Back to Journals » Journal of Inflammation Research » Volume 14
A Novel NGF Receptor Agonist B355252 Ameliorates Neuronal Loss and Inflammatory Responses in a Rat Model of Cerebral Ischemia
Authors Wang HK ![]() , Chen JS, Hsu CY, Su YT, Sung TC
, Chen JS, Hsu CY, Su YT, Sung TC ![]() , Liang CL, Kwan AL, Wu CC
, Liang CL, Kwan AL, Wu CC ![]()
Received 26 January 2021
Accepted for publication 13 May 2021
Published 1 June 2021 Volume 2021:14 Pages 2363—2376
DOI https://doi.org/10.2147/JIR.S303833
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Hao-Kuang Wang,1,2 Jui-Sheng Chen,1,3,4 Chien-Yu Hsu,1 Yu-Ting Su,5 Tzu-Ching Sung,2 Cheng-Loong Liang,1,6 Aij-Lie Kwan,3,7 Cheng-Chun Wu6
1Department of Neurosurgery, E-DA Hospital, I-Shou University, Kaohsiung, Taiwan; 2School of Medicine for International Students, College of Medicine, I-Shou University, Kaohsiung, Taiwan; 3Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan; 4Department of Neurosurgery, E-Da Dachang Hospital, I-Shou University, Kaohsiung, Taiwan; 5Department of Obstetrics and Gynecology, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, Taiwan; 6School of Medicine, College of Medicine, I-Shou University, Kaohsiung, Taiwan; 7Department of Neurosurgery, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan
Correspondence: Cheng-Chun Wu
School of Medicine, College of Medicine, I-Shou University, Kaohsiung, Taiwan
Tel +886-7-6151100-7961
Fax +886-7-6155150
Email [email protected]
Introduction: Cerebral ischemia is a leading cause of disability and death worldwide. However, an effective therapeutic approach for the condition remains undiscovered. The previously proposed growth factor-based therapy has been inefficient due to its inability to pass through the blood–brain barrier. B355252, a newly developed small molecule, exhibited a potential neuroprotective effect in vivo. However, its exact efficacy in cerebral ischemia remains unclear.
Methods: We adopt an endothelin-1 stereotaxic intracranial injection to induced cerebral ischemia in rat. We further conducted 2,3,5-triphenyltetrazolium chloride (TTC) staining, immunofluorescent staining, enzyme-linked immunosorbent assay (ELISA), and behavioral tests to evaluate the efficacy of B355252 in neuroprotection, anti-inflammation, and behavioral outcome improvements.
Results: We identified that B355252 could protect ischemic neurons from neuronal loss by attenuating DNA damage, reducing ROS production and the LDH level, and preventing neuronal apoptosis. Moreover, inflammatory responses in astrocytic and microglial gliosis, as well as IL-1β and TNF-α levels, were ameliorated. Consequently, the behavioral outcomes of ischemic rats in neurologic responses and fore paw function recovery were improved.
Discussion: Overall, our study verified the in vivo therapeutic potential of B355252. The study findings further support its application in the development of a therapeutic approach for stroke.
Keywords: stroke, NGF, B355252, stroke, DNA damage, inflammation, mNSS, cylinder test
Introduction
Cerebral ischemia is a leading cause of long-term disability and death worldwide. However, there are no effective treatment approaches for the condition.1 Recently, growth factor-based therapy has been proposed to prevent and treat neuronal degeneration. Growth factors have the potential to promote neural repair by regulating underlying mechanisms including angiogenesis, cell proliferation and differentiation, migration, survival and apoptosis, synaptic plasticity, and immunomodulation.2,3 Studies have focused on the use of brain-derived neurotrophic factor (BDNF),4 epidermal growth factor plus erythropoietin,5 and human chorionic gonadotropin plus erythropoietin6 to treat cerebral ischemia. However, the blood–brain barrier is a major hurdle for the delivery of peptide-based therapeutic agents. Moreover, poor pharmacokinetic behavior and bioavailability to the targeted region limit the use of peptide agents.7,8 Therefore, extensive research is under way to explore nonpeptidyl small-molecule neurotrophin mimics that potentially evoke expected neuroregenerative responses.9,10
Williams et al successfully synthesized B355252, a phenoxy thiophene sulfonamide small molecule, which could potentiate NGF-induced neurite outgrowth (the molecular structure please see Supplementary Figure 1).11 In addition, anti-apoptotic effects of B355252 in glutamate-induced excitotoxicity, as well as in a murine hippocampal cell line (HT22) model of Parkinson disease (PD), have been reported. Glutamate- and PD-associated oxidative stress was significantly ameliorated by B355252 treatment.12 Glutamate or PD excitotoxicity and reactive oxygen species (ROS) pathologic characteristics are observed in stroke. However, how B355252 mediates its effects on an animal model of ischemic stroke remains unelucidated.
Inflammatory responses following a stroke cause secondary damage to ischemic regions, exacerbating neuronal loss and functional recovery.13 NGF has been reported to be involved in anti-inflammation, but whether B355252 administration ameliorates ischemia-induced neuroinflammation and further promotes functional recovery after an ischemic insult is unclear. Accordingly, we attempted to examine the therapeutic potential of B355252 and characterize its role in stroke treatment.
In the present study, we evaluated B355252-mediated neuroprotective and anti-inflammatory effects on a rat model of endothelin-1 (ET-1)–induced cerebral ischemia. ET-1 has recently been used to induce focal ischemia which induces stroke and cell death after sustained vasoconstriction with reperfusion, leading to impaired executive memory function and to impaired pure-motor and sensorimotor behaviors that are dependent on the specific area of ischemic insult in rodents.14–19 Our data showed that B355252 protected neuronal loss due to stroke by attenuating neuronal apoptosis and inflammation. Behavioral outcomes in the animal seemed to recover, as was observed through improved neurological response and fore paw functional recovery. These findings support the neuroprotective effects of B355252 in vivo and its potential as a therapeutic candidate.
Materials and Methods
Experimental Animals and Drug Administration
The study was carried out in compliance with the ARRIVE guidelines and NIH Guide for the Care and Use of Laboratory Animals. All experiments were conducted under the approval of the Institutional Animal Care and Use Committee (IACUC) at E-Da Hospital, Taiwan. The experimental schedules and procedures were modified from our previous work.20 Adult male Sprague–Dawley rats were purchased from Lasco biotechnology company (Taipei, Taiwan). The rats were used for all experiments when they weighted 250–300g. Rats were allocated randomly to the following experimental groups: sham and ischemia insult with vehicle treatment, and ischemia insult with B355252 treatment. To induce the ischemia stroke model, the vasoconstrictor peptide intracranial injection was conducted. Briefly, total 3 μL of 100 pM endothelin-1 (Sigma, E7764; St Louis, MO) was stereotactic injected into the brain (AP 0, ML +2.5, DV −2.3; AP +2.3, ML+2.5, DV −2.3; AP +0.7, ML +3.8, DV −7.0).21 Endothelin-1 was dissolved in HBSS (Sigma, H6648). B355252 was purchased from Sigma (SML1007) and dissolved in DMSO. B355252 was daily intraperitoneal injected to the cerebral ischemia rats using 0.125 mg/kg. The first injection was conducted at 24 h after stroke (PSD1), 9:00 am, before the behavioral tests. The injections in the upcoming days were consisted to PSD 1 until last day that the rats were sacrificed. For sham surgery, sham rats were stereotactic injected with HBSS only. DMSO was used as vesicle control of drug injection.
ROS Assay, LDH Assay, ELISA
The brain lysates preparation followed the our previous description.20 To analyze protein expression levels following ischemia insult and drug treatment, extracts were prepared from the brain tissue (bregma: +3 to −1 mm). To prepare the sample, we used a brain slicer to cut the brain into slices in the ischemic hemisphere. ROS and LDH assay kits were bought from BioVision K936-100-250 and K726 (Milpitas, CA). ELISA kits for detection of IL-1β, and TNF-α were acquired from R&D RLB00 and RTA00 (Minneapolis, MN). All the procedures were conducted followed the manufacturer’s directions.
Immunofluorescent Staining
The detailed protocol for IF staining followed our previously paper.20,22 To prepare the tissue for IF staining, the rats were anesthetized and perfused transcardially by PBS and 4% paraformaldehyde. The brain of each rat was removed and immersed in a 4% PFA solution for 2 h and dehydrated by gradient concentrations of sucrose. The sections were collected from bregma +2 to −4 mm; the cryo-tissues were sliced at 10 μm per section; one of three sections was collected and attached on the slide; 6 sections were collected on a slide, and 10 slides were prepared from a single rat brain. The penumbra region was identified by the intensity of immunoreactivity; the ischemic core exhibited the most restricted immunoreaction. At least 20 image views were quantified across six sections. After preparing cryosections, the slides were incubated with a series of primary antibodies, including NeuN (Millipore, Catalog# MAB377), γH2AX (Millipore, Catalog# 05636), and caspase-3 (Cell signaling, Catalog# 9661S). The secondary antibodies used for the chromogenic reaction were AlexaFluor-conjugated secondary antibodies (Thermo, Waltham, MA). The sections were incubated with DAPI and mounted under coverslips with mounting medium (Dako, Glostrup, Denmark). All the pictures were acquired randomly from the penumbra region in the cortex and striatum. The representative figures were selected from the cortex of the penumbra region. The immunoreactive cells and positive area were quantified by ImageJ by setting threshold for the intensity of immunoreactivity.23
Behavioral Outcomes Assessment
In assessment of locomotor function, we used grid walk test to evaluate the ability in waking with foot fault or not that followed previous study`s instruction.24 In assessment of sensory function, we conducted von Frey test to evaluate the sensory function responding to mechanical stimuli that followed a previous report.25 In addition, a focal scoring system for neurological severity score (NSS)26 was used to evaluate neurological outcomes of experimental rats on PSDs 1, 3, and 7. The detail procedure was modified from our previous work.20 Three grade scores were designed to each animal, with functional measures including gait, body symmetry, climbing, turning behavior, fore limb extension, compulsory circling, and sensory response. Furthermore, cylinder test was conducted to evaluate forelimb deficits followed the previous paper.27 The animal is placed in a transparent cylinder and evaluated. When assessing behavior in the cylinder, the number of independent wall placements observed for the right forelimb and left forelimb are recorded. The ratio of using ill site fore limb was quantified as R/(L+R)*100%.
Statistics
All data are presented as the mean ± SEM. All data were normally distributed and were analyzed by one-way ANOVA with multiple-group comparison or two-way ANOVA with multiple-group comparison. Differences with P < 0.05 were considered statistically significant.
Results
B355252 Attenuates Infarct Volume in Rats with Cerebral Ischemia
To evaluate the potential of B355252 in neuroprotection after cerebral ischemia, we conducted triphenyl tetrazolium chloride (TTC) staining to determine the infarct volume at post surgery day (PSD) 3. The representative figure of TTC staining (Figure 1A) showed ET-1–cerebral injection induced significant infarct damage where presented in white, but B355252-treated rats displayed decreased damage with larger brain parenchyma presented in red. In the quantified data (Figure 1B), we found the infarct volume was significantly attenuated by B355252 treatment, suggesting its efficacy in neuroprotection following cerebral ischemia.
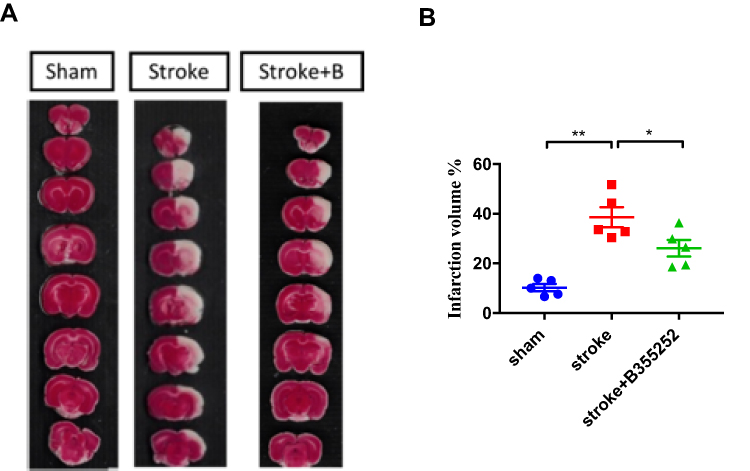 |
Figure 1 B355252 treatment improves the ischemic volume of rats with cerebral ischemia. (A) Representative figures of TTC staining from groups of sham, stroke, and stroke+B355252 at PSD 3. (B) Quantified data of TTC staining presented in ischemia volume (%). N=5 per group. * p< 0.05, ** p< 0.01 by one-way ANOVA. |
B355252 Administration Reveals Neuroprotection After Stroke
To understand the underlying mechanism of B355252-mediated improvement in the behavioral outcomes of ischemic rats, we further examined the poststroke neuronal loss through immunofluorescent staining for NeuN at PSD 7. ET-1–induced ischemic damage considerably reduced the number of neurons in the cortex and striatum, but this effect was reversed in cerebral ischemic rats treated with B355252 (Figure 2A). For data quantification, a whole semi-brain–based section screening was used. We found that the number of cells with immunoreactive NeuN was significantly reduced in cerebral ischemic rats and that B355252 treatment protected post-stroke neuronal loss (Figure 2B), suggesting the neuroprotective potential of the agent in stroke treatment.
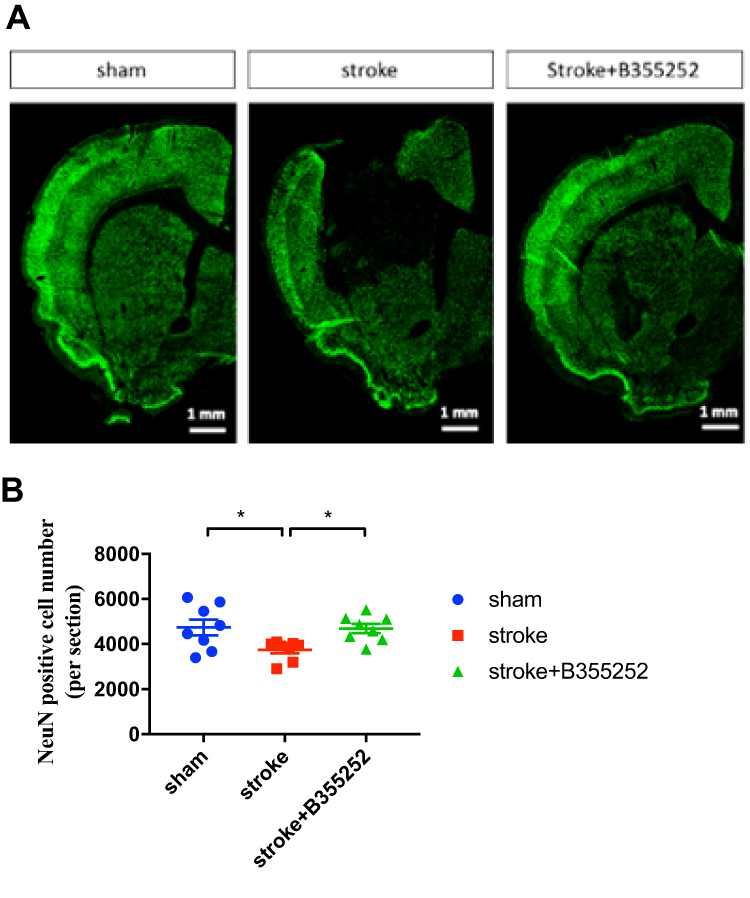 |
Figure 2 B355252 protects the brain from neuronal loss in rats with cerebral ischemia. (A) Immunofluorescent staining for NeuN. Data were showed in semibrain from ipsilaretal site of surgery at PSD 7. (B) Data quantification of cell with NeuN immunoreactivity from per semibrain. N=8 per group. * p< 0.05 by one-way ANOVA. |
B355252 Attenuates Neuronal Apoptosis After Stroke
After having confirmed the neuroprotective effect of B355252 in rats with cerebral ischemia, we further investigated the role of B355252 in neuronal apoptosis. Brain sections obtained at 3 days after stroke from ischemic rats and sham controls were submitted to immunofluorescent double staining for cleaved caspase-3 and NeuN (Figure 3A). Immunoreactivity of cleaved caspase-3 and cleaved caspase/NeuN double-positive cells was considerably increased in cerebral ischemic rats compared with sham controls. B355252 administration effectively reduced the number of cells demonstrating immunoreactive cleaved caspase and cleaved caspase-3/NeuN double-positive cells (Figure 3B and C). These data further support the ability of B355252 in preventing ischemic neurons from poststroke apoptosis.
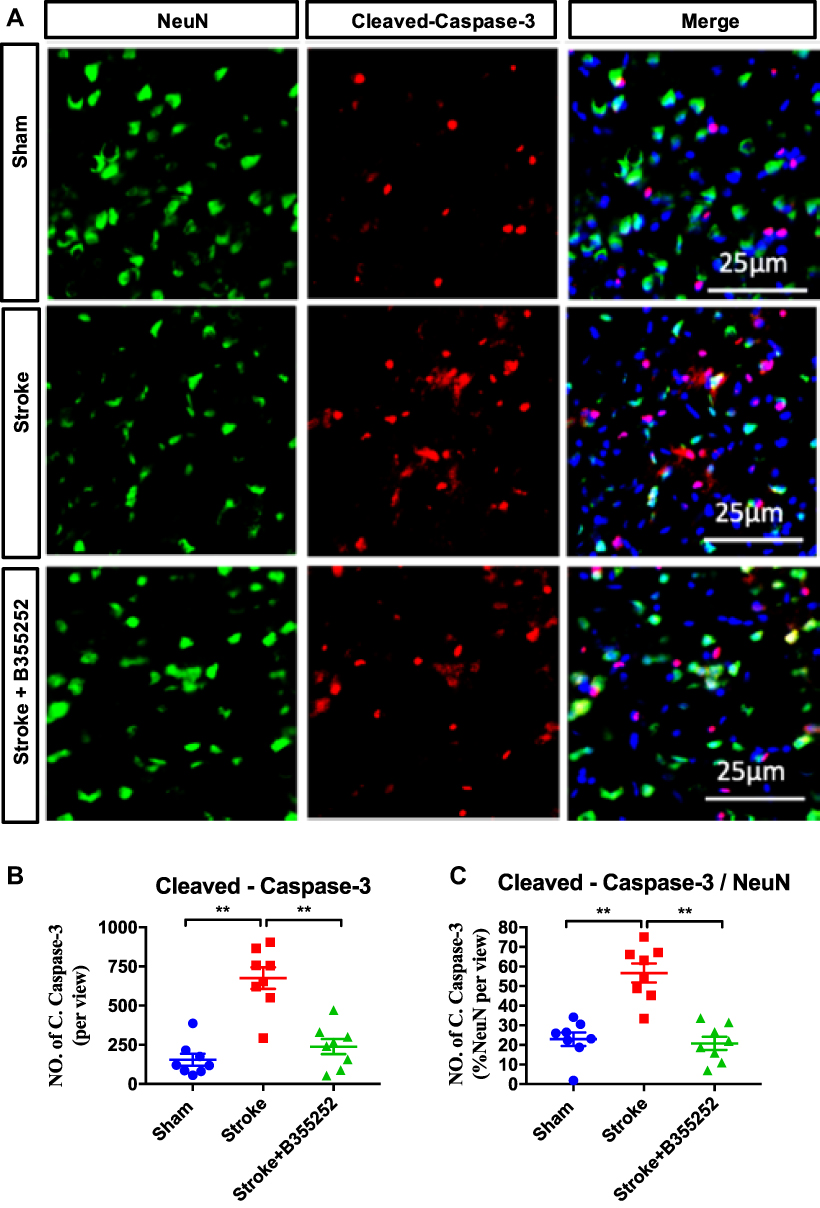 |
Figure 3 B355252 treatment ameliorates the level of cleaved-caspase-3 in the brain of cerebral ischemia. (A) Immunofluorescent staining for NeuN and cleaved-caspase-3 at PSD 3. (B) Data quantification for total cleaved-caspase-3 number and (C) NeuN and cleaved-caspase-3 double-positive cell number at PSD3. N=6 per group, ** p< 0.01 by one-way ANOVA. Bar: 25 μm. |
B355252 Reduces ROS Accumulation and DNA Damage Levels After Stroke
To further characterize the therapeutic potential of B355252, the level of neuronal DNA damage was determined using immunofluorescent quantification of γH2AX and NeuN28 (Figure 4A). Neuronal DNA damage in ET-1–induced cerebral ischemic rats was considerable compared with that in sham controls at 3 days after stroke. B355252 treatment could protect neurons from DNA damage, as was observed by the decreased number of cells with γH2AX immunoreactivity (Figure 4B). Furthermore, the level of reactive oxygen species from the tissue lysate of the cerebral ischemia brain was determined. B355252 treatment could reduce ROS levels at 3 days after stroke (Figure 4C). These findings suggest the ability of B355252 to protect the ischemic brain from ROS stress and DNA damage.
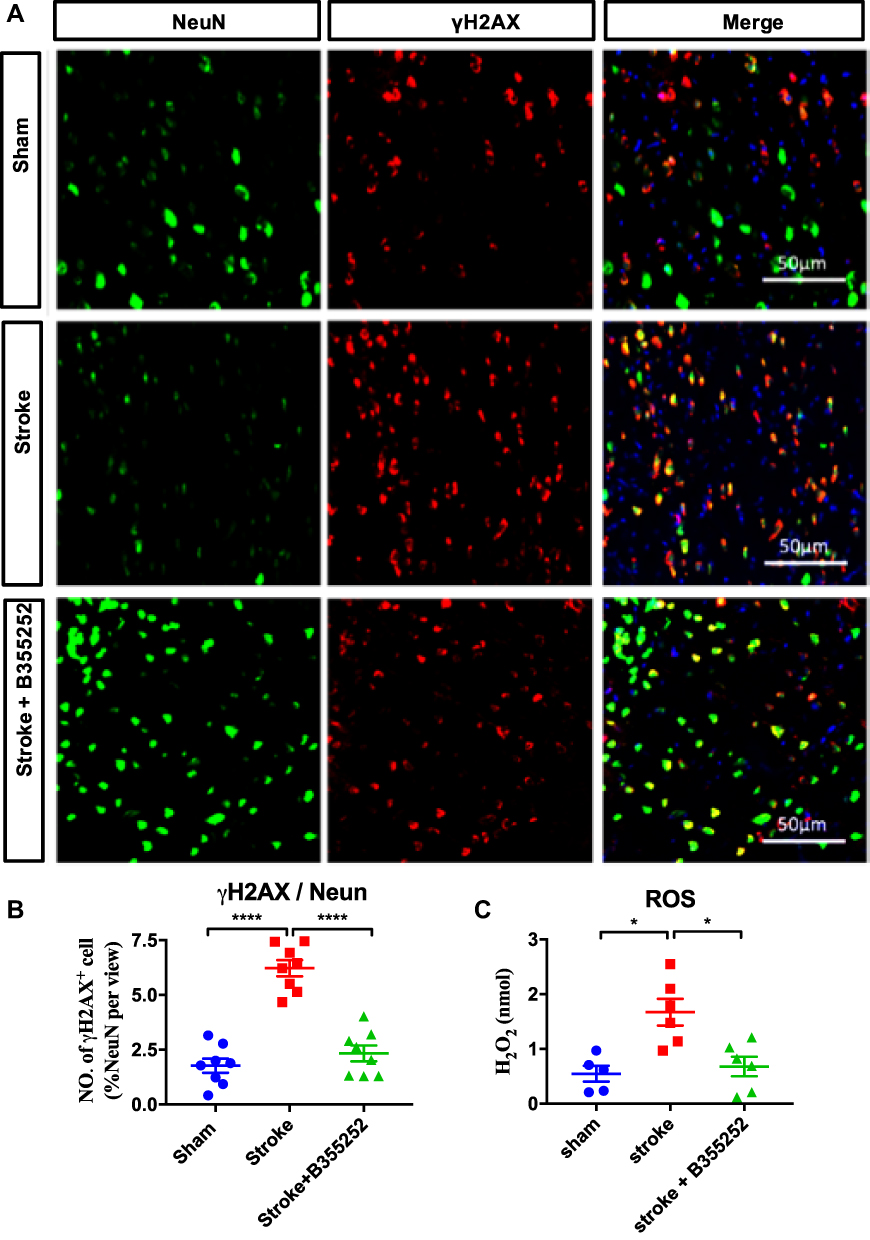 |
Figure 4 B355252 treatment protects neuron from DNA damage in the brain of cerebral ischemia. (A) Immunofluorescent staining for NeuN and γH2AX at PSD 3. (B) Data quantification for NeuN and γH2AX double-positive cell number at PSD3. (C) Reactive oxygen species assessment from experimental rats at PSD 3. N=6 per group, * p< 0.05, **** p< 0.0001 by one-way ANOVA. Bar: 50 μm. |
B355252 Attenuates Gliosis and Inflammation After Stroke
To understand B355252-mediated beneficial effects on stroke recovery, we examined the extent of gliosis at 3 days after stroke through immunofluorescent staining. For observing astrogliosis and microgliosis, we applied anti-glial filament associated protein (GFAP) and anti-Iba-1 antibodies (Figures 5A and 6A). In the quantified data, we found that gliosis was markedly increased in ET-1–induced cerebral ischemia, and B355252 treatment was able to reverse the elevated gliosis level. In addition, the number of cells demonstrating GFAP and Iba-1 immunoreactivity was significantly reduced following B355252 treatment (Figures 5B and 6B).
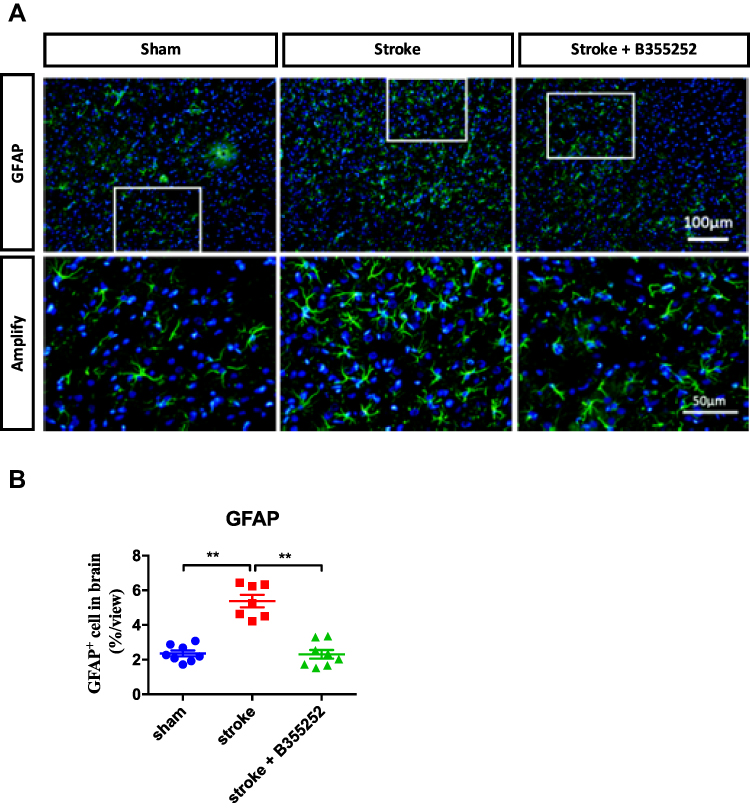 |
Figure 5 B355252 treatment decreases the level of astrocytes reactivation in the brain of cerebral ischemia. (A) Immunofluorescent staining for GFAP at PSD 3. (B) Data quantification for GFAP immunoreacted cell number. N=8 per group, ** p< 0.01 by one-way ANOVA. Bar: 100 μm (upper panel) and 50 μm (lower panel). |
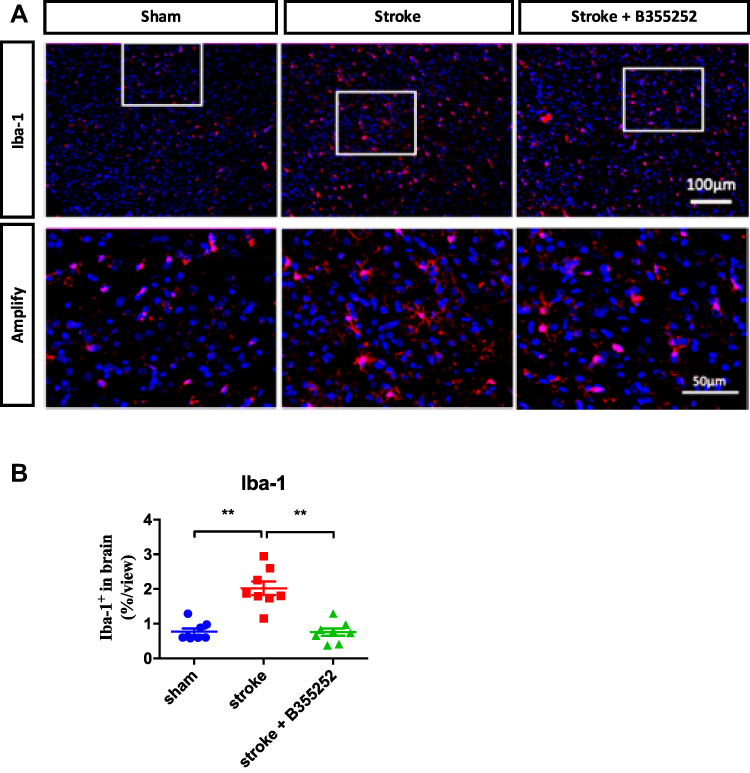 |
Figure 6 B355252 treatment decreases the level of microglial reactivation in the brain of cerebral ischemia. (A) Immunofluorescent staining for Iba-1 at PSD 3. (B) Data quantification for Iba-1 immunoreacted cell number. N=8 per group, ** p< 0.01 by one-way ANOVA. Bar: 100 μm (upper panel) and 50 μm (lower panel). |
B355252 Reduces Inflammatory Cytokine and LDH Levels
The anti-inflammatory effect of B355252 was further validated by determining IL-1β, IL-6, and TNF-α levels from tissue lysates obtained from the cerebral ischemic brain at 3 days after stroke. ET-1–induced cerebral ischemia markedly increased IL-1β and TNF-α, but not IL-6, levels (Figure 7A–C). B355252 treatment could reduce IL-1β and TNF-α levels (Figure 7A and C). In addition, LDH levels in the ischemic brain lysate were determined. Results revealed that B355252 could reduce ischemia-induced LDH levels (Figure 7D). Accordingly, these data favor the potential of B355252 treatment in improving inflammation and attenuating tissue damage.
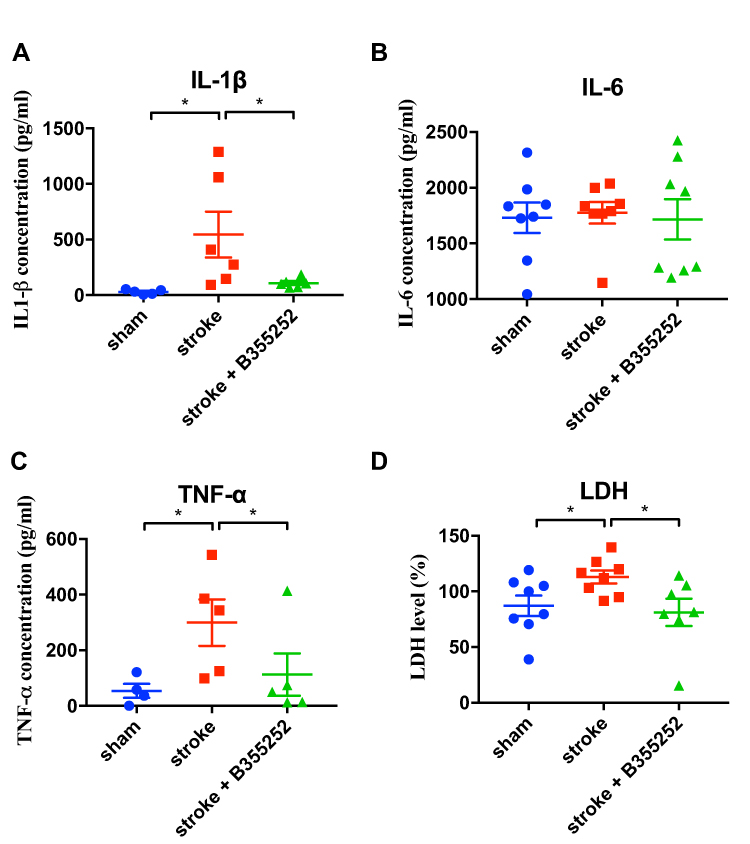 |
Figure 7 B355252 treatment decreases the levels of inflammatory cytokines in the brain of ischemia rats. ELISA assessments were conducted at PSD 3 for assessment of (A) IL-1β, (B) IL-6, (C) TNF-α, and (D) LDH. N=6 per group, * p< 0.05 by one-way ANOVA. |
B355252 Improves Behavioral Outcomes in Rats with Cerebral Ischemia
The therapeutic potential of B355252 after stroke was assessed in the rat stroke model by determining the foot fault ratio and reaction to mechanical stimuli for assessment of motor and sensory function at PSD 1 and 3 (Figure 8A and B). Our data showed ET-1 induced cerebral ischemia impaired the both functional assessments whereas B355252 treatment improved the outcomes in foot fault test (Figure 8A) and mechanical stimuli (Figure 8B). Furthermore, we evaluated neurological function by using the modified neurological severity score at PSD 1, 3, 7. The rats with the brain ischemic damage exhibited deficits in neuromuscular function following stroke, whereas B355252 treatment improved ischemia-induced neurological impairment (Figure 8C). In addition, a cylinder test was performed to evaluate the ability of cerebral ischemic rats for using contract lateral fore paw after stroke. Findings revealed that ET-1 markedly impaired the use of the fore paw, and this effect was attenuated with B355252 treatment. The treatment also retained the functioning of the fore paw of the contralateral side (Figure 8D). These data support the potential of B355252 to alleviate stroke-induced behavioral deficits.
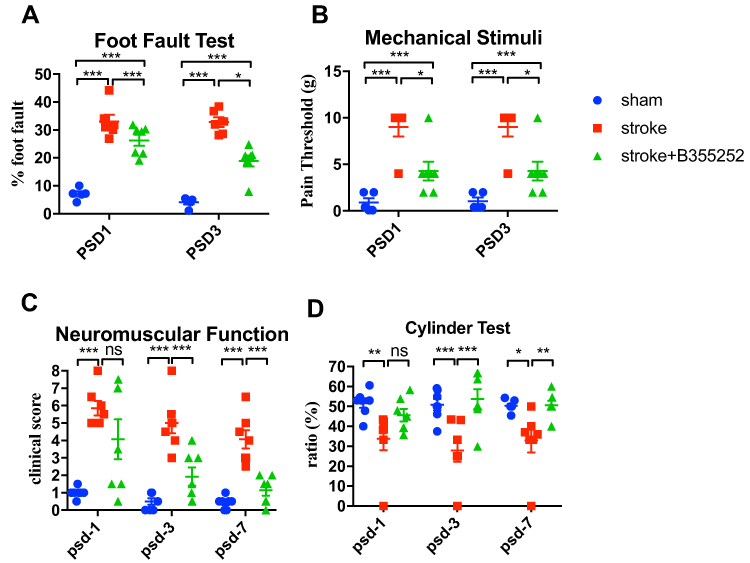 |
Figure 8 B355252 ameliorates behavioral outcomes of brain ischemia rats. (A) Foot fault test evaluated by grid walking at PSD 1 and 3. (B) Mechanical stimuli response was evaluated by von Frey test at PSD 1 and 3. (C) Data of mNSS from experimental rats measured at PSD 1, 3, 7. (D) Data of cylinder test from experimental rats measured at PSD 1, 3, 7. N=7 per group. ns: not significant change, * p< 0.05, ** p< 0.01, *** p< 0.001 by two-way ANOMA. |
Discussion
In this study, we verified the potential of B355252 in neuroprotection and anti-inflammation. B355252 treatment ameliorated neuronal loss, neuronal apoptosis, DNA damage, gliosis, and levels of inflammation cytokines. Importantly, behavioral outcomes in neuromuscular responses and functional recovery of cerebral ischemic rats were reversed. Therefore, our findings support the potential of B355252 in stroke therapy. This is the first study to validate the beneficial effects of B355252 in improving neuroprotection and reducing inflammation following cerebral ischemic damage in vivo.
Regarding the time course along stroke pathogenesis, The pathogenesis of ischemia brain damage is majorly characterized into early hyperacute (0–6 h), late hyperacute (6–24 h), acute (1 week), subacute (1–3 week), and chronic stages (more than 3 weeks).29 In the acute stage, neuronal necrosis appears immediately the following ischemia. The rest of the neurons in the penumbra region encounter other stress cascades, eg, gliosis, inflammation, excitotoxicity, mitochondrial dysfunction, superoxide, DNA damage, BBB disruption, cell apoptosis, etc. in the following days.30 These are critical events associated with neuronal survival. The neuronal loss starts from minutes after ischemia insult and the number is increased along the time course with a peak at 72 h after stroke and progressively decreased until about 1 week.31,32 In addition, data from clinical MRI also support that scans with maximum diffusion-weighted imaging lesion volume occurred at a mean of 70 hours.33 Accordingly, we selected PSD 3 and 7 as our observed time points.
Stroke is a leading cause of morbidity and mortality worldwide, but an effective therapeutic strategy is not available. Currently, tissue plasminogen activator (tPA) is the standard treatment for stroke. Promising therapeutic strategies include protecting the penumbra neurons from glutamate excitotoxicity, ROS, and inflammatory responses.34 Several potential drugs or compounds targeting these pathologic progresses are being studied in clinical trials. Some of these compounds include the NOX inhibitors apocynin and diphenyleneiodonium in the inhibition of ROS damage,35,36 hypoglycemic drugs thiazolidinediones and metformin in neuroprotection, and fingolimod or dimethyl in reducing central nervous system inflammation.37 However, an accurate therapeutic approach is limited by side effects, low bioavailability, and drug specificity. In this study, we found that B355252 as a small ligand target to NGF receptor ameliorated behavioral outcomes and protected neuronal damage from focal ischemia. Remarkably, the inflammatory level was decreased with B355252 treatment, suggesting its potential in stroke therapy.
NGF is a member of the family of neurotrophins, such as BDNF, NT-3, and NT-4/5, that function to support neuronal growth and survival.38,39 NGF binds the TrkA receptor,40 its intracellular downstream signal is able to protect neuronal cells from apoptosis and induce neuronal regeneration in vivo.41,42 Thus, the role of NGF and other neurotrophins in the prevention or reversal of cognitive decline and improvement of neurodegenerative diseases is established. The activation of TrkA intracellular signaling pathways can promote cell survival via PI3K/Akt/Bad pathway, promotes neurotrophins release, neurite outgrowth, neural survival via Ras, such as the Ras-ERK signaling by transcription factor-CREB activation.43 As an NGF receptor potentiator, B355252 could elicit neuroprotective effects in neutrophin withdrawal conditions,11 glutamate toxicity,12 6-OHDA toxicity,44 and CoCl2-induced hypoxia conditions.45 B35525 has been shown to enhance the ability of NGF-primed NS-1 cells to differentiate into a neuron-like phenotype with extensive networks of branched neurites.12,45 B35525 does not induce neurite outgrowth alone, but enhances the effect of sub-physiological concentrations of NGF on neurons in vitro. The potent neuroprotective effects of the agent included protection from cell apoptosis, activation of the MAPK-Erk intracellular signaling, attenuation of ROS insult, and regulation of the mitochondrial membrane potential. However, previous studies have been performed in neuron-like cells or hippocampal cell lines, such as NS-1, PC12, or HT22, not in the neurons. The underlying mechanisms of B355252 in neurons or in vivo remain to be validated. In the present study, we examined the therapeutic potential of B355252 either in behavioral outcomes or in neuroprotection, as well as anti-inflammation in the brain parenchyma, in a rat cerebral ischemic model. For instance, through mNSS and the cylinder test, we confirmed the potential of B355252 in improving functional recovery after stroke. Our data further support its effect in preventing neuronal loss through attenuation of DNA damage and neuronal apoptosis. Meanwhile, assessments of ROS and LDH levels also supported its effect on ischemia pathogenesis. Furthermore, we characterized that B355252 treatment could alleviate neuronal inflammation after stroke, reduce the levels of inflammatory cytokines such as IL-1β and TNF-α, and reduce the gliosis levels of reactive astrocytes and microglia. These data further shed light on the therapeutic effect of B355252 on stroke.
Regarding the underlying mechanism of the anti-inflammatory effect of B355252, we prospect the anti-inflammatory effect of B355252 can result from neuroprotective effect in the maintenance of neuronal survival and attenuation of gliosis in astrocyte and microglia reactivation. In the present study, our data support B355252 is essential to protect neurons from ischemia damage. Given cessation of cerebral blood flow leads to energy depletion and necrotic neuron death, which can trigger immune responses ultimately leading to inflammatory cell activation and infiltration.46 As neuroinflammation can be induced by neuronal damage or necrosis, when more nerves are protected from damage, the less inflammation response will be activated. In stroke pathogenesis, another critical role is ROS production and accumulation following ischemia damage and disruption of mitochondria respiratory trains.30 ROS can stimulate ischemic cells and neurons to secrete inflammatory cytokines and chemokines can cause adhesion molecule upregulation in the cerebral vasculature and peripheral leukocyte recruitment, leading to the major course of inflammatory response after ischemic damage.33 To this notion, B355252 was reported involving in the preservation of mitochondria membrane potential, attenuation of ROS production, inhibition of cytochrome C release, and modulation of JNK cascade in vitro studies.32 These findings are paralleled to our data in vivo and further explains how the ROS can be improved by B355252 treatment. In addition, previous data have revealed that in vivo NGF deprivation in rats with experimental allergic encephalomyelitis increased brain inflammation and led to more severe clinical features.47 Moreover, inhibition of the intracellular NGF exacerbated infiltration of neutrophils and macrophages and worsened the gut lesions in an animal model of colitis.48 Importantly, data from monocytes also indicated that inflammatory stimuli activated pro-inflammatory responses through Toll-like receptors (TLR), which also induced increased expression of the TrkA.49–52 Furthermore, NGF and TrkA binding attenuated the NF-κB nuclear translocation and reduced glycogen synthase kinase 3 (GSK3) activity, reducing the production of inflammatory cytokines.51,53,54 By contrast, TrkA activation induces the elevated TLR-induced activation in the intracellular PI3K/Akt pathway, which reduces TLR ligand-induced inflammatory responses.55 NGF administration regulated a balance between the pro- and anti-inflammatory pathways, improving the release of anti-inflammatory mediators such as IL-10.54–56 B355252 treatment can improve gliosis is resulting from its potential in the modulation of JNK cascade evaluated from a previous study.32 Because the gliosis and release of inflammation cytokine can be modulated by MAPK/JNK cascade, these previous findings can also further support the role of B355252 treatment in anti-inflammation. Consequently, B355252-induced TrkA activation may affect the physiological anti-inflammatory mechanism.
Regarding the underlying competence of brain barrier penetration of B355252, current no data can directly support its potential of BBB penetration. However, several lines of evidence showed (a) The molecular weight of B355252 is 514 Da, which belongs to a small molecular. Because molecules below 900 Da can diffuse into cells faster in the human body and the appropriate molecular weight of CNS drug is between 151 and 655 Da,57 we expect B355252 is feasible for diffuse in body and BBB penetration. (b) Given a study of comparative properties for BBB penetration, estimated pKa limits for penetration between 4 and 10.58 The pKa of B355252 is 10.16±0.50 (predicted), thus it exhibits the potential for BBB penetration. (c) Because hydrogen bonding is primarily associated with oxygen and nitrogen moieties in a molecule, if the sum of the nitrogen (N) and oxygen (O) atoms in the molecule is five or less, then the molecule has a high probability of entering the CNS.59 The N and O atoms number of B355252 is five, supporting its potential for BBB penetration.
Conclusion
In the present study, we identified the potentials of B355252 administration in vivo in improvement of DNA damage, ROS production, glial reactivation, and inflammation cytokines. Accordingly, our data support that B355252 is feasible to be considered as a therapeutic approach in cerebral ischemia.
Data Sharing Statement
The data used to support the findings of this study are included within the article, containing Figures 1–7. Other data that might be useful for the findings of this study will be supplied by the corresponding author (C.C. Wu) upon request.
Acknowledgments
This study was supported by E-DA Hospital and I-Shou University, Kaohsiung, Taiwan (EDAHP107001, EDAHP108025, EDAHP109029, EDAHP109039, ISU-109-01-16A). It was also partly supported by grants from the Ministry of Science and Technology, Taiwan (108-2314-B-182A-084, 109-2320-B-214-001) and Kaohsiung Chang Gung Memorial Hospital, Taiwan (CMRPG8G0091, CMRPG8G0092, CMRPG8G0093).
Disclosure
The authors declare that they have no conflict of interest for this work.
References
1. Cheng NT, Kim AS. Intravenous thrombolysis for acute ischemic stroke within 3 hours versus between 3 and 4.5 hours of symptom onset. Neurohospitalist. 2015;5(3):101–109. doi:10.1177/1941874415583116
2. Lanfranconi S, Locatelli F, Corti S, et al. Growth factors in ischemic stroke. J Cell Mol Med. 2011;15(8):1645–1687.
3. Oliveira SL, Pillat MM, Cheffer A, Lameu C, Schwindt TT, Ulrich H. Functions of neurotrophins and growth factors in neurogenesis and brain repair. Cytometry A. 2013;83(1):76–89. doi:10.1002/cyto.a.22161
4. Ren JM, Finklestein SP. Growth factor treatment of stroke. Curr Drug Targets CNS Neurol Disord. 2005;4(2):121–125. doi:10.2174/1568007053544101
5. Kolb B, Morshead C, Gonzalez C, et al. Growth factor-stimulated generation of new cortical tissue and functional recovery after stroke damage to the motor cortex of rats. J Cereb Blood Flow Metab. 2007;27(5):983–997. doi:10.1038/sj.jcbfm.9600402
6. Belayev L, Khoutorova L, Zhao KL, Davidoff AW, Moore AF, Cramer SC. A novel neurotrophic therapeutic strategy for experimental stroke. Brain Res. 2009;1280:117–123. doi:10.1016/j.brainres.2009.05.030
7. Thorne RG, Frey WH. Delivery of neurotrophic factors to the central nervous system: pharmacokinetic considerations. Clin Pharmacokinet. 2001;40(12):907–946. doi:10.2165/00003088-200140120-00003
8. Miller G. Drug targeting. Breaking down barriers. Science. 2002;297(5584):1116–1118. doi:10.1126/science.297.5584.1116
9. Saragovi HU, Gehring K. Development of pharmacological agents for targeting neurotrophins and their receptors. Trends Pharmacol Sci. 2000;21(3):93–98. doi:10.1016/S0165-6147(99)01444-3
10. Dago L, Bonde C, Peters D, et al. NS 1231, a novel compound with neurotrophic-like effects in vitro and in vivo. J Neurochem. 2002;81(1):17–24. doi:10.1046/j.1471-4159.2002.00803.x
11. Williams AL, Dandepally SR, Gilyazova N, Witherspoon SM, Ibeanu G. Microwave-assisted synthesis of 4-chloro-N-(naphthalen-1-ylmethyl)-5-(3-(piperazin-1-yl)phenoxy)thiophene-2-sulfo namide (B-355252): a new potentiator of Nerve Growth Factor (NGF)-induced neurite outgrowth. Tetrahedron. 2010;66(50):9577–9581. doi:10.1016/j.tet.2010.09.028
12. Gliyazova NS, Huh EY, Ibeanu GC. A novel phenoxy thiophene sulphonamide molecule protects against glutamate evoked oxidative injury in a neuronal cell model. BMC Neurosci. 2013;14:93. doi:10.1186/1471-2202-14-93
13. Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. 2019;16(1):142.
14. Fuxe K, Bjelke B, Andbjer B, Grahn H, Rimondini R, Agnati LF. Endothelin-1 induced lesions of the frontoparietal cortex of the rat. A possible model of focal cortical ischemia. Neuroreport. 1997;8(11):2623–2629. doi:10.1097/00001756-199707280-00040
15. Abeysinghe HC, Bokhari L, Dusting GJ, Roulston CL. Brain remodelling following endothelin-1 induced stroke in conscious rats. PLoS One. 2014;9(5):e97007. doi:10.1371/journal.pone.0097007
16. Cordova CA, Jackson D, Langdon KD, Hewlett KA, Corbett D. Impaired executive function following ischemic stroke in the rat medial prefrontal cortex. Behav Brain Res. 2014;258:106–111. doi:10.1016/j.bbr.2013.10.022
17. Blasi F, Whalen MJ, Ayata C. Lasting pure-motor deficits after focal posterior internal capsule white-matter infarcts in rats. J Cereb Blood Flow Metab. 2015;35(6):977–984. doi:10.1038/jcbfm.2015.7
18. Tennant KA, Jones TA. Sensorimotor behavioral effects of endothelin-1 induced small cortical infarcts in C57BL/6 mice. J Neurosci Methods. 2009;181(1):18–26. doi:10.1016/j.jneumeth.2009.04.009
19. Lee B, Clarke D, Al Ahmad A, et al. Perlecan domain V is neuroprotective and proangiogenic following ischemic stroke in rodents. J Clin Invest. 2011;121(8):3005–3023. doi:10.1172/JCI46358
20. Wu CC, Wang LC, Su YT, Wei WY, Tsai KJ. Synthetic alpha5beta1 integrin ligand PHSRN is proangiogenic and neuroprotective in cerebral ischemic stroke. Biomaterials. 2018;185:142–154. doi:10.1016/j.biomaterials.2018.09.014
21. Horie N, Maag AL, Hamilton SA, Shichinohe H, Bliss TM, Steinberg GK. Mouse model of focal cerebral ischemia using endothelin-1. J Neurosci Methods. 2008;173(2):286–290. doi:10.1016/j.jneumeth.2008.06.013
22. Wu CC, Wang IF, Chiang PM, Wang LC, Shen CJ, Tsai KJ. G-CSF-mobilized Bone Marrow Mesenchymal Stem Cells Replenish Neural Lineages in Alzheimer’s Disease Mice via CXCR4/SDF-1 Chemotaxis. Mol Neurobiol. 2016.
23. Wu CC, Lien CC, Hou WH, Chiang PM, Tsai KJ. Gain of BDNF Function in Engrafted Neural Stem Cells Promotes the Therapeutic Potential for Alzheimer’s Disease. Sci Rep. 2016;6:27358. doi:10.1038/srep27358
24. Schaar KL, Brenneman MM, Savitz SI. Functional assessments in the rodent stroke model. Exp Transl Stroke Med. 2010;2(1):13. doi:10.1186/2040-7378-2-13
25. Alamri FF, Shoyaib AA, Biggers A, Jayaraman S, Guindon J, Karamyan VT. Applicability of the grip strength and automated von Frey tactile sensitivity tests in the mouse photothrombotic model of stroke. Behav Brain Res. 2018;336:250–255. doi:10.1016/j.bbr.2017.09.008
26. Petullo D, Masonic K, Lincoln C, Wibberley L, Teliska M, Yao DL. Model development and behavioral assessment of focal cerebral ischemia in rats. Life Sci. 1999;64(13):1099–1108. doi:10.1016/S0024-3205(99)00038-7
27. Hua Y, Schallert T, Keep RF, Wu J, Hoff JT, Xi G. Behavioral tests after intracerebral hemorrhage in the rat. Stroke. 2002;33(10):2478–2484. doi:10.1161/01.STR.0000032302.91894.0F
28. Wu CC, Jin LW, Wang IF, et al. HDAC1 dysregulation induces aberrant cell cycle and DNA damage in progress of TDP-43 proteinopathies. EMBO Mol Med. 2020;12(6):e10622. doi:10.15252/emmm.201910622
29. Allen LM, Hasso AN, Handwerker J, Farid H. Sequence-specific MR imaging findings that are useful in dating ischemic stroke. Radiographics. 2015;32(5):1285–1297. doi:10.1148/rg.325115760
30. Rodrigo R, Fernandez-Gajardo R, Gutierrez R, et al. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 2013;12(5):698–714. doi:10.2174/1871527311312050015
31. Popa-Wagner A, Carmichael ST, Kokaia Z, Kessler C, Walker LC. The response of the aged brain to stroke: too much, too soon? Curr Neurovasc Res. 2007;4(3):216–227. doi:10.2174/156720207781387213
32. Zhou Z, Lu J, Liu WW, et al. Advances in stroke pharmacology. Pharmacol Ther. 2018;191:23–42.
33. Schwamm LH, Koroshetz WJ, Sorensen AG, et al. Time course of lesion development in patients with acute stroke: serial diffusion- and hemodynamic-weighted magnetic resonance imaging. Stroke. 1998;29(11):2268–2276.
34. Yang Q, Huang Q, Hu Z, Tang X. Potential Neuroprotective Treatment of Stroke: targeting Excitotoxicity, Oxidative Stress, and Inflammation. Front Neurosci. 2019;13:1036. doi:10.3389/fnins.2019.01036
35. Genovese T, Mazzon E, Paterniti I, Esposito E, Bramanti P, Cuzzocrea S. Modulation of NADPH oxidase activation in cerebral ischemia/reperfusion injury in rats. Brain Res. 2011;1372:92–102. doi:10.1016/j.brainres.2010.11.088
36. Barua S, Kim JY, Yenari MA, Lee JE. The role of NOX inhibitors in neurodegenerative diseases. IBRO Rep. 2019;7:59–69. doi:10.1016/j.ibror.2019.07.1721
37. Collino M, Aragno M, Mastrocola R, et al. Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol. 2006;530(1–2):70–80. doi:10.1016/j.ejphar.2005.11.049
38. Friedman WJ, Greene LA. Neurotrophin signaling via Trks and p75. Exp Cell Res. 1999;253(1):131–142. doi:10.1006/excr.1999.4705
39. Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10(3):381–391. doi:10.1016/S0959-4388(00)00092-1
40. Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11(3):272–280. doi:10.1016/S0959-4388(00)00208-7
41. Bibel M, Barde YA. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000;14(23):2919–2937. doi:10.1101/gad.841400
42. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi:10.1146/annurev.neuro.24.1.677
43. Nusser N, Gosmanova E, Zheng Y, Tigyi G. Nerve growth factor signals through TrkA, phosphatidylinositol 3-kinase, and Rac1 to inactivate RhoA during the initiation of neuronal differentiation of PC12 cells. J Biol Chem. 2002;277(39):35840–35846. doi:10.1074/jbc.M203617200
44. Gliyazova NS, Ibeanu GC. The Chemical Molecule B355252 is Neuroprotective in an In Vitro Model of Parkinson’s Disease. Cell Mol Neurobiol. 2016;36(7):1109–1122. doi:10.1007/s10571-015-0304-5
45. Chimeh U, Zimmerman MA, Gilyazova N, Li PA. B355252, a novel small molecule, confers neuroprotection against cobalt chloride toxicity in mouse hippocampal cells through altering mitochondrial dynamics and limiting autophagy induction. Int J Med Sci. 2018;15(12):1384–1396. doi:10.7150/ijms.24702
46. Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184(1–2):53–68. doi:10.1016/j.jneuroim.2006.11.014
47. Micera A, Properzi F, Triaca V, Aloe L. Nerve growth factor antibody exacerbates neuropathological signs of experimental allergic encephalomyelitis in adult Lewis rats. J Neuroimmunol. 2000;104(2):116–123. doi:10.1016/S0165-5728(99)00272-6
48. Reinshagen M, Rohm H, Steinkamp M, et al. Protective role of neurotrophins in experimental inflammation of the rat gut. Gastroenterology. 2000;119(2):368–376. doi:10.1053/gast.2000.9307
49. Ehrhard PB, Ganter U, Stalder A, Bauer J, Otten U. Expression of functional trk protooncogene in human monocytes. Proc Natl Acad Sci U S A. 1993;90(12):5423–5427. doi:10.1073/pnas.90.12.5423
50. Caroleo MC, Costa N, Bracci-Laudiero L, Aloe L. Human monocyte/macrophages activate by exposure to LPS overexpress NGF and NGF receptors. J Neuroimmunol. 2001;113(2):193–201. doi:10.1016/S0165-5728(00)00441-0
51. Prencipe G, Minnone G, Strippoli R, et al. Nerve growth factor downregulates inflammatory response in human monocytes through TrkA. J Immunol. 2014;192(7):3345–3354. doi:10.4049/jimmunol.1300825
52. la Sala A, Corinti S, Federici M, Saragovi HU, Girolomoni G. Ligand activation of nerve growth factor receptor TrkA protects monocytes from apoptosis. J Leukoc Biol. 2000;68(1):104–110.
53. Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13(11):460–469. doi:10.1016/j.molmed.2007.09.002
54. Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24(7):358–363. doi:10.1016/S1471-4906(03)00139-X
55. Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5(6):446–458. doi:10.1038/nri1630
56. Han J, Ulevitch RJ. Limiting inflammatory responses during activation of innate immunity. Nat Immunol. 2005;6(12):1198–1205. doi:10.1038/ni1274
57. Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2(4):541–553. doi:10.1602/neurorx.2.4.541
58. Palm K, Luthman K, Ros J, Grasjo J, Artursson P. Effect of molecular charge on intestinal epithelial drug transport: pH-dependent transport of cationic drugs. J Pharmacol Exp Ther. 1999;291(2):435–443.
59. Osterberg T, Norinder U. Prediction of polar surface area and drug transport processes using simple parameters and PLS statistics. J Chem Inf Comput Sci. 2000;40(6):1408–1411. doi:10.1021/ci000065l
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.