Back to Journals » OncoTargets and Therapy » Volume 14
A New Treatment Strategy for Early T‐Cell Precursor Acute Lymphoblastic Leukemia: A Case Report and Literature Review
Authors Mao J, Xue L, Wang H, Zhu Y ![]() , Wang J, Zhao L
, Wang J, Zhao L
Received 25 March 2021
Accepted for publication 3 June 2021
Published 18 June 2021 Volume 2021:14 Pages 3795—3802
DOI https://doi.org/10.2147/OTT.S312494
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Jianping Mao,1,* Lianguo Xue,1,* Haiqing Wang,2 Yuanxin Zhu,1 Juan Wang,3 Lidong Zhao1
1Department of Hematology, The First People’s Hospital of Lianyungang, The Affiliated Lianyungang Hospital of Xuzhou Medical University, The Affiliated Hospital of Kangda College of Nanjing Medical University, Lianyungang Clinical College of Nanjing Medical University, Lianyungang, People’s Republic of China; 2Department of Laboratory medicine, The First People’s Hospital of Lianyungang, The Affiliated Lianyungang Hospital of Xuzhou Medical University, The Affiliated Hospital of Kangda College of Nanjing Medical University, Lianyungang Clinical College of Nanjing Medical University, Lianyungang, People’s Republic of China; 3Department of Pediatrics, The First People’s Hospital of Lianyungang, The Affiliated Lianyungang Hospital of Xuzhou Medical University, The Affiliated Hospital of Kangda College of Nanjing Medical University, Lianyungang Clinical College of Nanjing Medical University, Lianyungang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Juan Wang
Department of Pediatrics, The First People’s Hospital of Lianyungang, The Affiliated Lianyungang Hospital of Xuzhou Medical University, The Affiliated Hospital of Kangda College of Nanjing Medical University, Lianyungang Clinical College of Nanjing Medical University, Lianyungang, People’s Republic of China
Email [email protected]
Lidong Zhao
Department of Hematology, The First People’s Hospital of Lianyungang, The Affiliated Lianyungang Hospital of Xuzhou Medical University, The Affiliated Hospital of Kangda College of Nanjing Medical University, Lianyungang Clinical College of Nanjing Medical University, Lianyungang, People’s Republic of China
Email [email protected]
Abstract: Early T‐cell precursor acute lymphoblastic leukemia (ETP-ALL) is an aggressive and extremely fatal subtype of T-cell acute lymphoblastic leukemia (T-ALL), characterized by the similar transcriptional and immunophenotypic profiles to those of early T-cell precursors and positive expressions of myeloid antigens. Besides, the gene expression profile in ETP-ALL is similar to that in myeloid malignancies. The clinical characteristics, treatments and prognoses of ETP-ALL are significantly heterogeneous. In the present study, we reported a 43-year-old female patient who lacked terminal deoxynucleotidyl transferase (TDT) expression in immunophenotype and displayed mutations of fms-like tyrosine kinase-internal tandem duplication (FLT3-ITD), paired-box domain 5 (PAX5) and SH2B adaptor protein 3 (SH2B3) (PAX5 and SH2B3, the genes critical to B cell identity and function), which represent myeloid and precursor B-lineage associated gene mutations, respectively. It was a rare T-ALL or T-lineage case. Because of multiple poor prognostic factors in this case, conventional induction regimens, like hyper-CVAD (cyclophosphamide, vincristine, doxorubicin, dexamethasone), were invalid. The patient showed inadequate response, suggesting that this treatment was not employed on the basis of the immunophenotype. FLAG-IDA regimen (fludarabine, cytarabine [Ara-C], granulocyte-colony stimulating factor [G-CSF] and idarubicin), which is usually applied to eliminate leukemia cells, was administered combining with sorafenib as an effective induction chemotherapy. The case achieved long-term survival following the allogeneic hematopoietic stem cell transplantation (allo-HSCT). We recommend that adult ETP-ALL patients can be treated with a myeloid-oriented chemotherapy (as frontline induction treatment) along with gene-targeting inhibitors, followed by allo-HSCT.
Keywords: early T-cell precursor acute lymphoblastic leukemia, FLAG-IDA, PAX5, TDT
Introduction
Early T‐cell precursor acute lymphoblastic leukemia (ETP-ALL) is a subtype of T-cell acute lymphoblastic leukemia (T-ALL) recognized by the World Health Organization (WHO) in 2016,1 accounting for 12.0–16.2% of childhood T-ALL2 and 17.0–22.1% of adulthood T-ALL.3 ETP cells originate from hematopoietic stem cells, a subset of cells that have just migrated from the bone marrow to the thymus, and therefore they retain a certain potential of multilineage differentiation into myeloid cells and T cells.2,4,5 ETP-ALL is mainly diagnosed with the immunophenotypes of leukemia cells. The expression of T-cell differentiation antigens is limited and the antigens of stem and myeloid cells are retained, turning immunophenotypes similar to those of hematopoietic stem cells. Typically, the immunophenotype of ETP-ALL is featured by limited T-cell differentiation antigens (CD1a, CD8 and CD5), and the expression of at least one stem cell antigen or myeloid antigen (CD34, CD117, HLA-DR, CD13, CD33, CD11b, CD65).2,4,6 Moreover, gene mutations in ETP cells are complicated with a high incidence of multiple mutations commonly found in hematopoietic stem cells and myeloid progenitor cells.4 Briefly, incidences of acute myeloid leukemia (AML)-related gene mutations, like FLT3, DNMT3A and NRAS/KRAS, are relatively high, whilst those commonly detected in T-ALL are less mutated in ETP-ALL cases.7–9 However, the ETP-ALL does not have a uniform or pathognomonic cytogenetic aberration profile.10 Owing to the multilineage differentiation potential, earlier-stage immunophenotypes and complicated gene mutations, ETP-ALL manifests high heterogeneous clinical features, aggressiveness, fatality, incurability and prognosis. In the present study, we reported an ETP-ALL case with a typical immunophenotype, deficiency of terminal deoxynucleotidyl transferase (TDT) expression, and mutations of myeloid genes, the B cell specific transcription factor paired-box domain 5 (PAX5) and SH2B adaptor protein 3 (SH2B3, a negative regulator in the pro-B progenitors), all serving as poor prognostic factors. Hyper-CVAD (cyclophosphamide, vincristine, doxorubicin, dexamethasone) was invalid to this case. After an induction chemotherapy combining an AML regimen and targeted drugs, allogeneic hematopoietic stem cell transplantation (allo-HSCT) achieved a favorable overall survival.
Case Presentation
A 43-year-old woman presented at a local hospital with the chief complaint of fatigue for a month. Routine peripheral blood examination showed white blood cell count (WBC) 2.61*10^9/L, red blood cell count (RBC) 2.36*10^12/L, hemoglobin level (Hb) 83 g/L, and blood platelet count (PLT) 147*10^9/L. But no diagnosis and treatment were made. The fatigue increased progressively for half year. Then the patient took a visit to the hospital and was recommended to the department of hematology. Physical examination results were unremarkable, with no hepatosplenomegaly or lymphadenopathy. Peripheral blood was WBC 11.86*10^9/L, Hb 58g/L, RBC 1.37*10^12/L and PLT 58*10^9/L. Peripheral smear showed 11% blasts. Bone marrow aspirate smears were hypercellular with 54% lymphoid blasts. The blasts exhibited round-like shape in medium sizes, with less cytoplasm and granules in the cytoplasm. Burr like or pseudopod like protrusions and tailing could be seen in most of the cytoplasm. Scattered plasma plasmids, round like nucleus and coarse granules of chromatin could be seen, and 1 to 2 clear nucleoli could be seen in some of the cytoplasm. Auer rods could not be seen in the blasts. Cytochemistry revealed these blasts were negative for POX (peroxidase) staining (Figure 1). In flow cytometry (FCM), the results were obtained by a FACSCanto II dual laser six color flow cytometer (Becton Dickinson, CA, USA) in a panel with 32 markers. The immunophenotypes of lymphoid blasts were displayed as CD7, CD117, CD34, CD33, CD38, CD3(dim), cytoplasmic CD3, and negative for TDT, CD123, CD36, HLA-DR, CD64, CD11b, CD13, CD16, CD14, CD15, CD64, CD11c, CD4, CD5, CD8, CD10, CD19, lambda, kappa, CD20, CD22, MPO, CD1a, CD99, CD2 (Figure 2). The FCM findings fulfilled ETP-ALL criteria. 4/10 of metaphase chromosomes had an abnormal karyotype with a +4; 6/10 of chromosome karyotypes were 46 XX; Breakpoint cluster/Abelson tyrosine kinase (BCR/ABL) fusion was not detected by Fluorescence in situ hybridization (FISH). The next generation sequencing (NGS) was based on Illumina nextseq high-throughput sequencing platform. A Ph-like gene panel was used for NGS (Table 1). The NGS demonstrated mutations in FLT3-ITD, PAX5, IL7R, SH2B3, and other genes were negative (Table 2). The patient received hyper-CVAD (six doses of intravenous (IV) 300 mg/m2 cyclophosphamide over 3 hours every 12 hours on days 1 through 3, 2 mg of IV vincristine on days 4 and 11, 50 mg/m2 IV doxorubicin on day 4, and 40 mg of dexamethasone daily on days 1 through 4 and 11 through 14) chemotherapy but the treatment course was complicated by neutropenic fever. The chemotherapy failed. The peripheral blood and bone marrow aspirate smears did not indicate complete remission. Bone marrow assessment at day +14 showed 25% blasts. Then, the patient was given the second cycle of chemotherapy with FLAG-IDA regimen (25 mg/m2 IV fludarabine x 4 doses daily, 2 g/m2 IV cytarabine [Ara-C] on day 1, 10 mg/m2 IV idarubicin x 3 doses, 5–10 μg/kg granulocyte-colony stimulating factor [G-CSF] daily on days 0 through 5]) was given, combined with FLT3 inhibitor sorafenib (400 mg po bid) from day +8. The patient showed complete response (CR) and minimal residual disease (MRD)-negative. Then the patient received prophylactic intrathecal methotrexate (MTX) and Ara-C. After 2 courses of FLAG-IDA, the MRD maintained negative and FLT3-ITD mutation became negative. Subsequently, sorafenib was continued until a myeloablative (human leukocyte antigen) HLA-matched sibling donor allo-HSCT was performed. The bone marrow still remained CR, MRD-negative, and FLT3-ITD negative at current 12 months after transplantation.
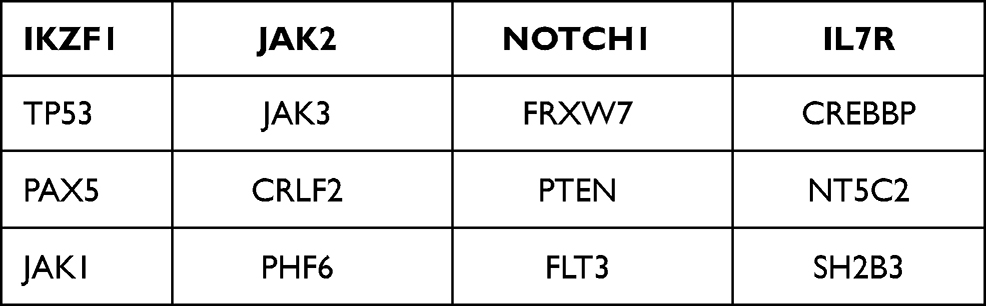 |
Table 1 The Ph-Like Gene Panel for NGS |
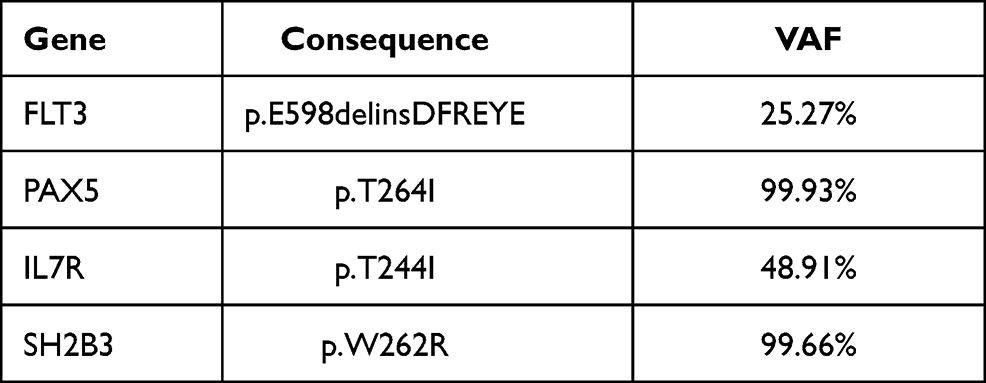 |
Table 2 Gene Mutations in This ETP-ALL Patient |
 |
Figure 1 Cellular morphology of bone marrow aspirate smear and chromosome karyotypes. (A, B) Bone marrow smear showing a proliferation of blasts (Giemsa-Wright stain). (A, B) The blasts exhibited medium sizes, round, round-like or irregularly shape, the cytoplasm were less with trailing and burr-like changes (100 × oil immersion). (C) The cytochemiscal staining was negative for POX (peroxidase). (D) Chromosome analysis demonstrated + 4 (arrow) in 4 of 10 metaphases examined. |
 |
Figure 2 Immunophenotypic analysis of the blasts cells exhibited ETP-ALL. Through the flow cytometry analysis, blast cells were gated and showed in blue (P3). Normal mature lymphocytes were showed in green for comparison (P2). The immunophenotype of blasts were CD7, CD117, CD34, CD33, CD38, CD3 (dim), cytoplasmic CD3, and negative for TDT, CD123, CD36, HLA-DR, CD64, CD11b, CD13, CD16, CD14, CD15, CD64, CD11c, CD4, CD5, CD8, CD10, CD19, lambda, kappa, CD20, CD22, MPO, CD1a, CD99, CD2. The phenotype was consistent with early T-cell precursor acute lymphoblastic leukemia (ETP-ALL). |
Discussion
The ETP‐ALL is a unique, lethal, rare type of leukemia that was first described by the WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues in 2016,1 with characteristics of distinct immunophenotypic and genetic features.2,4,11 In ETP-ALL, myeloid and hematopoietic stem-cell markers are expressed, such as CD13, CD33, CD34, CD117, HLA-DR, but MPO- ….2,12 Express CD7, dim CD5 (<75% positive cells), other T-lineage markers, such as CD1a and CD8, usually negative or dim.2,12 ETP-ALL shows diverse genetic features similar to those of acute myeloid leukemia, rather T lymphoid leukemia.1,13 The genomic mutations of ETP-ALL are enriched in transcriptional regulators (such as BCL11B, ETV6, RUNX1, WT1), epigenetic factors (histone modification, including PHF6, CTCF, EZH2, and SUZ12), and signaling genes (activation JAK-STAT, IL-7R and RAS signaling pathway, including JAK1, JAK3, IL7R, SH2B3, NRAS, KRAS, FLT3 and PTPN11).11 Compared with non-ETP-ALL, several high-frequency mutation genes have been found in ETP-ALL, such as FLT3-ITD, NOTCH1 and PTEN.14–17 In ETP-ALL, some genes show mutations in DNA methylation similar to those in non-ETP-ALL, including DNMT3A, IDH1, IDH2, TET2, TET3, and WT1.3,7,18 The three high frequency genes in ETP-ALL are FLT3, DNMT3A, and NOTCH1.9,18
The case in this report showed the similar immunophenotype with that in other studies,19 but the TDT of the ETP-ALL in this case was absent. TDT is usually expressed in lymphocyte precursors, as a marker that can be detected by flow cytometry in 95% of all T-acute lymphoblastic leukemia/lymphoma (T-ALL/LBL) patients.20,21 However, large-size studies on TDT-negative T-ALL/LBL are rare,22 and most of them are case reports.22–24 Compared with TDT-positive T-ALL/LBL, TDT-negative T-ALL/LBL is more likely to be found in ETP-ALL patients (12% versus 43%).22 TDT-negative T-ALL/LBL, especially ETP, has poor response to chemotherapy regimen containing prednisone, so it has been reported that the Hyper-CVAD regimen is ineffective.22,25 The patients without TDT expression in T-ALL/LBL demonstrate a significantly higher rate of relapse and a shorter overall survival, hence, TDT is an independent risk factor in ETP-ALL.22
Fms-like tyrosine kinase (FLT3) mutation is one of the most-common gene mutations in AML cases.26 Zhang et al4 identified a high frequency of FLT3 mutation and other mutations including IDH1, IDH2, DNMT3A, KRAS, NRAS, JAK1, JAK3, and IL7R associated with AML by whole genome sequencing. Abundant clinical evidences have supported the great significance of activation and mutation of FLT3 in the progression of AML, making it a biomarker of driving gene mutation and poor prognosis,27 FLT3-ITD+ AML is featured by the short survival, high recurrence rate and low remission rate of induction chemotherapy.28 Serving as a growth factor receptor that stimulates the proliferation and differentiation of hematopoietic stem-progenitor cells, FLT3 is usually expressed in CD34+ hematopoietic stem-progenitor cells of normal bone marrow, but rarely in other types of cells. So far, clinical study of FLT3 mutation in ALL cases is rarely reported, and the incidence of FLT3-ITD mutation is relatively low in ALL and T-ALL cases,29–31 but significantly high in ETP-ALL cases. M Neumann et al8 found a high rate of FLT3 mutation, as a poor prognostic factor, in ETP-ALL similar to that in myeloid cells, and its sensitivity to tyrosine kinase inhibitors.
Interleukin 7 (IL7) and its receptor (IL7R) are essential for normal lymphoid development and leukemia-initiating cell activity, pathogenesis of T-ALL.32,33 Besides the oncogenic role of mutant IL7R, excessive IL7R-mediated signaling can accelerate the course of lymphoid leukemia disease and enhance its resistance to chemotherapy.33 IL7R overexpression and mutation in ETP-ALL result in an aggressive ETP-like disease.4,34 Despite these, IL7R provides a potentiality of develop feasible and effective treatment strategies through targeting IL7R signaling pathway.35
SH2B adaptor protein 3 (SH2B3) is a negative regulator of JAK/STAT signaling in the homeostasis of hematopoietic stem cells and pro-B progenitors.36,37 But loss of SH2B3 increases the transducer and activator of transcription signaling, promotes lymphoid cell proliferation, and accelerates ALL development in a mouse model.36 In the genomic studies, somatic mutations in SH2B3 have been described at low frequency high-risk B-precursor ALL and early T-cell precursor ALL.4,38 Recently, Geet al have reported that high IL7R expression level and low SH2B3 expression level in B-ALL are high risk factors.39 Conversely, the association between IL7R and SH2B3 expression has not been found in T-ALL.35 In another study, T-cell development is unperturbed in SH2B3-deficient mice.40 The prognostic significance of SH2B3 mutation remains unclear and needs to be further clarified in Chinese patients with ETP-ALL.
PAX5 is a member of the paired box (PAX) family of transcription factor genes, which controls the identity and maintain the function of B lymphocytes throughout the development from pro-B to mature B-cell.41,42 PAX5 is the guardian of B cell identity and function.41 Hence, as an oncogene in B-cell malignancies, PAX5 is frequently mutated by translocations, deletions and mutations in human B-lineage leukemia.43 However, only few studies have demonstrated that Pax5 mutates in T-ALL or T-cell lymphoma.44–47 So far, mutation of PAX5 in ETP-ALL has not been reported. The loss-of-function of PAX5 can block B-cell differentiation at its early stages, which causes multi-lineage potential of T-cell differentiation and therefore may influence T-ALL pathogenesis.47–49 Pax5 inactivation in B-ALL has different effects on prognosis.50–52 Furthermore, when PAX5 mutates with IKZF1, FLT3 is considered as a poor prognostic factor.53,54 The co-mutation of PAX5 and FLT3 may indicate the poor prognosis in this case],51 but this indication needs to be further confirmed by relevant studies.
There is no uniform genetic lesion in ETP-ALL, but the presence of multiple lesions indicates a certain connection between mutations during the development and progression of leukemia.11 Therefore, patients can benefit from a combination of inhibitors targeting the mutations with chemotherapy.11 So far, there is no unified standard and optimal treatment for ETP-ALL.10,55–57 The present patient received induction chemotherapy according to hyper-CVAD protocol, however, bone marrow (BM) assessment showed 25% blasts at day +14; therefore, a different second induction regimen was given along with FLT3 inhibitors.
At present, the guidelines for ETP-ALL are unavailable.58 Heterogeneity exists in the treatment scheme for ETP-ALL.10,59 Because of ETP-ALL presents a unique immature immune phenotype, a characteristic gene expression profile, and an increased number and size of genomic lesions, ETP-ALL is more likely to develop into acute myeloid leukemia than lymphoid leukemia.19,55,60 Hence, ETP-ALL needs a AML (acute myeloid leukemia)-oriented and intensification chemotherapy regimens together with allo-HSCT.2,7,55,60 Novel strategies, such as target therapy and FLT3 inhibitors should be designed.5,11 ETP-ALL is sensitive to cytarabine therapy and high dose cytarabine may be a good regimen for ETP-ALL.61,62 FLAG-IDA regime may be a potential and beneficial schedule for ETP-ALL.10,60 Therefore, after the initial induction failed, we turned to FLAG-IDA combined with sorafenib, a treatment that achieved satisfactory outcomes.
Conclusion
ETP-ALL is a lethal subtype of ALL with a weak response to induction chemotherapy, and a poor prognosis. Diagnosis of ETP-ALL is mainly based on immunophenotyping, and its gene expression profile has also a similarity with that of myeloid lineage. Sensitive prognostic biomarkers in ETP-ALL have not been identified. In this ETP-ALL case with multiple poor prognostic factors, an intensive treatment combining targeted drugs and sequential Allo-HSCT proves effective.
Acknowledgments
The authors thank the patient and all of physicians and nurses who took care of the patient. The patient has provided written informed consent for the publication of case details and accompanying images. This case report does not require institutional approval to publish the case details. Jianping Mao and Lianguo Xue are co-first authors for this study.
Funding
This work was supported by the Foundation of the Health Commission of Lianyungang (No. 201907).
Disclosure
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
1. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi:10.1182/blood-2016-03-643544
2. Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10(2):147–156.
3. Bond J, Graux C, Lhermitte L, et al. Early Response-Based Therapy Stratification Improves Survival in Adult Early Thymic Precursor Acute Lymphoblastic Leukemia: a Group for Research on Adult Acute Lymphoblastic Leukemia Study. J Clin Oncol. 2017;35(23):2683–2691.
4. Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–163.
5. Booth CAG, Jacobsen SEW, Mead AJ. Origins of ETP leukemia. Oncoscience. 2018;5(11–12):271–272.
6. Inukai T, Kiyokawa N, Campana D, et al. Clinical significance of early T-cell precursor acute lymphoblastic leukaemia: results of the Tokyo Children’s Cancer Study Group Study L99-15. Br J Haematol. 2012;156(3):358–365.
7. Neumann M, Heesch S, Schlee C, et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood. 2013;121(23):4749–4752.
8. Neumann M, Heesch S, Gokbuget N, et al. Clinical and molecular characterization of early T-cell precursor leukemia: a high-risk subgroup in adult T-ALL with a high frequency of FLT3 mutations. Blood Cancer J. 2012;2(1):e55.
9. Neumann M, Greif PA, Baldus CD. Mutational landscape of adult ETP-ALL. Oncotarget. 2013;4(7):954–955.
10. Castaneda Puglianini O, Papadantonakis N. Early precursor T-cell acute lymphoblastic leukemia: current paradigms and evolving concepts. Ther Adv Hematol. 2020;11:2040620720929475.
11. Tavakoli Shirazi P, Eadie LN, Heatley SL, Hughes TP, Yeung DT, White DL. The effect of co-occurring lesions on leukaemogenesis and drug response in T-ALL and ETP-ALL. Br J Cancer. 2020;122(4):455–464.
12. Zuurbier L, Gutierrez A, Mullighan CG, et al. Immature MEF2C-dysregulated T-cell leukemia patients have an early T-cell precursor acute lymphoblastic leukemia gene signature and typically have non-rearranged T-cell receptors. Haematologica. 2014;99(1):94–102.
13. Maude SL, Dolai S, Delgado-Martin C, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125(11):1759–1767.
14. Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211–1218.
15. Neumann M, Coskun E, Fransecky L, et al. FLT3 mutations in early T-cell precursor ALL characterize a stem cell like leukemia and imply the clinical use of tyrosine kinase inhibitors. PLoS One. 2013;8(1):e53190.
16. Mansur MB, Hassan R, Barbosa TC, et al. Impact of complex NOTCH1 mutations on survival in paediatric T-cell leukaemia. BMC Cancer. 2012;12:9.
17. Noronha EP, Marques LVC, Andrade FG, et al. T-lymphoid/myeloid mixed phenotype acute leukemia and early T-cell precursor lymphoblastic leukemia similarities with NOTCH1 mutation as a good prognostic factor. Cancer Manag Res. 2019;11:3933–3943.
18. Ribeiro AF, Pratcorona M, Erpelinck-Verschueren C, et al. Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood. 2012;119(24):5824–5831.
19. Jain N, Lamb AV, O’Brien S, et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. Blood. 2016;127(15):1863–1869.
20. Seegmiller AC, Kroft SH, Karandikar NJ, McKenna RW. Characterization of immunophenotypic aberrancies in 200 cases of B acute lymphoblastic leukemia. Am J Clin Pathol. 2009;132(6):940–949.
21. Kaleem Z, Crawford E, Pathan MH, et al. Flow cytometric analysis of acute leukemias. Diagnostic utility and critical analysis of data. Arch Pathol Lab Med. 2003;127(1):42–48.
22. Zhou Y, Fan X, Routbort M, et al. Absence of terminal deoxynucleotidyl transferase expression identifies a subset of high-risk adult T-lymphoblastic leukemia/lymphoma. Mod Pathol. 2013;26(10):1338–1345.
23. Brcic I, Labar B, Peric-Balja M, Basic-Kinda S, Nola M. Terminal deoxynucleotidyl transferase negative T-cell lymphoblastic lymphoma in aleukemic patient. Int J Hematol. 2008;88(2):189–191.
24. Kidoguchi K, Yokoo M, Umino A, Aoki S, Kimura S. Terminal deoxynucleotidyl transferase (TdT) negative early T cell precursor acute lymphoblastic leukemia (ETP-ALL) with spontaneous acute kidney injury. Ann Hematol. 2020;99(4):885–886.
25. Tran T, Krause J. Early T-cell precursor acute lymphoblastic leukemia with KRAS and DNMT3A mutations and unexpected monosomy 7. Proc. 2018;31(4):511–513.
26. Metzeler KH, Herold T, Rothenberg-Thurley M, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686–698.
27. Khaled S, Al Malki M, Marcucci G. Acute Myeloid Leukemia: biologic, Prognostic, and Therapeutic Insights. Oncology. 2016;30(4):318–329.
28. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374(23):2209–2221.
29. Wellmann S, Moderegger E, Zelmer A, et al. FLT3 mutations in childhood acute lymphoblastic leukemia at first relapse. Leukemia. 2005;19(3):467–468.
30. Elyamany G, Awad M, Alsuhaibani O, et al. FLT3 Internal Tandem Duplication and D835 Mutations in Patients with Acute Lymphoblastic Leukemia and its Clinical Significance. Mediterr J Hematol Infect Dis. 2014;6(1):e2014038.
31. Grossmann V, Haferlach C, Weissmann S, et al. The molecular profile of adult T-cell acute lymphoblastic leukemia: mutations in RUNX1 and DNMT3A are associated with poor prognosis in T-ALL. Genes Chromosomes Cancer. 2013;52(4):410–422.
32. Oliveira ML, Akkapeddi P, Ribeiro D, Melao A, Barata JT. IL-7R-mediated signaling in T-cell acute lymphoblastic leukemia: an update. Adv Biol Regul. 2019;71:88–96.
33. Gonzalez-Garcia S, Mosquera M, Fuentes P, et al. IL-7R is essential for leukemia-initiating cell activity of T-cell acute lymphoblastic leukemia. Blood. 2019;134(24):2171–2182.
34. Treanor LM, Zhou S, Janke L, et al. Interleukin-7 receptor mutants initiate early T cell precursor leukemia in murine thymocyte progenitors with multipotent potential. J Exp Med. 2014;211(4):701–713.
35. Gianfelici V, Messina M, Paoloni F, et al. IL7R overexpression in adult acute lymphoblastic leukemia is associated to JAK/STAT pathway mutations and identifies patients who could benefit from targeted therapies. Leuk Lymphoma. 2019;60(3):829–832.
36. Perez-Garcia A, Ambesi-Impiombato A, Hadler M, et al. Genetic loss of SH2B3 in acute lymphoblastic leukemia. Blood. 2013;122(14):2425–2432.
37. Cheng Y, Chikwava K, Wu C, et al. LNK/SH2B3 regulates IL-7 receptor signaling in normal and malignant B-progenitors. J Clin Invest. 2016;126(4):1267–1281.
38. Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153–166.
39. Ge Z, Gu Y, Xiao L, et al. Co-existence of IL7R high and SH2B3 low expression distinguishes a novel high-risk acute lymphoblastic leukemia with Ikaros dysfunction. Oncotarget. 2016;7(29):46014–46027.
40. Takaki S, Sauer K, Iritani BM, et al. Control of B cell production by the adaptor protein lnk. Definition Of a conserved family of signal-modulating proteins. Immunity. 2000;13(5):599–609.
41. Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function. Nat Immunol. 2007;8(5):463–470.
42. Nasr MR, Rosenthal N, Syrbu S. Expression profiling of transcription factors in B- or T-acute lymphoblastic leukemia/lymphoma and burkitt lymphoma: usefulness of PAX5 immunostaining as pan-Pre-B-cell marker. Am J Clin Pathol. 2010;133(1):41–48.
43. Nebral K, Denk D, Attarbaschi A, et al. Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia. 2009;23(1):134–143.
44. Olsson L, Lundin-Strom KB, Castor A, et al. Improved cytogenetic characterization and risk stratification of pediatric acute lymphoblastic leukemia using single nucleotide polymorphism array analysis: a single center experience of 296 cases. Genes Chromosomes Cancer. 2018;57(11):604–607.
45. Zhang X, Lin Z, Kim I. Pax5 expression in non-Hodgkin’s lymphomas and acute leukemias. J Korean Med Sci. 2003;18(6):804–808.
46. Tzankov AS, Went PT, Munst S, Papadopoulos T, Jundt G, Dirnhofer SR. Rare expression of BSAP (PAX-5) in mature T-cell lymphomas. Mod Pathol. 2007;20(6):632–637.
47. Okuyama K, Strid T, Kuruvilla J, et al. PAX5 is part of a functional transcription factor network targeted in lymphoid leukemia. PLoS Genet. 2019;15(8):e1008280.
48. Cobaleda C, Jochum W, Busslinger M. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature. 2007;449(7161):473–477.
49. Rolink AG, Nutt SL, Melchers F, Busslinger M. Long-term in vivo reconstitution of T-cell development by Pax5-deficient B-cell progenitors. Nature. 1999;401(6753):603–606.
50. Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–480.
51. Hutter G, Kaiser M, Neumann M, et al. Epigenetic regulation of PAX5 expression in acute T-cell lymphoblastic leukemia. Leuk Res. 2011;35(5):614–619.
52. Schwab C, Nebral K, Chilton L, et al. Intragenic amplification of PAX5: a novel subgroup in B-cell precursor acute lymphoblastic leukemia? Blood Adv. 2017;1(19):1473–1477.
53. Stanulla M, Dagdan E, Zaliova M, et al. IKZF1(plus) Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J Clin Oncol. 2018;36(12):1240–1249.
54. Khalid A, Aslam S, Ahmed M, Hasnain S, Aslam A. Risk assessment of FLT3 and PAX5 variants in B-acute lymphoblastic leukemia: a case-control study in a Pakistani cohort. PeerJ. 2019;7:e7195.
55. Conter V, Valsecchi MG, Buldini B, et al. Early T-cell precursor acute lymphoblastic leukaemia in children treated in AIEOP centres with AIEOP-BFM protocols: a retrospective analysis. Lancet Haematol. 2016;3(2):e80–86.
56. Conter V, Bartram CR, Valsecchi MG, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115(16):3206–3214.
57. Schrappe M, Valsecchi MG, Bartram CR, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. 2011;118(8):2077–2084.
58. Rahman K, Singh P, Chandra D, Dev Yadav D, Gupta R, Gupta A. Early T-cell precursor acute lymphoblastic leukemia with Auer rods-A case report. Int J Lab Hematol. 2020;42(1):e27–e29.
59. Wolach O, Stone RM. How I treat mixed-phenotype acute leukemia. Blood. 2015;125(16):2477–2485.
60. Genesca E, Morgades M, Montesinos P, et al. Unique clinico-biological, genetic and prognostic features of adult early T-cell precursor acute lymphoblastic leukemia. Haematologica. 2020;105(6):e294–e297.
61. Wang XX, Wu D, Zhang L. Clinical and molecular characterization of early T-cell precursor acute lymphoblastic leukemia: two cases report and literature review. Medicine. 2018;97(52):e13856.
62. Bataller A, Garrote M, Oliver-Caldes A, et al. Early T-cell precursor lymphoblastic leukaemia: response to FLAG-IDA and high-dose cytarabine with sorafenib after initial refractoriness. Br J Haematol. 2019;185(4):755–757.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.