Back to Journals » Nature and Science of Sleep » Volume 8
A pilot study of serotonin-1A receptor genotypes and rapid eye movement sleep sensitivity to serotonergic/cholinergic imbalance in humans: a pharmacological model of depression
Authors Biard K, Douglass A, Robillard R ![]() , De Koninck J
, De Koninck J ![]()
Received 18 August 2015
Accepted for publication 19 October 2015
Published 18 December 2015 Volume 2016:8 Pages 1—8
DOI https://doi.org/10.2147/NSS.S94549
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Steven Shea
Kathleen Biard,1,2 Alan B Douglass,2,3 Rébecca Robillard,2 Joseph De Koninck1,2
1School of Psychology, University of Ottawa, 2University of Ottawa Institute for Mental Health Research, 3Royal Ottawa Mental Health Center, University of Ottawa Institute for Mental Health Research, Ottawa, ON, Canada
Rationale: The serotonergic and cholinergic systems are jointly involved in regulating sleep but this system is theorized to be disturbed in depressed individuals. We previously reported that cholinergic and serotonergic agents induce sleep changes partially consistent with monoamine models of sleep disturbances in depression. One potential cause of disturbed neurotransmission is genetic predisposition. The G(-1019) allele of the serotonin-1A (5-HT1A) receptor promoter region predicts an increased risk for depression compared to the wild-type C(-1019) allele.
Objective: The goal of this study was to investigate how serotonin-1A receptor genotypes mediate sleep sensitivity to pharmacological probes modeling the serotonergic/cholinergic imbalance of depression.
Methods: Seventeen healthy female participants homozygous for either C (n=11) or G (n=6) alleles aged 18–27 years were tested on four nonconsecutive nights. Participants were given galantamine (an anti-acetylcholinesterase), buspirone (a serotonergic agonist), both drugs together, or placebos before sleeping.
Results: As reported previously, buspirone significantly increased rapid eye movement (REM) latency (P<0.001), as well as awakenings, percentage of time spent awake, and percentage of time asleep spent in stage N1 (P<0.019). Galantamine increased awakenings, percentage of time spent awake, percentage of time asleep spent in stage N1, and percentage of time asleep spent in REM, and decreased REM latency and percentage of time asleep spent in stage N3 (P<0.019). Galantamine plus buspirone given together disrupted sleep more than either drug alone, lowering sleep efficiency and percentage of time asleep spent in stage N3 and increasing awakenings, percentage of time spent awake, and percentage of time asleep spent in stage N1 (P<0.019). There was no main effect of genotype nor was there a significant multivariate interaction between genotype and drug condition.
Conclusion: These findings are partially consistent with the literature about sleep in depression, notably short REM latency, higher percentage of total sleep time spent in REM, lower percentage of time asleep spent in stage N3, and increased sleep fragmentation. The C/G mutation in the serotonin-1A receptor promoter region does not appear to cause noticeable differences in the sleep patterns of a relatively small sample of healthy young females. Future studies with larger sample sizes are required.
Keywords: depression, sleep, serotonin, acetylcholine, buspirone, galantamine
Introduction
While it has long been known that genetics have some effect on the risk of mood disorders, recent genetic research has begun to pinpoint specific genes implicated in major depression. One such genetic risk factor is the allelic variation of the serotonin-1A (5-HT1A) receptor.1 The G(-1019) allele of the 5-HT1A receptor predicts an increased risk for depression compared to the wild-type C(-1019) allele. This effect appears to be even stronger for persons homozygous for the “G” allele while homozygosity for the “C” allele appears to confer some protection against depression. Pharmacological probes could be used to gain a clearer picture of the link between a serotonergic/cholinergic imbalance and abnormalities in sleep structure2,3 in individuals with different 5-HT1A receptor genotypes.
The gene that codes for the 5-HT1A receptor is found on chromosome 5q11.2-113.4 There are several low frequency single nucleotide polymorphisms (SNPs) that have been found on this gene but the more common C(-1019)G SNP has been associated with anxious and depressive traits as well as substance abuse, anxiety disorders, depression, psychosis, and suicidality.5–8 The C(-1019)G SNP is in an upstream, regulatory region of the gene and modulates 5-HT1A transcription; while the C allele correctly binds to repressing regulatory factors, the G allele insufficiently represses transcription of the 5-HT1A receptor, some of which are found in postsynaptic sites while others are autoreceptors on 5-HT neurons. An excess of autoreceptors would be expected to lead to overall reduced serotonergic neurotransmission.9,10
5-HT has been heavily implicated in the pathophysiology of depression and its effects on rapid eye movement (REM) sleep are also well known. REM sleep is inhibited by the serotonergic neurons originating from the dorsal raphe nucleus which project to the cholinergic cells of the pontine nuclei11 where rems are generated. Here, 5-HT acts primarily at the 5-HT1A receptor to suppress REM sleep and reduce percentage of time asleep spent in REM (REM%).12
Depression has also been linked to cholinergic hypersensitivity.13 Importantly, the cholinergic system is involved in the regulation of REM sleep.14 Cholinomimetics have been found to shorten REM latency (RL: the amount of time it takes to enter REM sleep after sleep onset) and increase REM density (RD: the number of rems per minute of REM sleep), effects that have been found to be stronger in depressed patients than in controls.15 Consequently, the sensitivity of REM sleep to cholinergic stimulus could be a trait marker for depression.
Using pharmacological probes in healthy adults, we recently reported that a serotonergic agonist increases RL, while a promoter of cholinergic activity shortens RL and increases REM sleep duration.16 These changes in REM sleep are well-aligned with the potential role of monoamine deregulation in the emergence of sleep disturbances in depression. Recent longitudinal studies have found that abnormal REM sleep measures such as shorter RL and elevated RD can predict the onset and course of depression in adolescents and adults.17,18 It is possible that these REM alterations are not necessarily caused by depressive episodes but may reflect an underlying neuropathology; the neural mechanisms governing sleep regulation are closely linked with those regulating cognitive and affective systems.19 However, not all patients with mood disorders display these REM sleep abnormalities. It may be postulated that specific genetic variations, such as the C(-1019)G SNP in the case of the 5-HT1A receptor, could modulate the association between REM alterations and the risk for depression.
The main objective of the present study was to determine whether 5-HT1A genotype may affect REM sleep sensitivity to cholinergic and serotonergic manipulations. We aimed to gain some insight into some of the possible genotypic and sleep-related pathophysiological mechanisms which could be linked to the risk for depression. We used a 5-HT1A agonist (buspirone) to suppress REM sleep and a drug that increases acetylcholine (galantamine) to increase REM sleep.
Buspirone is a 5-HT1A agonist most commonly used either as an anxiolytic or an antidepressant, and sometimes given in conjunction with other antidepressants to augment their effects.20,21 When it is administered orally, it suppresses REM sleep, suggesting that it acts more strongly at the postsynaptic 5-HT1A inhibitory receptors in the laterodorsal tegmental and pedunculopontine nuclei than at the autoreceptors in the dorsal raphe nucleus (which would be expected to reduce 5-HT transmission and increase REM).22 Buspirone also increases sleep fragmentation. Because it reliably suppresses REM and is well tolerated in human subjects, buspirone was chosen as the serotonergic drug for this study.
For our cholinergic drug, we used galantamine, commonly prescribed for the management of mild to moderate dementia in Alzheimer’s disease. Galantamine is a short-acting anti-acetylcholinesterase that enhances central cholinergic activity. Acetylcholine is an agonist at muscarinic receptors (M1 and M2) whereas galantamine is also an allosteric modulator of nicotinic receptors.14,23 Animal studies have found that tonic REM (electroencephalography (EEG) changes and atonia) is primarily mediated by muscarinic receptors while phasic ponto-geniculo-occipital waves (associated with rem) are mediated by nicotinic receptors.24 Previously, Riemann et al14 found that galantamine taken before sleeping shortened RL, increased RD, and suppressed slow-wave sleep in healthy participants, and recommended its use for future pharmacological challenge studies. Galantamine is now available in a long-acting form that is ideal for sleep research and was used as the cholinergic probe in this study.
To model the genetic predisposition for depression, we focused on healthy participants with two genotypes: the homozygous “protective” CC alleles and the homozygous “at-risk” GG alleles. Our hypothesis was that the GG participants would be less affected by the serotonergic drug due to impaired 5-HT neurotransmission and more strongly affected by the cholinergic drug. More specifically, we predicted that:
- Buspirone would have a stronger effect on REM sleep in the CC group than in the GG group due to a better serotonergic tone in the CC group.
- There would be an interaction effect of the two genotype groups with galantamine in that this drug would increase RD more prominently in the GG group than in the CC group, illustrating reduced serotonergic tone at the laterodorsal tegmental and pedunculopontine nuclei (the generator of bursts of rems) in the GG group.
- There would be an interaction effect of the two genotype groups with combined galantamine plus buspirone. In the combined drug condition, the GG group would have a higher RD, shorter RL, and an increased REM percentage compared to the CC group, illustrating their normal sensitivity to cholinergic drive but reduced serotonergic tone despite the administration of 5-HT1A agonist. This aimed to test the serotonergic/cholinergic balance theory of depression: participants with weaker serotonergic systems were expected to be more affected by the cholinergic challenge.
Methods
Participants
Seventeen female participants aged 18–27 years were recruited through the University of Ottawa’s online recruitment system, the Integrated System for Participation in Research, and through notices in local classified advertisements. Participants were chosen from the same sex and age group in order to reduce variance in the dependent variables. Participants were asked to fill out the Sleep Disorders Questionnaire25 as a prescreening measure for sleep disorders, as well as the Beck Depression Inventory 2 (BDI-2).26 Participants were also asked about their general health and their immediate family’s psychiatric history to determine eligibility. Exclusion criteria included a family history of major mental illness, a score above 12 on the BDI-2,27 current use of psychotropic drugs, significant health problems, or pregnancy/lactation. The 5-HT1A G(-1019)C polymorphism genotype was determined using Oragene-DNA (OG-250) saliva sample collection28 and only homozygous participants were included: eleven “CC” and six “GG” participants. The two groups did not significantly differ by Student’s t-test in age or BDI scores: the CC participants had a mean age of 20.5 years (standard deviation [SD] 2.4) and a BDI-2 of 5.4 (SD 3.5), while the GG group had a mean age of 19.5 years (SD 2.3) and a BDI-2 of 2.5 (SD 3.3).
Eligible participants were screened for psychiatric illness using the Structured Clinical Interview for DSM-IV, Non-Patient version.29 They were assessed by a physician prior to their participation via physical examination, measurement of blood pressure and pulse rate in the sitting position, and clinical laboratory tests (LDH, SGOT, bilirubin, serum creatinine, thyroid FT3, TSH, urinalysis, and CBC including platelets). Participants with normal results on these measures underwent a screening nocturnal polysomnogram, which ruled out sleep disorders such as sleep apnea or periodic limb movement disorder. Sleep EEG was recorded by a ten-electrode array (C3-A2, C4-A1, O1-A2, O2-A1, EOG, EKG (lead-II), and EMG), as well as a microphone to detect snoring, a motion sensor, finger pulse oximetry, respiration belts, pressure and temperature airflow sensors on the upper lip, and electrodes on the anterior tibial areas for leg movements. This nocturnal polysomnogram included 8 hours of EKG on two channels, so it also served to exclude any participants with cardiac arrhythmias or other EKG abnormalities.
The screening nocturnal polysomnogram also served as an acclimatization to the sleep laboratory to eliminate the “first-night effect” from the subsequent drug trial nights.30 Participants were asked not to use any drugs, alcohol, or caffeine (after noon) for the duration of the study. Strenuous physical activity was not permitted within 4 hours of bedtime on sleep study nights to avoid possible induction of sleep initiation disturbances.
Procedure
Participants were recorded for four nonconsecutive study nights in the sleep laboratory. On each of the study nights, participants received two identical capsules at bedtime in a counter-balanced randomized double-blind design: 1) one 16 mg capsule of galantamine (slow release) and one placebo (sugar pill); 2) one 15 mg capsule of buspirone and one placebo capsule; 3) one capsule of galantamine plus one of buspirone; or 4) two capsules of the placebo. The order of the drug conditions was randomized with a latin square design to avoid order effects of the drug conditions. The capsules were taken orally 1 to 2 hours before bedtime and at least 2 hours after the last meal for optimal timing and absorption.
Galantamine has in the past been sold commercially in both immediate-release and slow-release forms. The geriatric starting dose is either 8 or 16 mg for slow-release galantamine.31 Since our participants were all healthy young adults, the slow-release formula was selected in order that its effects on REM sleep would still be occurring in the latter part of the night and the 16 mg dose was chosen to maximize the potential response.14
Buspirone’s suggested therapeutic dose is 20–30 mg in two to three divided doses, mainly to avoid daytime sedation. Starting dose is suggested at 5 mg tid to avoid sedation.31 Since sedation is not an issue in a sleep study and since we needed to guarantee measureable effects throughout the night, a dose of 15 mg was selected.
The half-lives of the drugs (galantamine =8 hours, buspirone =2–10 hours with a mean of 2.8 hours) required a 48-hour washout period after each night in the sleep lab in order to allow five half-lives to elapse before the next test drug.31 Therefore, the participants slept for at least one night at home between each of the lab nights 2, 3, 4, and 5 (median number of nights at home between study nights was 7, mean 11.2, SD 12.4). Plasma concentrations of the drugs were not measured but the half-lives of the drugs are sufficiently long that the effects would persist for most of the night. Both drugs have been used in previous sleep studies and have demonstrated acute, one-time effects on sleep measures.
Participants were self-reported good sleepers with regular sleep schedules. To avoid circadian misalignment, they chose their bedtimes between 9 pm and midnight according to when they usually fell asleep at home and awoke between 6 am and 8 am according to their preferences and routines.
Sleep methodology
On nights two through five, EEG, EMG, EKG, and EOG electrodes were used, with an acquisition rate of 200 Hz. Sleep was recorded on the Somnologica® computerized sleep recording system (Embla Systems, Ottawa, Canada) using digital amplification and American Academy of Sleep Medicine recommended filters (EEG and EOG: low 0.3 Hz, high 35 Hz; ECG: low 0.3 Hz, high 70 Hz; EMG: low 10 Hz, high 100 Hz). Sleep stages were scored visually on 30 second epochs from the C3/A2 and O2/A1 electrode traces by a single experienced researcher (KB) using standard American Academy of Sleep Medicine criteria32 with the Stellate Harmonie® system, version 6.1 (Stellate Systems, Montreal, Canada). Harmonie uses published algorithms to identify individual rems.33
The analyzed sleep variables were: sleep onset latency, RL, RD, total sleep time, percentage of time spent awake (Wake%), percentage of time asleep spent in the non-REM stages N1, N2, and N3 (N1%, N2%, N3% respectively), percentage of time asleep spent in REM sleep (REM%), number of awakenings during the night (awakenings), and sleep efficiency (SE).
Ethics approval was granted by the Research Ethics Board of the Royal Ottawa Mental Health Center, the University of Ottawa Faculty of Social Sciences and Humanities, and experimental administration of the drugs was approved by Health Canada (REB#2008–019). Written, informed consent was obtained from all participants. The study ran from June 2010 to January 2014. Polysomnographic data for the entire sample of participants (irrespective of genotype) was previously published in a report on the effects of galantamine and buspirone on sleep.16
Statistical analysis
All variables were assessed for normal distribution via normality plots and measures of kurtosis and skew using the SPSS statistical program (IBM Corporation, Armonk, NY, USA). Variance stabilizing transformations were done if the plot showed significant deviation from a normal distribution (logit and natural log transforms were used) and if the z scores of the skew or kurtosis were higher than 1.96. The nonnormal variables were: sleep onset latency, Wake%, N1%, Awakenings, and SE. These variables were normalized with a natural log transform except for SE, which required a logit transform.
These data were submitted to a mixed design, repeated measures multivariate analysis of variance (MANOVA), with independent genotype groups and repeated measures over drug conditions. A second analysis was done using night order instead of drug condition to check for order effects.
Results
The main effects of drug conditions across the entire sample of participants (ie, irrespective of genotype) are published elsewhere.16 Averages and univariate results taking in account genotypes are presented in Table 1. In brief, consistent with previous analyses, the mixed design repeated-measures MANOVA (with repeated measures across drug conditions and between measures of genotype groups) used in the present study revealed a significant effect of drug condition on sleep (F=3.575, P<0.001). Compared to placebo, buspirone increased Awakenings, Wake%, N1%, and RL (all P<0.019), while galantamine increased Awakenings, Wake%, N1%, and REM%, and decreased percentage of time asleep spent in stage N3 and RL (all P<0.019). Compared to placebo, the combination of galantamine plus buspirone lowered SE and percentage of time asleep spent in stage N3, and increased Awakenings, Wake%, and N1% (all P<0.019). No order effects were found.
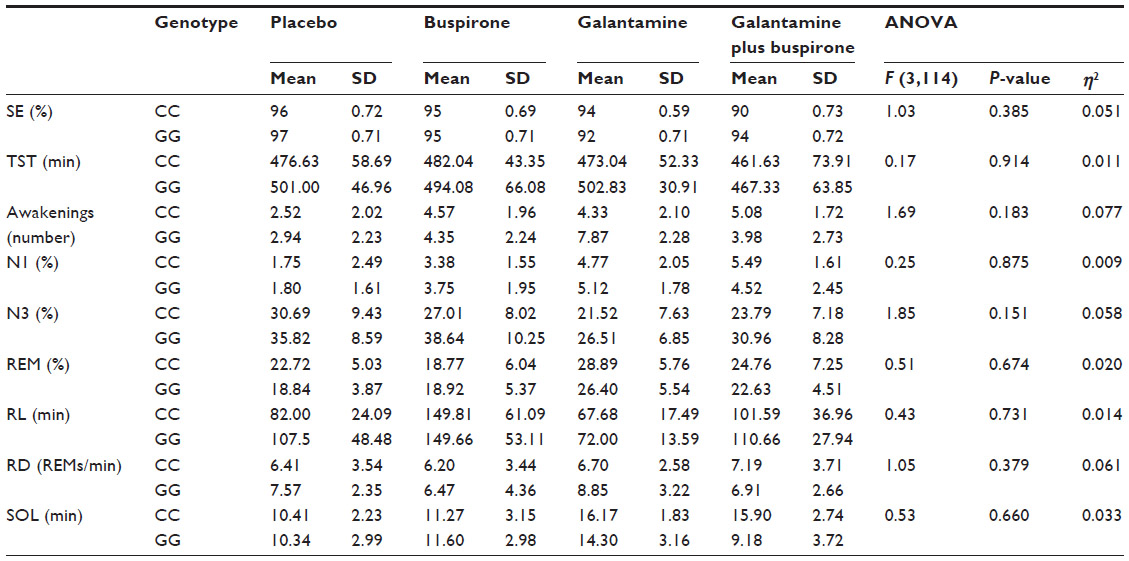 | Table 1 Results from the univariate genotype-by-drug interaction post hoc analyses |
The mixed design MANOVA showed no significant main effect of genotype on sleep measures (F=0.456, P=0.869). There was no significant interaction between drug condition and genotype at the multivariate level (F=1.053, P=0.409). At the univariate level, repeated-measure ANOVAs indicated a significant interaction between drug and genotype on N2% (P<0.01) and Wake% (P<0.05) as shown in Figures 1 and 2. N2% was significantly higher in the CC wild-type group compared to the GG group on both the buspirone night and the buspirone plus galantamine night; Wake% was significantly higher in the GG group than in the CC group on the galantamine night, but higher in the CC group on the galantamine plus buspirone night.
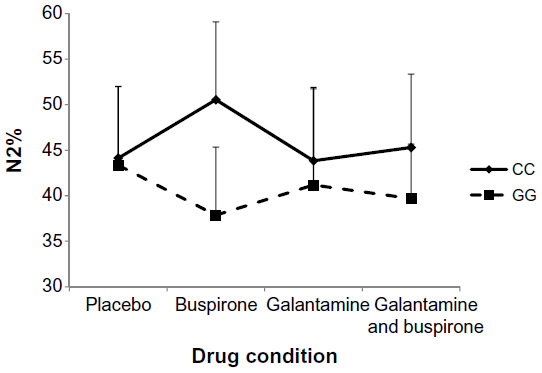 | Figure 1 Percentage of sleep spent in stage N2 in wild-type (CC) and mutant (GG) participants (interaction P=0.010, η2=0.215) with standard deviation error bars. |
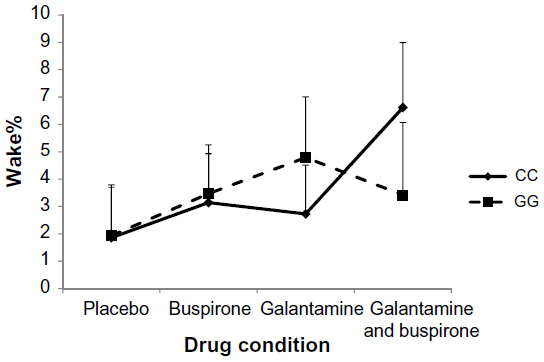 | Figure 2 Percentage of time in bed spent awake (Wake%) in wild-type (CC) and mutant (GG) participants (interaction P=0.045, ·2=0.117) with standard deviation error bars. |
Discussion
The current study identified no marked differences in sleep structure between 5-HT1A genotypes. This could be due to the small sample size and the expected subtlety of an effect of genotype. While it is possible that a larger sample might highlight small differences, it is also likely that the C/G mutation is not enough to cause noticeable differences in the sleep patterns of healthy young adults. For instance, sleep may be sensitive to combinations of genotypic factors with other risk factors or systemic stressors.
Similarly, we did not find any multivariate interaction between the drug conditions and genotypes. Two of the univariate post hoc ANOVAs indicated significant interactions: N2% and Wake%. Buspirone appeared to increase the proportion of sleep spent in stage N2 more in the CC wild-type group than in the GG group. While this could be interpreted as being due to buspirone more easily affecting the intact serotonergic system in the CC group and therefore having a stronger effect on those participants, it bears repeating that the overall multivariate interaction was not significant and this effect was not seen in other measures previously shown to be affected by buspirone (such as RL). The Wake% interaction is harder to interpret, with galantamine appearing to disrupt sleep in the GG group more than in the CC group, but the buspirone plus galantamine drug night having a stronger effect on the wild-type CC participants.
Buspirone appears to affect participants with the GG polymorphism in a similar way to those with CC polymorphism, or at least well enough that the difference in the effect on REM and slow-wave sleep structure is negligible. This is an encouraging finding: if buspirone had noticeably less effect on individuals with the GG genotype, this would have suggested that patients should be tested for their 5-HT1A receptor genotype before using buspirone as part of their treatment plan, to adjust dosage.
The effects of the drugs on sleep structure are discussed more in depth in our previous article,16 but it is worth noting here that both drugs affected sleep as predicted by current models of the serotonergic/cholinergic imbalance in depression. Our serotonergic agonist, buspirone, suppressed REM sleep by increasing RL. Our cholinergic agent, galantamine, led to shallow sleep as reflected by a higher proportion of time spent awake, more numerous Awakenings, and less stage N3 ‘deep’ slow-wave sleep.
Galantamine and buspirone given together tended to cancel out each other’s effects on REM sleep latency and REM% while their deleterious effects on sleep quality appeared to be additive: both drugs increased sleep fragmentation, leading to a higher percentage of time in bed spent awake. The higher cholinergic tone led to more REM sleep, similar to the finding that depressed patients have a higher percentage of total sleep time spent in REM. The way buspirone counteracted this effect was similar to how antidepressants act in depressed patients: by lowering the proportion of sleep spent in REM. With regard to the C(1019)G polymorphism, increased cholinergic drive on the galantamine plus buspirone night was expected to overwhelm impaired serotonergic systems in those participants with the GG polymorphism but this effect was not shown. This suggests that 5-HT1A receptors may possibly not be as integral to sleep regulation as other serotonergic and cholinergic mechanisms. Performing a similar push–pull experiment with cholinergic and serotonergic drugs in populations with different relevant mutations might prove to be more effective in finding an interaction between genotype and cholinergic/serotonergic imbalance effects on sleep.
In addition to the small sample size, one of this study’s limitations is the restricted demographic characteristics of the participants. They were all healthy young women, so these findings may not be generalizable to older, male, or patient populations. Future studies in this area should replicate this design in a clinical population, such as in patients with major depressive disorders, in order to see the effect of the drugs on participants whose sleep is endogenously affected by a serotonergic–cholinergic imbalance.
Conclusion
While the C(1019)G 5-HT1A receptor genotype is a risk factor for depression, it had limited effects on sleep structure in the healthy females in our experimental design. REM sleep parameters of mutant and wild-type participants did not differ when taking the 5-HT1A-targeting buspirone or when taking the cholinergic agent galantamine, and only univariate trends were found for other sleep variables. However, because this small pilot study had limited statistical power to assess the above sleep effects, it is possible that larger studies might find that the G/G mutation does have a small but statistically significant effect. Future research into genetic risk factors for mood disorders should investigate the effects on sleep structure of this and other mutations in the 5-HT system in order to better our understanding of biomarkers for depression.
Acknowledgment
This research was supported by the University of Ottawa Institute of Mental Health Research. The authors would like to acknowledge and thank Dr Paul Albert and technician Mireille Daigle from the Ottawa Hospital Research Institute (ON, Canada) for their expertise in genetic sequencing, Dr Roseanne Armitage for help in the preparation of this manuscript, and Lorelle Weiss and Elena Drozd for their help in collecting the data.
Disclosure
Dr Robillard received a postdoctoral training award from the Fonds de recherche du Québec – Santé. The authors have no other conflicts of interest to disclose.
References
Stockmeier CA, Shapiro LA, Dilley GE, Kolli TN, Friedman L, Rajkowska G. Increase in serotonin-1A autoreceptors in the midbrain of suicide victims with major depression-postmortem evidence for decreased serotonin activity. J Neurosci. 1998;18(18):7394–7401. | |
Janowsky DS, Davis JM, El-Yousef MK, Sekerke HJ. A cholinergic-adrenergic hypothesis of mania and depression. Lancet. 1972;300(7778):632–635. | |
Jouvet M. The role of monoamines and the acetylcholine-containing neurons in the regulation of the sleep-waking cycle. Rev Physiol Biochem Pharmacol. 1972;64:166–307. | |
Fargin A, Raymond JR, Lohse MJ, Kobilka BK, Caron MG, Lefkowitz RJ. The genomic clone G-21 which resembles a beta-adrenergic receptor sequence encodes the 5-HT1A receptor. Nature. 1988;335(6188):358–360. | |
Erdmann J, Shimron-Abarbanell D, Cichon S, et al. Systematic screening for mutations in the promoter and the coding region of the 5-HT1A gene. Am J Med Genet. 1995;60(5):393–399. | |
Nakhai B, Nielsen DA, Linnoila M, Goldman D. Two naturally occurring amino acid substitutions in the human 5-HT1A receptor: glycine 22 to serine 22 and isoleucine 28 to valine 28. Biochem Biophys Res Commun. 1995;210(2):530–536. | |
Kawanishi Y, Harada S, Tachikawa H, Okubo T, Shiraishi H. Novel mutations in the promoter and coding region of the human 5-HT1A receptor gene and association analysis in schizophrenia. Am J Med Genet. 1998;81(5):434–439. | |
Lesch KP, Gutknecht L. Focus on The 5-HT1A receptor: emerging role of a gene regulatory variant in psychopathology and pharmacogenetics. Int J Neuropsychopharmacol. 2004;7(4):381–385. | |
Lemonde S, Turecki G, Bakish D, et al. Impaired repression at a 5-hydroxytryptamine 1A receptor gene polymorphism associated with major depression and suicide. J Neurosci. 2003;23(258):8788–8799. | |
Koller G, Bondy B, Preuss UW, Zill P, Soyka M. The C(-1019)G 5-HT1A promoter polymorphism and personality traits: no evidence for significant association in alcoholic patients. Behav Brain Funct. 2006;2:7. | |
Jun L, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441(7093):589–594. | |
Palagini L, Baglioni C, Ciapparelli A, Gemignani A, Riemann D. REM sleep dysregulation in depression: state of the art. Sleep Med Rev. 2013;17:377–390. | |
Dilsaver SC. Cholinergic mechanisms in depression. Brain Res Rev. 1986;11(3):285–316. | |
Riemann D, Hohagen F, Bahro M, et al. Cholinergic neurotransmission, REM sleep and depression. J Psychosom Res. 1994;38(Suppl 1):15–25. | |
Riemann D, Hohagen F, Krieger S, et al. Cholinergic REM induction test: muscarinic supersensitivity underlies polysomnographic findings in both depression and schizophrenia. J Psychiat Res. 1994;28(3):195–210. | |
Biard K, Douglass AB, De Koninck J. The effects of galantamine and buspirone on sleep structure: implications for understanding sleep abnormalities in major depression. J Psychopharmacol. 2015;29(10):1106–1111. | |
Modell S, Ising M, Holsboer F, Lauer C. The Munich vulnerability study on affective disorders: premorbid polysomnographic profile of affected high-risk probands. Biol Psychiatry. 2005;58(9):694–699. | |
Augustinavicius J, Zanjani A, Zakzanis K, Shapiro C. Polysomnographic features of early-onset depression: a meta-analysis. J Affect Disord. 2014;158:11–18. | |
Riemann D, Berger M, Voderholzer U. Sleep and depression – results from psychobiological studies: an overview. Biol Psychol. 2001;57(1–3):67–103. | |
Altamura AC, Moliterno D, Paletta S, Maffini M, Mauri MC, Bareggi S. Understanding the pharmacokinetics of anxiolytic drugs. Expert Opin Drug Metab Toxicol. 2013;9(4):423–440. | |
Robinson DS, Rickels K, Feighner J, et al. Clinical effects of the 5-HT1A partial agonists in depression: a composite analysis of buspirone in the treatment of depression. J Clin Psychopharmacol. 1990;10(3 Suppl):67S–76S. | |
Wilson SJ, Bailey JE, Rich AS, et al. The use of sleep measures to compare a new 5HT1A agonist with buspirone in humans. J Psychopharmacol. 2005;19(6):609–613. | |
Jiang P, Kasarskis A, Winrow CJ, Renger JJ, Turek FW. A systems biology approach for uncovering the genetic landscape for multiple sleep-wake traits. In: Shaw P, Tafti M, Thorpy M, editors. Genetic Basis of Sleep and Sleep Disorders. Cambridge, England: Cambridge University press; 2013:104–118. | |
Gillin JC, Salin-Pascual R, Velazquez-Moctezuma J, Shiromani P, Zoltoski R. Cholinergic receptor subtypes and REM sleep in animals and normal controls. Prog Brain Res. 1993;98(46):379–387. | |
Douglass AB, Bornstein R, Nino-Murcia G, et al. The sleep disorders questionnaire I: creation and multivariate structure of SDQ. Sleep. 1994;17:160–167. | |
Beck AT, Steer RA, Ball R, Ranieri W. Comparison of Beck depression inventories-IA and -II in psychiatric outpatients. J Pers Assess. 1996;67(3):588–597. | |
Kjaergaard M, Wang C, Waterloo K, Jorde R. A study of the psychometric properties of the Beck depression inventory-II, the Montgomery and Asberg depression rating scale, and the hospital anxiety and depression scale in a sample from a healthy population. Scand J Psychol. 2014;55:83–89. | |
Nunes AP, Oliveira IO, Santos BR, et al. Quality of DNA extracted from saliva samples collected with the Orangene™ DNA self-collection kit. BMC Med Res Methodol. 2012;12(65):1–5. | |
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Association; 2000. | |
Israel B, Buysse DJ, Krafty RT, Begley A, Miewald J, Hall M. Short-term stability of sleep and heart rate variability in good sleepers and patients with insomnia: for some measures, one night is enough. Sleep. 2012;35(9):1285–1291. | |
CPHA. CPS: Compendium of Pharmaceuticals and Specialties. 43rd ed. Ottawa: Canadian Pharmacists Association; 2008. | |
Iber C, Ancoli-Israel S, Chesson A, Quan SF; for the American Academy of Sleep Medicine. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications. Westchester, IL: American Academy of Sleep Medicine; 2007. | |
Agarwal R, Gotman J. Computer-assisted sleep staging. IEEE Trans Biomed Eng. 2001;48(12):1412–1423. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.