22 and 44 μg treatment groups, respectively). Most injection-site reactions were mild, with only eight incidences of skin necrosis (>150,000 injections). Less common, but serious, adverse events include decreased numbers of white blood cells and platelets and elevated liver enzymes. Similar safety profiles were reported in 4-year follow-up studies. Over 4 years, there were three deaths reported none of which were related to the treatment.90,92
There were initially reports of IFNβ therapy causing an increase in the incidence of depression. Given the neurologic pathology of MS and its chronic progressive nature, the estimated lifetime prevalence of major depression in MS is as high as 50%.93,94 With such a high comorbidity rate of depression among MS patients,11 it is difficult to definitively link IFNβ therapy with an increased risk of depression. There was no significant difference among treatment groups in the PRISMS trial; depression was reported by 28% of patients receiving placebo, 21% of patients receiving 22 μg of IFNβ and 24% of patients receiving 44 μg of IFNβ.23 During the study, one patient in the placebo group committed suicide and three patients in each group reported attempting suicide or suicidal ideation. Furthermore, no difference in the incidence of depression has been found between patients receiving IFNβ and glatiramer acetate therapy.95
Neutralizing antibodies
Another concern associated with IFNβ-1a therapy is the production of neutralizing antibodies (NAbs). In the PRISMS study, 23.8% of participants receiving the 22 μg dose and 12.5% of participants receiving the 44 μg dose were positive for NAbs but concluded that NAbs had no impact on the mean relapse count in either intervention.23 The INCOMIN study reported more frequent production of NAbs with IFNβ-1b (30%) than with IFNβ-1a (7%) therapy, but again this did not appear to negatively impact treatment efficacy.31 In contrast, the EVIDENCE study with IFNβ-1a reported a higher prevalence of NAbs within the 44 μg SC TIW (26%) compared to the 30 μg IM QW (3%) intervention and noted that NAbs developed earlier among the high-dose intervention.32 Disease activity was significantly greater in participants receiving 44 μg SC TIW who were positive for NAbs compared to those who were negative for NAbs; however, NAb+ve and NAb−ve participants had less MRI activity than their 30 μg IM QW counterparts.
The clinical implications of NAbs in IFNβ therapy remain unclear. High titers of NAbs appear to be related to the formulation, dosing regimen and route of administration.31,32,96–98 NAb levels often peak within 6–12 months of starting therapy and then decline, with ~10% of patients developing persistent NAbs.98 At persistently high titers, NAbs can reduce biological activity99,100 and negatively impact long-term drug therapy.97,99,101–103 The most recent treatment recommendations provide little consensus on how to approach this issue:21 current practices include periodic testing of NAb levels, testing for NAbs only when considering switching therapies or opting to base treatment decisions solely on clinical outcomes.
Adherence
Frequent IFNβ administration has been shown more effective for managing RRMS;31,32 however, given that the disease is chronic and progressive, this dosing regimen may not be amenable for many patients. Overall, adherence rates of injectable DMTs requiring frequent administration (i.e., ≥QW) have been estimated at 27–76%,104–110 with decreased adherence associated with an increased risk of relapse.105,106 The most commonly reported reasons for discontinuation were lack of efficacy, tolerability (i.e., adverse effects such as flu-like symptoms and injection-site reactions) and patients’ decision (i.e., both practical issues and mental exhaustion).111–115 Discontinuation due to adverse effects typically occurs within the first year of treatment,114,115 but this can be mitigated by dose titration, prophylactic treatment of flu-like symptoms and injection-site reactions, increased patient education and the use of autoinjectors.116,117 Another viable approach has been drug modification to increase pharmacokinetic profile and prolong bioavailability to reduce frequency of administration, such as the pegylation of IFNβ-1a.44
Pegylated interferon β
Pegylation is the process by which molecules of polyethylene glycol (PEG) are chemically conjugated to a biological product. The addition of PEG increases the size of a macromolecule, generally increasing its solubility, stability and mobility in solution.118,119 Pegylation decreases the rate of renal clearance and may also reduce receptor- or antibody-mediated clearance and proteolytic degradation.44,118 This results in increased exposure, half-life and serum concentrations of the therapeutic agent. Other potential benefits of pegylation are reduced antigenicity and immunogenicity, as PEG could potentially mask recognition of epitopes.44,118 Addition of large PEG molecules can often decrease binding to target through steric hindrance,119 yet the overall goal is to balance pharmacodynamic and pharmacokinetic properties to improve treatment efficacy.118 These attributes are ideal for a biomolecule such as IFNs, which are rapidly cleared without any modifications.62 For example, pegylated forms of IFNα-2a (Pegasys®; Hoffman La Roche) and IFNα-2b (PegIntron®; Schering-Plough) have been used in the management of chronic hepatitis C for over a decade.119,120 The development of PEG-IFNβ-1a, a 20 kDa linear methoxy PEG molecule conjugated to the alpha amino group of the N terminal amino acid residue of IFNβ, demonstrated a desirable pharmacokinetic and pharmacodynamic profile to decrease the frequency of administration for the management of MS.44
Dosage and administration
PEG-IFNβ-1a is marketed as Plegridy® and currently indicated for the treatment of RRMS. PEG-IFNβ-1a is administered SC every 2 weeks (Q2W) at a dose of 125 µg.
Mechanisms of action
The mechanisms of action of PEG-IFNβ-1a are presumably similar to its parent compound. The N-terminus of IFNβ, which is known not to be critical for its activity, was chosen for the site of attachment.44 Although predicted not to affect ligand-binding interactions, a size-dependent decrease in antiviral activity was observed with increasing size of PEG molecules. As smaller PEG molecules did not convey the desired pharmacokinetic properties, 20 kDa was chosen for the pegylation of IFNβ-1a.44 In healthy controls, PEG-IFNβ-1a and IFNβ-1a administration resulted in similar changes in cytokine gene expression favoring an anti-inflammatory profile.121 To our knowledge, the anti-inflammatory properties of PEG-IFNβ-1a have not yet been directly investigated or reported in clinical trials or animal models of MS.
Clinical trials
To date, there has been one randomized, double-blind, placebo-controlled clinical trial investigating PEG-IFNβ-1a in RRMS. The ADVANCE trial25,122,123 was a 2-year study, consisting of a 48-week placebo-controlled phase and 48-week crossover phase, comparing the efficacy of 125 μg PEG-IFNβ-1a administered by SC injection Q2W or every 4 weeks (Q4W; Table 2). At 48 weeks, the primary outcome of ARRs showed a benefit for both PEG-IFNβ therapy regimens compared to placebo.25 The ARRs were 0.256 (95% CI 0.206–0.318) for the Q2W intervention and 0.288 (95% CI 0.234–0.355) for the Q4W intervention compared to 0.397 for placebo (95% CI 0.328–0.481), corresponding to a 36% and 28% reduction in the Q2W and Q4W, respectively. MRI outcomes25,123 showed a decrease in the number of new or newly enlarging T2 lesions in both PEG-IFNβ treatment groups but only a significant decrease in the number of Gd-enhancing and T1 hypointense lesions in the Q2W group compared to placebo. Total T2 lesion volume increased in the placebo group by 0.77 cm3 and Q4W treatment group by 0.06 cm3 and decreased in the Q2W treatment group by 0.26 cm3. The proportion of patients achieving “no evidence of disease activity” (NEDA), defined as absence of both clinical and MRI disease activity, was 33.9% in the PEG-IFNβ Q2W, 21.5% in the PEG-IFNβ Q4W and 15.1% in the placebo group.123 These data demonstrate a benefit of both PEG-IFNβ treatments but suggest an advantage of a more frequent dosing regimen to minimize disease activity.
Upon completion of the first year, participants receiving placebo were re-randomized to one of the treatment interventions and participants already receiving drug intervention remained on the same dosing regimen.122 Both continuous intervention groups showed a reduction in the ARR compared to the corresponding delayed-treatment groups, 37% reduction and 17% reduction in the Q2W and Q4W dosing, respectively. The ARR decreased among participants receiving continuous PEG-IFNβ Q2W from 0.230 (95% CI 0.183–0.291) to 0.178 (95% CI 0.136–0.233), while the ARR was roughly maintained among participants receiving continuous PEG-IFNβ Q4W (0.286 [95% CI 0.231–0.355] and 0.291 [95% CI 0.231–0.368]). Also, the number of new or newly enlarging T2 lesions continued to decline for both the continuous Q2W and Q4W groups. When directly comparing the two dosing regimens, there was a 24% reduction in ARR and 60% reduction in the number of new or newly enhancing T2 lesions in the Q2W compared to the Q4W. Subgroup analysis indicated that the efficacy of PEG-IFNβ was similar across different populations of RRMS patients.124 A recent preliminary report from the ATTAIN study, an extension of the ADVANCE trial, indicates that the efficacy of each treatment was maintained over 3 years.45 This suggests, similar to other IFNβ formulations, that early intervention, higher cumulative dosage and increased exposure time are advantageous.
Therapeutic considerations
Safety profile
Reported side effects of PEG-IFNβ were similar to those previously reported for both SC and IM injections of IFNβ. These included mild-to-moderate adverse events such as injection-site reactions, influenza-like illness, fever, chills, headache and myalgia. In year 1, the incidence of discontinuation of the study due to adverse events was 6% in both treatment groups and 1% in the placebo group, the most commonly reported reason being influenza-like illness. The rate of severe adverse events was similar among the three study groups: 11% in the placebo, 18% for the every 2-week intervention and 16% for the every 4-week intervention.25 Participants in both intervention groups had decreased number of blood cells and elevated liver enzymes, but neither was determined to be clinically significant and the majority returned to normal ranges within 2 years. The incidence of abnormal white blood cell counts remained low (≤10% of participants) across both intervention groups.25,122 Over the 2-year study, nine deaths occurred; however, an independent safety-monitoring board reported that these events were not likely related to the study drug and did not change the risk–benefit profile of PEG-IFN.122 A 3-year follow-up from the ATTAIN study indicates that PEG-IFN continues to show a similar safety profile.125 As with other pegylated pharmaceuticals, adverse side effects are associated primarily with the active drug not the PEG moiety.119
Neutralizing antibodies
As stated previously, traditional SC IFNβ-1a therapy can result in persistently high levels of NAbs (~10% of patients), which may negatively impact the treatment efficacy. Development of IFNβ antibodies was <1% in both interventions, whereas development of antibodies against PEG was reported at 6% in Q2W and 8% in Q4W interventions.122 The appearance of NAbs was determined not to impact efficacy or safety of treatment, but due to low rate of incidence in this study, data should be interpreted with caution.126
Adherence
The use of PEG-IFNβ-1a has been estimated to reduce the number of injections by >90% compared to other first-line therapies.37 In the ADVANCE study, 88% of the participants completed the first year25 and 90% of continuing patients completed the second year.122 Tolerability and adverse events, such as flu-like symptoms and injection-site reactions, were still major factors leading to discontinuation; however, management strategies similar to standard IFN therapies can be used to mitigate impact.127 Adherence to treatment, defined by the number of doses received divided by the number of doses expected to receive, was >99% in each group.25 It should be noted though that adherence in controlled trials is usually greater than in a clinical setting.116 While reduced frequency of administration is expected to increase adherence,44,45,47,49 this will need to be further evaluated as PEG-IFN therapy is integrated into clinical practice.
Interferon βas versus pegylated interferon β
A comparison of the efficacy, safety profile and pharmacokinetics data for PEG-IFNβ-1a and its parent compound IFNβ-1a is reported in Table 3. A considerable amount of data are available regarding the pharmacokinetic properties of different IFNβ formulations, dosages and routes of administration; the only factor that substantially alters the pharmacokinetic profile is pegylation.62 PEG-IFNβ-1a administration by IM injection showed a fourfold increase in drug exposure, assessed by the area under the curve (AUC), compared to an equal dosage of IFNβ-1a.121 PEG-IFNβ and IFNβ administration both resulted in altered cytokine expression profiles, but the pharmacological response with PEG-IFNβ had a more rapid onset and was sustained for a longer period.121 Only one study has done a direct comparison between standard therapeutic doses of IFNβ-1a and PEG-IFNβ-1a administered by SC injection over the course of 2 weeks.128 When healthy participants were evaluated for accumulative drug exposure with six doses of 44 μg IFNβ-1a SC compared to a single dose of 125 μg PEG-IFNβ-1a, the AUC was found to be 60% higher for the PEG-IFNβ dosage regime. This corresponds to approximately fourfold increase in Cmax, approximately sixfold increase in Tmax and approximately twofold increase in half-life. Thus, pegylation of the IFNβ-1a molecule results in an increased serum concentration and greater drug exposure compared to the traditional IFNβ-1a therapy. The tolerability of PEG-IFNβ was favorable with a lower incidence of adverse events compared to the IFNβ dosing regimen.128 This was presumably due to the need for less frequent injections with PEG-IFNβ.
To date, there are no published head-to-head clinical trials comparing PEG-IFNβ to the alternative formulations of IFNβ or any other DMT. In the PRISMS study23 and the ADVANCE study,25 similar efficacy was observed between IFNβ-1a and PEG-IFNβ-1a interventions when comparing % relative reduction in relapse rate. Both studies also reported favorable MRI outcomes, including a decrease in disease activity (T1- and/or T2-weighted) and burden of disease (total lesion volume) with treatment. However, due to different methodology and reporting measures, direct comparisons of study outcomes are difficult. Although meta-analysis of efficacy and safety profiles37 and cost-effectiveness analysis129 tend to favor PEG-IFNβ-1a, these are relatively premature. Full results from the ATTAIN study will provide further data on the long-term efficacy on safety of PEG-IFNβ therapy. In addition to clinical trial data, longitudinal studies with IFNβ-1a treatment also suggest positive effects on cognitive function130–132 and long-term benefits of disease progression,55,90 but these types of data sets are not yet available for PEG-IFNβ-1a. Ultimately, head-to-head clinical trials and observational studies from clinical practice will continue to inform us about the role of PEG-IFNβ in MS disease management.
What are future therapeutic possibilities for PEG-IFN?
The conjugation of PEG to IFNβ-1a has expanded the therapeutic options for the management of MS. By prolonging the half-life of IFNβ-1a, increasing its exposure and decreasing its elimination,44,62 this novel formulation provides a safe and effective first-line therapy with a decreased frequency of administration. This new PEG-IFNβ formulation has also created a platform upon which other immunomodulatory molecules may be added (Figure 1). This may serve to enhance the efficacy of IFNβ by 1) providing opportunity to develop combination therapies and 2) targeting the IFNβ to specific locations within the body. For example, by specifically targeting VCAM-1, expressed on activated endothelial cells, the anti-VCAM-PEG-IFN molecule could more effectively block activated T cells from crossing the BBB. Theoretically, this would also increase the local concentration of IFNβ to the BBB possibly directing its immunomodulatory effects to this compartment and reduce non-specific systemic effects. Whether these hypothetical modifications would enhance IFN activity has yet to be determined. However, if successful it may lead to targeted delivery of IFNβ with potentially greater efficacy and a reduction in frequency of side effects.
Conclusion
IFNβ, a naturally occurring cytokine, was the first DMT approved for MS therapy. Clinical evidence has consistently demonstrated a decrease in relapse rates and MRI activity; however, the inherent physical properties require it to be frequently administered in order to maintain therapeutic concentrations. The conjugation of PEG molecules to IFNβ alters the pharmacokinetic properties of the parent drug, thereby decreasing the frequency of administration. This also provides a novel platform for combination therapy approach.
ADVANCE is currently the only clinical trial with published results on the efficacy of PEG-IFNβ-1a compared to placebo in the treatment of RRMS; to date, there are no head-to-head trials comparing PEG-IFNβ-1a to other approved DMTs. Furthermore, there are no treatment guidelines for practitioners that include initiating or switching to PEG-IFNβ therapy. Here, we have made an effort to compile currently available information regarding IFNβ therapy options with respect to various formulations. All available data suggest that the efficacy and safety profile of PEG-IFNβ-1a is comparable to other IFNβ therapies, with the added benefit of a decreased dosing schedule. This provides patients and practitioners with a viable alternate first-line therapy in the treatment of MS.
Acknowledgments
Funding from Canadian Institutes of Health Research, Saskatchewan Health Research Foundation and Natural Sciences and Engineering Research Council of Canada is gratefully acknowledged. KLF is supported by the endMS Postdoctoral Fellowship from the Multiple Sclerosis Society of Canada. We thank Dr. Katherine Knox for review of an earlier draft of the manuscript. This manuscript is dedicated to our colleague Dr. J. Ronald Doucette who passed away last year.
Disclosure
The authors report no conflicts of interest in this work.
References
Multiple Sclerosis International Federation (MSIF) [homepage on the Internet]. Atlas of MS: Mapping Multiple Sclerosis Around the World. 2013. Available from: www.msif.org/atlas. Accessed January 14, 2017. | ||
Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15(9):545–558. | ||
Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–1517. | ||
Lipsy RJ, Schapiro RT, Prostko CR. Current and future directions in MS management: key considerations for managed care pharmacists. J Manag Care Pharm. 2009;15(9 suppl A):S2–S15. quiz S16–7. | ||
Coles A. Multiple sclerosis. Pract Neurol. 2009;9(2):118–126. | ||
Induruwa I, Constantinescu CS, Gran B. Fatigue in multiple sclerosis – a brief review. J Neurol Sci. 2012;323(1–2):9–15. | ||
Chalah MA, Riachi N, Ahdab R, Creange A, Lefaucheur JP, Ayache SS. Fatigue in multiple sclerosis: neural correlates and the role of non-invasive brain stimulation. Front Cell Neurosci. 2015;9:460. | ||
Penner IK. Evaluation of cognition and fatigue in multiple sclerosis: daily practice and future directions. Acta Neurol Scand. 2016;134(suppl 200):19–23. | ||
Rao SM, Leo GJ, Bernardin L, Unverzagt F. Cognitive dysfunction in multiple sclerosis. I. frequency, patterns, and prediction. Neurology. 1991;41(5):685–691. | ||
Rocca MA, Amato MP, De Stefano N, et al. Clinical and imaging assessment of cognitive dysfunction in multiple sclerosis. Lancet Neurol. 2015;14(3):302–317. | ||
Marrie RA, Reingold S, Cohen J, et al. The incidence and prevalence of psychiatric disorders in multiple sclerosis: a systematic review. Mult Scler. 2015;21(3):305–317. | ||
Annibali V, Mechelli R, Romano S, et al. IFN-beta and multiple sclerosis: from etiology to therapy and back. Cytokine Growth Factor Rev. 2015;26(2):221–228. | ||
Ebers G. Interactions of environment and genes in multiple sclerosis. J Neurol Sci. 2013;334(1–2):161–163. | ||
Golan D, Staun-Ram E, Miller A. Shifting paradigms in multiple sclerosis: from disease-specific, through population-specific toward patient-specific. Curr Opin Neurol. 2016;29(3):354–361. | ||
Carlson RJ, Doucette JR, Knox K, Nazarali AJ. Pharmacogenomics of interferon-beta in multiple sclerosis: what has been accomplished and how can we ensure future progress? Cytokine Growth Factor Rev. 2015;26(2):249–261. | ||
Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–286. | ||
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302. | ||
McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50(1):121–127. | ||
Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain. 2006;129(pt 3):606–616. | ||
Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444–1452. | ||
Freedman MS, Selchen D, Arnold DL, et al. Treatment optimization in MS: Canadian MS working group updated recommendations. Can J Neurol Sci. 2013;40(3):307–323. | ||
IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. 1993;43(4):655–661. | ||
PRISMS Study Group. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. Lancet. 1998;352(9139):1498–1504. | ||
Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The multiple sclerosis collaborative research group (MSCRG). Ann Neurol. 1996;39(3):285–294. | ||
Calabresi PA, Kieseier BC, Arnold DL, et al. Pegylated interferon beta-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. Lancet Neurol. 2014;13(7):657–665. | ||
Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The copolymer 1 multiple sclerosis study group. Neurology. 1995;45(7):1268–1276. | ||
Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–1097. | ||
Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. | ||
Confavreux C, O’Connor P, Comi G, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(3):247–256. | ||
O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293–1303. | ||
Durelli L, Barbero P, Clerico M; INCOMIN Trial Study Group. A randomized study of two interferon-beta treatments in relapsing-remitting multiple sclerosis. Neurology. 2006;67(12):2264. author reply 2264–5. | ||
Schwid SR, Panitch HS. Full results of the evidence of interferon dose-response-European North American comparative efficacy (EVIDENCE) study: a multicenter, randomized, assessor-blinded comparison of low-dose weekly versus high-dose, high-frequency interferon beta-1a for relapsing multiple sclerosis. Clin Ther. 2007;29(9):2031–2048. | ||
Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs glatiramer acetate in relapsing MS disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008;7(10):903–914. | ||
O’Connor P, Filippi M, Arnason B, et al. 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol. 2009;8(10):889–897. | ||
Vermersch P, Czlonkowska A, Grimaldi LM, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler. 2014;20(6):705–716. | ||
Roskell NS, Zimovetz EA, Rycroft CE, Eckert BJ, Tyas DA. Annualized relapse rate of first-line treatments for multiple sclerosis: a meta-analysis, including indirect comparisons versus fingolimod. Curr Med Res Opin. 2012;28(5):767–780. | ||
Tolley K, Hutchinson M, You X, et al. A network meta-analysis of efficacy and evaluation of safety of subcutaneous pegylated interferon beta-1a versus other injectable therapies for the treatment of relapsing-remitting multiple sclerosis. PLoS One. 2015;10(6):e0127960. | ||
Tramacere I, Del Giovane C, Salanti G, D’Amico R, Filippini G. Immunomodulators and immunosuppressants for relapsing-remitting multiple sclerosis: a network meta-analysis. Cochrane Database Syst Rev. 2015;9:CD011381. | ||
Calabresi PA, Radue EW, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(6):545–556. | ||
Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. | ||
Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910. | ||
Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1819–1828. | ||
Coles AJ, Fox E, Vladic A, et al. Alemtuzumab versus interferon beta-1a in early relapsing-remitting multiple sclerosis: post-hoc and subset analyses of clinical efficacy outcomes. Lancet Neurol. 2011;10(4):338–348. | ||
Baker DP, Pepinsky RB, Brickelmaier M, et al. PEGylated interferon beta-1a: meeting an unmet medical need in the treatment of relapsing multiple sclerosis. J Interferon Cytokine Res. 2010;30(10):777–785. | ||
Bhargava P, Newsome SD. An update on the evidence base for peginterferon β1a in the treatment of relapsing–remitting multiple sclerosis. Ther Adv Neurol Disord. 2016;9(6):483–490. | ||
Cocco E, Marrosu MG. Profile of PEGylated interferon beta in the treatment of relapsing-remitting multiple sclerosis. Ther Clin Risk Manag. 2015;11:759–766. | ||
Howley A, Kremenchutzky M. Pegylated interferons: a nurses’ review of a novel multiple sclerosis therapy. J Neurosci Nurs. 2014;46(2):88–96. | ||
Hoy SM. Peginterferon beta-1a: a review of its use in patients with relapsing-remitting multiple sclerosis. CNS Drugs. 2015;29(2):171–179. | ||
Kieseier BC, Calabresi PA. PEGylation of interferon-beta-1a: a promising strategy in multiple sclerosis. CNS Drugs. 2012;26(3):205–214. | ||
Reuss R. PEGylated interferon beta-1a in the treatment of multiple sclerosis – an update. Biologics. 2013;7:131–138. | ||
Nallar SC, Kalvakolanu DV. Interferons, signal transduction pathways, and the central nervous system. J Interferon Cytokine Res. 2014;34(8):559–576. | ||
Severa M, Rizzo F, Giacomini E, Salvetti M, Coccia EM. IFN-beta and multiple sclerosis: cross-talking of immune cells and integration of immunoregulatory networks. Cytokine Growth Factor Rev. 2015;26(2):229–239. | ||
Buzzard KA, Broadley SA, Butzkueven H. What do effective treatments for multiple sclerosis tell us about the molecular mechanisms involved in pathogenesis? Int J Mol Sci. 2012;13(10):12665–12709. | ||
Jacobs L, Johnson KP. A brief history of the use of interferons as treatment of multiple sclerosis. Arch Neurol. 1994;51(12):1245–1252. | ||
Runkel L, Meier W, Pepinsky RB, et al. Structural and functional differences between glycosylated and non-glycosylated forms of human interferon-beta (IFN-beta). Pharm Res. 1998;15(4):641–649. | ||
Kappos L, Kuhle J, Multanen J, et al. Factors influencing long-term outcomes in relapsing-remitting multiple sclerosis: PRISMS-15. J Neurol Neurosurg Psychiatry. 2015;86(11):1202–1207. | ||
Goodin DS, Reder AT, Ebers GC, et al. Survival in MS: a randomized cohort study 21 years after the start of the pivotal IFNbeta-1b trial. Neurology. 2012;78(17):1315–1322. | ||
Shirani A, Zhao Y, Karim ME, et al. Association between use of interferon beta and progression of disability in patients with relapsing-remitting multiple sclerosis. JAMA. 2012;308(3):247–256. | ||
Goodin DS, Reder AT, Cutter G. Treatment with interferon beta for multiple sclerosis. JAMA. 2012;308(16):1627. author reply 1627-8. | ||
Greenberg BM, Balcer L, Calabresi PA, et al. Interferon beta use and disability prevention in relapsing-remitting multiple sclerosis. JAMA Neurol. 2013;70(2):248–251. | ||
Derfuss T, Kappos L. Evaluating the potential benefit of interferon treatment in multiple sclerosis. JAMA. 2012;308(3):290–291. | ||
Hegen H, Auer M, Deisenhammer F. Pharmacokinetic considerations in the treatment of multiple sclerosis with interferon-beta. Expert Opin Drug Metab Toxicol. 2015;11(12):1803–1819. | ||
Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162(1):1–11. | ||
Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118(5):1680–1690. | ||
Durelli L, Conti L, Clerico M, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol. 2009;65(5):499–509. | ||
Zhang L, Yuan S, Cheng G, Guo B. Type I IFN promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS One. 2011;6(12):e28432. | ||
Kozovska ME, Hong J, Zang YC, et al. Interferon beta induces T-helper 2 immune deviation in MS. Neurology. 1999;53(8):1692–1697. | ||
Ramgolam VS, Sha Y, Jin J, Zhang X, Markovic-Plese S. IFN-beta inhibits human Th17 cell differentiation. J Immunol. 2009;183(8):5418–5427. | ||
de Andres C, Aristimuno C, de Las Heras V, et al. Interferon beta-1a therapy enhances CD4+ regulatory T-cell function: an ex vivo and in vitro longitudinal study in relapsing-remitting multiple sclerosis. J Neuroimmunol. 2007;182(1–2):204–211. | ||
Ramgolam VS, Sha Y, Marcus KL, et al. B cells as a therapeutic target for IFN-beta in relapsing-remitting multiple sclerosis. J Immunol. 2011;186(7):4518–4526. | ||
Rizzo F, Giacomini E, Mechelli R, et al. Interferon-beta therapy specifically reduces pathogenic memory B cells in multiple sclerosis patients by inducing a FAS-mediated apoptosis. Immunol Cell Biol. 2016;94(9):886–894. | ||
Saraste M, Irjala H, Airas L. Expansion of CD56Bright natural killer cells in the peripheral blood of multiple sclerosis patients treated with interferon-beta. Neurol Sci. 2007;28(3):121–126. | ||
Shinohara ML, Kim JH, Garcia VA, Cantor H. Engagement of the type I interferon receptor on dendritic cells inhibits T helper 17 cell development: role of intracellular osteopontin. Immunity. 2008;29(1):68–78. | ||
Mirandola SR, Hallal DE, Farias AS, et al. Interferon-beta modifies the peripheral blood cell cytokine secretion in patients with multiple sclerosis. Int Immunopharmacol. 2009;9(7–8):824–830. | ||
Liu Z, Pelfrey CM, Cotleur A, Lee JC, Rudick RA. Immunomodulatory effects of interferon beta-1a in multiple sclerosis. J Neuroimmunol. 2001;112(1–2):153–162. | ||
Krakauer M, Sorensen P, Khademi M, Olsson T, Sellebjerg F. Increased IL-10 mRNA and IL-23 mRNA expression in multiple sclerosis: interferon-beta treatment increases IL-10 mRNA expression while reducing IL-23 mRNA expression. Mult Scler. 2008;14(5):622–630. | ||
Ozenci V, Kouwenhoven M, Huang YM, et al. Multiple sclerosis: levels of interleukin-10-secreting blood mononuclear cells are low in untreated patients but augmented during interferon-beta-1b treatment. Scand J Immunol. 1999;49(5):554–561. | ||
Calabresi PA, Tranquill LR, McFarland HF, Cowan EP. Cytokine gene expression in cells derived from CSF of multiple sclerosis patients. J Neuroimmunol. 1998;89(1–2):198–205. | ||
Chen M, Chen G, Nie H, et al. Regulatory effects of IFN-beta on production of osteopontin and IL-17 by CD4+ T cells in MS. Eur J Immunol. 2009;39(9):2525–2536. | ||
Bernal F, Elias B, Hartung HP, Kieseier BC. Regulation of matrix metalloproteinases and their inhibitors by interferon-beta: a longitudinal study in multiple sclerosis patients. Mult Scler. 2009;15(6):721–727. | ||
Boz C, Ozmenoglu M, Velioglu S, et al. Matrix metalloproteinase-9 (MMP-9) and tissue inhibitor of matrix metalloproteinase (TIMP-1) in patients with relapsing-remitting multiple sclerosis treated with interferon beta. Clin Neurol Neurosurg. 2006;108(2):124–128. | ||
Avolio C, Filippi M, Tortorella C, et al. Serum MMP-9/TIMP-1 and MMP-2/TIMP-2 ratios in multiple sclerosis: relationships with different magnetic resonance imaging measures of disease activity during IFN-beta-1a treatment. Mult Scler. 2005;11(4):441–446. | ||
Calabresi PA, Pelfrey CM, Tranquill LR, Maloni H, McFarland HF. VLA-4 expression on peripheral blood lymphocytes is downregulated after treatment of multiple sclerosis with interferon beta. Neurology. 1997;49(4):1111–1116. | ||
Muraro PA, Leist T, Bielekova B, McFarland HF. VLA-4/CD49d downregulated on primed T lymphocytes during interferon-beta therapy in multiple sclerosis. J Neuroimmunol. 2000;111(1–2):186–194. | ||
Muraro PA, Liberati L, Bonanni L, et al. Decreased integrin gene expression in patients with MS responding to interferon-beta treatment. J Neuroimmunol. 2004;150(1–2):123–131. | ||
Biernacki K, Antel JP, Blain M, Narayanan S, Arnold DL, Prat A. Interferon beta promotes nerve growth factor secretion early in the course of multiple sclerosis. Arch Neurol. 2005;62(4):563–568. | ||
Caggiula M, Batocchi AP, Frisullo G, et al. Neurotrophic factors in relapsing remitting and secondary progressive multiple sclerosis patients during interferon beta therapy. Clin Immunol. 2006;118(1):77–82. | ||
Lindquist S, Hassinger S, Lindquist JA, Sailer M. The balance of pro-inflammatory and trophic factors in multiple sclerosis patients: effects of acute relapse and immunomodulatory treatment. Mult Scler. 2011;17(7):851–866. | ||
Li DK, Paty DW. Magnetic resonance imaging results of the PRISMS trial: a randomized, double-blind, placebo-controlled study of interferon-beta1a in relapsing-remitting multiple sclerosis. prevention of relapses and disability by interferon-beta1a subcutaneously in multiple sclerosis. Ann Neurol. 1999;46(2):197–206. | ||
PRISMS Study Group. PRISMS-4: long-term efficacy of interferon-beta-1a in relapsing MS. Neurology. 2001;56(12):1628–1636. | ||
Uitdehaag B, Constantinescu C, Cornelisse P, et al. Impact of exposure to interferon beta-1a on outcomes in patients with relapsing-remitting multiple sclerosis: exploratory analyses from the PRISMS long-term follow-up study. Ther Adv Neurol Disord. 2011;4(1):3–14. | ||
Gold R, Rieckmann P, Chang P, Abdalla J; PRISMS Study Group. The long-term safety and tolerability of high-dose interferon beta-1a in relapsing-remitting multiple sclerosis: 4-year data from the PRISMS study. Eur J Neurol. 2005;12(8):649–656. | ||
Siegert RJ, Abernethy DA. Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry. 2005;76(4):469–475. | ||
Patten SB, Francis G, Metz LM, Lopez-Bresnahan M, Chang P, Curtin F. The relationship between depression and interferon beta-1a therapy in patients with multiple sclerosis. Mult Scler. 2005;11(2):175–181. | ||
Schippling S, O’Connor P, Knappertz V, et al. Incidence and course of depression in multiple sclerosis in the multinational BEYOND trial. J Neurol. 2016;263(7):1418–1426. | ||
Bertolotto A, Malucchi S, Sala A, et al. Differential effects of three interferon betas on neutralising antibodies in patients with multiple sclerosis: a follow up study in an independent laboratory. J Neurol Neurosurg Psychiatry. 2002;73(2):148–153. | ||
Boz C, Oger J, Gibbs E, Grossberg SE; Neurologists of the UBC MS Clinic. Reduced effectiveness of long-term interferon-beta treatment on relapses in neutralizing antibody-positive multiple sclerosis patients: a Canadian multiple sclerosis clinic-based study. Mult Scler. 2007;13(9):1127–1137. | ||
Hegen H, Schleiser M, Gneiss C, et al. Persistency of neutralizing antibodies depends on titer and interferon-beta preparation. Mult Scler. 2012;18(5):610–615. | ||
Pachner AR, Cadavid D, Wolansky L, Skurnick J. Effect of anti-IFN{beta} antibodies on MRI lesions of MS patients in the BECOME study. Neurology. 2009;73(18):1485–1492. | ||
Pachner AR, Warth JD, Pace A, Goelz S; INSIGHT investigators. Effect of neutralizing antibodies on biomarker responses to interferon beta: the INSIGHT study. Neurology. 2009;73(18):1493–1500. | ||
Paolicelli D, D’Onghia M, Pellegrini F, et al. The impact of neutralizing antibodies on the risk of disease worsening in interferon beta-treated relapsing multiple sclerosis: a 5 year post-marketing study. J Neurol. 2013;260(6):1562–1568. | ||
Francis GS, Rice GP, Alsop JC, PRISMS Study Group. Interferon beta-1a in MS: results following development of neutralizing antibodies in PRISMS. Neurology. 2005;65(1):48–55. | ||
Petkau AJ, White RA, Ebers GC, et al. Longitudinal analyses of the effects of neutralizing antibodies on interferon beta-1b in relapsing-remitting multiple sclerosis. Mult Scler. 2004;10(2):126–138. | ||
Reynolds MW, Stephen R, Seaman C, Rajagopalan K. Persistence and adherence to disease modifying drugs among patients with multiple sclerosis. Curr Med Res Opin. 2010;26(3):663–674. | ||
Steinberg SC, Faris RJ, Chang CF, Chan A, Tankersley MA. Impact of adherence to interferons in the treatment of multiple sclerosis: a non-experimental, retrospective, cohort study. Clin Drug Investig. 2010;30(2):89–100. | ||
Tan H, Cai Q, Agarwal S, Stephenson JJ, Kamat S. Impact of adherence to disease-modifying therapies on clinical and economic outcomes among patients with multiple sclerosis. Adv Ther. 2011;28(1):51–61. | ||
Wong J, Gomes T, Mamdani M, Manno M, O’Connor PW. Adherence to multiple sclerosis disease-modifying therapies in Ontario is low. Can J Neurol Sci. 2011;38(3):429–433. | ||
Evans C, Marrie RA, Zhu F, et al. Adherence and persistence to drug therapies for multiple sclerosis: a population-based study. Mult Scler Relat Disord. 2016;8:78–85. | ||
Lafata JE, Cerghet M, Dobie E, et al. Measuring adherence and persistence to disease-modifying agents among patients with relapsing remitting multiple sclerosis. J Am Pharm Assoc (2003). 2008;48(6):752–757. | ||
Hansen K, Schussel K, Kieble M, et al. Adherence to disease modifying drugs among patients with multiple sclerosis in Germany: a retrospective cohort study. PLoS One. 2015;10(7):e0133279. | ||
Giovannoni G, Southam E, Waubant E. Systematic review of disease-modifying therapies to assess unmet needs in multiple sclerosis: tolerability and adherence. Mult Scler. 2012;18(7):932–946. | ||
Rinon A, Buch M, Holley D, Verdun E. The MS choices survey: findings of a study assessing physician and patient perspectives on living with and managing multiple sclerosis. Patient Prefer Adherence. 2011;5:629–643. | ||
Treadaway K, Cutter G, Salter A, et al. Factors that influence adherence with disease-modifying therapy in MS. J Neurol. 2009;256(4):568–576. | ||
O’Rourke KE, Hutchinson M. Stopping beta-interferon therapy in multiple sclerosis: an analysis of stopping patterns. Mult Scler. 2005;11(1):46–50. | ||
Tremlett HL, Oger J. Interrupted therapy: stopping and switching of the beta-interferons prescribed for MS. Neurology. 2003;61(4):551–554. | ||
Girouard N, Theoret G. Management strategies for improving the tolerability of interferons in the treatment of multiple sclerosis. Can J Neurosci Nurs. 2008;30(4):18–25. | ||
Bayas A. Improving adherence to injectable disease-modifying drugs in multiple sclerosis. Expert Opin Drug Deliv. 2013;10(3):285–287. | ||
Fishburn CS. The pharmacology of PEGylation: balancing PD with PK to generate novel therapeutics. J Pharm Sci. 2008;97(10):4167–4183. | ||
Turecek PL, Bossard MJ, Schoetens F, Ivens IA. PEGylation of biopharmaceuticals: a review of chemistry and nonclinical safety information of approved drugs. J Pharm Sci. 2016;105(2):460–475. | ||
Aghemo A, Rumi MG, Colombo M. Pegylated interferons alpha2a and alpha2b in the treatment of chronic hepatitis C. Nat Rev Gastroenterol Hepatol. 2010;7(9):485–494. | ||
Hu X, Miller L, Richman S, et al. A novel PEGylated interferon beta-1a for multiple sclerosis: safety, pharmacology, and biology. J Clin Pharmacol. 2012;52(6):798–808. | ||
Kieseier BC, Arnold DL, Balcer LJ, et al. Peginterferon beta-1a in multiple sclerosis: 2-year results from ADVANCE. Mult Scler. 2015;21(8):1025–1035. | ||
Arnold DL, Calabresi PA, Kieseier BC, et al. Effect of peginterferon beta-1a on MRI measures and achieving no evidence of disease activity: results from a randomized controlled trial in relapsing-remitting multiple sclerosis. BMC Neurol. 2014;14:240. | ||
Newsome SD, Kieseier BC, Arnold DL, et al. Subgroup and sensitivity analyses of annualized relapse rate over 2 years in the ADVANCE trial of peginterferon beta-1a in patients with relapsing-remitting multiple sclerosis. J Neurol. 2016;263(9):1778–1787. | ||
Kremenchutzky M, Liu S, Cui Y, Hung S, Seddighzadeh A, Evilevitch V. Long-term safety and tolerability of peginterferon beta-1a: interim analysis from ATTAIN, a phase 3 extension study. Neurology. 2015;84(14):S4.002. | ||
White JT, Newsome SD, Kieseier BC, et al. Incidence, characterization, and clinical impact analysis of peginterferon beta1a immunogenicity in patients with multiple sclerosis in the ADVANCE trial. Ther Adv Neurol Disord. 2016;9(4):239–249. | ||
Halper J, Centonze D, Newsome SD, et al. Management strategies for flu-like symptoms and injection-site reactions associated with peginterferon beta-1a: obtaining recommendations using the delphi technique. Int J MS Care. 2016;18(4):211–218. | ||
Hu X, Shang S, Nestorov I, et al. COMPARE: pharmacokinetic profiles of subcutaneous peginterferon beta-1a and subcutaneous interferon beta-1a over 2 weeks in healthy subjects. Br J Clin Pharmacol. 2016;82(2):380–388. | ||
Hernandez L, Guo S, Kinter E, Fay M. Cost-effectiveness analysis of peginterferon beta-1a compared with interferon beta-1a and glatiramer acetate in the treatment of relapsing-remitting multiple sclerosis in the united states. J Med Econ. 2016;19(7):684–695. | ||
Mori F, Kusayanagi H, Buttari F, et al. Early treatment with high-dose interferon beta-1a reverses cognitive and cortical plasticity deficits in multiple sclerosis. Funct Neurol. 2012;27(3):163–168. | ||
Patti F, Morra VB, Amato MP, et al. Subcutaneous interferon beta-1a may protect against cognitive impairment in patients with relapsing-remitting multiple sclerosis: 5-year follow-up of the COGIMUS study. PLoS One. 2013;8(8):e74111. | ||
Cinar BP, Kosehasanogullari G, Yigit P, Ozakbas S. Cognitive dysfunction in patients with multiple sclerosis treated with first-line disease-modifying therapy: a multi-center, controlled study using the BICAMS battery. Neurol Sci. Epub 2016 Nov 24. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.