Back to Journals » Drug Design, Development and Therapy » Volume 10
A novel imidazopyridine derivative, X22, attenuates sepsis-induced lung and liver injury by inhibiting the inflammatory response in vitro and in vivo
Authors Ge X, Feng Z, Xu T, Wu B, Chen H, Xu F, Fu L, Shan X, Dai Y, Zhang Y, Liang G
Received 28 November 2015
Accepted for publication 4 February 2016
Published 24 June 2016 Volume 2016:10 Pages 1947—1959
DOI https://doi.org/10.2147/DDDT.S101449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Xiangting Ge,1,2,* Zhiguo Feng,1,* Tingting Xu,2 Beibei Wu,3 Hongjin Chen,1 Fengli Xu,3 Lili Fu,1 Xiaoou Shan,3 Yuanrong Dai,2 Yali Zhang,1 Guang Liang1
1Chemical Biology Research Center, School of Pharmaceutical Sciences, 2Department of Pulmonary Medicine, The 2nd Affiliated Hospital, 3Department of Pediatrics, The 2nd Affiliated Hospital, Wenzhou Medical University, Wenzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: Sepsis remains a leading cause of death worldwide. Despite years of extensive research, effective drugs to treat sepsis in the clinic are lacking. In this study, we found a novel imidazopyridine derivative, X22, which has powerful anti-inflammatory activity. X22 dose-dependently inhibited lipopolysaccharide (LPS)-induced proinflammatory cytokine production in mouse primary peritoneal macrophages and RAW 264.7 macrophages. X22 also downregulated the LPS-induced proinflammatory gene expression in vitro. In vivo, X22 exhibited a significant protection against LPS-induced death. Pretreatment or treatment with X22 attenuated the sepsis-induced lung and liver injury by inhibiting the inflammatory response. In addition, X22 showed protection against LPS-induced acute lung injury. We additionally found that pretreatment with X22 reduced the inflammatory pain in the acetic acid and formalin models and reduced the dimethylbenzene-induced ear swelling and acetic acid-increased vascular permeability. Together, these data confirmed that X22 has multiple anti-inflammatory effects and may be a potential therapeutic option in the treatment of inflammatory diseases.
Keywords: LPS, imidazopyridine derivative, sepsis, acute lung injury, inflammation
Introduction
Sepsis, a whole-body inflammatory response triggered by an infection, remains a critical health problem and a major cause of death even in many modern intensive care units.1,2 Research efforts in the field of sepsis have focused primarily on the innate immune system, and typically have conceptually viewed sepsis as a syndrome of hyperinflammation.3,4 Reports pointed out cercal ligation and puncture (CLP) and lipopolysaccharide (LPS) were used for the establishment of sepsis animal model.5–7 Although CLP may greatly mimic the progression and characteristics of human sepsis, the high consistency and reproducibility of CLP-induced septic animal were seriously influenced by the little different procedures of CLP.6,8 However, Gram-negative bacteria are among the most important pathogens of sepsis and their LPS content is regarded as an important stimulator of the mammalian innate immune system that triggers the systemic inflammatory reaction.9 In sepsis, the large amount of cytokines produced causes edema, cellular metabolic stress, and, ultimately, tissue necrosis.10,11 Cytokines also induce vasodilatation and transient increase in capillary permeability producing extravasation of plasma proteins.12 The proinflammatory cytokines, such as tumor necrosis factor (TNF)-α and interleukin (IL)-6, are primarily involved in promoting inflammatory processes and play an important role in sepsis.10,13 The inhibitory targeting of cytokines has been identified as an important potential strategy for treating sepsis.
Many efforts have been made to explore anti-inflammatory drugs; however, there are no effective drugs to treat sepsis in a clinical setting. Activated drotrecogin alfa, originally marketed for severe sepsis, has not been found to be useful, and was withdrawn from the market in 2011.14 Eritoran, a synthetic tetraacylated lipid A, showed positive results in Phase I and Phase II clinical trials of severe sepsis.15 However, a Phase III clinical study for severe sepsis failed.16 Additionally, TAK242 was developed to inhibit the early stage of LPS signaling in host cells, but a clinical trial of TAK242 failed to suppress the cytokine storm in patients with severe sepsis.17 Thus, the development of novel anti-inflammatory agents for treating sepsis is urgent.
Benzimidazole and imidazopyridine have been shown to have anti-inflammatory activity in previous reports.18 Accordingly, we designed and synthesized a series of novel derivatives based on the structures of benzimidazole and imidazopyridine and evaluated their anti-inflammatory activity in vitro.19 X22, one of the imidazopyridine derivatives (the chemical structure is listed in Figure 1), exhibited its inhibitory activity on the LPS-induced production of TNF-α and IL-6 in RAW 264.7 macrophages.19 In addition, X22 was found to attenuate the high fat diet-induced arterial injuries and prevent the retinal ischemia-reperfusion injury through its anti-inflammatory effects.20,21 Based on these results, we wanted to find out whether X22 has the potential to treat sepsis. Herein, we will use LPS to induce mice sepsis for evaluating the anti-inflammatory activity of X22 in vitro and the protective effects of X22 on an endotoxin shock in vivo model.
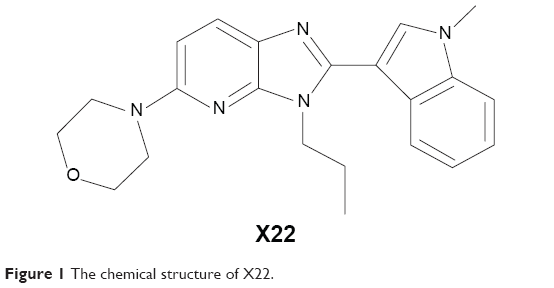 | Figure 1 The chemical structure of X22. |
Materials and methods
Animals
Institute of Cancer Research (ICR) mice and male C57BL/6 mice weighing 18–22 g were obtained from the Animal Center of Wenzhou Medical College (Wenzhou, People’s Republic of China). The animals were housed at a constant room temperature with a 12:12 hour light–dark cycle, and fed a standard rodent diet and water. The animals were acclimatized to the laboratory for at least 3 days before being used in experiments. Protocols involving the use of animals were approved by the Wenzhou Medical College Animal Policy and Welfare Committee (approval documents: wydw2013-0167). All animal care and experiments were performed in accordance with the approved protocols and the ‘The Detailed Rules and Regulations of Medical Animal Experiments Administration and Implementation’ (Document No. 1998–55, Ministry of Public Health, People’s Republic of China).
Reagents
LPS, acetic acid, and formalin solution were purchased from Sigma (Sigma-Aldrich Co., St Louis, MO, USA). Saline was prepared as a 0.9% NaCl solution. X22 was synthesized and characterized as previously described.16 The murine IL-6 and TNF-α enzyme-linked immunosorbent assay (ELISA) Kits were obtained from eBioscience (eBioScience, San Diego, CA, USA). Trizol-reagent and the two-step M-MLV Platinum SYBR Green quantitative polymerase chain reaction SuperMix-UDG kit were purchased from Invitrogen (Invitrogen, Carlsbad, CA, USA) Biotechnology (Santa Cruz, CA, USA).
Primary cell preparation and culture
Mouse peritoneal macrophage (MPM) preparation was conducted as previously described.22 Briefly, ICR mice were stimulated by an intraperitoneal injection of 2 mL thioglycollate solution per mouse and kept in pathogen-free conditions for 3 days before peritoneal macrophage isolation. Total peritoneal macrophages were harvested by washing the peritoneal cavity with Roswell Park Memorial Institute (RPMI)-1640 medium (8 mL per mouse), centrifuged, then the pellet was resuspended in RPMI-1640 medium with 10% fetal calf serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin. Peritoneal macrophages were cultured on 35 mm plates and maintained at 37°C in 5% CO2-humidified air. Nonadherent cells were removed by washing with medium at 4 hours after seeding. Experiments were undertaken after the cells adhered firmly to the culture plates.
Methyl thiazolyl tetrazolium assay
Mouse peritoneal macrophages were seeded into 96-well plates at a density of 5,000 cells per well in RPMI-1640 medium. Cells were incubated at 37°C in 5% CO2 for 24 hours. Cells were cultured with 40, 20, 10, 5, and 2.5 μM tested X22 or an equal volume of saline for 24 hours before the MTT assay. A fresh solution of methyl thiazolyl tetrazolium (MTT) (5 mg/mL) prepared in phosphate-buffered solution (PBS) was added to each single well. The plate was then incubated in an incubator with 5% CO2 for 4 hours. Afterwards, cells were dissolved with 150 μL dimethyl sulfoxide (DMSO), and the optical density was read at 490 nm. Viability was defined as the ratio (expressed as a percentage) of absorbance of treated cells to DMSO treated cells.
Determination of TNF-α and IL-6
The TNF-α and IL-6 levels in the medium, serum, and bronchoalveolar lavage fluid (BALF) were determined by ELISA analysis following the instructions of the manufacturer of the kit. Mouse peritoneal macrophages and RAW 264.7 macrophages were seeded into 6-well plates at a density of 40,000 cells per well in an RPMI-1640 medium. Cells were incubated at 37°C in 5% CO2 for 24 hours. Cells were cultured with a series of concentrations (40, 20, 10, 5, and 2.5 μM) of X22 or an equal volume of saline for 30 minutes, which was followed by the treatment of 0.5 μg/mL LPS. After treatment, the cells were incubated for 24 hours. The media were collected to measure the amount of TNF-α and IL-6. The total amount of the inflammatory factors in the culture medium was normalized to the total protein quantity of the viable cell pellets.
Real-time quantitative polymerase chain reaction
Total RNA was extracted from cells, lung, or liver tissues (50–100 mg) using TRIZOL according to the manufacturer’s protocol. Both reverse transcription and quantitative polymerase chain reaction were carried out using a two-step M-MLV Platinum SYBR Green quantitative polymerase chain reaction SuperMix-UDG kit. The Eppendorf Mastercycler ep realplex detection system was used for real-time quantitative polymerase chain reaction (RT-qPCR) analysis. The primers for the genes selected for the analysis, namely, inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), TNF-α, IL-6, IL-1β, and β-actin were synthesized by Invitrogen. The primer sequences used are shown as follows: mouse TNF-α sense primer: 5′-TGGAACTGGCAGAAGAGG-3′, mouse TNF-α antisense primer: 5′-AGACAGAAGAGCGTGGTG-3′; mouse IL-6 sense primer: 5′-GAGGATACCACTCCCAACAGACC-3′, mouse IL-6 antisense primer: 5′-AAGTGCATCATCGTTGTTCATACA-3′; mouse COX-2 sense primer: 5′-TGGTGCCTGGTCTGATGATG-3′, mouse COX-2 antisense primer: 5′-GTGGTAACCGCTCAGGTGTTG-3′; mouse iNOS sense primer: 5′-CAGCTGGGCTGTACAAACCTT-3′, mouse iNOS antisense primer: 5′-CATTGGAAGTGAAGCGTTTCG-3′; mouse IL-1β sense primer: 5′-ACTCCTTAGTCCTCGGCCA-3′, mouse IL-1β antisense primer: 5′-CCATCAGAGGCAAGGAGGAA-3′; mouse β-actin sense primer: 5′-TGGAATCCTGTGGCATCCATGAAAC-3′, mouse β-actin antisense primer: 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′. The amount of each gene was determined and normalized to the amount of β-actin.
Acute toxicity test
In the acute toxicity test, 30 C57BL/6 mice (aged 8 weeks) were randomly divided into three groups (five animals/sex/group). The vehicle group was injected 0.2 mL saline by tail vein, while the X22 groups were injected a single dose of 25 mg/kg and 50 mg/kg of X22 in 0.2 mL saline. All animals had free diet and water for 7 days and their body weight was recorded every 24 hours.
LPS-induced septic mortality and sepsis in C57BL/6 mice
As mice injected with X22 at 50 mg/kg showed no obvious toxicity, we used 20 mg/kg as the maximum concentration for further in vivo study. For septic mortality experiment, male C57BL/6 mice, weighing 18–22 g, were treated with 20 mg/kg of an X22 solution by tail vein injection 15 minutes before a 20 mg/kg LPS injection. Negative control animals received only an equal volume of saline. After the LPS injection, mice were monitored every 24 hours for 7 days; we set explicitly death as standard for mortality and recorded the mortality for 7 days. For sepsis experiment, male C57BL/6 mice, weighing 18–22 g, were treated with 10 or 20 mg/kg of an X22 solution by tail vein injection 15 minutes before a 20 mg/kg LPS injection, or a 20 mg/kg X22 solution by tail vein injection 15 minutes after 20 mg/kg LPS injection. Negative control animals received only an equal volume of saline. Mice were anesthetized and sacrificed 6 hours after LPS injection. Blood samples were collected from the right ventricle orbital veniplex using a heparinized syringe with a needle. Lung and liver tissues were harvested.
Lung and liver histopathology and immunohistochemistry analysis
Lung and liver tissues were fixed in 4% paraformaldehyde solution, embedded in paraffin, and sectioned at 5 μm. After dehydration, sections were stained with hematoxylin and eosin according to the previously reported methods. A pathologist blindly scored each lung injury according to the following four categories: alveolar congestion, hemorrhage, neutrophil infiltration into the airspace or vessel wall, and thickness of alveolar wall/hyaline membrane formation. Each category was graded on a 0- to 4-point scale: 0= no injury; 1= injury up to 25% of the field; 2= injury up to 50% of the field; 3= injury up to 75% of the field; and 4= diffuse injury.23 Liver histological scoring was performed according to Adawi et al with slight modification.24 The degree of hepatocellular necrosis and inflammatory cell infiltration was quantitatively assessed blindly at the Department of Pathology as follows: 1, minimal; 2, moderate; 3, submassive; and 4, massive. The immunohistochemistry analysis was performed following the staining protocol for the anti-CD68 antibody.
LPS-induced acute lung injury (ALI)
Male C57BL/6 mice, weighing 18–22 g, were treated with 10 or 20 mg/kg of an X22 solution by tail vein injection 15 minutes before 5 mg/kg of LPS was administered by intratracheal instillation, or 20 mg/kg of an X22 solution by tail vein injection 15 minutes after 5 mg/kg of LPS was administered by intratracheal instillation. For the LPS intratracheal instillation, the mice were anesthetized by ether. Their necks were then dissected to visualize the trachea. Mice were then intubated with a 25-gauge catheter. LPS was instilled through the intratracheal catheter at a dose of 5 mg/kg, and diluted to a total volume of 1 mL/kg with saline to induce ALI. Control mice were instilled with the same volume of saline (1 mL/kg) intratracheally. After LPS instillation, the neck was sutured with nylon thread and the mice were left to recover. Six hours after LPS administration, animals were euthanized to collect the BALF and lung tissue samples. Collection of the BALF was performed three times through a tracheal cannula with autoclaved physiological saline, instilled up to a total volume of 3 mL.
Acetic acid-induced vascular permeability assay in ICR mice
Model animals received 200 μL each of 10% azovan blue solution in saline injected intravenously and 300 μL of acetic acid (0.7%) injected intraperitoneally. Following 30 minutes after the acetic acid injection, the peritoneal fluid was collected in a sterile and heparinized PBS (1 mL) solution. The total peritoneal fluid was evaluated by an optical density assay (620 nm).
Dimethylbenzene-induced ear swelling in mice
Ear swelling was induced by smearing of dimethylbenzene (20 μL/ear) in the right ear, while the left ear was smeared with an equal volume of saline as a negative control. The thickness of both ears was measured by a Vernier caliper after swelling, in each mouse.
Chemically-induced inflammatory abdominal constrictions in mice
Abdominal constrictions caused by the intraperitoneal injection of acetic acid (0.6%) were monitored. Male ICR mice weighing 18–22 g were pretreated with X22 (20 mg/kg, tail vein injection) 15 minutes before the injection of acetic acid. After the challenge, pairs of mice were placed in separate boxes, and the number of abdominal constrictions was cumulatively counted.
Formalin-induced inflammatory nociception in mice
Mice were injected under their paw surface of the right hind paw with 20 μL of 2.5% formalin in PBS. The mice were observed simultaneously from 0 to 30 minutes following the injection of formalin. The amount of time spent licking the injected paw was timed with a chronometer and was considered indicative of pain.
Statistical analysis
The results are presented as standard error of mean ( ± SEM). The statistical significance of differences between groups was obtained by Student’s t-test. A P-value of less than 0.05 (P<0.05) was considered indicative of significance.
Results
Cytotoxicity
Before our study, we tested the cytotoxicity of X22 in mouse peritoneal macrophages by MTT after 24 hours of treatment of the cells with different concentrations (2.5, 5, 10, 20, and 40 μM) of X22. The treatment with X22 displayed no obvious toxicity in MPMs, as shown in Figure 2, indicating that X22, even at 40 μM, was relatively safe.
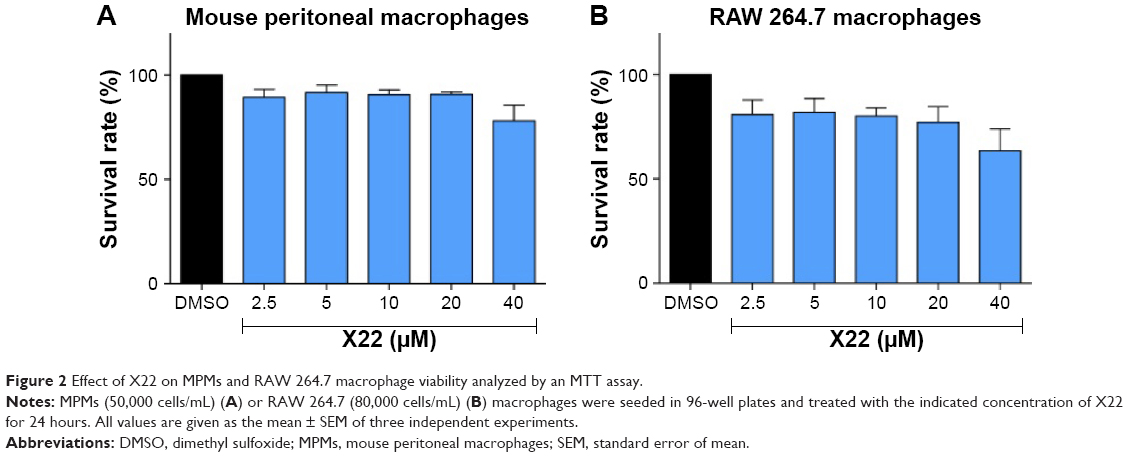 | Figure 2 Effect of X22 on MPMs and RAW 264.7 macrophage viability analyzed by an MTT assay. |
X22 inhibited LPS-induced production of proinflammatory cytokines in MPMs
Subsequently, to determine the anti-inflammatory effect of X22, the level of the proinflammatory cytokines, IL-6, and TNF-α, induced by LPS, was monitored in both MPMs and RAW 264.7 macrophages. The MPMs and RAW 264.7 macrophages were treated with LPS (0.5 μg/mL) in the presence or absence of X22 for 24 hours and then the medium was examined for the production of proinflammatory cytokines by ELISA analysis. The data presented in Figure 3A and B revealed that pretreatment with X22 could dose-dependently decrease the LPS-induced production of TNF-α and IL-6 in MPMs. Specifically, the results indicated that the LPS-induced expression of inflammatory cytokines was inhibited the most by X22 at 20 and 40 μM. Similar results were observed in RAW 264.7 macrophages, where X22 also inhibited the LPS-induced TNF-α and IL-6 secretion (Figure 3C and D). After considering the results of the cytotoxicity and the anti-inflammatory activities, the concentration of 20 μM was selected for further studies.
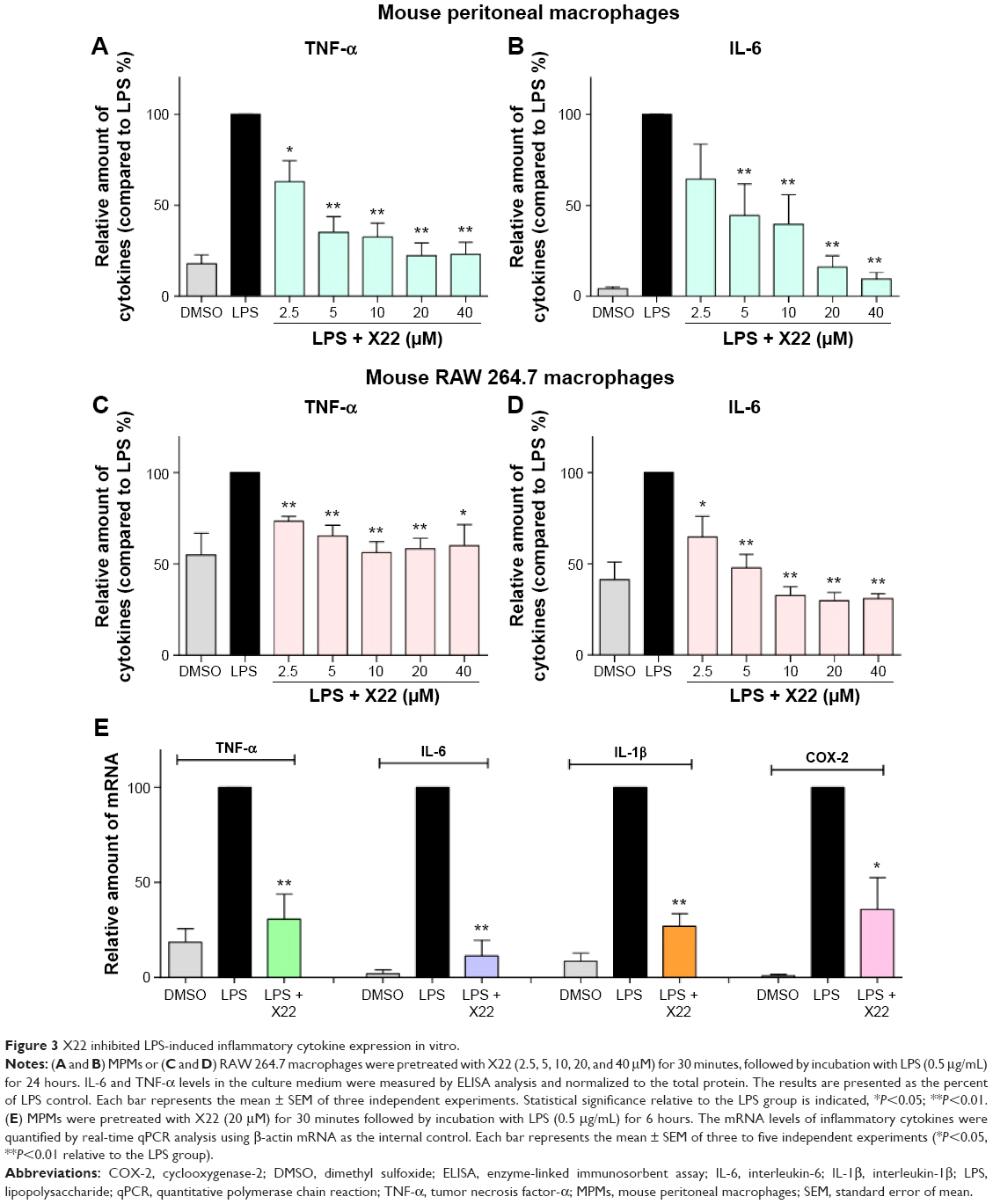 | Figure 3 X22 inhibited LPS-induced inflammatory cytokine expression in vitro. |
Next, we examined whether the compound X22 exerts any inhibitory effects on the messenger RNA (mRNA) expression of TNF-α, IL-6, IL-1β, and COX-2 by real-time quantitative polymerase chain reaction analysis. MPMs were treated with LPS (0.5 μg/mL) in the presence or absence of X22 for 6 hours and LPS significantly induced the expression of proinflammatory genes (Figure 3E). The results show that pretreatment with X22 at 20 μM obviously inhibited the LPS-induced TNF-α, IL-6, IL-1β, and COX-2 mRNA expression.
X22 effectively protects mice from LPS-induced mortality
The body weight of the X22-treated groups (25 and 50 mg/kg, tail vein injection) was not different from that of the control group (Figure 4A), which indicates that X22, even at the high dose of 50 mg/kg, is not toxic in mice. Male C57BL/6 mice injected with LPS (20 mg/kg) in the presence of X22 pretreatment (tail vein injection) were monitored for 7 days to assess their survival rates. The data presented in Figure 4B revealed that LPS alone caused 80% of mice death within 48 hours, whereas pretreatment with X22 at 20 mg/kg prior to LPS injection significantly improved the survival rate compared with the LPS-treated group alone.
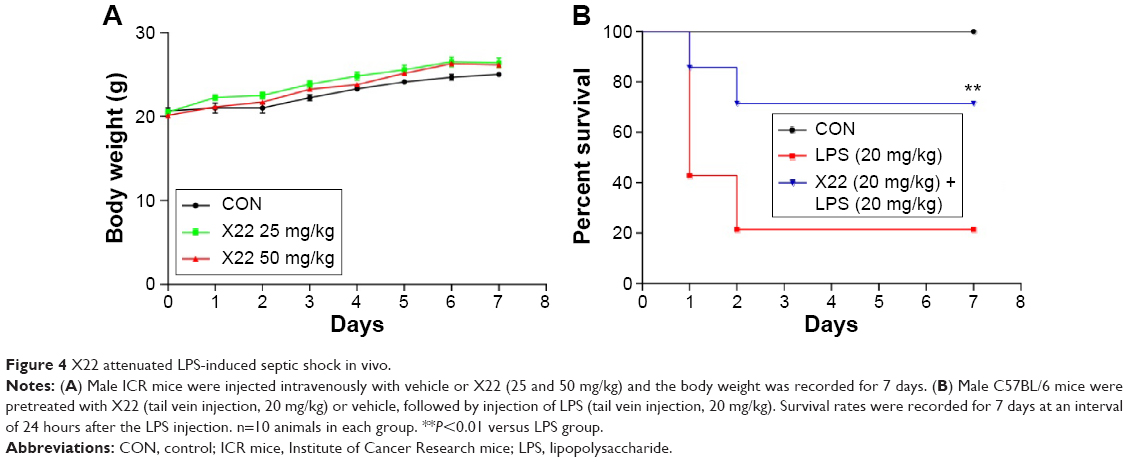 | Figure 4 X22 attenuated LPS-induced septic shock in vivo. |
X22 decreases TNF-α expression in serum
X22 was further evaluated for its anti-inflammatory benefits to animals. Mice were treated with X22 at 20 or 10 mg/kg before being administered LPS at 20 mg/kg or treated with X22 at 20 mg/kg after being administered LPS at 20 mg/kg. Mice in the negative control group were treated with vehicle (saline) and those in the positive control group were injected with LPS alone at 20 mg/kg. All mice were sacrificed 6 hours after LPS injection and the plasma TNF-α level was determined by ELISA analysis. The result revealed that the serum level of TNF-α both in the X22 pre-LPS challenged groups and X22 post-LPS challenged group was significantly decreased compared to the LPS alone group, as shown in Figure 5. This result indicated that X22 has an obvious anti-inflammatory activity in vivo.
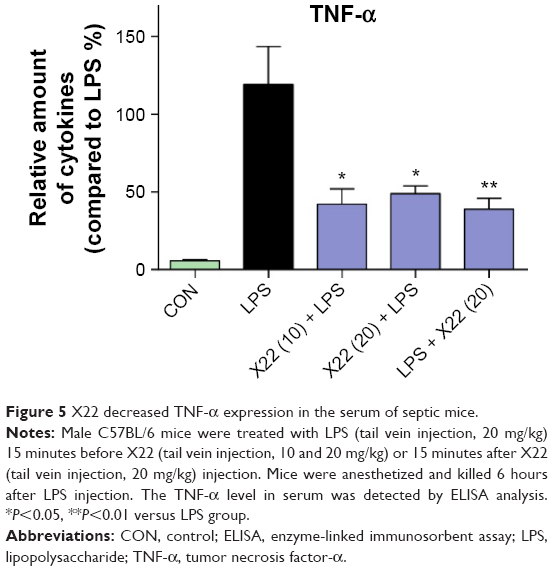 | Figure 5 X22 decreased TNF-α expression in the serum of septic mice. |
Effect of X22 on reducing sepsis-induced inflammation and histopathological changes in lung and liver tissues
Lung is one of the primary organs affected in septic shock. Real-time polymerase chain reaction analysis was used to verify the profiles of the selected genes TNF-α, IL-1β, IL-6, and iNOS, which were found to be involved in the inflammatory response in mouse lung after the LPS challenge. The mRNA levels of these four genes in the lung were significantly induced by LPS and suppressed by X22 both in X22 pre-LPS challenged groups and X22 post-LPS challenged group (Figure 6A–D). These results suggested that X22 significantly inhibits the transcription of the LPS-induced inflammatory genes in lung. Furthermore, LPS injection induced structural change and inflammatory cells infiltration in lung tissues compared with the control group (Figure 6E and G). Figure 6F shows the semiquantitative lung injury score, which further reveals the protective effects of X22. Compared with the LPS group, the X22 pre-LPS challenged groups and X22 post-LPS challenged group had normal lung structure, very little histopathological changes, and macrophage infiltration.
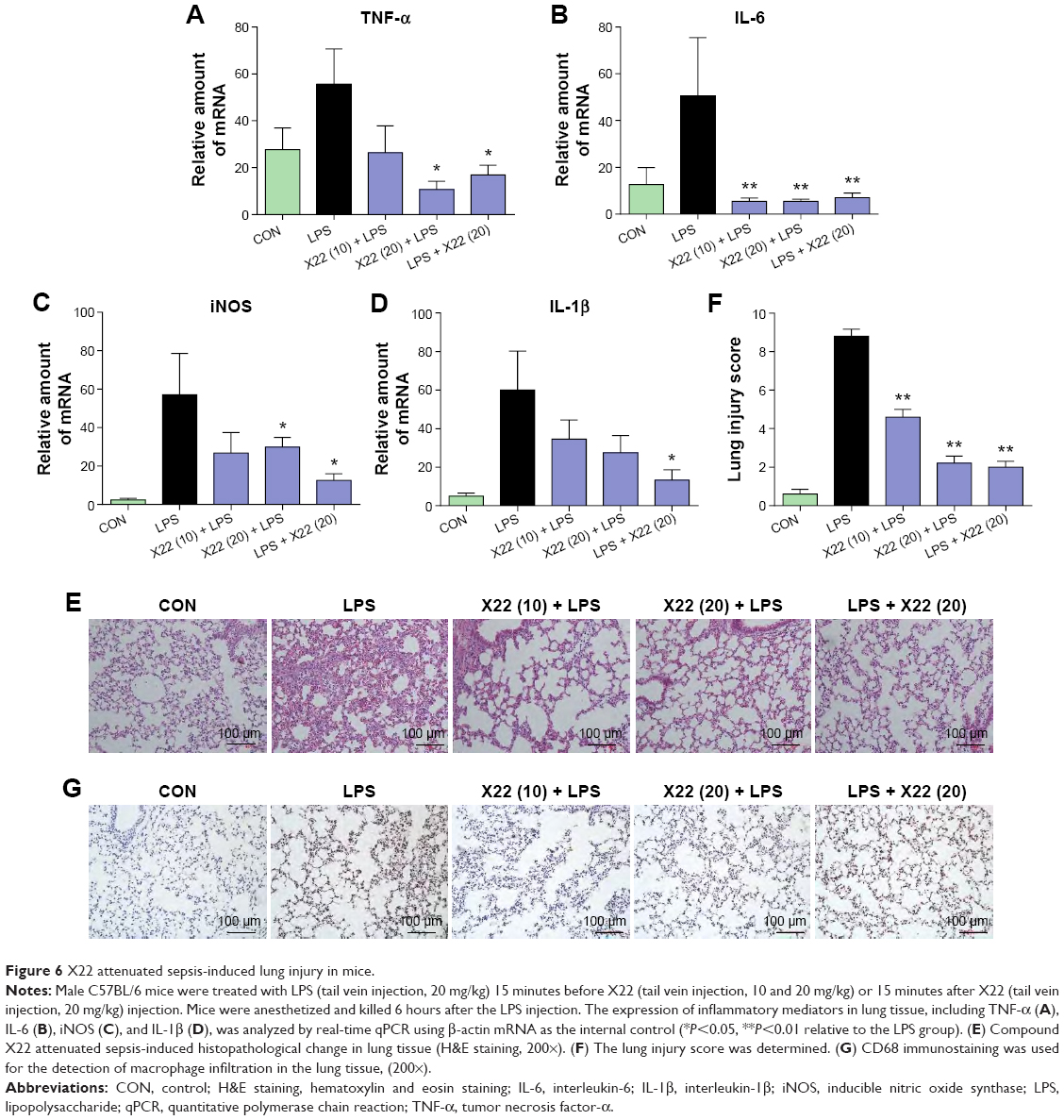 | Figure 6 X22 attenuated sepsis-induced lung injury in mice. |
Similar results were observed in the liver tissue as well. As shown in Figure 7A–D, compound X22 significantly inhibited the sepsis-induced upregulation of TNF-α, IL-6, iNOS, and IL-1β transcripts in the liver tissue of both the X22 pre-LPS challenged groups and X22 post-LPS challenged group. In addition, we also observed the effect of compound X22 on the changes of liver function. In particular, sepsis clearly induced the increase of alanine aminotransferase and aspartate transaminase, the markers of liver function, whereas X22 attenuated such changes of liver function (Figure 7E and F). Staining with hematoxylin and eosin showed that the liver sections from the sepsis group underwent liver structural changes and inflammatory cell infiltration, whereas in the negative control, X22 pre-LPS challenged groups, and X22 post-LPS challenged group, such changes were not evident (Figure 7G). Figure 7H shows the semiquantitative liver injury score, which further reveals the protective effects of X22. Macrophage infiltration was detected by CD68 immunohistochemical staining analysis. The results of this analysis revealed that, as shown in Figure 7I, compared with the LPS group, X22 pre-LPS challenged groups and X22 post-LPS challenged group reduce the sepsis-induced large amount of macrophage infiltration.
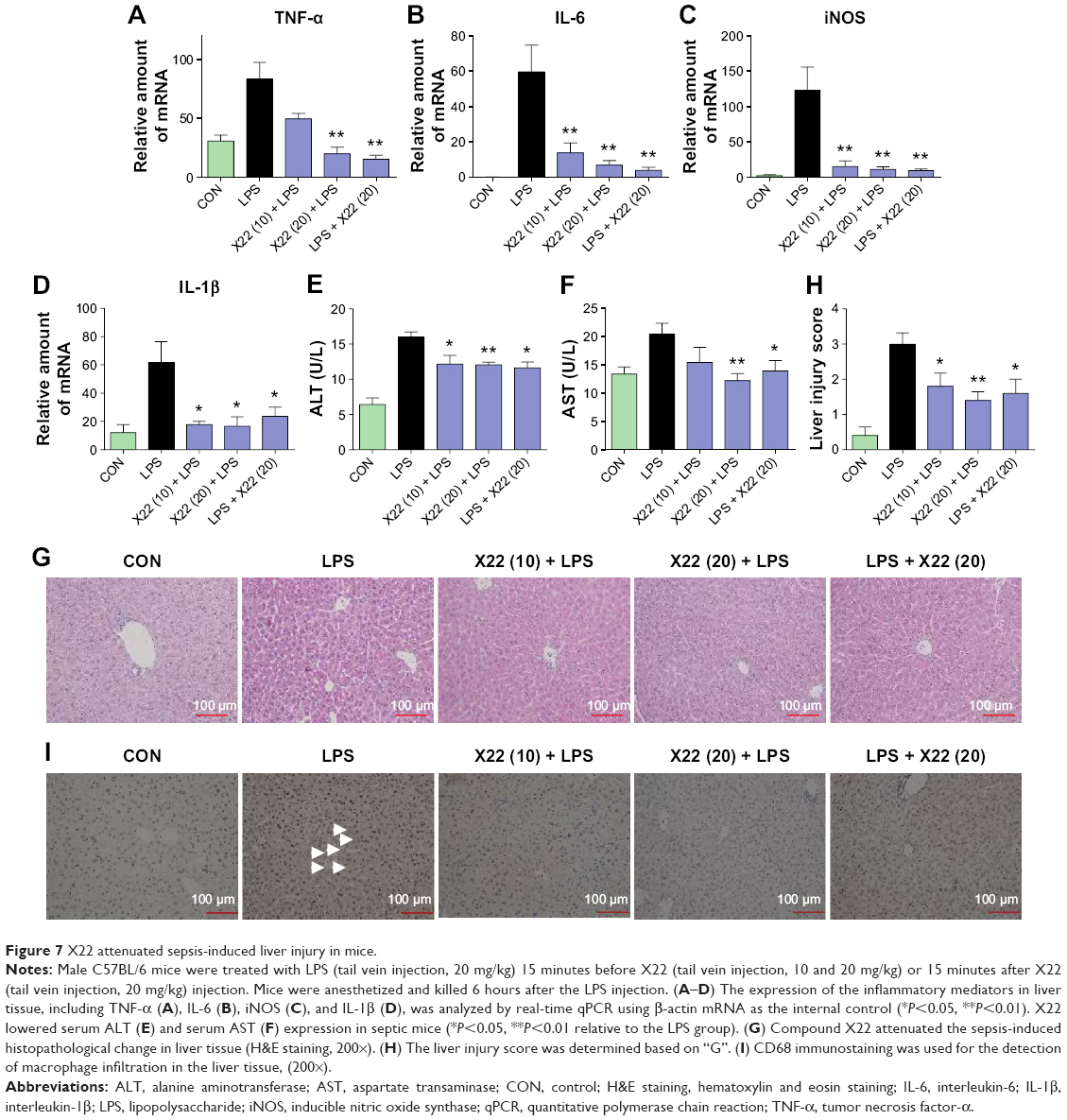 | Figure 7 X22 attenuated sepsis-induced liver injury in mice. |
Effect of X22 on LPS-induced ALI
To further characterize the anti-inflammatory effects of X22 in vivo, we assessed LPS-induced ALI in C57BL/6 mice. Treatment with X22 markedly attenuated ALI as indicated by the observed amelioration of histological changes (Figure 8A) and Figure 8B shows the semiquantitative lung injury score. Treatment with X22 also reduced the ALI-induced increase of total number of cells in BALF (Figure 8C). Meanwhile, X22 also inhibited the LPS-induced expression of TNF-α and IL-6 in BALF (Figure 8D and E), and TNF-α in serum (Figure 8F). We next analyzed the proinflammatory gene expression in lung tissue. The data, presented in Figure 8G–I, indicate that pretreatment of mice with X22 dose-dependently reduced the ALI-induced IL-6, IL-1β, and COX-2 mRNA transcription. In particular, the X22 post-LPS challenged group exhibited similar effects as the X22 pre-LPS challenged groups. Taken together, our results revealed that X22 has the ability to attenuate ALI by inhibiting the inflammatory response.
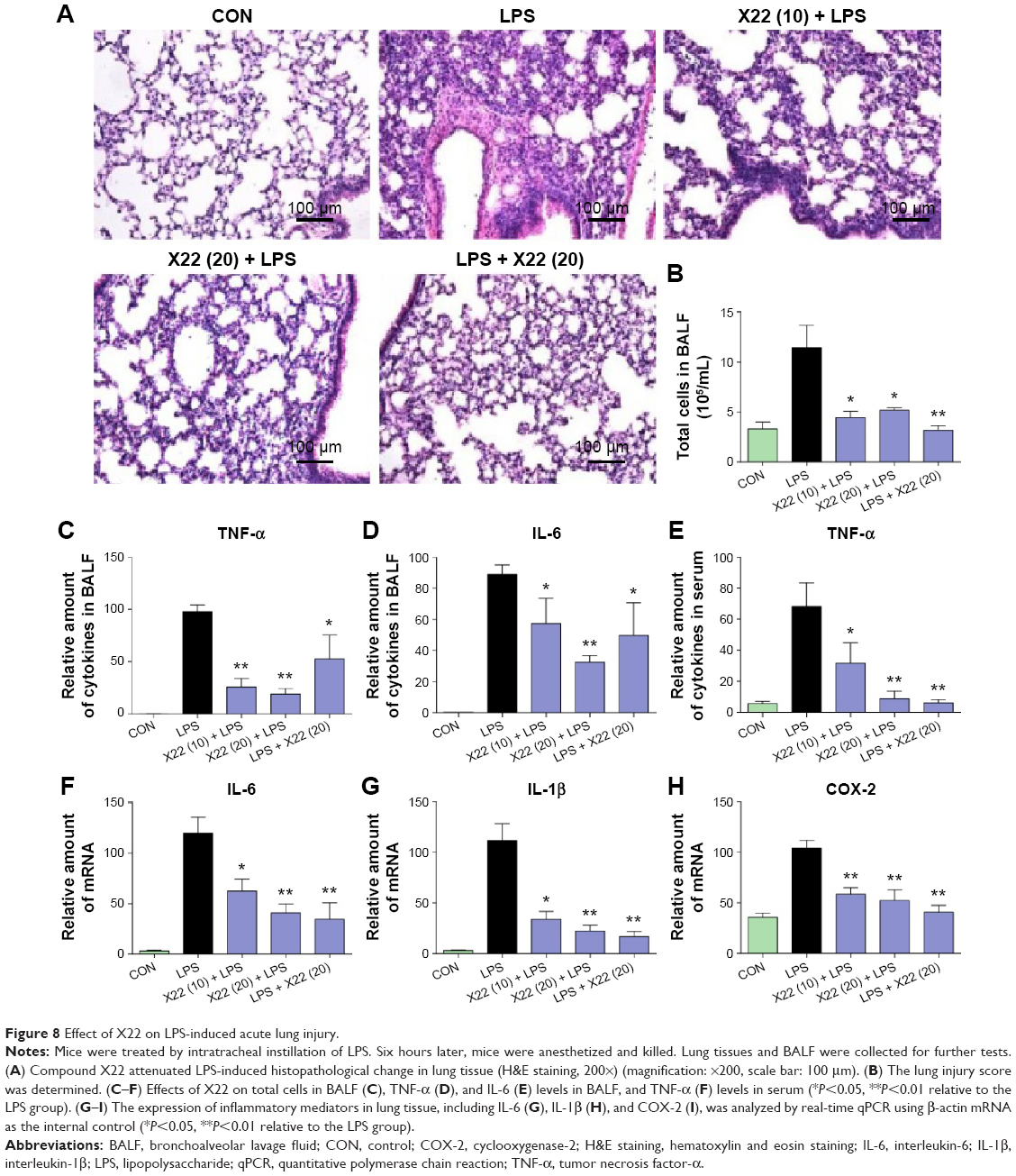 | Figure 8 Effect of X22 on LPS-induced acute lung injury. |
Treatment with X22 inhibited inflammation-related reactions in chemically-induced inflammatory models
We further examined the beneficial effects of X22 on inflammation-related models challenged by other stimuli. Dimethylbenzene-induced ear swelling has been widely used as an inflammation model in vivo.25 We found that compared to the dimethylbenzene group, pretreatment with X22 (20 mg/kg, tail vein injection) could significantly reduce the ear swelling, as shown in Figure 9A. Furthermore, we found that ICR mice pretreated with X22 (20 mg/kg, tail vein injection) 15 minutes before intraperitoneal injection of acetic acid showed less writhing number (Figure 9B). Additionally, pretreatment with X22 also reduced the acetic acid-induced vascular permeability using azovan blue dye (Figure 9C).
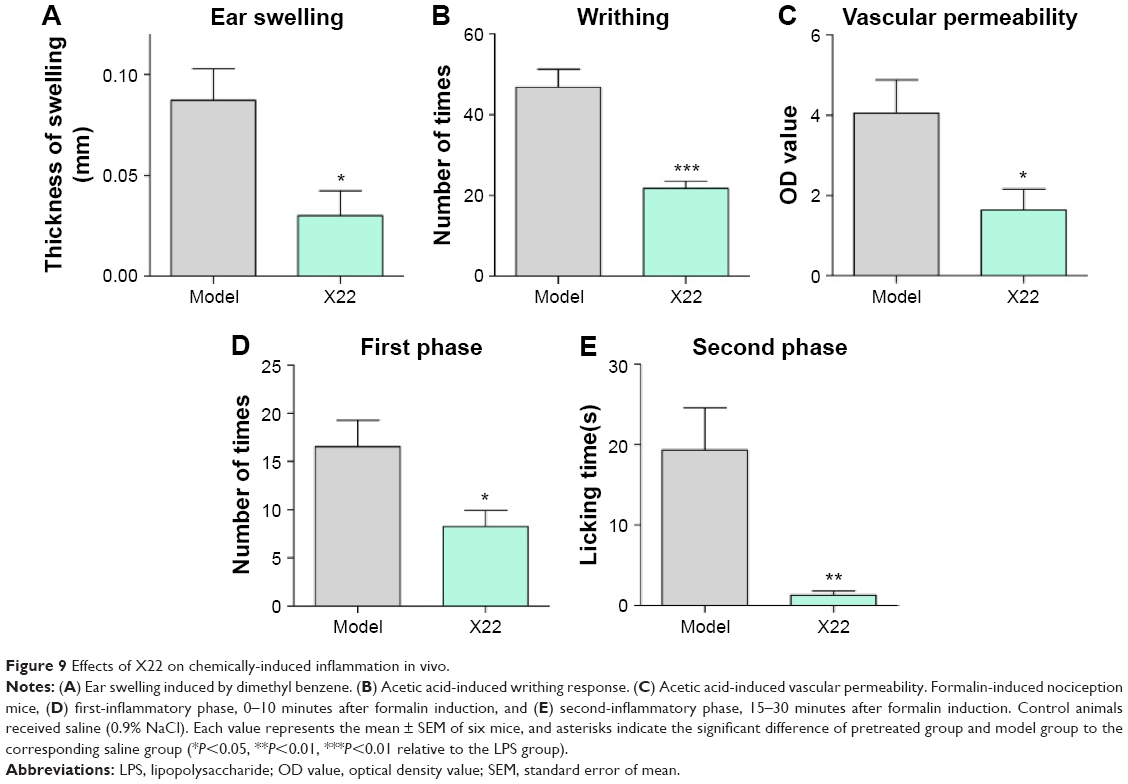 | Figure 9 Effects of X22 on chemically-induced inflammation in vivo. |
We also evaluated the inhibitory effects of X22 on formalin-induced inflammatory nociception (both first and second phase) in ICR mice. The results show that pretreatment with X22 could reduce the licking times of the mouse which was stimulated by formalin, with an inhibition of 50.2% in the first phase (0–10 minutes after formalin injection) and an inhibition of 95.3% in the second phase (15–30 minutes after formalin injection), as illustrated in Figure 9D and E. The results strongly suggest that X22 is able to effectively inhibit the inflammatory response induced by chemical stimuli, such as acetic acid and formalin, in vivo.
Discussion
Numerous preclinical and clinical studies show that patients with conditions, such as trauma and burns, are highly prone to infection induced by sepsis which often causes septic shock and multiorgan failure.26–29 At present, the current treatments for sepsis include antimicrobial and non-antimicrobial therapies.30 Leone et al30 thought the empiric use of new cephalosporin or tygecycline should not be recommended in patients because of the high risk of multidrug resistant pathogen carriage. The inefficacy of antibiotics against key pathogens makes their use in severe sepsis unsafe.31 Among non-antimicrobial therapy, anti-inflammatory therapy is the main strategy. Steroids and nonsteroidal anti-inflammatory drugs have long been used as major therapeutic anti-inflammatory agents.32,33 High doses of steroids have failed to show a benefit in severe sepsis and whether a low dose of hydrocortisone should be used in severe sepsis still needs to be confirmed by ongoing studies.30 The substantial body of accumulated evidence from cellular and animal models and the trends from human studies suggest that nonsteroidal anti-inflammatory drugs may have beneficial effects in sepsis and further study is warranted.34 Low-dose of acetyl salicylic acid appears to be beneficial in the prevention and treatment of sepsis and systemic inflammatory response syndrome.35 However, the serious side effects of acetyl salicylic acid, including renal impairment and increased bleeding, cannot be ignored by us.36,37 Accordingly, the need to find novel and effective therapeutic approaches for sepsis is all the more urgent.
In our previous studies, we synthesized many mono-carbonyl curcumin analogs and evaluated their anti-inflammatory activity, and some exhibited promising pharmacological effects and are currently undergoing preclinical evaluation.38,39 However, the water insoluble activity of these active compounds limits their usefulness in the treatment of sepsis. X22, which was synthesized based on the structure of imidazopyridine, exhibited obvious anti-inflammatory activity19 and water solubility. Thus, we considered X22 as a promising anti-inflammatory candidate for treating sepsis. Here, we show that X22 dose-dependently inhibited LPS-induced TNF-α and IL-6 production in MPMs and RAW 264.7 macrophages (Figure 3A–D). Although X22 exhibits similar inhibitory rates against TNF-α and IL-6 to previously reported compounds, the water solubility of X22 gives it more potential for clinical study. Upon LPS stimulation, various inflammatory mediators, such as cytokines and enzymes, are induced at varying magnitudes.40 The mRNA levels of TNF-α, IL-6, IL-1β, and COX-2 were shown to be upregulated by LPS induction. X22, at 20 μM, reduced the expression of inflammatory genes in LPS treated MPMs (Figure 3E).
As a major endotoxin, LPS from Gram-negative bacteria has been implicated as a major cause of sepsis. In vivo, 20 mg/kg LPS was employed to induce sepsis in animals, which can cause a variety of organ failures, such as lung and liver tissue.41 After LPS injection, the peak levels of inflammatory cytokines induced by LPS were at between 1.5 and 4 hours and tissue injury began from 6 hours,5 so we choose 6 hours to evaluate the tissue protective effects of X22 in sepsis. As expected, animals receiving X22 either prior to LPS or after LPS were protected from LPS-induced septic death improving the survival rate by 50% and they exhibited attenuated lung and liver injuries (Figures 4–7). However, Kim et al report their novel synthetic histone deacetylase inhibitor, 9a (20 mg/kg), improved survival rate by 30%.42 Mohanty et al reported that TAF-5 played a protective effect in lethal shock. With the oral dose of 10 and 30 mg/kg, the animals’ survival rate were 20% and 30%, respectively.43 These results elucidated X22 has the potential to be a candidate for reducing the mortality of sepsis.
To elucidate the anti-inflammatory mechanism of X22 in vitro and in vivo, we did a lot of work. Several reports pointed out that imidazopyridine derivatives were inhibitors of COX-2;44 we have detected COX-2 activity. However, X22 has no effect on inhibiting the activity of COX-2 (data not shown). In response to LPS stimulation, MAPKs are phosphorylated and NF-κB are activated.45 In our two previous studies, we found that X22 attenuates obesity-induced arterial injuries by inhibiting NF-κB activation and prevents the retinal ischemia-reperfusion injury via the inhibition of MAPKs.20,21 However, we found no effects of X22 on the phosphorylation of MAPKs and NF-κB activation in both MPMs and RAW 264.7 macrophages (data not shown). Unfortunately, our study does not shed any light on the anti-inflammatory mechanism of X22 in LPS-induced sepsis. In future work, we will explore the anti-inflammatory mechanism of X22.
Conclusion
In summary, the main findings of the present study, both in vitro and in vivo, demonstrate the preventive role of X22 against the LPS-induced inflammatory response, sepsis-induced lung and liver injuries, and septic mortality. Although continued research is required to examine the underlying anti-inflammatory mechanism of X22, these results clearly suggest the therapeutic potential of X22 in the treatment of sepsis. In addition, we also found that X22 has the ability to prevent LPS-induced ALI, dimethylbenzene-induced ear swelling, acetic acid-caused writhing response and vascular permeability, and formalin-induced inflammatory nociception. Taken together, we found that X22 may be a potential agent for treating inflammatory diseases, including sepsis.
Acknowledgments
Financial support was provided by the National Natural Science Funding of China (81503123, 81570027, and 21272179), Zhejiang Provincial Natural Science Funding (LQ14H310003), and Funding for Scientific Research of Wenzhou Medical University (QTJ15010).
Author contributions
GL, YZ, and XG conceived the project and wrote the paper; XG, TX, BW, HC, and FX performed the biological experiments; ZF and LF synthesized the compound X22; XS and YD revised the paper. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Martin GS. Sepsis, severe sepsis and septic shock: changes in incidence, pathogens and outcomes. Expert Rev Anti Infect Ther. 2012;10(6):701–706. | ||
Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS international sepsis definitions conference. Crit Care Med. 2003;31(4):1250–1256. | ||
Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013;13(3):260–268. | ||
Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420(6917):885–891. | ||
Remick DG, Newcomb DE, Bolgos GL, Call DR. Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation find puncture. Shock. 2000;13(2):110–116. | ||
Dejager L, Pinheiro I, Dejonckheere E, Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 2011;19(4):198–208. | ||
Willenberg I, Rund K, Rong S, Shushakova N, Gueler F, Schebb NH. Characterization of changes in plasma and tissue oxylipin levels in LPS and CLP induced murine sepsis. Inflamm Res. 2016;65: 133–142. | ||
Rittirsch D, Hoesel LM, Ward PA. The disconnect between animal models of sepsis and human sepsis. J Leuko Biol. 2007;81(1): 137–143. | ||
Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327(5963):291–295. | ||
Schulte W, Bernhagen J, Bucala R. Cytokines in sepsis: potent immunoregulators and potential therapeutic targets – an updated view. Mediators Inflamm. 2013;2013:165974. | ||
Lichtenstern C, Brenner T, Bardenheuer HJ, Weigand MA. Predictors of survival in sepsis: what is the best inflammatory marker to measure? Curr Opin Infect Dis. 2012;25(3):328–336. | ||
Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol. 2009;77(12):1763–1772. | ||
Kothari N, Bogra J, Abbas H, et al. Tumor necrosis factor gene polymorphism results in high TNF level in sepsis and septic shock. Cytokine. 2013;61(2):676–681. | ||
Poole D, Bertolini G, Garattini S. Withdrawal of ‘Xigris’ from the market: old and new lessons. J Epidemiol Community Health. 2012;66(7):571–572. | ||
Tidswell M, Tillis W, Larosa SP, et al. Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit Care Med. 2010;38(1):72–83. | ||
Opal SM, Laterre PF, Francois B, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309(11):1154–1162. | ||
Rice TW, Wheeler AP, Bernard GR, et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. 2010;38(8):1685–1694. | ||
Grice CA, Tays KL, Savall BM, et al. Identification of a potent, selective, and orally active leukotriene a4 hydrolase inhibitor with anti-inflammatory activity. J Med Chem. 2008;51(14):4150–4169. | ||
Chen G, Liu Z, Zhang Y, et al. Synthesis and anti-inflammatory evaluation of novel benzimidazole and imidazopyridine derivatives. ACS Med Chem lett. 2013;4(1):69–74. | ||
Bian Y, Ren L, Wang L, et al. A novel imidazopyridine derivative, X22, prevents the retinal ischemia-reperfusion injury via inhibition of MAPKs. Exp Eye Res. 2015;135:26–36. | ||
Li W, Wang L, Huang W, et al. Inhibition of ROS and inflammation by an imidazopyridine derivative X22 attenuate high fat diet-induced arterial injuries. Vascular Pharmacol. 2015;72:153–162. | ||
Wu J, Li J, Cai Y, et al. Evaluation and discovery of novel synthetic chalcone derivatives as anti-inflammatory agents. J Med Chem. 2011;54(23):8110–8123. | ||
Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol. 2007;179(3):1855–1863. | ||
Adawi D, Kasravi FB, Molin G, Jeppsson B. Effect of Lactobacillus supplementation with and without arginine on liver damage and bacterial translocation in an acute liver injury model in the rat. Hepatology. 1997;25(3):642–647. | ||
Li Q, Yang S, Yang S, Xin F, Wang M. Anti-inflammatory activity of phlomisoside F isolated from Phlomis younghusbandii Mukerjee. Int Immunopharmacol. 2015;28(1):724–730. | ||
Gustot T. Multiple organ failure in sepsis: prognosis and role of systemic inflammatory response. Curr Opin Crit Care. 2011;17(2):153–159. | ||
Bohannon JK, Luan L, Hernandez A, et al. Role of G-CSF in monophosphoryl lipid A-mediated augmentation of neutrophil functions after burn injury. J Leukoc Biol. Epub 2015 Nov 4. | ||
Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):U104–U115. | ||
Patil NK, Parajuli N, MacMillan-Crow LA, Mayeux PR. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. Am J Physiol Renal Physiol. 2014;306(7):F734–F743. | ||
Leone M, Textoris J, Michel F, Wiramus S, Martin C. Emerging drugs in sepsis. Expert Opin Emerg Drugs. 2010;15(1):41–52. | ||
Savaris RF, de Moraes GS, Cristovam RA, Braun RD. Are antibiotics necessary after 48 hours of improvement in infected/septic abortions? A randomized controlled trial followed by a cohort study. Am J Obstet Gynecol. 2011;204(4):301.e301–e305. | ||
Kessel L, Tendal B, Jorgensen KJ, et al. Post-cataract prevention of inflammation and macular edema by steroid and nonsteroidal anti-inflammatory eye drops: a systematic review. Ophthalmology. 2014;121(10):1915–1924. | ||
Jaturapatporn D, Isaac MG, McCleery J, Tabet N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst Rev. 2012;2:CD006378. | ||
Eisen DP. Manifold beneficial effects of acetyl salicylic acid and nonsteroidal anti-inflammatory drugs on sepsis. Intensive Care Med. 2012;38(8):1249–1257. | ||
Eisen DP, Reid D, McBryde ES. Acetyl salicylic acid usage and mortality in critically ill patients with the systemic inflammatory response syndrome and sepsis. Crit Care Med. 2012;40(6):1761–1767. | ||
Berg KJ. Acute effects of acetylsalicylic acid in patients with chronic renal insufficiency. Eur J Clin Pharmacol. 1977;11(2):111–116. | ||
Dale J, Myhre E, Loew D. Bleeding during acetylsalicylic acid and anticoagulant therapy in patients with reduced platelet reactivity after aortic valve replacement. Am Heart J. 1980;99(6):746–752. | ||
Wu J, Zhang Y, Cai Y, et al. Discovery and evaluation of piperid-4-one-containing mono-carbonyl analogs of curcumin as anti-inflammatory agents. Bioorg Med Chem. 2013;21(11):3058–3065. | ||
Zhang Y, Jiang X, Peng K, et al. Discovery and evaluation of novel anti-inflammatory derivatives of natural bioactive curcumin. Drug Des Devel Ther. 2014;8:2161–2171. | ||
Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117(14):3720–3732. | ||
Bayraktar O, Tekin N, Aydin O, Akyuz F, Musmul A, Burukoglu D. Effects of S-allyl cysteine on lung and liver tissue in a rat model of lipopolysaccharide-induced sepsis. Naunyn Schmiedebergs Arch Pharmacol. 2015;388(3):327–335. | ||
Kim SJ, Baek KS, Park HJ, et al. 9a, a novel synthetic histone deacetylase inhibitor, protects against septic injury by suppressing MAPK signaling in mice. Br J Pharmacol. 2015; Epub ahead of print. | ||
Mohanty S, Gautam Y, Maurya AK, et al. Indenes and tetralenes analogues attenuates lipopolysaccharide-induced inflammation: an in-vitro and in-vivo study. Chem Biol Interact. 2016;245:12–19. | ||
Movahed MA, Zarghi A. Design and synthesis of new imidazopyridine derivatives as selective COX-2 inhibitors. Res Pharm Sci. 2012;7(5):S531. | ||
Lu Y-C, Yeh W-C, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145–151. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.