Back to Journals » International Journal of Nanomedicine » Volume 17
A Tissue-Tended Mycophenolate-Modified Nanoparticle Alleviates Systemic Lupus Erythematosus in MRL/Lpr Mouse Model Mainly by Promoting Local M2-Like Macrophagocytes Polarization
Authors Jiang B, Zhang Y, Li Y ![]() , Chen Y, Sha S, Zhao L, Li D, Wen J, Lan J, Lou Y, Su H, Zhang C, Zhu J, Tao J
, Chen Y, Sha S, Zhao L, Li D, Wen J, Lan J, Lou Y, Su H, Zhang C, Zhu J, Tao J ![]()
Received 16 March 2022
Accepted for publication 20 July 2022
Published 28 July 2022 Volume 2022:17 Pages 3251—3267
DOI https://doi.org/10.2147/IJN.S361400
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Biling Jiang,1,* Yamin Zhang,1,* Yuce Li,2 Yu Chen,2 Shanshan Sha,1 Liang Zhao,1 Danqi Li,1 Jingjing Wen,1 Jiajia Lan,1 Yuchen Lou,1 Hua Su,3 Chun Zhang,3 Jintao Zhu,2 Juan Tao1
1Hubei Engineering Research Center of Skin Disease Theranostics and Health, Department of Dermatology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology (HUST), Wuhan, People’s Republic of China; 2Hubei Engineering Research Center for Biomaterials and Medical Protective Materials, School of Chemistry and Chemical Engineering, HUST, Wuhan, People’s Republic of China; 3Department of Nephrology, Union Hospital, Tongji Medical College, HUST, Wuhan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Juan Tao; Jintao Zhu, Email [email protected]; [email protected]
Background: Mycophenolate mofetil (MMF), for which the bioactive metabolite is mycophenolic acid (MPA), is a frequently used immunosuppressant for systemic lupus erythematosus (SLE). However, its short half-life and poor biodistribution into cells and tissues hinder its clinical efficacy. Our dextran mycophenolate-based nanoparticles (MPA@Dex-MPA NPs) have greatly improved the pharmacokinetics of MMF/MPA. We here tested the therapeutic efficacy of MPA@Dex-MPA NPs against SLE and investigated the underlying mechanism.
Methods: The tissue and immune cell biodistributions of MPA@Dex-MPA NPs were traced using live fluorescence imaging system and flow cytometry, respectively. Serological proinflammatory mediators and kidney damage were detected to assess the efficacy of MPA@Dex-MPA NPs treatments of MRL/lpr lupus-prone mice. Immune cell changes in the kidney and spleen were further analyzed post-treatment via flow cytometry. Bone marrow-derived macrophages were used to investigate the potential mechanism.
Results: MPA@Dex-MPA NPs exhibited superior therapeutic efficacy and safety in the MRL/lpr mice using significantly lower administration dosage (one-fifth) and frequency (once/3 days) compared to MMF/MPA used in ordinary practice. The overall prognosis of the mice was improved as they showed lower levels of serological proinflammatory mediators. Moreover, kidney injury was alleviated with reduced pathological signs and decreased urine protein-creatinine ratio. Further investigations of the underlying mechanism revealed a preferential penetration and persistent retention of MPA@Dex-MPA NPs in the spleen and kidney, where they were mostly phagocytosed by macrophages. The macrophages were found to be polarized towards a CD206+ M2-like phenotype, with a downregulation of surface CD80 and CD40, and reduced TNF-α production in the spleen and kidney and in vitro. The expansion of T cells was also significantly inhibited in these two organs.
Conclusion: Our research improved the efficacy of MPA for MRL/lpr mice through synthesizing MPA@Dex-MPA NPs to enhance its tissue biodistribution and explored the possible mechanism, providing a promising strategy for SLE therapy.
Keywords: mycophenolate, nanoparticles, macrophage polarization, tissue damage, systemic lupus erythematosus
Graphical Abstract:

Introduction
Systemic lupus erythematosus (SLE) is a refractory autoimmune disease.1–3 In recent years, many novel biologics and small-molecule inhibitors have been tested in clinical trials to treat SLE but with little success.4 Belimumab has been the only biological agent approved so far by the Food and Drug Administration for SLE as an adjuvant therapy.5 However, its efficacy is limited and no more than 50% of patients seem to benefit from it.5–7 Hence, glucocorticoids, antimalarial agents and immunosuppressants are still the mainstay as SLE therapies.
Local inflammation, triggered and exacerbated mainly by innate immune cells, particularly macrophages, is the leading cause of tissue damage in SLE.1,8 The resolution of tissue inflammation is essential and is therefore one of the primary goals of SLE management. Lupus nephritis (LN) is frequently developed in SLE patients and mycophenolate mofetil (MMF) is the primary immunosuppressant used to treat it.9–11 Mycophenolic acid (MPA), the biologically active metabolite of MMF, was released through hydrolysis in vivo.9 Although MMF has become the first-line treatment for LN and an alternate choice for other tissue damage in SLE, there are still some problems in its clinical application. It is easily metabolized and eliminated from the body.12,13 Moreover, its biodistribution into tissues and cells is very poor. Hence, for better efficacy in resolving tissue inflammation, relatively large doses of MMF at a high frequency are usually utilized in clinical practice. However, the risk of unwanted side effects thereby increases, greatly impairing the long-term prognosis. Modification of MMF/MPA characteristics in the following way is therefore urgently needed: (1) long action time; (2) a direct inhibition of inflammation in targeted tissues; and (3) a satisfactory safety profile.
Nanotechnology has been applied to protect and precisely deliver MMF/MPA to target tissues and cells to treat inflammatory diseases and has shown promise.14–20 For example, a triglyceride mimetic prodrug of MPA has been developed for targeted delivery to mesenteric lymph nodes and successfully suppressed the immune reaction to an oral ovalbumin antigen challenge in mice.17 In addition, MMF-loaded PEG-PLGA nanoparticles (NPs) have been perfused into a donor mouse heart prior to transplantation and successfully inhibited post-transplantation inflammation.18 SLE is one of the main clinical indications for MMF/MPA therapy, but little research has been conducted on MMF/MPA nanomedicine for SLE. A type of MPA-encapsulated nanogel has been fabricated for the treatment of SLE.21 The core of this preparation contains cyclodextrins, which significantly enhance the aqueous solubilization and encapsulation of hydrophobic MPA. However, the MPA drug-loading efficiency of this nanogel was still found to be low (<1%, w/w), with a short release duration in vitro (within several hours) and elimination from most organs within 24 h.21 As a result, the interaction between MPA and tissue cells was not sufficient with this delivery method, and the resulting efficacy against SLE, tested in NZB/W F1 mice, was unsatisfactory. Hence, it could be inferred that optimizing the modification of MMF/MPA to elevate drug loading and long tissue retention may be a promising treatment strategy for SLE.
Recently, we successfully fabricated dextran (Dex) mycophenolate-based NPs (MPA@Dex-MPA NPs).22 With a combined chemical conjugation (~ 50%) and physical encapsulation (~ 20%) of the drug, MPA@Dex-MPA NPs reached a high MPA loading content of >70% in total.22 The loading efficiency of encapsulated MPA was as high as 85.95%. Besides, drug release profile was also investigated in vitro through a dialysis method at 37°C. The initial release concentration of MPA was high and more than 80% of MPA was released gradually within eight days. As indicated by the results, encapsulated MPA could thereby be released quickly and act effectively in vivo and in vitro, while the conjugation guaranteed a long-term effect through a sustained release. The MPA@Dex-MPA NPs displayed spherical shape and the mean diameter of these NPs was ~ 100 nm, enabling them to be easily taken up by phagocytes such as macrophages and dendritic cells (DCs).22,23 In this current study, we tested the clinical therapeutic efficacy of MPA@Dex-MPA NPs using a lupus-prone mouse model and also investigated the mechanisms involved. We found that MPA@Dex-MPA NPs tended to accumulate and stay in the spleen and kidney in the MRL/lpr lupus-prone mice. Local inflammation was successfully and mainly suppressed by these particles through the induction of M2-like macrophages and inhibition of T cell expansion. MPA@Dex-MPA NPs treatment thus relieved tissue damage and improved the overall prognosis of the MRL/lpr mice, with a lower dosage and frequency of treatment required, and a satisfactory long-term safety profile achieved. This treatment strategy thus has promise as a future SLE therapy.
Materials and Methods
Preparation of MPA@Dex-MPA NPs
MPA (Aladdin, Shanghai, China) was connected to Dex (Sigma-Aldrich, St Louis, MO) through an esterification reaction between carboxyl group in MPA and hydroxyl groups in Dex to form MPA-Dex, which was catalyzed by 1-ethyl-3-(3-(dimethylamino) propyl)-carbodiimide hydrochloride (EDC)/4-dimethylaminopyridine (DMAP). MPA@Dex-MPA NPs were then synthesized mainly through an ultrasonic emulsification and solution evaporation method (Figure 1A). Further details of this method can be found in our previous study.22
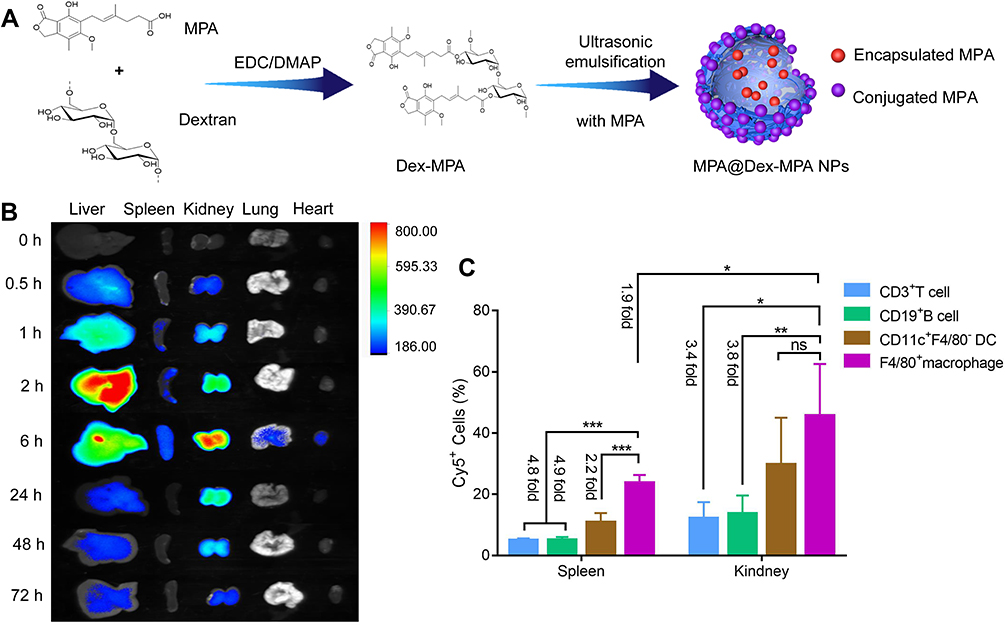 |
Figure 1 Biodistribution of MPA@Dex-MPA NPs among the tissues and immune cells of MRL/lpr mice. (A) Preparation procedure for MPA@Dex-MPA NPs. (B) Fluorescence microscopy images of the liver, spleen, kidney, lung and heart of MRL/lpr mice at 0, 0.5, 1, 2, 6, 24, 48 and 72 h post intraperitoneal injection of Cy7-labeled MPA@Dex-MPA NPs. (C) Cellular internalization of Cy5-labeled MPA@Dex-MPA NPs in the spleen and kidney. The proportions of T cells, B cells, DCs and macrophages that internalized the Cy5-labeled MPA@Dex-MPA NPs were analyzed at 6 h post intraperitoneal injection through flow cytometry analysis (n = 5/group). Error bars represent the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviation: ns, not significant. |
Animals
Female MRL/lpr lupus-prone mice, aged 8 weeks and weighing 25–30 g, were purchased from Cavens Laboratory Animal Co., Ltd. (Changzhou, China). Female C57BL/6 mice, aged 6–8 weeks, were purchased from the Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). These mice were all raised in the Animal Center of the Huazhong University of Science and Technology, under specific pathogen-free conditions. All animal experiments met the requirements of the Guide for the Care and Use of Laboratory Animals of Huazhong University of Science and Technology and approved by the Institutional Animal Care and Use Committee, Tongji Medical College, Huazhong University of Science and Technology.
In vivo Treatment Study
After being raised to 12 weeks of age, MRL/lpr mice were randomly divided into five groups (n = 8/group) based on the MPA dosage as follows: PBS control, dextran, 10 mg/kg free MPA (fMPA), 50 mg/kg fMPA and 10 mg/kg MPA@Dex-MPA. Drugs were administered by intraperitoneal injection in a 250 μL volume every 3 days for 12 weeks. During this process, the survival rates were recorded. Peripheral blood was collected from the orbital angular vein after the mice were anesthetized to obtain serum every four weeks. The titer of the autoantibodies against dsDNA in the serum was determined by ELISA (Cusabio, Wuhan, China). The serum levels of several cytokines (including IL-6, TNF-α, IL-17A and IFN-γ) were measured using cytometric bead array cytokine kit (BD, Franklin Lakes, New Jersey). Urine was obtained by bladder massage. Urine protein and creatinine were detected by Coomassie brilliant blue G-250 dyeing method and creatinine assay kits (Sigma-Aldrich, St. Louis, Missouri), respectively. After being sacrificed at the end of the treatment period, the spleen and one kidney from the mice were collected for flow cytometry analysis of immune cells. The other kidney and internal organs (including the heart, liver and lung) were fixed (using 4% paraformaldehyde) and embedded in paraffin.
Flow Cytometry Analysis of Splenocytes and Renal Cells
The mouse spleens were firstly weighed, photographed, and then processed into single cell suspension through grinding gently. Trimmed of extra fat, kidneys were carefully minced and digested in 1640-RPMI (Gibco, Grand Island, NY) containing 10% fetal calf serum (FBS) (Gibco, Grand Island, NY) and 1 mg/mL collagenase I (Gibco, Grand Island, NY) for about 1 h at 37°C. Red blood cells (RBCs) were eliminated by lysis solution (Sigma-Aldrich, St. Louis, Missouri). Dead cells were identified using a fixable viability dyeing (Biolegend, San Diego, CA). An anti-CD16/32 blocking antibody (Biolegend, San Diego, CA) was used to reduce non-specific binding before staining with other antibodies including anti-mouse CD45 (I3/2.3), CD3e (145–2C11), CD19 (6D5), CD4 (GK1.5), CD8a (53–6.7), CD138 (281–2), F4/80 (BM8), CD11b (M1/70), CD11c (N418), MHC-II (25-9-17), CD86 (GL-1), CD40 (3/23), CD80 (16–10A1) and their isotype controls (all from Biolegend, San Diego, CA, USA). Precision count beads (Biolegend, San Diego, CA) were utilized to obtain actual cell numbers, in accordance with the manufacturer’s instructions. Stained cell suspensions were all analyzed by flow cytometry (BD LSR II, Franklin Lakes, New Jersey).
Immunofluorescence
The deposition of IgG and complement 3 (C3) was detected by immunofluorescence staining (IgG, Abcam, Cambridge, UK; C3, Santa Cruz Biotechnology, Santa Cruz, CA). Renal T cells and macrophages were also detected through immunofluorescence staining of kidney sections (CD3, Abcam, Cambridge, UK; F4/80, Cell Signaling Technology, MA). The fluorescence intensity of IgG and C3 was graded (0–3).24,25
Histological Analysis of the Mouse Kidney
Periodic acid-Schiff (PAS) staining of kidney sections was used to assess pathological damage to the kidney. Pathological change of glomerulus was scored (0–3) as described in previous studies by experienced nephrologists.24–26
In vivo Tissue Biodistribution
Cy7-labeled MPA@Dex-MPA NPs (10 mg/kg) were intraperitoneally injected into 12-week-old MRL/lpr mice. Organs (including liver, spleen, kidney, lung and heart) were harvested after 0, 0.5, 1, 2, 6, 24, 48 or 72 h post injection. Cy7 fluorescence intensity was detected using an imager (In vivo MS FX PRO; Bruker, Massachusetts). Cy5-labeled MPA@Dex-MPA NPs (10 mg/kg) were used to investigate the cellular biodistribution of the NPs. Immune cells from the mouse spleens and kidneys were collected and analyzed by flow cytometry at 6 h after intraperitoneal injection.
Generation and Culture of Bone Marrow-Derived Macrophages (BMDMs)
1640-RPMI (Gibco, Grand Island, NY) containing 10% FBS and 1% penicillin-streptomycin solution (Gibco, Grand Island, NY) was prepared as complete medium. The bone marrow cells were extracted from the lower limbs of female C57BL/6 mice aged 6–8 weeks. After the removal of RBCs, bone marrow cells were seeded and cultured in 100 mm Petri dishes in complete medium added with 50 ng/mL macrophage colony-stimulating factor (Peprotech, Rocky Hill, New Jersey) at 37°C for a continuous 7 days to obtain mature BMDMs.
NP Uptake Assay for BMDMs in vitro
BMDMs were seeded into 24-well plates at a density of 2.5×105 cells/well. The cells were then incubated with Nile Red-MPA@Dex-MPA NPs (containing 1, 10 or 50 μg/mL MPA) for 0.5, 1 or 2 h in the complete medium mentioned above. The cells were then removed from the plate surfaces using a cell dissociation reagent (Gibco, Grand Island, NY) and collected. Flow cytometry was then used to detect the Nile Red fluorescence. For confocal laser scanning microscopy imaging (CLSM, Nikon, Tokyo, Japan), cell nuclei were stained with DAPI (Sigma-Aldrich, St. Louis, Missouri) for 5 min after being treated with the NPs (10 μg/mL) for 2 h in the confocal dishes.
Effects of MPA@Dex-MPA NPs on BMDMs in vitro
After being seeded into 96-well (5×104 cells/well) or 48-well (2×105 cells/well) plates, BMDMs were treated with either PBS, dextran, fMPA or MPA@Dex-MPA NPs, respectively. The MPA dose was equivalent in the fMPA and MPA@Dex-MPA NPs groups (~ 10 μg/mL), as was the dose of dextran in the dextran and MPA@Dex-MPA NPs group (~ 4.29 μg/mL). The viability and apoptosis status of the BMDMs were assessed by CCK-8 and apoptosis detection kits (Dojindo, Shanghai, China), respectively, after an incubation period of 24, 48 or 72 h. To study the immune-related effects of MPA@Dex-MPA NPs on BMDMs, BMDMs were pretreated with PBS, dextran, fMPA or MPA@Dex-MPA NPs for 24 h and then further stimulated by 10 ng/mL lipopolysaccharide (Sigma-Aldrich, St. Louis, Missouri) combined with 10 ng/mL IFN-γ (Biolegend, San Diego, CA) for 12 h. The surface molecules on the BMDMs were analyzed by flow cytometry and the cytokines secreted into the culture supernatant were assayed using cytometric bead array cytokine kit (BD, Franklin Lakes, New).
Long-Term Toxicity Study
C57BL/6 normal female mice, aged 8 weeks, were randomly grouped (n = 5/group) and were administered with indicated drugs as in animal treatment experiment for 4 weeks. The body weights of the mice were recorded each week. At the end of the toxicity study, blood samples were collected in tubes with or without EDTA for routine examination (Sysmex 1800i, Kobe, Japan) and serum separation, respectively. The serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN) and serum creatinine were measured (Hitachi 7600, Tokyo, Japan). After sacrifice of the animals, the visceral organs (heart, liver, spleen, lung and kidney) were harvested, and fixed and embedded as mentioned above for hematoxylin-eosin (H&E) staining.
Statistical Analysis
Statistical analysis was conducted using GraphPad Prism 7 software. Quantitative data were expressed as a mean value ± standard error of the mean (SEM). An unpaired Student’s t-test was performed to analyze the statistical significance between two groups and one-way ANOVA was used to do so among multiple groups. A log-rank (Mantel-Cox) test was used to compare survival rates. P < 0.05 was considered to indicate statistical significance. *P < 0.05, **P < 0.01 and ***P < 0.001.
Results and Discussion
Biodistribution of MPA@Dex-MPA NPs in Tissues and Immune Cells of MRL/Lpr Lupus-Prone Mice
MPA@Dex-MPA NPs were synthesized using a template method as shown in Figure 1A, following the procedure described in our previous report.22 First, Dex-MPA conjugates were prepared through an esterification reaction using fMPA and dextran, catalyzed by EDC/DMAP. Ultrasonic emulsification-solution evaporation of Dex-MPA solution containing fMPA was then conducted to yield MPA@Dex-MPA NPs (Figure 1A).22 In our previous report, we carefully characterized MPA@Dex-MPA NPs and confirmed their improved pharmacokinetics.22 Their plasma protein binding rate was found to be low and drug release could be sustained for more than 7 days in vitro. Moreover, the half-life of MPA was prolonged from 12 h to more than 24 h in the peripheral circulation.22 Yet, the distribution and retention of the MPA@Dex-MPA NPs in tissues and immune cells in the lupus model mice was still unclear. To this end, we here employed fluorescence microscopy imaging to trace Cy7-labeled MPA@Dex-MPA NPs in the major organs of MRL/lpr mice. Our results showed that MPA@Dex-MPA NPs tended to accumulate mainly in the liver, spleen and kidney shortly after intraperitoneal injection (0.5–1 h), where they gradually reached a peak level at ~ 2, 6 and 6 h, respectively. Moreover, the retention of MPA@Dex-MPA NPs in the kidney was as long as 72 h (Figure 1B).
Since the spleen and kidney are among the most important organs related to, and affected by, systemic immunity, respectively, we further detected the biodistribution of Cy5-labeled MPA@Dex-MPA NPs among immune cells in these tissues at 6 h post intraperitoneal injection using flow cytometry. Immune cell populations were segregated according to the gating scheme presented in Figure S1. The results showed that the F4/80+ macrophages exhibited the highest NPs uptake rate in the kidney of the MRL/lpr mouse (45.33 ± 9.95%), followed by CD11c+ DCs (29.43 ± 7.79%). The uptake rate of macrophages was over 3-fold higher than that of B or T lymphocytes (13.35 ± 2.79% and 11.89 ± 2.77%, respectively). Similar results were obtained for the mouse spleen (Figure 1C). However, the NP uptake rates among immune cell populations in the kidney were all higher than the corresponding cells in the spleen, among which the uptake rate by kidney macrophages was almost 2-fold higher than those in the spleen (Figure 1C).
MPA@Dex-MPA NPs Improve the Overall Prognosis and Relieve Kidney Injury in MRL/Lpr Mice at a Lower Dosage and Injection Frequency
Clinically, MMF is currently administered as a twice-daily dosage, 1–3 g/day in SLE patients. In prior animal studies, the treatment dosage of MMF ranged from 30 to 100 mg/kg/day.21,27,28 However, MPA@Dex-MPA NPs have been shown in our prior report to exhibit a longer half-life in the peripheral circulation (more than 24 h)22 and also more persistent retention in the kidney (above 72 h). Hence, we adopted a treatment plan for our model mice involving a 10 mg/kg dose of MPA@Dex-MPA NPs that was intraperitoneally injected every 3 days from the age of 12 to 24 weeks (Figure 2A). The subsequent survival rate of the MRL/lpr mice in the 10 mg/kg MPA@Dex-MPA treatment group (75%) was higher than that of the PBS control, dextran or 10 mg/kg fMPA groups (50%), and was comparable to that of the 50 mg/kg fMPA group (75%) (Figure 2B). However, the difference of survival curves was not yet significant (P = 0.58).
 |
Figure 2 MPA@Dex-MPA NPs treatment improve the survival and reduce the levels of serological proinflammatory mediators in MRL/lpr mice. (A) Schema for the animal experiment (n = 8/group). (B) Survival curves for the MRL/lpr mice during the experiment (n = 8/group). (C) Serum levels of anti-dsDNA IgG in the MRL/lpr mice from age of 12 to 24 weeks (n = 4–8/group). (D and E) Serum concentrations of anti-dsDNA IgG (D) and cytokines (IL-6, IL-17A, TNF-α, IFN-γ) (E) at the age of 24 weeks (n = 4–6/group). Error bars represent the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviation: ns, not significant. |
The serum level of anti-dsDNA IgG, an important indicator of diagnosis and disease activity of SLE, was monitored during the experiment. The 10 mg/kg MPA@Dex-MPA NPs and 50 mg/kg fMPA treatments both effectively suppressed the increase in anti-dsDNA IgG in the MRL/lpr mice, which was especially evident after the age of 20 weeks, compared with the other groups (Figure 2C). At the age of 24 weeks, the anti-dsDNA IgG serum level among the mice in the 10 mg/kg MPA@Dex-MPA group (22.45 ± 1.62 ng/mL) was found to be significantly lower than that of PBS control group (126.2 ± 37.42 ng/mL), and even lower than that in the 50 mg/kg fMPA treatment group (35.16 ± 3.92 ng/mL) (Figure 2D).
The serum levels of some proinflammatory cytokines (eg, IL-6 and IL-17A) are closely related to the severity of SLE and can provide important clinical information regarding treatment choices.29 We thus further detected the concentrations of several cytokines in the serum from MRL/lpr mice. The MPA@Dex-MPA NPs treatments significantly reduced the serum level of proinflammatory cytokines, including IL-6, IL-17A, TNF-α and IFN-γ, compared with the PBS control group (2.46 ± 0.57 pg/mL vs 23.04 ± 3.89 pg/mL, 2.92 ± 0.69 pg/mL vs 6.80 ± 0.96 pg/mL, 14.96 ± 0.91 pg/mL vs 30.16 ± 3.20 pg/mL and 38.27 ± 1.85 pg/mL vs 72.53 ± 17.73 pg/mL, respectively; Figure 2E). The 10 mg/kg fMPA group and dextran group showed little reduction in these proinflammatory cytokines. Decreases in IL-6 and IL-17A were also observed in the 50 mg/kg fMPA group (6.67 ± 1.93 pg/mL and 3.35 ± 0.36 pg/mL, respectively) but TNF-α and IFN-γ were not at a significantly lower level in this group (18.35 ± 5.23 pg/mL, 51.78 ± 3.47 pg/mL) compared with the PBS control group. Furthermore, the serum level of the anti-inflammatory cytokine IL-10 showed an increased tendency in the treatment groups containing MPA, although without significance (Figure S2). The above results indicated the potential of MPA@Dex-MPA NPs to alleviate the production of anti-dsDNA autoantibodies and the imbalance of serum proinflammatory/anti-inflammatory cytokines.
Cyclosporine or MMF, in conjunction with glucocorticoids, is now the first-line therapy for LN.10 However, LN is still one of the leading causes of mortality among SLE patients.30–32 To assess kidney function after these treatments in the mouse, we measured the urine protein and creatinine levels to calculate the urine protein-creatinine ratio (UPCR), which reflects protein loss through the urine. Both the 10 mg/kg MPA@Dex-MPA and the 50 mg/kg fMPA treatment could successfully relieve the increase of the UPCR during the animal experiment (Figure 3A). And at the end of the animal experiment, the UPCR of the mice in the 10 mg/kg MPA@Dex-MPA group (715.90 ± 125.00 μg/mg) was significantly lower than that of the 10 mg/kg fMPA and PBS control groups (1222.00 ± 34.07 μg/mg and 1244.00 ± 86.06 μg/mg, respectively). The UPCR of the 10 mg/kg MPA@Dex-MPA group was also lower than that of the 50 mg/kg fMPA group (993.30 ± 75.97 μg/mg), although this difference was not statistically significant (P = 0.056; Figure 3B). In addition, the BUN of the mice in the 10 mg/kg MPA@Dex-MPA (23.97 ± 1.37 mg/dL) and 50 mg/kg fMPA (24.10 ± 1.25 mg/dL) groups was decreased compared with the PBS controls (32.44 ± 2.33 mg/dL) (Figure 3B). Furthermore, our PAS staining of the mouse kidney tissues revealed abnormalities such as glomerular hypercellularity, mesangial expansion and mesangial deposits. However, as seen in Figure 3C and D, these pathological manifestations were much milder in both the 10 mg/kg MPA@Dex-MPA and 50 mg/kg fMPA groups. Immune complexes and complements tend to deposit in the kidney and have the strong potential to promote tissue inflammation, and are therefore key factors that trigger and aggravate the pathological damage seen in LN.33 Hence, we also performed immunofluorescence staining of IgG and C3 deposition in the glomeruli. The 10 mg/kg MPA@Dex-MPA NPs could significantly reduce IgG and C3 deposition (Figure 3E–G) compared with that of the 10 mg/kg fMPA and PBS control groups, which may be partially attributable to a lower serum level of anti-dsDNA IgG.
 |
Figure 3 MPA@Dex-MPA NPs improve renal function and alleviate the pathological signs in the kidneys of the MRL/lpr mice. (A) The UPCR was obtained by measuring the urine protein and creatinine concentrations during the animal experiment (n = 4–8/group). (B) The UPCR and the BUN level was determined at the end of animal experiment (n = 4–6/group). (C) The representative micrographs of glomeruli stained by PAS. (D) The glomerulonephritis score was assessed. (E) The representative immunofluorescence micrographs of the deposition of IgG and complement 3 in the glomeruli (green, IgG; red, C3; blue, nucleus; n = 4–6/group). The fluorescence intensity of (F) IgG and (G) C3 was assessed. Scale bar, 20 μm. Error bars represent the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviation: ns, not significant. |
The above results indicated that MPA@Dex-MPA NPs therapy can effectively reduce kidney pathology and preserve its function at a significantly lower dosage and treatment frequency than MMF is currently used in the clinic. We suggest from this that the potential efficacy of MPA@Dex-MPA NPs against LN, compared with fMPA, is likely due to its direct and persistent retention in the kidney for three days. Consistent with our current findings, a prior study has reported that nephrotropic micelle-forming dexamethasone prodrug can remain in the kidney for four days and thereby showed a greatly improved efficacy in NZW/B F1 lupus mice when compared with an equivalent dose of dexamethasone.34,35 Taken together, the cumulative evidence indicates that tissue-specific drug delivery is a promising therapeutic strategy to prevent organ damage in SLE.
MPA@Dex-MPA NPs Exert Immunosuppressive Effects Mainly by Promoting M2-Like Macrophage Polarization in the Kidney and Spleen
As they accumulate in the kidney, MPA@Dex-MPA NPs can interact at a sufficient concentration with renal cells, among which the immune cells were of particular concern due to their central roles in the onset and progression of LN.33 We thus further analyzed the changes that manifested in the kidney immune cells after MPA@Dex-MPA NPs exposure to better elucidate the possible mechanisms by which these NPs can alleviate kidney damage.
We found from these analyses that the total amount of CD45+ kidney-infiltrating immune cells was markedly lower following the treatments containing MPA compared with the PBS control ((1.71 ± 0.06) × 106) and dextran ((1.65 ± 0.13) × 106) groups, especially the 10 mg/kg MPA@Dex-MPA ((0.93 ± 0.06) × 106) and 50 mg/kg fMPA ((1.04 ± 0.12) × 106) groups (Figure 4A). Of note in particular, the numbers of T cells and myeloid cells in the 10 mg/kg MPA@Dex-MPA group were found to have reduced to about 42% and 67% of their levels in PBS control group, respectively, and a comparable result was found in the 50 mg/kg fMPA group (Figure 4B and C and Figure S3). Furthermore, the number of kidney macrophages was reduced (Figure 4D and Figure S3) and their phenotype was affected by the MPA@Dex-MPA NPs treatment. Specifically, the proportion of CD80+ M1-like macrophages was significantly decreased in 10 mg/kg MPA@Dex-MPA group alone (Figure 4E), while the CD206+ M2-like macrophage levels were elevated in both the 10 mg/kg MPA@Dex-MPA group and 50 mg/kg fMPA group, compared with those of the PBS control group (Figure 4F). However, the CD40 and CD86 expression levels on kidney macrophages remained unaffected (Figure S4A). The number of DCs in the kidney and the expression of surface co-stimulatory molecules on these cells (CD40 and CD80) were also decreased in the 10 mg/kg MPA@Dex-MPA group (Figure S4B and C). These results indicated that MPA@Dex-MPA NPs can suppress the number of kidney-infiltrating immune cells and modulate the polarization of macrophages, an effect that was comparable or even more potent than observed on the 50 mg/kg fMPA group. This likely explains the reduced nephritis observed in the MRL/lpr mice in the MPA@Dex-MPA treatment group.
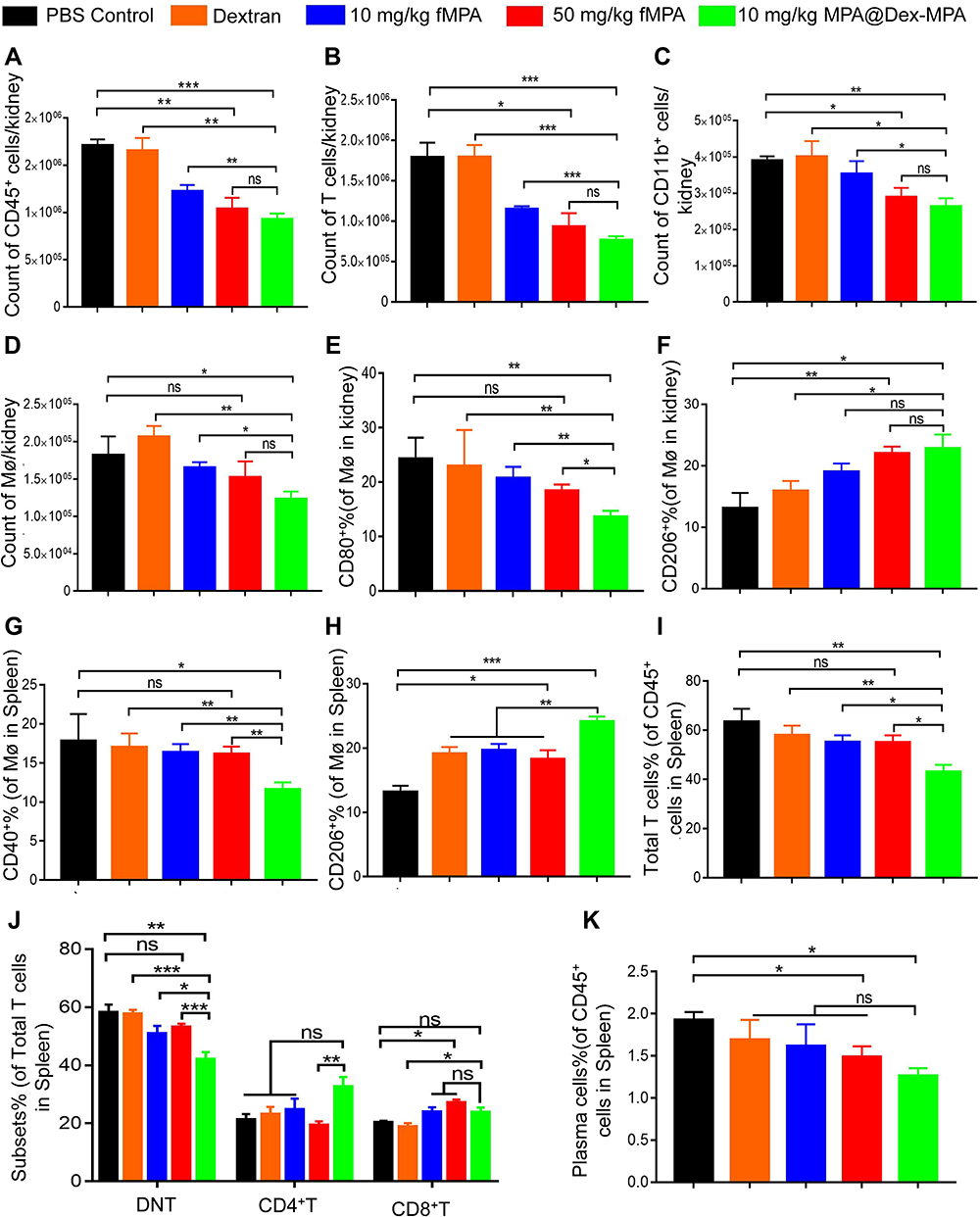 |
Figure 4 Changes of the immune cells in the kidney and spleen of MRL/lpr mouse after MPA@Dex-MPA NPs therapy. Counts were taken of infiltrating cells per kidney including the following: (A) CD45+ immune cells; (B) T cells; (C) CD11b+ myeloid cells; and (D) F4/80+ macrophages. These calculations were made using precision count beads and flow cytometry. (E and F) The percentages of CD80+ macrophages (E) and CD206+ M2-like macrophages (F) among the renal macrophages were also calculated. (G and H) The expressions of CD40 (G) and CD206 (H) among the splenic F4/80+ macrophages were assayed using flow cytometry. (I) Flow cytometric analysis of the percentage of splenic T cells among the total immune cell population. (J) Proportions of CD4+ T cells, CD8+ T cells and CD4−CD8− DNT cells among the total T cell population. (K) Flow cytometric analysis of the percentage of splenic CD138+ plasma cells among the CD45+ immune cells. Error bars represent the mean ±SEM, n = 4–6/group. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: ns, not significant; Mø, macrophage. |
The spleen is the major immune organ and was also where MPA@Dex-MPA NPs accumulation was observed (Figure 1B). As shown in Figure S5A, splenomegaly was only mildly relieved after this treatment. To better understand the influence of MPA@Dex-MPA NPs on systemic immunity in vivo, we further conducted flow cytometry analysis of splenic immune cells. Similar to the results observed in the mouse kidney, the phenotype of the macrophages in the spleen was also influenced (Figure 4G). The expression of the costimulatory molecule CD40 on these macrophages was significantly downregulated and the proportion of CD206+ M2-like macrophages was elevated in the 10 mg/kg MPA@Dex-MPA group, compared with the other four treatment groups (Figure 4H). However, the expression of other costimulatory molecules, CD80 and CD86, on the spleen macrophages were not significantly impacted (Figure S5B). These results indicated that MPA@Dex-MPA NPs therapy promotes the polarization of macrophages towards an M2-like state both in the kidney and spleen.
MPA@Dex-MPA NPs treatment was also found to inhibit the proliferation of T cells in the spleen in MRL/lpr mice. The percentage of total T cells among the splenic immune cells in the 10 mg/kg MPA@Dex-MPA group (43.06 ± 2.87%) was significantly reduced, compared with that of PBS control group (63.43 ± 5.30%), the equal-dose fMPA treatment group (55.25 ± 2.70%) and even lower than that of the 50 mg/kg fMPA group (55.00 ± 2.95%) (Figure 4I). Specifically, the expansion of the double-negative T (DNT) cell population (CD3+CD4−CD8−), which is frequently observed in autoimmune diseases, including SLE, aplastic anemia and Sjogren’s syndrome,36 was suppressed most significantly (Figure 4J). The proportion of DNT cells among the total T cell population in the MPA@Dex-MPA group (42.13 ± 2.41%) decreased by approximately 27% compared with that of PBS control group (58.25 ± 2.67%) (Figure 4J). DNT cells play an important role in the pathological mechanisms underlying SLE and are one of the primary sources of IL-17A.37 The decrease in DNT cells may thus have contributed to the lower serum IL-17A that we detected, as described above. Moreover, the percentage of CD138+ plasma cells among the splenic immune cells in the 10 mg/kg MPA@Dex-MPA group (1.26 ± 0.09%) was also significantly lower than that of the PBS control group (1.92 ± 0.10%), and even slightly lower than that of the 50 mg/kg fMPA treatment group (1.48% ± 0.13%) (Figure 4K). The activation of DCs was also slightly inhibited by the MPA@Dex-MPA NPs (Figure S5C).
MPA is known as a potent, reversible and noncompetitive inhibitor of inosine monophosphate dehydrogenase, the rate-limiting enzyme of the de novo pathway synthesizing the guanine nucleotides required for cell proliferation.13 Inhibiting the proliferation of activated lymphocytes in circulation, which solely rely on the de novo pathway, is the best known immunosuppressive mechanism of MMF.13 With its high plasma albumin binding rate (>97%), MMF exerts its immunosuppressive effects mainly in the circulation, rather than in local tissue.38 However, tissue damage, which is closely related to an SLE patient’s prognosis, is mainly driven by local inflammation.33 In recent years, the essential pathogenic roles of macrophages in the damage to vital tissues have been widely recognized in SLE.8 These cells initiate and amplify inflammation through multiple ways, including a failure to deal with apoptotic cells, aberrant activation, and an imbalanced polarization towards M1-like proinflammatory macrophages.39,40 By contrast, M2-like macrophages are considered anti-inflammatory as they can secrete immunosuppressive cytokines and provide insufficient costimulatory signals to lymphocytes to induce immune tolerance.39 Treatments aiming to induce M2-like macrophage production have been previously proved to be effective for SLE.41,42 Different from fMPA, MPA@Dex-MPA NPs in our current experiments exhibited tissue-targeting properties in relation to the kidney and spleen where they accumulated for a persistent period. They were found to be then mostly phagocytosed by macrophages in both organs, leading to the polarization of the macrophages towards an anti-inflammatory M2-like state. Hence, we speculated that MPA@Dex-MPA NPs mainly exerted their therapeutic effects against SLE through local macrophages. We could not exclude the possibility however that a small fraction of MPA was also released from the MPA@Dex-MPA NPs in vivo and then functioned through a classical action pathway.22 Hence, the inhibition of T cell proliferation may have been a direct effect of the MPA@Dex-MPA NPs and the fMPA released from them, or the indirect consequence of weakened stimulating signals from macrophages after they had phagocytosed the NPs.
MPA@Dex-MPA NPs Impact the Viability and Function of BMDMs in vitro
According to the above results, MPA@Dex-MPA NPs therapy greatly influenced the phenotype and number of macrophages in vivo. We further tested the direct effect of MPA@Dex-MPA NPs on macrophages in vitro. BMDMs were successfully induced and then treated with varied concentrations of Nile Red-MPA@Dex-MPA NPs (1, 10, 50 μg/mL) for different time duration (0.5, 1.0, 2.0 h). As shown in Figure 5A and Figure S6, NPs were phagocytosed by BMDMs in time- and concentration-dependent manners, demonstrating their ability to enter BMDMs.
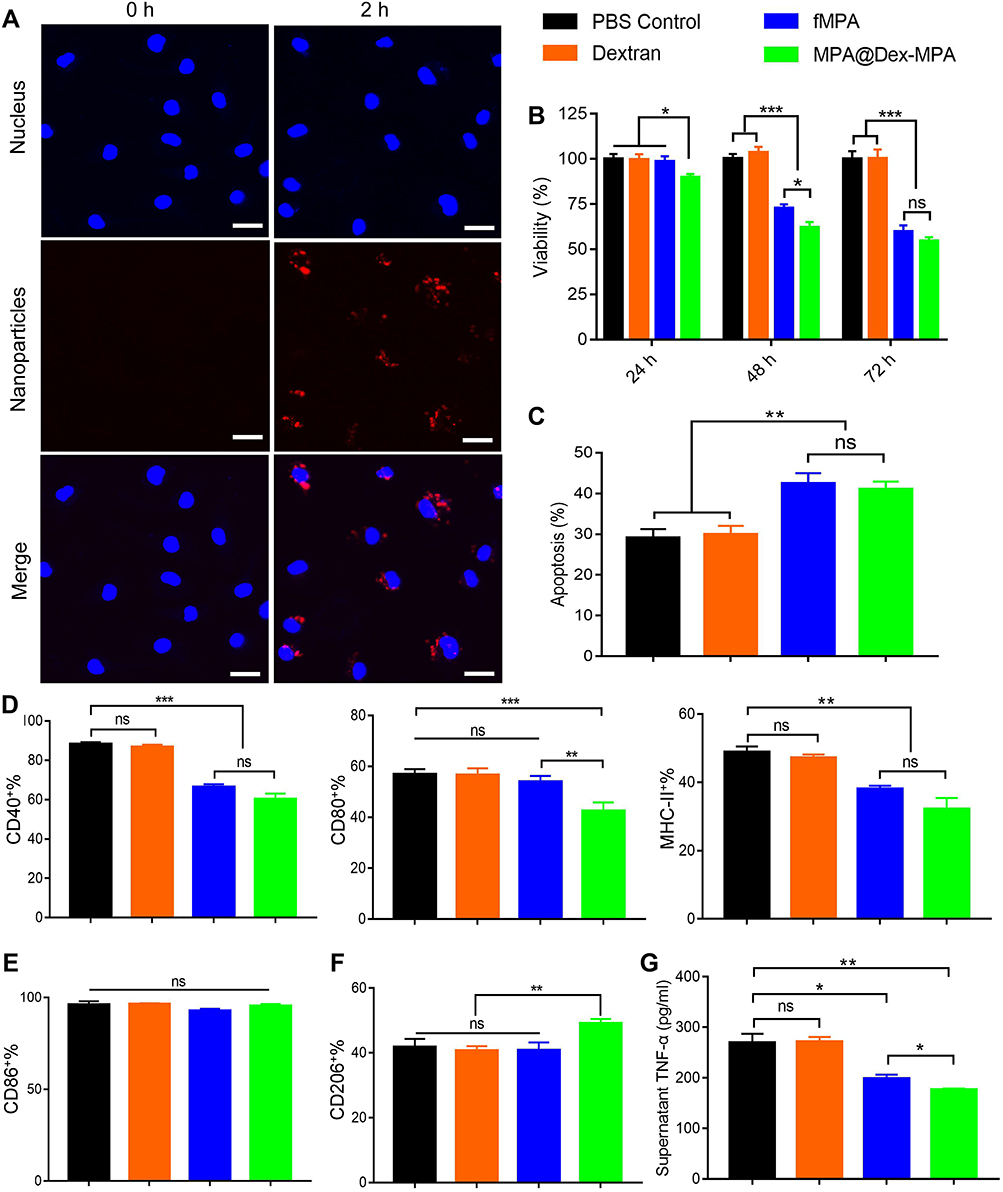 |
Figure 5 MPA@Dex-MPA NPs inhibit the viability and inflammatory function of BMDMs in vitro. (A) Representative confocal microscope images of the BMDM uptake of Nile Red-MPA@Dex-MPA NPs at 2 h post-treatment. Blue, nucleus; red, NPs. Scale bar, 20 μm. (B) CCK-8 analysis of BMDM viability after treatment with dextran, fMPA or MPA@Dex-MPA NPs for 24, 48 or 72 h, relative to the PBS control group. (C) Apoptotic response of BMDMs measured at 72 h by flow cytometry analysis. (D–F) Flow cytometry analysis of the percentages of CD40-, CD80-, and MHC-II- (D), or CD86-positive (E) BMDMs, and also of CD206+ M2-like BMDMs (F) after 24 h-pretreatment of the cells and further stimulation with lipopolysaccharide and IFN-γ for 12 h. (G) Supernatant level of TNF-α. Error bars represent the mean ± SEM, n = 5/group. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviation: ns, not significant. |
Based on prior studies, a 10 μg/mL concentration of MPA was chosen for our current in vitro studies on BMDMs.22,43 The viability of the BMDMs was firstly confirmed using cell count kit-8 (CCK-8) after treatment of the cells with PBS, dextran, fMPA or MPA@Dex-MPA NPs for 24, 48 and 72 h, respectively. As early as 24 h, the cell viability of the BMDMs in the MPA@Dex-MPA group (89.80 ± 1.78%) was significantly lower than that of the other three groups. The cell viability of the fMPA group (59.71 ± 3.56%) did not decrease to a similar level to that of MPA@Dex-MPA group (54.66 ± 2.09%) until 72 h (Figure 5B), indicating that MPA@Dex-MPA NPs worked faster than fMPA in vitro. At 72 h, the apoptosis rates of BMDMs in the MPA@Dex-MPA (41.03 ± 1.91%) and fMPA (42.47 ± 2.56%) groups were significantly elevated compared with the PBS (29.16 ± 2.12%) and dextran (29.94 ± 2.10%) groups (Figure 5C and Figure S7). Moreover, the viability of BMDMs also decreased along with drug dose increased (Figure S8).
We then further verified the effects of MPA@Dex-MPA NPs on the phenotype and function of BMDMs in vitro. The cells were pretreated with PBS, dextran, fMPA or MPA@Dex-MPA NPs for 24 h prior to a 12 h stimulation with lipopolysaccharide and IFN-γ. The expression of the surface molecules CD40, CD80, CD86 and MHC-II was then detected using flow cytometry. The upregulation of CD40 and MHC-II after stimulation was suppressed by fMPA and MPA@Dex-MPA NPs, while the expression of CD80 on BMDMs was downregulated solely by MPA@Dex-MPA NPs (Figure 5D). However, in animal experiments with MPA@Dex-MPA NPs therapy, the downregulation of CD40 and CD80 was only observed on splenic and renal macrophages, respectively. This inconsistency may be due to the varied MPA exposure levels among the organs and the high heterogeneity of macrophages themselves, which could lead to their different sensitivities to MPA. Consistent with the above in vivo findings, the expression of CD86 on BMDMs was unaffected (Figure 5E). In addition, the percentage of CD206+ M2-like macrophages was elevated by MPA@Dex-MPA NPs, compared with other groups (Figure 5F). Moreover, the supernatant level of TNF-α was significantly reduced by MPA@Dex-MPA NPs, compared with the other three groups (Figure 5G).
These in vitro investigations further confirmed that MPA@Dex-MPA NPs can decrease the number of macrophages and modulate their phenotype towards an M2-like state, as is the case in vivo. Hence, our nanomodification process represents a new and potent mechanism of administering MPA for SLE treatment.
MPA@Dex-MPA NPs Exhibit Excellent Long-Term Biocompatibility in vivo
As the long-term use of MMF is usually required in a clinical setting, we evaluated the safety of MPA@Dex-MPA NPs exposure in C57BL/6 mice over 4 weeks. During this period, the mice gained weight normally in all of the treatment groups (Figure 6A). The serum levels of ALT, AST, BUN and creatinine were all within physiological ranges at the end of the month (Figure 6B and C). Routine blood work was also conducted. The platelet count and the hemoglobin level were comparable among the groups (Figure 6D and E). By contrast, as shown in Figure 6F, the total white blood cell (WBC) count was significantly reduced after treatments containing MPA, mainly for the lymphocytes (Figure 6F). The decrease of WBCs and lymphocytes in our MPA@Dex-MPA NPs group was milder than that found in the 50 mg/kg fMPA group, indicating a probable lower risk of infection. Moreover, H&E staining of the major mouse organs, including the heart, liver, spleen, lung and kidneys, indicated no obvious pathological manifestation in any of the treatment groups (Figure 7). These results demonstrated a good long-term safety profile for the MPA@Dex-MPA NPs, which was an important issue for SLE patients.
 |
Figure 6 MPA@Dex-MPA NPs treatments exhibit an excellent long-term safety profile in C57BL/6 mice. (A) Body weight gaining curves for the mice during the 4 weeks’ treatment period. (B) Serum levels of ALT and AST detected by biochemical analyzer (n = 5/group). (C) BUN and serum creatinine levels, also detected by biochemical analyzer (n = 5/group). (D–F) Hemoglobin level (D), platelets count (E), and total white blood cell and lymphocyte counts (F) were determined using a hematology analyzer (n = 5/group). Error bars represent the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviation: ns, not significant. |
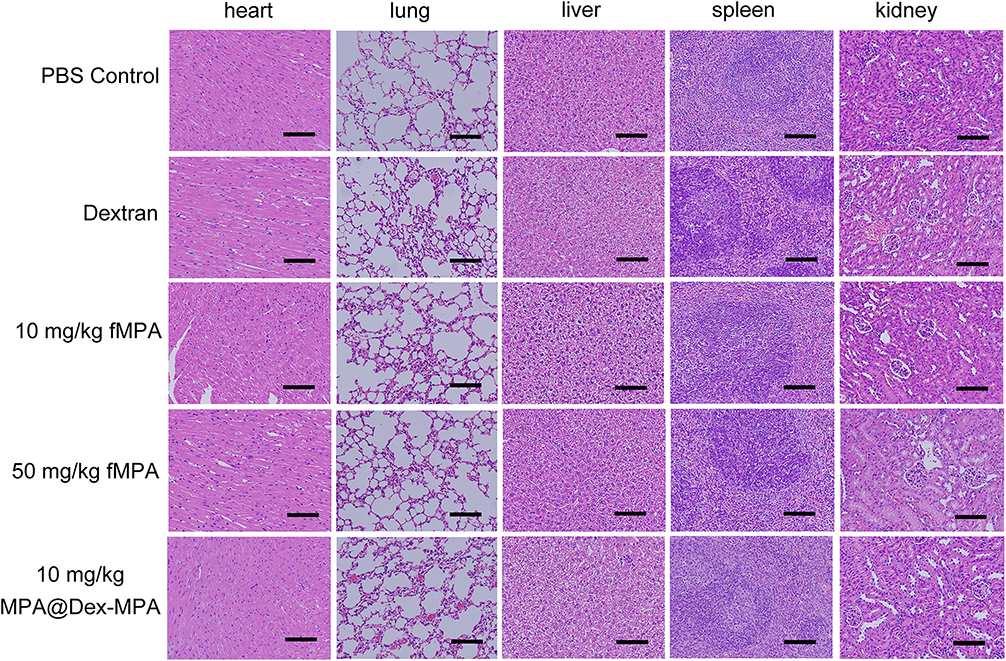 |
Figure 7 H&E staining of the major organs in the C57BL/6 mice indicated no obvious pathological manifestations after a 4 week’s MPA@Dex-MPA NPs treatment regimen. Representative H&E staining images are shown of the heart, liver, spleen, lung and kidney (n = 5/group). Scale bar, 50 μm. |
Conclusion
Our mycophenolate-modified NP system exhibits a superior therapeutic efficacy and better long-term safety profile in MRL/lpr mice using a lower dosage and treatment frequency. The better distribution and longer retention of these NPs in the kidney and spleen diversified the ways in which they interacted with dysfunctional immune cells, ie, both directly in these tissues as well as through a classical pathway, to inhibit the proliferation of lymphocytes. In this present study, we found that MPA@Dex-MPA NPs were phagocytosed mostly by macrophages in the mouse spleen and kidney. The number of macrophages was significantly reduced and they showed a predominantly M2-like phenotype with a lowered expression of costimulatory molecules and inflammatory cytokines, thereby contributing to the relief of tissue damage. Hence, our present study describes a promising new treatment strategy for SLE with the potential for clinical application in the near future.
Abbreviations
ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMDM, bone marrow-derived macrophage; BUN, blood urea nitrogen; C3, complement 3; DCs, dendritic cells; Dex, dextran; DMAP, 4-dimethylaminopyridine; DNT, double negative T; EDC, 1-ethyl-3-(3-(dimethylamino) propyl)-carbodiimide hydrochloride; fMPA, free mycophenolic acid; H&E, hematoxylin and eosin; LN, lupus nephritis; MMF, mycophenolate mofetil; MPA, mycophenolic acid; NPs, nanoparticles; PAS, periodic acid-Schiff; RBC, red blood cell; SEM, standard error of the mean; SLE, systemic lupus erythematosus; UPCR, urine protein-creatinine ratio.
Acknowledgments
The authors sincerely acknowledge the financial support of the National Natural Science Foundation of China (grant numbers 82003366 and 81573047).
Disclosure
The authors report no conflicts of interest in relation to this study.
References
1. Tsokos GC. Autoimmunity and organ damage in systemic lupus erythematosus. Nat Immunol. 2020;21(6):605–614. doi:10.1038/s41590-020-0677-6
2. Fanouriakis A, Tziolos N, Bertsias G, Boumpas DT. Update on the diagnosis and management of systemic lupus erythematosus. Ann Rheum Dis. 2021;80(1):14–25. doi:10.1136/annrheumdis-2020-218272
3. Kiriakidou M, Ching CL. Systemic lupus erythematosus. Ann Intern Med. 2020;172(11):Itc81–itc96. doi:10.7326/AITC202006020
4. Murphy G, Isenberg DA. New therapies for systemic lupus erythematosus - past imperfect, future tense. Nat Rev Rheumatol. 2019;15(7):403–412. doi:10.1038/s41584-019-0235-5
5. Blair HA, Duggan ST. Belimumab: a review in systemic lupus erythematosus. Drugs. 2018;78(3):355–366. doi:10.1007/s40265-018-0872-z
6. Wise LM, Stohl W. Belimumab and rituximab in systemic lupus erythematosus: a tale of two B cell-targeting agents. Front Med. 2020;7:303. doi:10.3389/fmed.2020.00303
7. Allen ME, Rus V, Szeto GL. Leveraging heterogeneity in systemic lupus erythematosus for new therapies. Trends Mol Med. 2021;27(2):152–171. doi:10.1016/j.molmed.2020.09.009
8. Saferding V, Blüml S. Innate immunity as the trigger of systemic autoimmune diseases. J Autoimmun. 2020;110:102382. doi:10.1016/j.jaut.2019.102382
9. Broen JCA, van Laar JM. Mycophenolate mofetil, azathioprine and tacrolimus: mechanisms in rheumatology. Nat Rev Rheumatol. 2020;16(3):167–178. doi:10.1038/s41584-020-0374-8
10. Fanouriakis A, Kostopoulou M, Cheema K, et al. 2019 update of the joint European league against rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of lupus nephritis. Ann Rheum Dis. 2020;79(6):713–723. doi:10.1136/annrheumdis-2020-216924
11. Fanouriakis A, Kostopoulou M, Alunno A, et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. 2019;78(6):736–745. doi:10.1136/annrheumdis-2019-215089
12. Abd Rahman AN, Tett SE, Staatz CE. Clinical pharmacokinetics and pharmacodynamics of mycophenolate in patients with autoimmune disease. Clin Pharmacokinet. 2013;52(5):303–331. doi:10.1007/s40262-013-0039-8
13. Naffouje R, Grover P, Yu H, et al. Anti-tumor potential of IMP dehydrogenase Inhibitors: a century-long story. Cancers. 2019;11(9):1346. doi:10.3390/cancers11091346
14. Al-Lawati H, Aliabadi HM, Makhmalzadeh BS, Lavasanifar A. Nanomedicine for immunosuppressive therapy: achievements in pre-clinical and clinical research. Expert Opin Drug Deliv. 2018;15(4):397–418. doi:10.1080/17425247.2018.1420053
15. Rizzuto MA, Salvioni L, Rotem R, et al. Are nanotechnological approaches the future of treating inflammatory diseases? Nanomedicine. 2019;14(17):2379–2390. doi:10.2217/nnm-2019-0159
16. Wang H, Zhou Y, Sun Q, et al. Update on nanoparticle-based drug delivery system for anti-inflammatory treatment. Front Bioeng Biotechnol. 2021;9:630352. doi:10.3389/fbioe.2021.630352
17. Kochappan R, Cao E, Han S, et al. Targeted delivery of mycophenolic acid to the mesenteric lymph node using a triglyceride mimetic prodrug approach enhances gut-specific immunomodulation in mice. J Control Release. 2021;332:636–651. doi:10.1016/j.jconrel.2021.02.008
18. Uehara M, Bahmani B, Jiang L, et al. Nanodelivery of mycophenolate mofetil to the organ improves transplant vasculopathy. ACS Nano. 2019;13(11):12393–12407. doi:10.1021/acsnano.9b05115
19. Teng J, Zang L, Li L, Qiu X, Liu Y, Sun F. Overall condition improvement in a rat model of nephrotic syndrome treated with CellCept nanoliposomes. Artif Cells Nanomed Biotechnol. 2017;45(1):128–134. doi:10.3109/21691401.2016.1138484
20. Shirali AC, Look M, Du W, et al. Nanoparticle delivery of mycophenolic acid upregulates PD-L1 on dendritic cells to prolong murine allograft survival. Am J Transplant. 2011;11(12):2582–2592. doi:10.1111/j.1600-6143.2011.03725.x
21. Look M, Stern E, Wang QA, et al. Nanogel-based delivery of mycophenolic acid ameliorates systemic lupus erythematosus in mice. J Clin Invest. 2013;123(4):1741–1749. doi:10.1172/JCI65907
22. Li Y, Lou Y, Chen Y, et al. Polysaccharide mycophenolate-based nanoparticles for enhanced immunosuppression and treatment of immune-mediated inflammatory diseases. Theranostics. 2021;11(8):3694–3709. doi:10.7150/thno.52891
23. Boraschi D, Italiani P, Palomba R, et al. Nanoparticles and innate immunity: new perspectives on host defence. Semin Immunol. 2017;34:33–51. doi:10.1016/j.smim.2017.08.013
24. Zhao J, Wang H, Dai C, et al. P2X7 blockade attenuates murine lupus nephritis by inhibiting activation of the NLRP3/ASC/caspase 1 pathway. Arthritis Rheum. 2013;65(12):3176–3185. doi:10.1002/art.38174
25. Fu R, Xia Y, Li M, et al. Pim-1 as a therapeutic target in lupus nephritis. Arthritis Rheumatol. 2019;71(8):1308–1318. doi:10.1002/art.40863
26. Muraoka M, Hasegawa H, Kohno M, et al. IK cytokine ameliorates the progression of lupus nephritis in MRL/lpr mice. Arthritis Rheum. 2006;54(11):3591–3600. doi:10.1002/art.22172
27. Lee YF, Cheng CC, Lan JL, et al. Effects of mycophenolate mofetil on cutaneous lupus erythematosus in (NZB × NZW) F1 mice. J Chin Med Assoc. 2013;76(11):615–623. doi:10.1016/j.jcma.2013.07.010
28. Lee HK, Kim KH, Kim HS, et al. Effect of a combination of prednisone or mycophenolate mofetil and mesenchymal stem cells on lupus symptoms in MRL.Fas(lpr) mice. Stem Cells Int. 2018;2018:4273107. doi:10.1155/2018/4273107
29. Ruchakorn N, Ngamjanyaporn P, Suangtamai T, et al. Performance of cytokine models in predicting SLE activity. Arthritis Res Ther. 2019;21(1):287. doi:10.1186/s13075-019-2029-1
30. Maria NI, Davidson A. Protecting the kidney in systemic lupus erythematosus: from diagnosis to therapy. Nat Rev Rheumatol. 2020;16(5):255–267. doi:10.1038/s41584-020-0401-9
31. Anders HJ, Saxena R, Zhao MH, Parodis I, Salmon JE, Mohan C. Lupus nephritis. Nat Rev Dis Primers. 2020;6(1):7. doi:10.1038/s41572-019-0141-9
32. Li W, Titov AA, Morel L. An update on lupus animal models. Curr Opin Rheumatol. 2017;29(5):434–441. doi:10.1097/BOR.0000000000000412
33. Flores-Mendoza G, Sansón SP, Rodríguez-Castro S, Crispín JC, Rosetti F. Mechanisms of tissue injury in lupus nephritis. Trends Mol Med. 2018;24(4):364–378. doi:10.1016/j.molmed.2018.02.003
34. Jia Z, Wang X, Wei X, et al. Micelle-forming dexamethasone prodrug attenuates nephritis in lupus-prone mice without apparent glucocorticoid side effects. ACS Nano. 2018;12(8):7663–7681. doi:10.1021/acsnano.8b01249
35. Zhao Z, Jia Z, Foster KW, et al. Dexamethasone prodrug nanomedicine (ZSJ-0228) treatment significantly reduces lupus nephritis in mice without measurable side effects - A 5-month study. Nanomedicine. 2021;31:102302. doi:10.1016/j.nano.2020.102302
36. Moulton VR, Suarez-Fueyo A, Meidan E, Li H, Mizui M, Tsokos GC. Pathogenesis of human systemic lupus erythematosus: a cellular perspective. Trends Mol Med. 2017;23(7):615–635. doi:10.1016/j.molmed.2017.05.006
37. Li H, Adamopoulos IE, Moulton VR, et al. Systemic lupus erythematosus favors the generation of IL-17 producing double negative T cells. Nat Commun. 2020;11(1):2859. doi:10.1038/s41467-020-16636-4
38. Dasgupta A. Therapeutic drug monitoring of mycophenolic acid. Adv Clin Chem. 2016;76:165–184. doi:10.1016/bs.acc.2016.04.001.
39. Ma C, Xia Y, Yang Q, Zhao Y. The contribution of macrophages to systemic lupus erythematosus. Clin Immunol. 2019;207:1–9. doi:10.1016/j.clim.2019.06.009
40. Xu N, Li J, Gao Y, et al. Apoptotic cell-mimicking gold nanocages loaded with LXR agonist for attenuating the progression of murine systemic lupus erythematosus. Biomaterials. 2019;197:380–392. doi:10.1016/j.biomaterials.2019.01.034
41. Horuluoglu B, Bayik D, Kayraklioglu N, Goguet E, Kaplan MJ, Klinman DM. PAM3 supports the generation of M2-like macrophages from lupus patient monocytes and improves disease outcome in murine lupus. J Autoimmun. 2019;99:24–32. doi:10.1016/j.jaut.2019.01.004
42. Han S, Zhuang H, Shumyak S, et al. Liver X receptor agonist therapy prevents diffuse alveolar hemorrhage in murine lupus by repolarizing macrophages. Front Immunol. 2018;9:135. doi:10.3389/fimmu.2018.00135
43. Kannegieter NM, Hesselink DA, Dieterich M, et al. The Effect of tacrolimus and mycophenolic acid on CD14+ monocyte activation and function. PLoS One. 2017;12(1):e0170806. doi:10.1371/journal.pone.0170806
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Mannosamine-Engineered Nanoparticles for Precision Rifapentine Delivery to Macrophages: Advancing Targeted Therapy Against Mycobacterium Tuberculosis

Luan H, Peng C, Yasin P, Shang Q, Xiang W, Song X
Drug Design, Development and Therapy 2025, 19:2081-2102
Published Date: 19 March 2025
Nanomaterial-Based Drug Delivery Systems Targeting Functional Cells for Osteoarthritis Treatment: Mechanisms, Challenges and Future Prospects

Kong H, He Q, Han J, Zhang XA
International Journal of Nanomedicine 2025, 20:5291-5320
Published Date: 25 April 2025
The Regulatory Network of Transcription Factors in Macrophage Polarization
Liu J, Wang M, Zhao Y
ImmunoTargets and Therapy 2025, 14:555-575
Published Date: 6 June 2025
ZnO NPs Attenuate LPS-Induced Inflammation in RAW264.7 Macrophages by Inhibiting NF-κB and JAK1-STAT1/STAT3 Pathways and Reducing ROS
Tang H, Zhang L, Deng C, Li Y, Ren G, Sun Z, Chang L, Xu M, Xiao D, Zhang H
Journal of Inflammation Research 2025, 18:14075-14096
Published Date: 9 October 2025