Back to Journals » International Journal of Nanomedicine » Volume 18
A Targeted and pH-Responsive Nano-Graphene Oxide Nanoparticle Loaded with Doxorubicin for Synergetic Chemo-Photothermal Therapy of Oral Squamous Cell Carcinoma
Authors Li R ![]() , Liu C
, Liu C ![]() , Wan C, Liu T
, Wan C, Liu T ![]() , Zhang R, Du J, Wang X
, Zhang R, Du J, Wang X ![]() , Jiao X, Gao R, Li B
, Jiao X, Gao R, Li B ![]()
Received 24 January 2023
Accepted for publication 6 June 2023
Published 17 June 2023 Volume 2023:18 Pages 3309—3324
DOI https://doi.org/10.2147/IJN.S402249
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Ran Li,1,2,* Chen Liu,1,2,* Chaoqiong Wan,1,2 Tiantian Liu,1,2 Rongrong Zhang,1,2 Jie Du,1 Xiangyu Wang,1,2 Xiaofeng Jiao,1,2 Ruifang Gao,1,2 Bing Li1
1Shanxi Province Key Laboratory of Oral Diseases Prevention and New Materials, Shanxi Medical University School and Hospital of Stomatology, Taiyuan, Shanxi, People’s Republic of China; 2Department of Pediatric and Preventive Dentistry, Shanxi Medical University School and Hospital of Stomatology, Taiyuan, Shanxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ruifang Gao; Bing Li, Email [email protected]; [email protected]
Purpose: Oral squamous cell carcinoma (OSCC) is a malignant disease with serious impacts on human health and quality of life worldwide. This disease is traditionally treated through a combination of surgery, radiotherapy, and chemotherapy. However, the efficacy of traditional treatments is hindered by systemic toxicity, limited therapeutic effects, and drug resistance. Fibroblast activation protein (FAP) is a membrane-bound protease. Although FAP has limited expression in normal adult tissues, it is highly expressed in the tumor microenvironment of many solid cancers – a characteristic that makes it an ideal target for anticancer therapy. In this study, we constructed a nano-drug delivery system (NPF@DOX) targeting FAP to increase the therapeutic efficiency of synergistic chemo-photothermal therapy against OSCC.
Methods: We utilized PEGylated nano-graphene oxide (NGO) to link doxorubicin (DOX) and fluorescently-labeled, FAP-targeted peptide chains via hydrogen bonding and π–π bonding to enhance the targeting capability of NPF@DOX. The synthesis of NPF@DOX was analyzed using UV–Vis and FT–IR spectroscopy and its morphology using transmission electron microscopy (TEM). Additionally, the drug uptake efficiency in vitro, photo-thermal properties, release performance, and anti-tumor effects of NPF@DOX were evaluated and further demonstrated in vivo.
Results: Data derived from FT–IR, UV–Vis, and TEM implied successful construction of the NPF@DOX nano-drug delivery system. Confocal laser scanning microscopy images and in vivo experiments demonstrated the targeting effects of FAP on OSCC. Furthermore, NPF@DOX exhibited a high photothermal conversion efficiency (52.48%) under near-infrared radiation. The thermogenic effect of NPF@DOX simultaneously promoted local release of DOX and apoptosis based on a pH-stimulated effect. Importantly, FAP-targeted NPF@DOX in combination with PTT showed better tumor suppression performance in vivo and in vitro than did either therapy individually.
Conclusion: NPF@DOX can precisely target OSCC, and combined treatment with chemical and photothermal therapy can improve the therapeutic outcomes of OSCC. This method serves as an efficient therapeutic strategy for the development of synergistic anti-tumor research.
Keywords: fibroblast activation protein, nano-graphene oxide, photothermal therapy, oral squamous cell carcinoma, targeted combination therapy
Introduction
Oral squamous cell carcinoma (OSCC) is an aggressive cancer that accounts for 80–90% of oral malignancies.1 Surgery combined with chemoradiotherapy remains the primary treatment.2 While chemotherapy is necessary, it has several severe limitations, such as systemic toxicity, cancer recurrence, and developed drug resistance. This is mainly due to the untargeted systemic administration of chemotherapies, which can collect in non-cancerous tissues before reaching the tumor site. Therefore, it is necessary to develop an alternative targeted therapy.3,4
Recently, researchers have shown an interest in the tumor microenvironment as a new target for chemotherapy.5,6 Cancer-associated fibroblasts (CAFs) in the tumor microenvironment promote tumor growth and invasion.7 Fibroblast activation protein (FAP) is overexpressed in CAFs.8 FAP is a type II integrated membrane glycoprotein and a member of the dipeptidyl peptidase IV family, owing to its ability to cut Pro-XXX amino acid chains, which inhibit serine endopeptidases in the tumor microenvironment by acting on FAP.9 Furthermore, many studies have shown that FAP may be a promising target for the design of tumor-targeting drugs and imaging agents.10 Therefore, altering the tumor microenvironment of OSCC with FAP-targeted therapy may be an effective strategy to disrupt interactions within the microenvironment and selectively kill cancer cells.
Over the past 20 years, tremendous efforts have been made to synthesize biomarkers that bind to nanoscale drug delivery systems to reach the disease sites in vivo. These systems are based on nanomaterials and combine several technologies to enable accurate, effective, and more efficient tumor treatments.11,12 However, ongoing, low-dose chemotherapy can increase the risk of metastasis and drug resistance.13
To overcome this deficiency, hyperthermia combined with chemotherapy can lead to superadditive or synergistic effects.14,15 Tumors can be penetrated by near-infrared (NIR) laser radiation, which has a wavelength range of 650–900 nm and does not harm surrounding tissue.16 Using this approach, tumor-targeting carbon-based nanoparticles gather at the tumor site, absorb NIR, and induce tumor cell death through hyperthermia.17 Furthermore, heat stress generated by NIR irradiation can induce chemosensitization through accelerated release at tumor targets, resulting in increased local drug concentrations. Drug release in combination with hyperthermia therapy can kill cancer cells in a specific tumor region, thereby minimizing damage to healthy tissues.18,19 This method significantly improves therapeutic efficacy against cancer cells and reduces side effects.
Nanoparticle-based nanotechnology offers viable nanomaterials for combining hyperthermia with tailored chemotherapies. As a promising nanomaterial, graphene has been widely used in photothermal therapy (PTT) and drug delivery in recent years.20 Graphene-based drug carriers can prolong drug blood circulation time, increase drug load, and control drug release.21 They are also designed to increase drug concentration at tumor sites through the enhanced permeability and retention (EPR) effect.22–24 By conjugating proteins, fluorophores, drugs, and protective coatings to their surface functional groups, graphene-based nanoparticles have potential for improving drug delivery.25–27 Nano-graphene oxide (NGO) can be introduced at hydrophilic moieties, such as hydroxyl groups and carboxylic acids, and is well-dispersed in aqueous solutions, reducing toxicity and increasing biocompatibility,28 making it a promising drug carrier material. Additionally, polyethylene glycol (PEG) can be used as a supplementary material for graphene. PEG can activate macrophages and has the potential to enhance anti-tumor immunity.29 As a material for light-assisted therapy, their combination (NGO–PEG) can reduce drug resistance by combining PTT with chemotherapeutic drugs. This makes NGO–PEG (NP) an efficient drug carrier, one that has been previously researched for drug delivery applications.30,31 Doxorubicin (DOX) is a fluorescent anticancer drug that has high therapeutic efficiency and activity against solid tumors.32 However, owing to severe side effects, such as cardiotoxicity, its clinical use is limited.33,34 Targeted nanocarriers have been developed to improve DOX efficacy and reduce its side effects. Therefore, we believe that a FAP-targeting peptide can be combined with NP to construct a new tumor-targeting drug vector (NPF) that can carry DOX. FAP-targeting combined with photothermal therapy may reduce systemic toxicity and acquired resistance as well as avoid insufficient drug accumulation issues due to its tumor localization, which altogether improves efficacy. In this study, we utilized the specific activity of FAP to successfully construct a drug delivery system NPF@DOX and demonstrated the efficacy of targeted FAP chemotherapy in combination with PPT for OSCC in vivo and in vitro. By concentrating in the tumor, NPF@DOX improved PTT and chemotherapy effects simultaneously. It provides a simple therapeutic strategy for promoting the synergistic treatment of OSCC.
Materials and Methods
Materials
NGO powder was purchased from Nanjing XFNANO Materials Co., Ltd. (XF004; Nanjing, China). PEG and EDC-HCL (1-(3-dimethyl aminopropyl)-3-ethyl carbodiimide hydrochloride) were bought from Shanghai Aladdin Biochemical Technology Co. (Shanghai, China). Peptide chains with FAP recognition sequences and functionalized with FITC and CY5.5 fluorescent dye were obtained from the Chinese Peptide Co. (997935; Hangzhou, China). Dalian Meilun Biotechnology, Co., Ltd. provided the DOX, trypsin-EDTA, penicillin–streptomycin, and calcein AM/PI Living and Dead Cell Stain Kit (Dalian, China). FAP antibody was purchased from Abcam (Cambridge, UK). Dulbecco’s Modified Eagle Medium (DMEM), Roswell Park Memorial Institute (RPMI) 1640 medium, and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). A DAB Immunohistochemistry Color Development Kit was purchased from Beijing Zhongshanjinqiao Biotechnology Co., Ltd (Beijing, China).
Cells Culture
CAL-27 cells (human tongue squamous cell carcinoma cell line) and HOK cells (human oral keratinocyte cell line) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). CAL-27 cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin. HOK cells were cultured in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin–streptomycin. All cells were cultured in a humidified incubator at 37 °C under 5% CO2 and dissociated with 0.25% trypsin–EDTA solution for passage once they reached 80% confluence.
Synthesis of NPF
The NGO suspension (1.0 mg/mL) was sonicated for 30 min to obtain a clear suspension. This solution and PEG were dissolved in stirred deionized water at a weight ratio of 4:1. The resulting solution was rinsed and purified in PBS repeatedly. We catalyzed the reaction with 40 mg/mL of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride with continuous stirring at 37 °C.35,36 The suspension was dialyzed (MWCO = 100 kD), and the unbound fraction was removed with pure water in the dark for 24 h to obtain the desired NP. Fluorescein dye (FITC)-labeled NPF nanoparticles were obtained by combining NP and FITC-His-Gly-Arg-Lys at a 5:1 weight ratio with deionized water. The mixture was stirred vigorously in the dark at room temperature for 24 h. The suspension was centrifuged at 10,000 rpm for 15 min and washed three times with deionized water to obtain NPF.
Preparation and Characterization of NPF@DOX
DOX was loaded onto NP and NPF nanoparticles, which were prepared as redox-loaded NP@DOX and NPF@DOX. The products were purified three times with deionized water after centrifugation at 10,000 rpm for 15 min. NP@DOX and NPF@DOX were extracted using lyophilization and stored in a low-temperature, dark environment. To estimate the loading rate of DOX, 1.0 mL of 1.0 mg/mL DOX aqueous solution was mixed with 1.0 mg of NPF. The mixture was stirred for 24 h at room temperature in darkness. The resulting clear suspension was centrifuged at 10,000 rpm for 15 min to remove the unreacted DOX. A UV–Vis spectrophotometer (490 nm) was used to measure the drug-loading efficiency of DOX on NPF.
Transmission electron microscopy (TEM) (JEM-2100F; JEOL, Ltd., Tokyo, Japan) was used to characterize the particle size and morphology of NPF@DOX. The absorbance spectra of NPF@DOX were characterized using UV–Vis spectroscopy (Lambda 35; Perkin Elmer, Waltham, MA, USA) and FT–IR spectroscopy (Tensor 27; Bruker, Billerica, MA, USA). Fluorescence imaging was obtained using a Bruker In-vivo Xtreme imaging system (Bruker). The photothermal conversion efficiency of NPF@DOX was evaluated using an infrared thermal camera (FLIR T540sc; Teledyne FLIR, Wilsonville, OR, USA) to examine its potential for use in PTT.
DOX Release
DOX release from NPF@DOX was investigated in PBS at pH 5.5 and 7.4. NPF@DOX was suspended in PBS and shaken at 150 rpm and room temperature. At different time intervals (2, 6, 12, 24, 48, and 72 h), 2.0 mL aliquots were obtained. The amount of released DOX was determined using UV–Vis spectrophotometry at 490 nm. To investigate the NIR stimuli-responsive behavior, NPF@DOX (0.5 mg) suspended in 0.5 mL of PBS buffer (pH 5.5, 7.4) was irradiated for 5 min at predetermined time intervals using an 808 nm NIR laser (1.0 W/cm2). Following irradiation, equal volumes of NPF suspension were collected, centrifuged at 10,000 rpm for 10 min, and supernatants collected for UV–Vis spectrophotometric analysis.
Immunohistochemistry
All subjects provided written, informed consent, and all experiments followed the Declaration of Helsinki. The experimental techniques and methods were in compliance with the pertinent regulations. This study was approved by the Ethics Committee of Shanxi Medical University (2021SLL092). OSCC and normal tissues were collected from the Shanxi Medical University School and Hospital of Stomatology. Surgical specimens and normal tissues were observed and analyzed using immunohistochemistry.
In this experiment, the tissues were fixed and embedded, dewaxed with xylene and gradient ethanol, blocked with 2% BSA, incubated with primary and secondary antibodies, and stained with DAB. In addition, the difference between FAP expression levels in CAL-27 and HOK cells was further verified through cellular immunochemistry. Dedicated cell slides were placed in 24-well plates, and CAL-27 (2 × 105 cells/well) and HOK cells (1.5 × 105 cells/well) were seeded in their respective plates and cultured at 37 °C under 5% CO2 for 24 h. When cells were attached, they were mounted on slides and fixed with 4% paraformaldehyde, and then permeabilized with 0.5% Triton-x100 at room temperature. Hydrogen peroxide (3%) was applied to inactivate endogenous enzymes, BSA (2%) was used to block non-specific binding, and the slides were incubated with primary antibody (FAP) at 4 °C overnight. Biotin-labeled anti-rabbit/mouse IgG was added the next day after incubation at room temperature. After blocking non-specific binding sites with SABC antibody, hematoxylin re-staining, ethanol gradient dehydration, and neutral glue blocking, the staining results were observed under a microscope.
Cellular Uptake
The cellular uptake of NP@DOX and NPF@DOX by CAL-27 cells was examined using confocal microscopy. Briefly, CAL-27 cells were seeded on confocal dishes (Lab-Tek; Nagle Nunc, Rochester, NY, USA) at 2×105 cells/well in 200 μL of medium. After 24 h, the medium was replaced with medium containing NP@DOX or NPF@DOX. After 3 h, the cells were washed three times with PBS and fixed for 20 min in a 4% paraformaldehyde solution. These cells were examined using a confocal laser scanning microscope (CLSM, Leica TCS SP5; Wetzlar, Germany) after staining with DAPI (Boster Bio, Pleasanton, CA, USA). To examine NIR chemosensitization in vitro, NP@DOX and NPF@DOX were incubated with CAL-27 cells for 3 h and then washed twice with PBS. Cells in the irradiated groups were further treated with a NIR laser (808 nm, 1.0 W/cm2, 5 min). Finally, cellular uptake and apoptosis in all treated cells were observed via CLSM.
In vitro Cytotoxicity Assay and Calcein–AM/PI Staining
A CCK-8 assay was used to evaluate the cytotoxicity of the nano-drug delivery system in vitro. CAL-27 cells were selected as an experimental model. The cells (8000 cells/well) were seeded in 96-well plates and cultured at 37 °C under 5% CO2 for 24 h. After cells were adherent, cells were treated with NFP, NP@DOX, NPF@DOX, NFP-NIR, NP@DOX-NIR, or NPF@DOX-NIR, and cells receiving no nanoparticle treatment served as controls. The experimental groups exposed to NIR were further irradiated with an 808 nm laser (1.0 W/cm2) for 5 min. Finally, cells from all experimental groups were washed three times with PBS, and 100 mL of freshly prepared CCK-8 solution was added into each well. The cells were then incubated in the dark for 1 h. A microplate reader (Multiskan FC; Thermo Fisher Scientific) was used to measure absorbance at 450 nm, and cell viability was calculated. The average percentage of viable cells was calculated as compared with that among untreated cells.
To investigate the biological safety of PTT and chemotherapy, we performed cytotoxicity and photothermal cytotoxicity assays on HOK cells under 808 nm laser irradiation and non-irradiation. HOK cells were seeded in 96-well plates at 6000 cells/well and treated with the above methods, and the survival rate of cells was observed.
To further evaluate the efficacy of synergistic therapy and cytotoxicity, we conducted a live/dead cell assay. CAL-27 cells (2 × 105 per well) were seeded in their respective 12-well plates, and the medium was replaced after 24 h with medium containing NPF, NP@DOX, or NPF@DOX. The incubated cells were then irradiated with NIR (808 nm) for 5 min or not irradiated. Cells from all groups were stained with acridine orange and propidium iodide in PBS, and fluorescence microscopy (ECLIPSE Ti; Nikon, Tokyo, Japan) was used to observe the results.
Animal Models and Biodistribution
Male BALB/c mice aged 6–8 weeks were purchased from Charles River Laboratories (Chicago, IL, USA). These animals were housed in air-conditioned environments with a natural light/dark cycle and a relative humidity of 50%. The Institutional Ethical Committee for Animal Experimentation at Shanxi Medical University approved all animal procedures. Governmental and international regulations on animal testing were followed for all procedures. For the mouse xenograft tumor model, CAL-27 cells (2 × 106 cells) suspended in 50 μL normal saline (NS) were injected into the right hind thighs of the mice. Tumors were imaged when their volumes reached 60–100 mm3. Mice were imaged using a Bruker In-vivo X-ray imaging system 1, 3, 6, 9, 12, and 24 h after intravenous injection of 200 μL of fluorescence nanoparticles (NPF with CY5.5 fluorescence dye) via the tail vein. All mice were sacrificed after 24 h, tumors and major organs (heart, liver, spleen, lungs, and kidneys) were excised, and fluorescence was assessed ex vivo using the Bruker In-vivo imaging system.
In vivo Antitumor Effect
For treatment, the mice were randomly divided into different groups: (1) Control; (2) NPF-NIR; (3) NP@DOX; (4) NPF@DOX; (5) NP@DOX-NIR; and (6) NPF@DOX-NIR. Tumor-bearing mice were intravenously injected with solutions of nanoparticles carrying equivalent DOX contents at 5.0 mg/kg body weight on day 0 through their tail veins. PTT (808 nm, 1.0 W/cm2, 5 min) was performed 6 h after tail vein administration. An infrared thermal camera recorded temperature increases during PTT. Tumor size and mouse weight were measured daily. Following the experiment, the mice were sacrificed via cervical dislocation under anesthesia (12 days post-injection). Tumor tissues and the main organs (heart, liver, spleen, lungs, and kidneys) were harvested for histological analysis. Morphological changes were observed under a microscope.
Results
Nanocarrier Synthesis and Characterization
Figure 1 shows the synthetic route and function of the NPF@DOX nanocarrier. The UV–Vis experiments (Figure S1) showed that NGO had a maximum absorption peak at 230 nm, which is the typical absorption peak of C=O.37 NGO–PEG showed a slightly higher absorbance than NGO. This phenomenon might be attributed to partial changes in the microstructure of the NGO nanoparticles caused by PEG conjugation. Previous studies have also shown that chemical modification of NGO using polyethylene glycol increases NGO thickness.29 The FITC signature peak indicates that fluorescent peptides containing FITC and a FAP recognition sequence were added to the NP surface via π–π stacking.38 DOX loading also occurred via π–π stacking. Direct evidence for DOX adherence was provided by the characteristic absorption peak of DOX at 490 nm in the UV–Vis spectra of NPF@DOX. The stretching vibration of hydroxyl (-OH) is represented by the absorption peak at 3390 cm−1 in the FT–IR spectrum of NPF@DOX (Figure S2), whereas the stretching vibration of C-O-C is represented by the high absorption peak at 1100 cm−1.39 Thus, it can be concluded that NGO and PEG were successfully grafted. Compared to the spectrum of the NP alone, these characteristic peaks were observed for both NPF and NPF@DOX.40 Figure 2A shows that compared to NGO nanoparticles alone, NP had a rough surface with some protuberances due to PEG attaching to the NGO nanoparticles. Small particles, likely FAP-targeting peptides, are also observed on the surface. Furthermore, the π–π stacking bond between the aromatic ring of DOX and the nano-graphene oxide nanoparticles makes the structure approximately porous. The drug loading efficiency of NPF@DOX was calculated to be 44.5 ± 2.1% according to the DOX standard curve (Figure S3). NP possessed comparable properties at this loading ratio because of its structural similarity to NPF. An infrared imager was used to observe the photothermal effect of NPF@DOX and evaluate its photothermal conversion efficiency to verify its potential application in PTT.
 |
Figure 1 The synthetic route and function of the NPF@DOX nanocarriers. The release of DOX was accelerated in the low-pH microenvironment, and the amount of DOX released increased with temperature. |
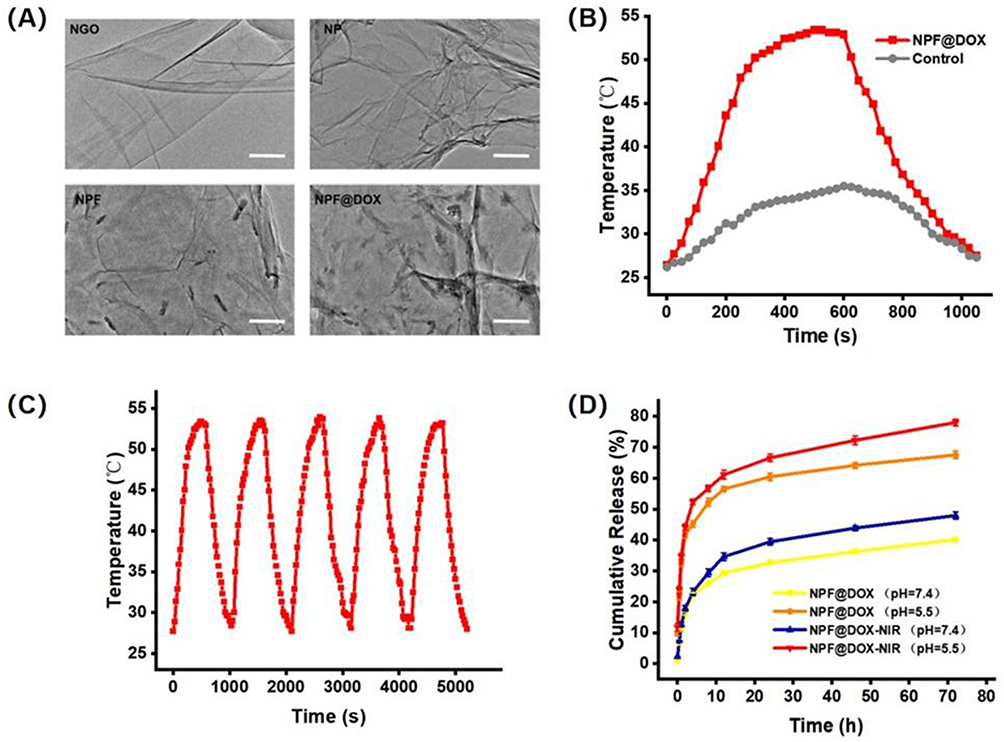 |
Figure 2 Nanocarrier preparation, characterization, and measurements of photothermal effects and stability. (A) TEM images of NGO, NP, NPF, and NPF@DOX nanocarriers. Scale bar: 200 µm. (B) Photothermic heating curves of NPF@DOX under 808 nm irradiation (1.0 W/cm2) followed by cooling to room temperature. (C) Photothermal properties of NPF@DOX throughout five cycles of laser irradiation at 808 nm (1.0 W/cm2). (D) Drug release curves of nanoparticles under pH 7.4 or 5.5 conditions with or without 808 nm NIR. |
As expected, NPF@DOX mediated a concentration-dependent increase in the temperature profile compared to that of NS (Figure S4). Figure 2B shows the temperature changes of NPF@DOX and NS in aqueous solutions under laser irradiation. NPF@DOX reached saturation in 10 min, and the temperature dropped exponentially to nearly normal after irradiation was stopped. However, NS treatment showed no significant increase in temperature. The results demonstrated that the nanocarriers retained their photothermal transfer effect and NGO stability under laser irradiation. The corresponding infrared images are shown in Figure S5, where the temperature of NPF@DOX increased with irradiation time and reached a maximum of about 50 °C after 5 min, which was sufficient to ablate tumor cells for efficient deep-tissue PTT,41 while the temperature of the NS controls only rose 5.8 °C. According to previously reported methods, the photothermal conversion efficiency of NPF@DOX was determined to be approximately 52.48% (Figure S6). No appreciable attenuation was observed in the temperature increase after five cycles of heating and cooling, demonstrating that NPF@DOX is photothermally stable (Figure 2C). To evaluate the photothermal capability of NPF in vivo, tumor-bearing mice were irradiated 6 h after injection of nanoparticles. The mice and temperature variations in the tumors with infrared imaging are shown in Figure S7. The tumor temperature of mice injected with NPF@DOX nanoparticles rose to approximately 49.5 °C after 300 s, which caused local hyperthermia. In contrast, the tumor temperature in the NS controls increased by only 1.6 °C.
We also used UV–Vis spectroscopy to assess the drug release characteristics of DOX-containing NPF. The effects of temperature and pH on drug release were investigated to assess the performance of the thermal/pH dual-stimulus response of the NPF@DOX nanocarriers. Normal tissue and blood are pH 7.4, whereas tumor cells/tissues are a lower pH, while endosomes are pH 5.5–6.0 and lysosomes pH 4.5–5.0.42 Figure 2D depicts the drug release curve in PBS, and NPF@DOX exhibited sustained and slow drug release. At pH 7.4, only approximately 34% of DOX was released from NPF@DOX within 72 h. At pH 5.5, the cumulative DOX released within 72 h was 67%, indicating that the acidic solution increased drug release. The partial dissociation of hydrogen bonds between NGO, DOX, and the -OH and -NH2 groups at pH 5.5 may have accelerated drug release.43 To hydrolyze the adhesive linkage of DOX and control its release behavior, we elevated the temperature of NPF@DOX using NIR irradiation. This addition accelerated DOX release at different pH levels in vitro. As shown in Figure 2D, laser irradiation resulted in an intense increase in DOX release from NPF@DOX after 5 min compared to that from NPF@DOX without NIR irradiation. At pH 7.4 and pH 5.5, 48% and 78% of DOX, respectively, was released within 72 h.
Immunohistochemistry Assay Shows High FAP Expressions in OSCC
FAP expression was notably increased in OSCC tissues compared to levels in controls, and FAP was expressed in both stromal cells and cancer cells near the stromal boundary (Figure 3A). Furthermore, higher FAP expression was observed in CAL-27 cells than in normal cells (Figure 3B). This finding suggests that targeting FAP could improve the specificity of therapy.
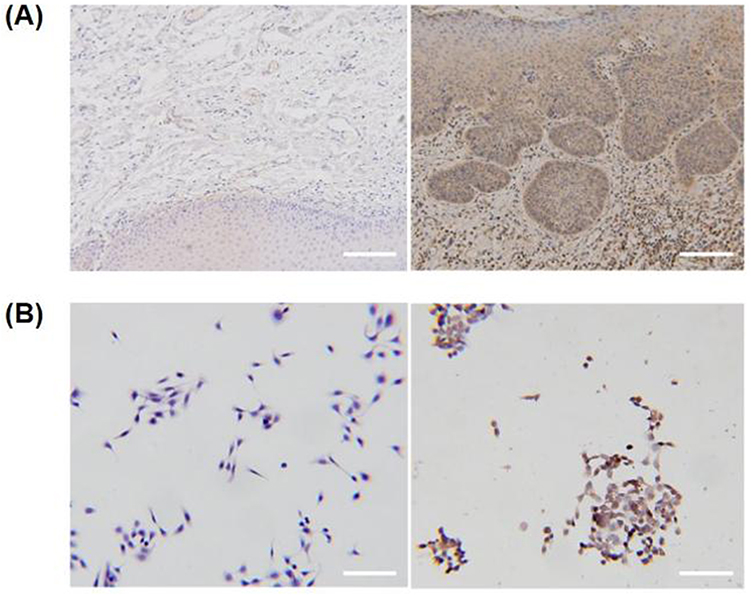 |
Figure 3 FAP is highly expressed in OSCC. (A) Representative immunohistochemistry images of FAP staining in normal and OSCC tissues. Scale bar: 100 µm. (B) Representative immunohistochemistry images of FAP staining in HOK and CAL-27 cells. Scale bar: 50 µm. |
In vitro Cellular Uptake and Drug Release
Effective cellular uptake of nanoparticles is critical for effective therapy. The cellular uptake of NPF functionalized with FITC fluorescent dye was investigated using CLSM. NPF was homogenously distributed in the cytoplasm of CAL-27 cells, indicating nanoparticle uptake. Because DOX is a fluorophore, it can be used to assess the intracellular activity of nanoparticles. Figure 4 shows nanoparticle and DOX accumulation after the cells were incubated with NP@DOX and NPF@DOX. The cells treated with NPF@DOX showed a lower fluorescence intensity, because a small amount of DOX was released into the cells through passive diffusion. In contrast, the cells treated with NP@DOX had significantly more DOX accumulation through targeted endocytosis mediated by FAP-targeting peptides. Cells that received laser irradiation exhibited stronger fluorescence than those that did not. NPF@DOX demonstrated increased cytotoxicity by combining chemotherapy and photothermal therapy than that achieved by chemotherapy alone. Therefore, NPF@DOX was imported into CAL-27 cells through FAP-dependent methods.
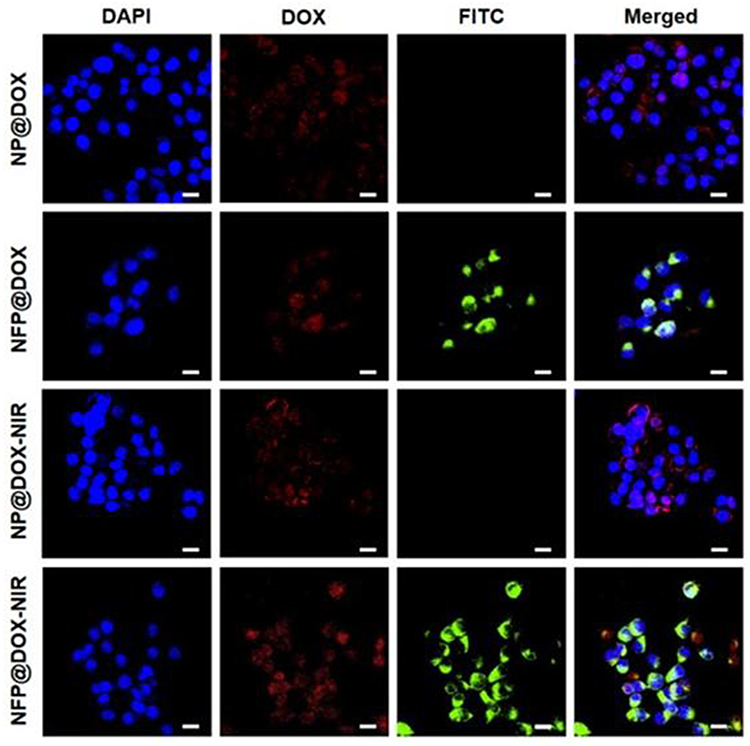 |
Figure 4 In vitro cellular uptake and drug release. CLSM images of CAL-27 cells treated with NP@DOX or NPF@DOX with or without 808 nm NIR irradiation (1.0 W/cm2) to evaluate cellular uptake. Scale bar: 20 µm. |
In vitro Combination Therapy
To study the synergistic anti-tumor effect of chemotherapy and photothermal therapy, the cytotoxicity of NPF@DOX to Cal-27 cells was examined using a CCK-8 assay. As Figure 5A shows, cell survival rates in the NPF-treated group remained above 90%, indicating negligible toxicity of NPF toward the cells. CAL-27 cells treated with NPF were irradiated with an 808 nm laser, and the inhibitory effect was partially enhanced. The results show that NPF has a certain photothermal treatment effect.
 |
Figure 5 Cytotoxicity and in vitro combination therapy. (A) Viability of CAL-27 cells after treatment with control, NPF, NPF-NIR, NP@DOX, NPF@DOX, NP@DOX-NIR, and NPF@DOX-NIR. Data are presented as means ± SD, n=3 (***P<0.001). (B) CAL-27 cells stained with calcein AM (green) and propidium iodide (red) following various treatments were analyzed for live/dead status. Scale bar: 50 µm. |
In addition, the cell survival rates of the NP@DOX-NIR and NPF@DOX-NIR groups decreased to 54% and 42%, respectively, when Cal-27 cells were irradiated, indicating that the antitumor effect of NP@DOX or NPF@DOX combined with NIR was superior to that of chemotherapy alone. In addition, we found that NPF@DOX-NIR showed more notable cytotoxicity than did NPF-NIR. These results suggested that chemotherapy and PTT can more effectively kill cancer cells together.
To investigate the biological safety of the combined PTT and chemotherapy, HOK cells were irradiated with an 808-nm laser or not irradiated, and as shown in Figure S8, the NPF-treated group did not produce obvious toxic effects in the cells. NP@DOX and NPF@DOX had similar cytotoxic effects, which indicated that the cytotoxicity of the FAP-targeting peptides was negligible. Compared with levels in the un-irradiated group, NIR irradiation significantly increased the cytotoxicity of both NPF@DOX and NP@DOX, possibly because the photothermal effect increased DOX release from NP@DOX and NPF@DOX.
To further determine the efficacy of the synergistic treatment, living and dead cells were labeled with green-emitting calcein AM and red-emitting propidium iodide (PI), respectively (Figure 5B). The results above all demonstrate the efficacy of NPF@DOX and NIR in eliminating tumor cells in vitro and the efficacy of combination therapy.
Tumor-Specific Infiltration Using Nanocarrier Fluorescence
Nanocarrier fluorescence was used to image tumor xenografts in vivo and confirm tumor-specific infiltration. The fluorescence images from Cy5.5-loaded nanoparticles (Figure 6A and B) revealed accumulating signals in tumor areas during the observation period. Because this occurred 6 h after administration, we applied radiation at this time point in future experiments. Additionally, we observed fluorescence from renal tissues due to excretion pathways for drug clearance.44 Fluorescence imaging was used to confirm the biological distribution of nanoparticles in vivo and in organs ex vivo. Fluorescence remained at tumor sites 24 h after administration. While fluorescence was also present in the kidneys and hepatic system, it was not found in the lungs, spleen, or heart, indicating a lack of deposition in other tissues.
 |
Figure 6 Tumor-specific infiltration using nanocarrier fluorescence. (A) In vivo imaging of CAL-27 tumor-bearing mice 1, 3, 6, 9, 12, and 24 h after intravenous injection of NPF-Cy5.5 or NS. (B) Ex vivo fluorescence images of NPF in the tumors and major organs/tissues after intravenous injection. |
Synergetic Chemo-Photothermal Therapy in vivo
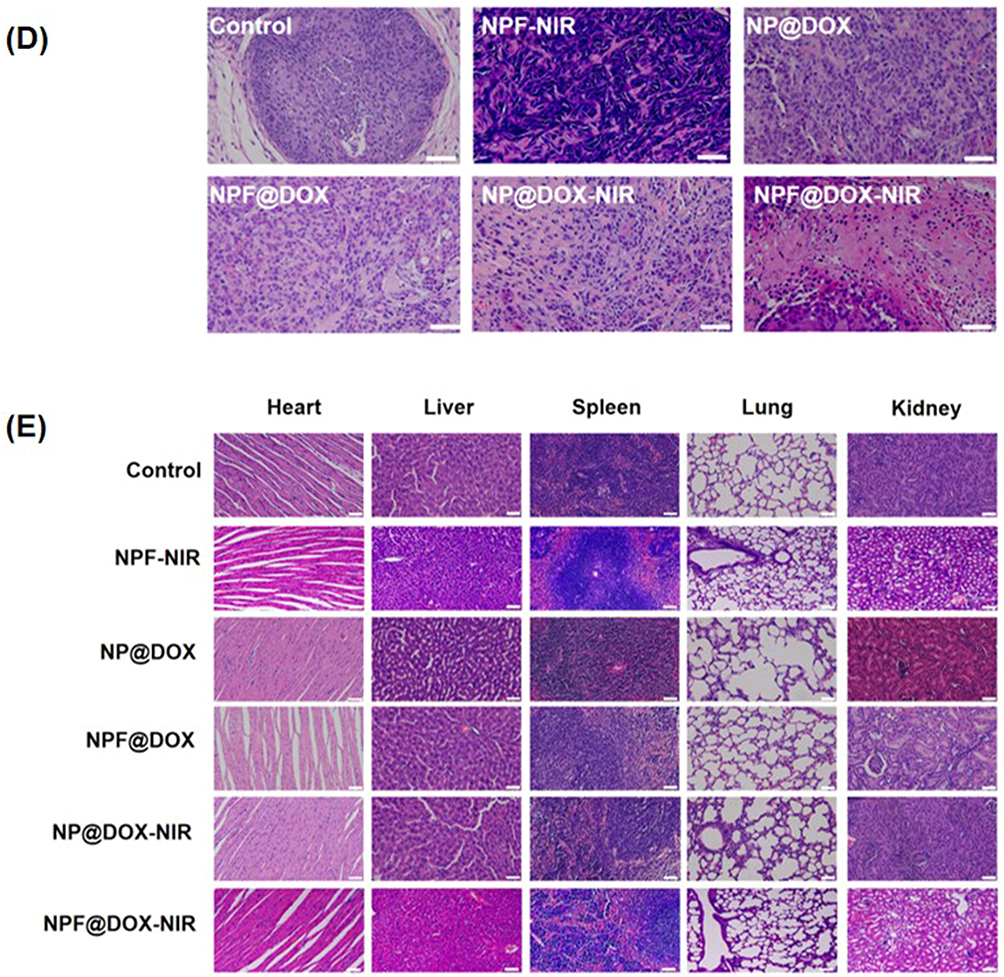
This study was divided into six groups (n = 3) of tumor-bearing mice treated with: NS, NPF with NIR, NP@DOX with and without NIR, and NPF@DOX with and without NIR. For the groups receiving NIR irradiation, the tumor areas were exposed 6 h after injection. Figure 7A and B shows tumor volume changes over time. On day 12, the relative tumor volume increased approximately 4-fold in the NS group. In comparison, mice treated with NPF@DOX showed a degree of inhibited CAL-27 tumor growth. Furthermore, PTT increased the inhibitory effects of NP@DOX and NPF@DOX. On day 12, NPF@DOX nanoparticles plus laser irradiation resulted in nearly complete growth inhibition of CAL-27 tumors. However, the antitumor effect of either NPF@DOX or NPF-nir alone was limited. Body weight change is a primary indicator of treatment side-effects. As shown in Figure 7C, the NPF@DOX nanoparticles had no significant effect on body weight compared with that in the control group. H&E staining was used for histological examination to further investigate the anti-tumor effect of NPF@DOX and evaluate its systemic safety. As illustrated in Figure 7D, tumor tissues treated with NPF@DOX followed by irradiation showed extensive fragmentation and nuclear constriction, demonstrating tumor eradication. Notably, the anti-tumor effects of NPF@DOX and NPF-NIR were far smaller than that of NPF@DOX-NIR. Other nanoparticle-treated, non-laser-irradiated mice, notably in treatment groups lacking FAP-targeting peptides, demonstrated only local tumor destruction and small areas of necrosis, indicating limited therapeutic effects. Figure 7E also shows the H&E staining results from the heart, liver, spleen, lungs, and kidneys after treatment of the control and nanoparticle groups under laser irradiation. The results showed that NPF@DOX nanoparticles had strong stability in circulation. Additionally, the nanoparticles had a strong eradicating effect on tumor tissues and little if any adverse effects on organs.
Figure 7 Continued. Figure 7 In vivo antitumor efficacy of NPF@DOX in BALB/c mice and biosafety analysis. (A) Representative images of tumor-bearing mice taken 12 d post-treatment. The arrows in the images points to the location of the tumor. (B) Tumor volume growth after various treatments. Data are presented as means ± SD, n=3 (***P<0.001) (C) Body weight comparisons of mice following intravenous administration of different treatments in tumor-bearing BALB/c nude mice (mean ± SD) with 3 mice in each group. (D) H&E staining images of excised tumors at the end of treatment. Scale bar: 50 µm. (E) H&E staining images of hearts, livers, spleens, lungs, and kidneys of mice in each group after tail vein injection of nanocarriers. Scale bar: 50 μm.
Discussion
Currently, OSCC is one of the most common oral malignancies. With high morbidity and mortality rates, it remains a major public health problem worldwide.45 Traditional surgery combined with chemoradiotherapy remains the main treatment.2 However, its five-year survival rate is still low because of invasion, metastasis, and recurrence. Moreover, conventional treatment leaves many patients suffering from facial defects, language issues, and difficulty swallowing.46 Therefore, the search for alternative treatments has become of interest among stomatologists. Chemotherapy plays an important role in treating OSCC.2 However, chemotherapies are prone to systemic side effects owing to a lack of tumor-targeting mechanisms.47 Multimodal therapy can compensate for the shortcomings of monotherapies. Additionally, significant progress has been made in developing nanomaterials for medical applications, such as diagnosis, therapy, and imaging-guided treatments.48,49
NGO, a promising nanomaterial, has been widely used in light-assisted therapy and drug delivery in recent years.20,50 Compared with other drug carriers, NGO is capable of increasing drug concentration in tumor sites due to the EPR effect; additionally, the surface area (2600 m2/g) of NGO is four orders of magnitude higher than that of any other nanomaterial used for drug delivery.51 Furthermore, NGO possesses unique optical absorption properties that can convert near-infrared light into heat energy for PTT.52 Taking advantage of this advantage, NGO-DOX-mediated chemo-photothermal therapy in the treatment of cancer has been studied.53 Our previous research54 has successfully applied NGO-based nano-drug delivery systems to kill OSCC epithelial cells in vitro. Based on previous research, we further functionalized NGO with PEG in the present study to increase the solubility and biocompatibility of the nanoparticles,29 while maintaining the photothermal properties of NGO. In this study, NGO accumulates in tumors and absorbs NIR to raise the temperature of the surrounding environment and induce apoptosis of cancer cells. In addition, the covalently bonded PEG coating on NGO also remarkably increased the NIR absorbance of NGO by at least 6-fold, and chemical reduction restored the aromatic and conjugated properties of NGO.55 The results showed that NPF@DOX accumulated in tumor tissue and increased the temperature above the tumor threshold. Therefore, it can be used as a powerful photothermic agent to perform effective tumor ablation in a mouse model to achieve effective synergistic chemo-photothermic therapy. Additionally, the size of a drug-delivery system affects the biodistribution, metabolism, and systemic toxicity of drugs. Small nanomaterials can be cleared after intravenous injection into tumor-bearing mice and show low systemic toxicity. In the present study, NPF@DOX was found to be a promising drug carrier. TEM results demonstrated that the NPF@DOX nanoparticles were small and well dispersed. It has also been reported that PEGylated nano-graphene oxide is a very small sheet with an area of less than 20 nm.56 This indicates that such particles realize the formation of thin-walled nanomaterials through electrostatic interactions and ultrasonic treatment of the final polymer.
In contrast to previously published papers,54 the receptor chosen in the present study was fibroblast activation protein (FAP), a type II integrated membrane glycoprotein. It belongs to the serine proteolytic enzyme family, and has dipeptidase and collagenase activities. FAP plays an important role in the growth, invasion, and metastasis of tumors. FAP is also associated with tumor size, lymph node metastasis, and shortened overall survival.57 In addition, the cytotoxic side effects of DOX, the chemotherapeutic agent, limit its clinical success, while a FAP-activated DOX prodrug minimized such off-target effects.58 It has been shown that FAP is highly expressed in cells of OSCC and this characteristic has led to cell proliferation and metastasis of OSCC via PTEN-PI3K-AKT and Ras-ERK signaling pathways or by inducing epithelial-mesenchymal transition.59,60 In addition, other studies have also shown that high FAP expression in OSCC can decrease expression of the epithelial marker E-cadherin and increase expression of the interstitial marker vimentin, which has similarly promoted the occurrence and development of OSCC.61 In contrast, decreased FAP expression inhibits the growth and metastasis of tumors.62 The immunohistochemistry results of the present study demonstrated that FAP is highly expressed in both the epithelial and interstitial cells of OSCC, suggesting its potential as a target in OSCC diagnosis and treatment. In this study, we observed that the nanoparticle NPF@DOX targets FAP and is effectively endocytosed by cancer cells in vitro. Notably, complete tumor ablation was observed in mice in vivo. This phenomenon could be due to the following factors: (1) more NPF nanoparticles accumulated at tumor sites and tumor cells as a result of the EPR effect and active FAP targeting, while blood vessel fenestration hampered the removal of NPF nanoparticles and extravasation from tumor tissue;63 (2) as a powerful photothermic agent, NPF mediated photoablation of tumor tissue; (3) the heat-stimulated response of NPF@DOX that promoted DOX release and the direct correlation between temperature and tumor cell membrane permeability both increased chemotherapy susceptibility resulting in synergistic chemo-photothermal treatment;64,65 and (4) the lower pH in the tumor microenvironment increased DOX release from NPF carriers and further enhanced the anti-tumor effects of the nanoparticles.
Conclusion
In summary, we demonstrated that FAP is overexpressed in both epithelial cancer cells and cancer-associated stroma cells of OSCC, and it can be used to judge surgical margins more accurately and has certain reference values for early diagnosis, intraoperative navigation, and prognosis prediction. Furthermore, we designed and synthesized a new nano drug delivery system (NPF@DOX), which can be used in synergistic chemo-photothermal treatment. We proved that these nanocarriers had a high drug-loading rate, selectively accumulated in CAL-27 tumor cells, demonstrated controlled drug release, and displayed no visible toxicity in vitro and in vivo at the doses used. The cytotoxic effects induced by FAP-targeted chemotherapy and NIR indicate the potential of NPF@DOX as a targeted delivery system in combination therapy to promote tumor eradication. NPF@DOX represents a simple therapeutic strategy for enhancing synergistic anti-tumor research, and this study indicates its applicability to OSCC treatment.
Abbreviations
CAFs, Cancer-associated fibroblasts; CCK8, Cell Counting Kit-8; DMEM, dulbecco’s modified Eagle’s medium; DOX, doxorubicin; EPR, enhanced permeability and retention; FAP, fibroblast activation protein; HE, hematoxylin and eosin; OSCC, oral squamous-cell carcinoma; PEG, polyethylene glycolNGO, Nano-graphene oxide; PTT, Photothermal therapy; NIR, near-infrared; NS, normal saline.
Acknowledgments
This work was partially supported by Shanxi Province Basic Research Program (202103021223235), Science and Technology Innovation Leader and Key Talent Team Project of Shanxi Province (202204051002034), Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province (20220020), Teaching Reform and Innovation Programs of Higher Education Institutions in Shanxi (J20220404).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Huang SH, O’Sullivan B. Oral cancer: current role of radiotherapy and chemotherapy. Med Oral Patol Oral Cir Bucal. 2013;18(2):e233–e240. doi:10.4317/medoral.18772
3. Peer D, Karp JM, Hong S, et al. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. doi:10.1038/nnano.2007.387
4. Cao M, Shi E, Wang H, et al. Personalized targeted therapeutic strategies against oral squamous cell carcinoma. an evidence-based review of literature. Int J Nanomedicine. 2022;17:4293–4306. doi:10.2147/IJN.S377816
5. Wang G, Zhang M, Cheng M, et al. Tumor microenvironment in head and neck squamous cell carcinoma: functions and regulatory mechanisms. Cancer Lett. 2021;507:55–69. doi:10.1016/j.canlet.2021.03.009
6. Gu F, Hu C, Cao W, et al. Tumor microenvironment multiple responsive nanoparticles for targeted delivery of doxorubicin and CpG against triple-negative breast cancer. Int J Nanomedicine. 2022;17:4401–4417. doi:10.2147/IJN.S377702
7. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–598. doi:10.1038/nrc.2016.73
8. Brennen WN, Isaacs JT, Denmeade SR. Rationale behind targeting fibroblast activation protein-expressing carcinoma-associated fibroblasts as a novel chemotherapeutic strategy. Mol Cancer Ther. 2012;11(2):257–266. doi:10.1158/1535-7163.Mct-11-0340
9. Zhao L, Chen J, Pang Y, et al. Fibroblast activation protein-based theranostics in cancer research: a state-of-the-art review. Theranostics. 2022;12(4):1557–1569. doi:10.7150/thno.69475
10. Feng X, Wang Q, Liao Y, et al. A synthetic urinary probe-coated nanoparticles sensitive to fibroblast activation protein alpha for solid tumor diagnosis. Int J Nanomedicine. 2017;12:5359–5372. doi:10.2147/IJN.S139039
11. Chen WH, Lecaros RL, Tseng YC, et al. Nanoparticle delivery of HIF1α siRNA combined with photodynamic therapy as a potential treatment strategy for head-and-neck cancer. Cancer Lett. 2015;359(1):65–74. doi:10.1016/j.canlet.2014.12.052
12. Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12(11):991–1003. doi:10.1038/nmat3776
13. Fan W, Yung B, Huang P, et al. Nanotechnology for multimodal synergistic cancer therapy. Chem Rev. 2017;117(22):13566–13638. doi:10.1021/acs.chemrev.7b00258
14. Yan F, Duan W, Li Y, et al. NIR-laser-controlled drug release from DOX/IR-780-loaded temperature-sensitive-liposomes for chemo-photothermal synergistic tumor therapy. Theranostics. 2016;6(13):2337–2351. doi:10.7150/thno.14937
15. Li Y, Nie J, Dai J, et al. pH/redox dual-responsive drug delivery system with on-demand RGD exposure for photochemotherapy of tumors. Int J Nanomedicine. 2022;17:5621–5639. doi:10.2147/IJN.S388342
16. Olejniczak J, Carling CJ, Almutairi A. Photocontrolled release using one-photon absorption of visible or NIR light. J Control Release. 2015;219:18–30. doi:10.1016/j.jconrel.2015.09.030
17. Melamed JR, Edelstein RS, Day ES. Elucidating the fundamental mechanisms of cell death triggered by photothermal therapy. ACS Nano. 2015;9(1):6–11. doi:10.1021/acsnano.5b00021
18. Zou L, Wang H, He B, et al. Current approaches of photothermal therapy in treating cancer metastasis with nanotherapeutics. Theranostics. 2016;6(6):762–772. doi:10.7150/thno.14988
19. Navarro-Palomares E, Garcia-Hevia L, Galan-Vidal J, et al. Shiga toxin-B targeted gold nanorods for local photothermal treatment in oral cancer clinical samples. Int J Nanomedicine. 2022;17:5747–5760. doi:10.2147/IJN.S381628
20. Zhou F, Wang M, Luo T, et al. Photo-activated chemo-immunotherapy for metastatic cancer using a synergistic graphene nanosystem. Biomaterials. 2021;265:120421. doi:10.1016/j.biomaterials.2020.120421
21. Zhou T, Zhou X, Xing D. Controlled release of doxorubicin from graphene oxide based charge-reversal nanocarrier. Biomaterials. 2014;35(13):4185–4194. doi:10.1016/j.biomaterials.2014.01.044
22. Liu J, Cui L, Losic D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013;9(12):9243–9257. doi:10.1016/j.actbio.2013.08.016
23. Sattari S, Adeli M, Beyranvand S, et al. Functionalized graphene platforms for anticancer drug delivery. Int J Nanomedicine. 2021;16:5955–5980. doi:10.2147/IJN.S249712
24. Gurunathan S, Han JW, Park JH, et al. Reduced graphene oxide-silver nanoparticle nanocomposite: a potential anticancer nanotherapy. Int J Nanomedicine. 2015;10:6257–6276. doi:10.2147/IJN.S92449
25. Peng C, Hu W, Zhou Y, et al. Intracellular imaging with a graphene-based fluorescent probe. Small. 2010;6(15):1686–1692. doi:10.1002/smll.201000560
26. Yang K, Feng L, Shi X, et al. Nano-graphene in biomedicine: theranostic applications. Chem Soc Rev. 2013;42(2):530–547. doi:10.1039/c2cs35342c
27. Gurunathan S, Kim JH. Synthesis, toxicity, biocompatibility, and biomedical applications of graphene and graphene-related materials. Int J Nanomedicine. 2016;11:1927–1945. doi:10.2147/IJN.S105264
28. Kiew SF, Kiew LV, Lee HB, et al. Assessing biocompatibility of graphene oxide-based nanocarriers: a review. J Control Release. 2016;226:217–228. doi:10.1016/j.jconrel.2016.02.015
29. Liu Z, Robinson JT, Sun X, et al. PEGylated nanographene oxide for delivery of water-insoluble cancer drugs. J Am Chem Soc. 2008;130(33):10876–10877. doi:10.1021/ja803688x
30. Yang HW, Lu YJ, Lin KJ, et al. EGRF conjugated PEGylated nanographene oxide for targeted chemotherapy and photothermal therapy. Biomaterials. 2013;34(29):7204–7214. doi:10.1016/j.biomaterials.2013.06.007
31. Thapa RK, Byeon JH, Ku SK, et al. Easy on-demand self-assembly of lateral nanodimensional hybrid graphene oxide flakes for near-infrared-induced chemothermal therapy. NPG Asia Mater. 2017;9(8):e416–e416. doi:10.1038/am.2017.141
32. Egorin MJ, Hildebrand RC, Cimino EF, et al. Cytofluorescence localization of Adriamycin and daunorubicin. Cancer Res. 1974;34(9):2243–2245.
33. Primeau AJ, Rendon A, Hedley D, et al. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin Cancer Res. 2005;11(24 Pt 1):8782–8788. doi:10.1158/1078-0432.Ccr-05-1664
34. Chlebowski RT. Adriamycin (doxorubicin) cardiotoxicity: a review. West J Med. 1979;131(5):364–368.
35. Rong P, Yang K, Srivastan A, et al. Photosensitizer loaded nano-graphene for multimodality imaging guided tumor photodynamic therapy. Theranostics. 2014;4(3):229–239. doi:10.7150/thno.8070
36. Tian B, Wang C, Zhang S, et al. Photothermally enhanced photodynamic therapy delivered by nano-graphene oxide. ACS Nano. 2011;5(9):7000–7009. doi:10.1021/nn201560b
37. Eda G, Chhowalla M. Chemically derived graphene oxide: towards large-area thin-film electronics and optoelectronics. Adv Mater. 2010;22(22):2392–2415. doi:10.1002/adma.200903689
38. Wang H, Zhang Q, Chu X, et al. Graphene oxide-peptide conjugate as an intracellular protease sensor for caspase-3 activation imaging in live cells. Angew Chem Int Ed Engl. 2011;50(31):7065–7069. doi:10.1002/anie.201101351
39. Wang Y, Liu K, Luo Z, et al. Preparation and tumor cell model based biobehavioral evaluation of the nanocarrier system using partially reduced graphene oxide functionalized by surfactant. Int J Nanomedicine. 2015;10:4605–4620. doi:10.2147/ijn.S82354
40. Madhusudhan A, Reddy GB, Venkatesham M, et al. Efficient pH dependent drug delivery to target cancer cells by gold nanoparticles capped with carboxymethyl chitosan. Int J Mol Sci. 2014;15(5):8216–8234. doi:10.3390/ijms15058216
41. Zhang Y, Song T, Feng T, et al. Plasmonic modulation of gold nanotheranostics for targeted NIR-II photothermal-augmented immunotherapy. Nano Today. 2020;35:100987. doi:10.1016/j.nantod.2020.100987
42. Quader S, Liu X, Toh K, et al. Supramolecularly enabled pH- triggered drug action at tumor microenvironment potentiates nanomedicine efficacy against glioblastoma. Biomaterials. 2021;267:120463. doi:10.1016/j.biomaterials.2020.120463
43. Wang Y, Li L, Li J, et al. Stable and pH-responsive polyamidoamine based unimolecular micelles capped with a zwitterionic polymer shell for anticancer drug delivery. RSC Adv. 2016;6(21):17728–17739. doi:10.1039/c5ra25505h
44. Choi HS, Liu W, Misra P, et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25(10):1165–1170. doi:10.1038/nbt1340
45. Gharat SA, Momin M, Bhavsar C. Oral squamous cell carcinoma: current treatment strategies and nanotechnology-based approaches for prevention and therapy. Crit Rev Ther Drug Carrier Syst. 2016;33(4):363–400. doi:10.1615/CritRevTherDrugCarrierSyst.2016016272
46. Rivera C. Essentials of oral cancer. Int J Clin Exp Pathol. 2015;8(9):11884–11894.
47. Hammann F, Gotta V, Conen K, et al. Pharmacokinetic interaction between taxanes and amiodarone leading to severe toxicity. Br J Clin Pharmacol. 2017;83(4):927–930. doi:10.1111/bcp.13155
48. Lungu II, Grumezescu AM, Volceanov A, et al. Nanobiomaterials used in cancer therapy: an up-to-date overview. Molecules. 2019;24(19):3547. doi:10.3390/molecules24193547
49. Teleanu DM, Chircov C, Grumezescu AM, et al. Contrast agents delivery: an up-to-date review of nanodiagnostics in neuroimaging. Nanomaterials. 2019;9(4):542. doi:10.3390/nano9040542
50. Hoseini-Ghahfarokhi M, Mirkiani S, Mozaffari N, et al. Applications of graphene and graphene oxide in smart drug/gene delivery: is the world still flat? Int J Nanomedicine. 2020;15:9469–9496. doi:10.2147/IJN.S265876
51. Yang K, Zhang S, Zhang G, et al. Graphene in mice: ultrahigh in vivo tumor uptake and efficient photothermal therapy. Nano Lett. 2010;10(9):3318–3323. doi:10.1021/nl100996u
52. Mirrahimi M, Alamzadeh Z, Beik J, et al. A 2D nanotheranostic platform based on graphene oxide and phase-change materials for bimodal CT/MR imaging, NIR-activated drug release, and synergistic thermo-chemotherapy. Nanotheranostics. 2022;6(4):350–364. doi:10.7150/ntno.64790
53. Zhang X, Luo L, Li L, et al. Trimodal synergistic antitumor drug delivery system based on graphene oxide. Nanomedicine. 2019;15(1):142–152. doi:10.1016/j.nano.2018.09.008
54. Li R, Gao R, Zhao Y, et al. pH-responsive graphene oxide loaded with targeted peptide and anticancer drug for OSCC therapy. Front Oncol. 2022;12:930920. doi:10.3389/fonc.2022.930920
55. Robinson JT, Tabakman SM, Liang Y, et al. Ultrasmall reduced graphene oxide with high near-infrared absorbance for photothermal therapy. J Am Chem Soc. 2011;133(17):6825–6831. doi:10.1021/ja2010175
56. Sun X, Liu Z, Welsher K, et al. Nano-graphene oxide for cellular imaging and drug delivery. Nano Res. 2008;1(3):203–212. doi:10.1007/s12274-008-8021-8
57. Wang H, Wu Q, Liu Z, et al. Downregulation of FAP suppresses cell proliferation and metastasis through PTEN/PI3K/AKT and Ras-ERK signaling in oral squamous cell carcinoma. Cell Death Dis. 2014;5(4):e1155. doi:10.1038/cddis.2014.122
58. Huang S, Fang R, Xu J, et al. Evaluation of the tumor targeting of a FAPalpha-based doxorubicin prodrug. J Drug Target. 2011;19(7):487–496. doi:10.3109/1061186X.2010.511225
59. Wang Y, Jing Y, Ding L, et al. Epiregulin reprograms cancer-associated fibroblasts and facilitates oral squamous cell carcinoma invasion via JAK2-STAT3 pathway. J Exp Clin Cancer Res. 2019;38(1):274. doi:10.1186/s13046-019-1277-x
60. Wu QQ, Zhao M, Huang GZ, et al. Fibroblast Activation Protein (FAP) overexpression induces Epithelial-Mesenchymal Transition (EMT) in oral squamous cell carcinoma by Down-Regulating Dipeptidyl Peptidase 9 (DPP9). Onco Targets Ther. 2020;13:2599–2611. doi:10.2147/ott.S243417
61. Nijkamp MM, Span PN, Hoogsteen IJ, et al. Expression of E-cadherin and vimentin correlates with metastasis formation in head and neck squamous cell carcinoma patients. Radiother Oncol. 2011;99(3):344–348. doi:10.1016/j.radonc.2011.05.066
62. Bughda R, Dimou P, D’Souza RR, et al. Fibroblast activation protein (FAP)-Targeted CAR-T cells: launching an attack on tumor stroma. Immunotargets Ther. 2021;10:313–323. doi:10.2147/itt.S291767
63. Shi Y, van der Meel R, Chen X, et al. The EPR effect and beyond: strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics. 2020;10(17):7921–7924. doi:10.7150/thno.49577
64. Wang H, Zhou J, Fu Y, et al. Deeply infiltrating iRGD-graphene oxide for the intensive treatment of metastatic tumors through PTT-mediated chemosensitization and strengthened integrin targeting-based antimigration. Adv Healthc Mater. 2021;10(16):e2100536. doi:10.1002/adhm.202100536
65. Bardajee GR, Hooshyar Z, Farsi M, et al. Synthesis of a novel thermo/pH sensitive nanogel based on salep modified graphene oxide for drug release. Mater Sci Eng C Mater Biol Appl. 2017;72:558–565. doi:10.1016/j.msec.2016.11.109
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.