Back to Journals » International Journal of Nanomedicine » Volume 13
A high bioavailability and sustained-release nano-delivery system for nintedanib based on electrospray technology
Authors Liu H, Du K, Li D, Du Y, Xi J, Xu Y, Shen Y, Jiang T, Webster TJ ![]()
Received 21 July 2018
Accepted for publication 5 November 2018
Published 10 December 2018 Volume 2018:13 Pages 8379—8393
DOI https://doi.org/10.2147/IJN.S181002
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Hongfei Liu, 1,2 Kunyu Du, 1 Dongli Li, 2 Yi Du, 2 Jumei Xi, 3 Ying Xu, 1 Yan Shen, 4 Tao Jiang, 5 Thomas J Webster 6
1School of Pharmacy, Jiangsu University, Zhenjiang 212013, China; 2School of Biotechnology and Health Sciences, Wuyi University, Jiangmen 529020, China; 3Health of Nepstar Drugstore Chain Company Limited, Wuxi 214000, China; 4State Key Laboratory of Natural Medicines, Department of Pharmaceutics, China Pharmaceutical University, Nanjing 210009, China; 5The Pharmacy Department, Jiangsu Cancer Hospital and Jiangsu Institute of Cancer Research and The Affiliated Cancer Hospital of Nanjing Medical University, Nanjing 210009, China; 6Department of Chemical Engineering, Northeastern University, Boston, MA 02115, USA
Background: Nintedanib is a new tyrosine kinase inhibitor and growth factor antagonist. It can be used to treat idiopathic pulmonary fibrosis diseases. Nintedanib has poor solubility in the intestinal tract environment, which leads to low bioavailability of just 4.7%.
Methods: In this study, a nintedanib solid dispersion was prepared by electrospray technology with an optimized formula (nintedanib:PVPK30:Soybean lecithin=1:5:0.25) and electrospray parameters (21 kV voltage, 18 cm receiving distance, 0.3 mL/h solution flow rate, 0.5 mm pinhole inner diameter).
Results: The accumulative release rate of the optimized solid dispersion was more than 60% in 30 minutes and 100% in 60 minutes. The size distribution was uniform and the surface observed with scanning electron microscopy (SEM) was smooth. The DSC and X-ray diffraction results showed that nintedanib existed in the solid dispersion through an amorphous form. Nintedanib solid dispersion sustained-release capsules were prepared to prolong drug release, improve patient compliance and reduce side effects. The accumulative release rate from the sustained release capsules was 35.17%, 54.78%, 70.58%, and 93.93% after 2 h, 6 h, 8 h, and 12 h, respectively, having obvious sustained release effects in vitro. The release behavior of solid dispersion sustained-release capsules in vitro was in accordance with the Ritger-Peppas model. The in vivo studies of nintedanib soft capsules, solid dispersion and nintedanib sustained release capsules in SD rats were investigated; the results showed that the Tmax of the soft capsule, solid dispersion and sustained release capsules were 3 h, 2 h, and 6 h, respectively. The Cmax were 2.945 mg/mL, 5.32 mg/mL, and 3.75 mg/mL, respectively, while the AUC 0– 24 h was 15.124 mg·h/mL, 23.438 mg·h/mL, and 24.584 mg·h/mL, respectively. The relevant bioavailability of the sustained-release capsules was 162.55% compared to the nintedanib soft capsule and 104.89% compared to the nintedanib solid dispersion.
Conclusion: The results suggested superior bioavailability and a sustained-release effect from nintedanib sustained-release capsules, as compared to the reference (commercial nintedanib soft capsule).
Keywords: nintedanib, electrospray technology, solid dispersion, sustained release capsule, pharmacokinetics
Corrigendum for this paper has been published
Introduction
Electrospray technology is a unique approach using electrostatic forces to produce fibers or spheres from polymer solutions.1–3 By adjusting various parameters of an electrospray device, micro- or nanoscale particles or fibers can be prepared. Electrospray technology has the advantages of simple operation and easy industrialization,4–6 high encapsulation efficiency and drug loading ratio, but not affecting the drug activity.7–9
Electrospray technology has gained more and more interest and attention in recent years due to its potential for applications in drug delivery systems.10–13 Xue14 used poly (lactic-co-glycolic acid) (PLGA) as a carrier to prepare PLGA-protein microspheres by using an electrostatic spray method. The particle size was as small as 1.2–1.5 μm, the particle size was uniform, and the loading capacity was up to 12%. Zhou et al15 prepared electrospun polyphosphazene microspheres and investigated the influence of various parameters on the morphology and particle size. The results showed that when the drug concentration was 0.25 g/mL, the microsphere particle size was 1–2 μm, approximately spherical.
Nintedanib was successfully developed by Boehringer Ingelheim and then approved by the Food and Drug Administration (FDA) in 2014. The dose of the nintedanib commercial soft capsule is 120 mg. It is a tyrosine kinase inhibitor and growth factor antagonist.16–18 It is used to treat idiopathic pulmonary fibrosis diseases which can only be treated through lung transplantation at present.19–23 However, due to its poor solubility, the bioavailability is reported as low as 4.7% which can lead to a higher dose.24,25 At present, the only commercial formulation for nintedanib is a soft capsule, as little research has been carried out on novel nintedanib delivery systems. Based on that, nintedanib is a new breakthrough in the treatment of idiopathic pulmonary fibrosis with lower bioavailability, and it is necessary to develop a preparation to increase the solubility and improve its bioavailability to further develop a sustained release delivery system to improve the patients’ adaptability and prolong its effect with time in the body.
A dispersion is a solid substance formed by highly dispersed insoluble drugs in solid carrier materials. Solid dispersion technology can effectively improve the dissolution of insoluble drugs.26,28 With the development of nanotechnology, a new preparation technology of a solid dispersion system – electrostatic spray – has been developed. Its simple operation and efficient preparation results have aroused the attention of researchers. Yu et al29 prepared ketoprofen solid disperions using electrostatic spray technology. When the receiving distance was 15 cm and the velocity was 2 mL/h with a voltage of 10 kV, the dissolution rate of a ketoprofen solid dispersion was 30 times faster than the drug substance, probably due to higher dispersion and larger surface to volume ratios, so that the drug is more likely to release.30 Ketoprofen solid dispersion phase identification results showed that ketoprofen was in amorphous form.
A sustained release dosage form with a suitable formulation can delay or control the rate of release of the drug in the body and maintain an effective concentration of the drug in the body to prolong drug action.31 Compared with the conventional preparation, the sustained release preparation can prolong drug release, improving patient compliance and reducing the frequency of administration.32,33
The purpose of this study was to establish a high bioavailability and sustained release delivery system for nintedanib based on electrospray technology. The nintedanib solid dispersion was prepared by using electrospray technology to improve its solubility. In this study, nintedanib was prepared as a solid dispersion and the drug was highly dispersed in the soluble carrier polyvinylpyrrolidone (PVP). Because of its high dispersion and larger specific surface area, the drug is easier to dissolve and release in vitro. Furthermore, nintedanib sustained release capsules were also prepared based on the solid dispersion to prolong drug release, improving patient compliance and reducing the frequency of administration. Finally, the bioavailability of nintedanib solid dispersion sustained release formulations in Sprague Dawley (SD) rats was investigated in this study.
Materials
Nintedanib was purchased from Jiangsu Aosaikang Pharmaceutical Co., Ltd. (Nanjing, China). Nintedanib soft capsules were purchased from Boehringer Ingelheim (Ingelheim, Germany). Nintedanib solid dispersions and nintedanib sustained release capsules were self-made. Nintedanib soft capsules (reference dosage form) were successfully developed by Boehringer Ingelheim and then approved by the FDA in 2014. PVP-K30 was purchased from BASF SE (Germany). Granulesten was purchased from Tywei Pharmaceutic Co., Ltd. (Shanghai, China). Three grades of hydroxypropyl methylcellulose (HPMC): MethocelTM K100M, K15M, and K4M premium CR were purchased from Colorcon (Shanghai, China). Starch was purchased from Anhui Sanhe Pharmaceutical Excipients Co., Ltd. (Anhui, China). Microcrystalline cellulose (MCC) was purchased from Shanghai Chineway Pharmaceutical Tech. Co., Ltd. (Shanghai, China). All other reagents and solvents used were of analytical grade. Male Sprague-Dawley rats with an average body weight of 180–220 g was provided by Jiangsu University Experimental Animal Research Center (Zhenjiang, China). A HL-2B Digital Display Constant Current Pump was purchased from Shanghai Huxi Analytical Instrument Factory (Shanghai, China). A HH-S2 digital display constant temperature water bath was purchased from Jintan Zhongda Instrument Factory (Jintan, China).
Methods
Fabrication of nintedanib solid dispersion by electrospraying technology
Nintedanib solid dispersions were prepared by an electrospraying method34–37 as follows: nintedanib, PVP, and phospholipid were dissolved in methanol by ultrasound, then the homogeneous solution was injected through a syringe (Model No: KDS-250-CE; Geneq Inc., Baoding, China) under a DC electric field (electrospraying process). With the basic parameters of a voltage at 21 kV (ES3OP-10W/DAM; Gamma High Voltage Research, China), a receiving distance of 15 cm, a flow rate of 0.5 mL/h, and a pinhole diameter of 0.5 mm, the solid dispersion was prepared under a dried environment (t=25°C, relative humidity [RH] =30%).
Optimization of the electrospray process parameters and solid dispersion formula
The parameters of the electrospraying process were investigated as follows with the morphology and drug release of the solid dispersion as the index: the voltage of the spray device (19, 21, 25 kV), the flow rate of the nintedanib solution (0.3, 0.5, 0.8, 1 mL/h), the needle inner diameter (0.3, 0.5, 0.9 mm), and the receiving distance (10, 15, 18 cm).
The solid dispersion formulas were investigated as follows with the morphology and drug release of the solid dispersion as the index: nintedanib/PVP ratios (1:1, 1:3, and 1:5) and nintedanib/PVP/phospholipid ratios (1:5:0.15 and 1:5:0.25).
Morphology and particle size distribution of the solid dispersions
Morphology of the solid dispersions was determined using scanning electron microscopy (SEM; Hitachi High-Technologies Corporation, Tokyo, Japan), after the samples were vacuum-coated with gold. Freeze-dried solid dispersions were suspended in 500 mL of deionized water and measured by a particle size analyzer (Foqels; Brookhaven Instruments Co., Austin, TX, USA). We measured the particle size according to the standard procedure of the instrument and measured it three times in parallel.
In vitro drug release studies
Solid dispersions containing 120 mg nintedanib were suspended in 900 mL of a pH 6.8 PBS solution (containing 0.5% w/v Tween 80) at 37°C by using the basket method. The release medium was withdrawn at certain time points (10, 20, 30, 45, 60, 90, and 120 minutes). The samples were filtered through a 0.45 μm membrane filter, and the amount of drug released was measured by UV spectrophotometry at 385 nm. The cumulative release was calculated as follows:
|
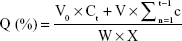
where Ct is the drug concentration (mg/mL) in the release medium, W is the weight of the solid dispersion, V0 is the volume of the release medium, V is the sample volume at each time, and X is the drug loading.
Three batches of nintedanib solid dispersions were prepared via the optimized preparation process, and the similarity of the drug release for the three batches of solid dispersions was investigated using the similarity factor (f2) method. f2 was calculated as follow:
|
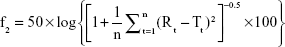
where Rt and Tt are the cumulative release of two batches of solid dispersions at time t; n is the sampling number. The value of f2 ranges from 0 to 100. The higher the f2, the more similar the two curves. The two release curves are considered to be similar if the f2 of the two batches is greater >50.
In vivo intestinal circulation absorption studies of solid dispersion
The ten SD rats were randomly divided into two groups (nintedanib active pharmaceutical ingredient [API] group and nintedanib solid dispersion group). Rats were intraperitoneally injected with pentobarbital sodium anesthesia (4 mg/100 g). A small incision was cut at each end of the intestinal segment (about 10 cm) to be examined, and a glass tube with a diameter of 0.3 cm was inserted. The hose of the circulating pump was connected to the glass tube and ligated by surgical line to form a circulation loop. At the beginning of the experiment, the physiological saline at 37°C was circulated at a rate of 5 mL/min to rinse the contents of the intestines. Then the test solution was circulated in the hose at a rate of 5 mL/min at 37°C to eliminate the adsorption of the drug by the hose until the outlet of the test solution was consistent with the test solution. Then the flow rate was adjusted by the circulating pump to 2.5 mL/min, samples were taken at 0.5, 1, 1.5, 2, 2.5, 3, and 3.5 h respectively. The samples were then centrifuged at 10,000 rpm for 10 minutes in a centrifuge tube, the supernatant was taken, and the concentration was determined according to the nintedanib in vivo drug concentration determination method. The absorption of the drug in different intestinal segments was judged by calculating the percentage of drug absorption P and the absorption rate constant Ka.38,39
|
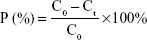
|
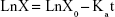
where C0 is the concentration of nintedanib in 0 h intestinal circulation fluid; Ct is the nintedanib concentration in a 3 h enteric circulation solution; X0 is the 0 h concentration in intestinal circulation fluid; and LnX is the logarithm of residual nintedanib in intestinal fluid at each time point value.
Preparation of nintedanib sustained release capsule
The nintedanib solid dispersion, HPMC, and other excipients were accurately weighed, then completely mixed and sieved by a 60-mesh sieve thrice. Then the granules were prepared with a 5% PVP solution as an adhesive and sieved by a 20-mesh sieve; the granules were dried for 2 hours at 55°C, sieved by a 16-mesh sieve, and then filled into the capsule.
Optimization of the formulation
The influence of the HPMC viscosity (K4M, K15M, and K100M), the amount of HPMC (5%, 10%, and 15%), and diluent agent (MCC, starch, and lactose) on the release behavior of the sustained release capsules were optimized.
Three batches of nintedanib sustained release capsules were prepared via the optimized formulation, and the similarity of drug release for the three batches was investigated using the similarity factor (f2) method. f2 was calculated as shown above. The in vitro release mechanism and model fitting of the nintedanib sustained release capsule were investigated.
In vivo study
The in vivo evaluation was performed by administering the prepared samples in 18 male SD rats (weighing 140–150 g). They fasted overnight for at least 12 hours, with free access to water. During the course of the experiment, water was not given until 6 hours after administration of the three preparations. The SD rats were divided into three groups randomly. Each rat was orally administered with 50 mg/kg of nintedanib. The three preparations were: 1) nintedanib soft capsule, 2) nintedanib solid dispersion, and 3) nintedanib sustained-release capsule. All studies were conducted in accordance with the Principles of Laboratory Animal Care (NIH publication no 85–23, revised in 1985) and were approved by the Department of Laboratory Animal Research at Jiangsu University. Blood samples were taken immediately before administering the drug and at the following times for each treatment respectively: 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, and 24 hours, and then were placed in the centrifuge tube with heparin solution in advance. Plasma samples were assayed for nintedanib concentration using high performance liquid chromatography (HPLC).40
Pharmacokinetics study
The pharmacokinetic parameters including the maximum plasma concentration (Cmax) and time of the maximum plasma concentration (Tmax) were observed from the plasma concentration–time curve. The areas under the serum concentration–time curve (AUC0–24 h) were calculated by the trapezoidal method.
Statistical analysis
Data were obtained at least in triplicate and expressed as mean ± SD. Statistical differences were determined by a Student’s two-tailed t-test. Differences were considered statistically significant at P<0.05. Results from the three preparations (nintedanib soft capsule, nintedanib solid dispersion, and nintedanib sustained-release capsule) were analyzed with the SPSS statistical package using analysis of variance to assess any significant (P<0.05) difference.
Results and discussion
Optimization of the electrospray process parameters and solid dispersion formula
Influence of PVP amount
The drug dissolution rate from the solid dispersions with different amounts of PVP (nintedanib:PVP =1:1, 1:3, and 1:5, respectively) are shown in Figure 1A, and the similarity of the drug dissolution from the different formulas is also shown in Figure 1A by the similarity factor (f2) method.
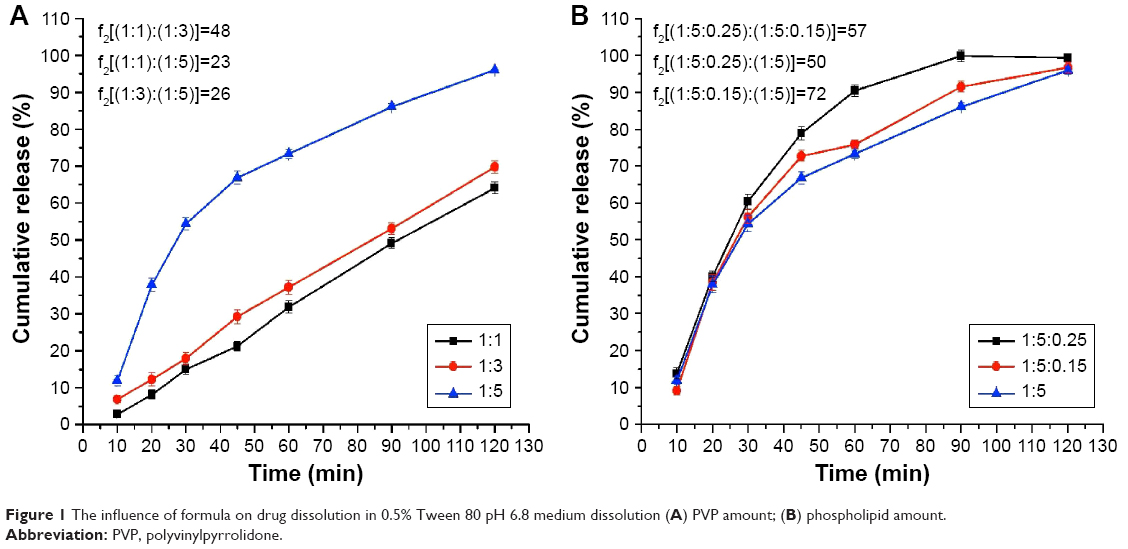 | Figure 1 The influence of formula on drug dissolution in 0.5% Tween 80 pH 6.8 medium dissolution (A) PVP amount; (B) phospholipid amount. |
From the dissolution curves of different ratios of solid dispersions, we could find that the cumulative release rate of nintedanib increases with the increase of PVP-K30 ratio. When the ratio was 1:1, 1:3, and 1:5, drug release ratios from the nintedanib solid dispersion at 2 hours were 64.10%, 69.78%, and 96%, respectively, while the cumulative release rate of the drug substance was only 50.41%. From the trend of the dissolution curves, we could clearly conclude that with the increase of the PVP amount, the drug released faster from the solid dispersion.
Therefore, the formulation of the nintedanib solid dispersion was further prepared by selecting a prescription drug loading ratio of 1:5.
The influence of phospholipid amount
The drug dissolution rate from the solid dispersions with different amounts of phospholipid (nintedanib:PVP:phospholipid =1:5:0, 1:5:0.15, and 1:5:0.25) is shown in Figure 1B, and the similarity of drug dissolution from the different formulas is also shown in Figure 1B by the similarity factor (f2) method.
Figure 1B shows that the addition of phospholipids accelerated the release of nintedanib from solid dispersions in vitro, especially the later release of nintedanib from the solid dispersion increased. When the amount of phospholipid was 0.25, the release rate from the solid dispersion was obviously greater than that of 0.15. It was shown that the dissolution of solid dispersions with 0.25 phospholipid was up to 90% after 60 minutes, while the dissolution rate of the solid dispersion was only 75% with 0.15 phospholipid; therefore, 0.25 phospholipid was chosen to continue with latter parameter optimization.
Influence of feed rate
The drug dissolution rate from the solid dispersions made with different parameters of the electrospraying process (feed rate: 0.3, 0.5, 0.8, and 1.0 mL/h, respectively) is shown in Figure 2A, and the similarity of the drug dissolution from the different parameters is also shown in Figure 2A by the similarity factor (f2) method.
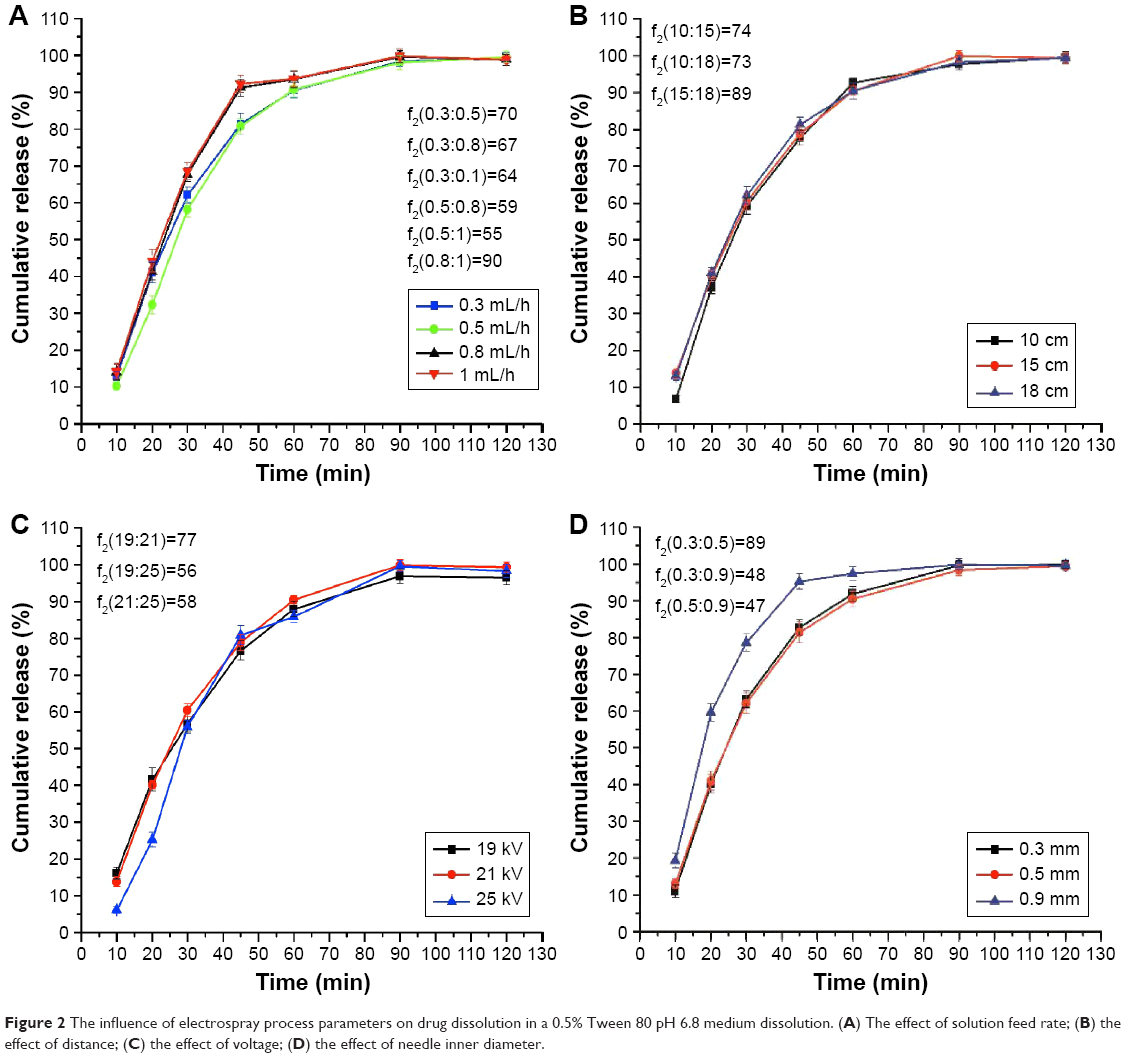 | Figure 2 The influence of electrospray process parameters on drug dissolution in a 0.5% Tween 80 pH 6.8 medium dissolution. (A) The effect of solution feed rate; (B) the effect of distance; (C) the effect of voltage; (D) the effect of needle inner diameter. |
Figure 2A shows that the feeding rate had little effect on the dissolution of the nintedanib solid dispersion. When the liquid flow rate was 0.8 and 1 mL/h, the drug dissolution between 30 and 45 minutes was faster than when the liquid flow rate was 0.3 and 0.5 mL/h, probably because the higher liquid flow led to a residual solvent of methanol in the solid dispersion, thereby promoting drug dissolution.
In Figure 3A, when the feeding rate was 0.3 mL/h (Figure 3A(i)), the particles had good dispersion, a smooth surface, and smaller size without adhesion. When the feeding rate was 0.5 mL/h (Figure 3A(ii)), the morphology of the particles was fibrous and spherical in size. When the feeding rate was 0.8 mL/h (Figure 3A(iii)), the morphology of the particles was elliptical or columnar. When the feeding rate was 1 mL/h (Figure 3A(iv)), the particle shape was in disorder and the particle size was not uniform. With the increase of the liquid flow, the particle size distribution widened and the size of the large particle size increased. The above phenomenon may be due to the increase in the liquid flow rate resulting in stretching in the process of solvent volatilization to form uneven particles and fibers.41 Under comprehensive consideration of the dissolution curve, the particle size and morphology of the solid dispersions prepared with a different feeding rate of 0.3 mL/h was chosen as the optimized feeding rate.
 | Figure 3 The effect of electrospray process parameters on the morphology of solid dispersion: (A) flow rate (i: 0.3 mL/h; ii: 0.5 mL/h; iii: 0.8 mL/h; iv: 1 mL/h), (B) distance (i: 10 cm; ii: 15 cm; iii: 18 cm), (C) voltage (i: 19 kV; ii: 21 kV; iii: 25 kV), and (D) needle inner diameter (i: 0.3 mm; ii: 0.5 mm; iii: 0.9 mm). |
The influence of receiving distances
The drug dissolution rate from the solid dispersions with different parameters of the electrospraying process (receiving distances: 10, 15, and 18 cm) is shown in Figure 2B, and the similarity of the drug dissolution from the different parameters is also shown in Figure 2B by the similarity factor (f2) method.
The distance had little effect on the dissolution of the nintedanib solid dispersion. When the receiving distance was 10 cm (Figure 3B(i)), the electric field strength was higher than that of 15 cm (Figure 3B(ii)) or 18 cm (Figure 3B(iii)), and the particle size of the solid dispersion was larger, leading to a slight slower dissolution.
In Figure 3B, we could see that when the distance was 10 cm (Figure 3B(i)), the diameter of the particles was between 200 and 800 nm. Because at the short receiving distance, the flying time of the droplet was reduced, the organic solvent could not fully evaporate before hitting the collector, also the droplets could not be well separated, resulting in the uneven distribution of the particle sizes. When the distance was 15 cm (Figure 3B(ii)), the diameter of the particle was between 200 and 350 nm, and the particle size was relatively uniform. When the distance increased to 18 cm (Figure 3B(iii)), the particle surface was smoother and rounder than that produced at other distances, and the particle size was smaller and relatively uniform. After considering the dissolution curve, the particle size, and the morphologies of the solid dispersions, 18 cm was chosen as optimal receiving distance.
Influence of voltage
The drug dissolution rate from the solid dispersions with different voltages in the electrospraying process (voltage: 19, 21, and 25 kV) is shown in Figure 2C, and the similarity of drug dissolution from the different parameters is also shown in Figure 2C by the similarity factor (f2) method.
The results showed that the voltage had little influence on the drug dissolution of the nintedanib solid dispersion. When the voltage was 21 kV, the overall dissolution of the solid dispersion was a little faster than that of the solid dispersion prepared by the voltage of 19 and 25 kV and the dissolution curve shows a smooth and steady growth trend.
The SEM image shows that when the voltage was 19 kV (Figure 3C(i)), the particle diameter was about 200 nm, and the particles were not separated, which was due to the low voltage, and the solvent was not fully volatile.37 When the voltage was 21 kV (Figure 3C(iii)), the particle diameter was about 200–350 nm, with a relatively uniform distribution. When the voltage increased to 25 kV (Figure 3C(iii)), the particle diameter was about 400–800 nm. This could be explained by the fact that higher voltage increases the traveling speed of the droplet and reduces the time for the organic solvent to evaporate and disperse, leading to a larger unevenly distributed particle size.
After considering the dissolution curve, the particle size and morphology of the solid dispersions prepared with a different voltage, 21 kV was chosen as the optimized voltage.
Influence of needle inner diameter
The drug dissolution rate from the solid dispersions with different needle inner diameters from the electrospraying process (needle inner diameter: 0.3, 0.5, and 0.9 mm) is shown in Figure 2D, and the similarity of the drug dissolution from the different parameters is also shown in Figure 2D by the similarity factor (f2) method.
Figure 2D shows that the drug release is similar between the solid dispersion made from a 0.3 and 0.5 mm needle inner diameter. When the inner diameter was increased to 0.9 mm, the drug release increased about 10%–15%, probably because the solvent was not completely evaporated, increasing the drug release. The SEM images of solid dispersions are shown in Figure 3D. When the needle inner diameter was 0.3 mm (Figure 3D(i)), the solid dispersion particles were irregular spheres. When the needle inner diameter was 0.9 mm (Figure 3D(ii)), the solid dispersion particles were clumped together. When the needle inner diameter was 0.5 mm (Figure 3D(iii)), the solid dispersions were uniform and separated. Hence, 0.5 mm should be chosen as the optimized needle inner diameter.
In summary, the optimal composition of the nintedanib solid dispersion is the mixture of nintedanib:PVP-K30:soybean lecithin at the ratio of 1:5:0.25. The optimal parameters of the electrospraying process were as follows: 21 kV voltage, 18 cm receiving distance, 0.3 mL/h feed rate of the solution, 0.5 mm needle inner diameter, and stable operating environments (t=25°C, RH =30%).
Characterization of the nintedanib solid dispersions prepared by optimized formulation and electrospraying method
In vitro drug dissolution studies
As shown in Figure 4A, when the in vitro cumulative release amount of the solid dispersions prepared by the optimized formulation and electrospraying method reached 90.4%±0.9% at the first hour, almost all of the nintedanib was released from the solid dispersions within 2 hours, meanwhile the crude nintedanib released only about 50% within 2 hours. In this study, nintedanib was prepared as a solid dispersion, and the drug was highly dispersed in the soluble carrier PVP. Because of its high dispersion and larger specific surface area, the drug is easier to dissolve and release in vitro. Therefore, after oral administration, the solid dispersion will be quickly dissolved in the intestine and absorbed by the gastrointestinal tract in the form of a solution, thus increasing the bioavailability, improving the efficacy and reducing the side effects in vivo.
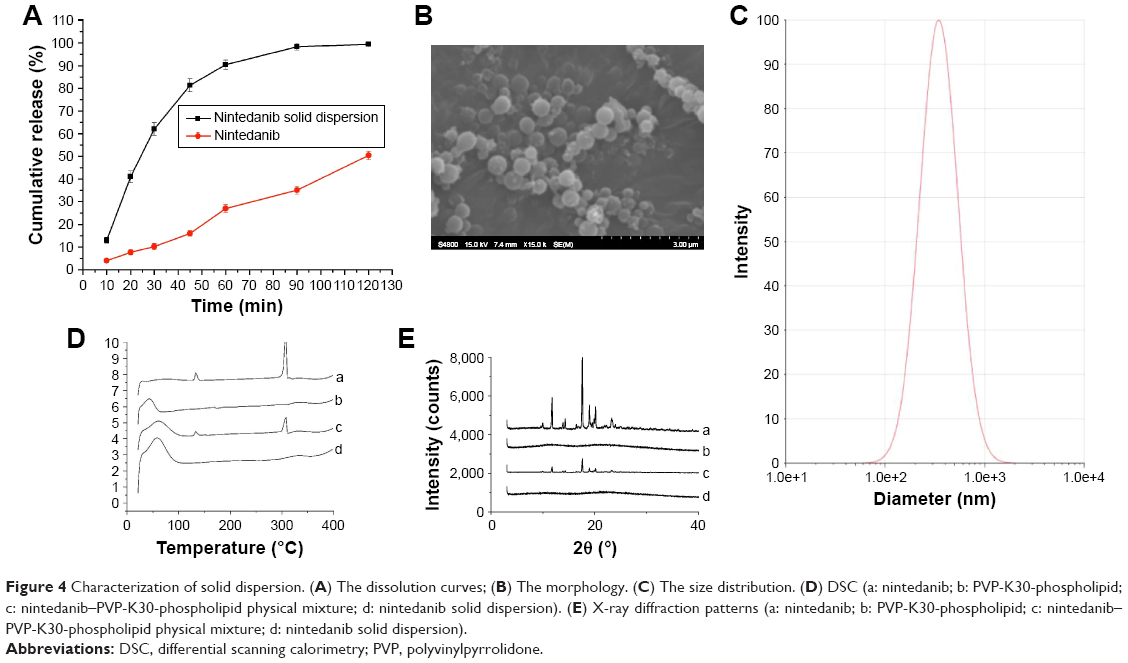 | Figure 4 Characterization of solid dispersion. (A) The dissolution curves; (B) The morphology. (C) The size distribution. (D) DSC (a: nintedanib; b: PVP-K30-phospholipid; c: nintedanib–PVP-K30-phospholipid physical mixture; d: nintedanib solid dispersion). (E) X-ray diffraction patterns (a: nintedanib; b: PVP-K30-phospholipid; c: nintedanib–PVP-K30-phospholipid physical mixture; d: nintedanib solid dispersion). |
Morphology and size distribution of solid dispersions
As shown in Figure 4B, the smooth and round nintedanib solid dispersion particles were fabricated with uniform morphology. In Figure 4C, the solid dispersions had a narrow distribution with an average diameter of 340±51 nm, and the polydispersity index (PDI) was 0.121.
Differential scanning calorimetry (DSC)
In Figure 4D, nintedanib API had sharp endothermic characteristic peaks between 130°C–150°C and 300°C–320°C. PVP-K30 and phospholipid had no significant endothermic peaks. The endothermic peak of nintedanib decreased in the physical mixture of nintedanib and drug carriers, but nintedanib still exists in the form of crystals. It was shown that the simple physical mixture cannot change the existence of nintedanib. The absorption peaks disappear completely in the nintedanib solid dispersion. For the preparation of nintedanib solid dispersions using electrospray, the interaction between nintedanib and PVP-K30-phospholipid eliminates the crystal structure, but it is stable in amorphous form.
X-ray diffraction (XRD) pattern
As shown in Figure 4E, in the range of 10°C–25°C, nintedanib has a more apparent crystallization peak as compared with PVP-K30 and phospholipid, the crystallization peak of nintedanib weakened after simple mixing of the drug and drug carriers, but it did not completely disappear. However, the crystallization peak of nintedanib disappeared completely in the Nintedanib solid dispersion, indicating that Nintedanib was highly dispersed in the solid dispersion. The X-ray diffraction results showed that nintedanib existed in the solid dispersion in amorphous form, which was proved to be a solid dispersion, not in the form of API. In summary, the DSC and XRD patterns validate the successful preparation of the nintedanib solid dispersion system.
In vivo intestinal circulation absorption studies of nintedanib API and solid dispersion
According to Table 1, the results showed that the order of absorption rate and volume of nintedanib API and nintedanib solid dispersion in different intestinal segments were: jejunum > duodenum > ileal > colon. It showed that nintedanib was absorbed in all intestinal segments, with no significant difference (P>0.05).
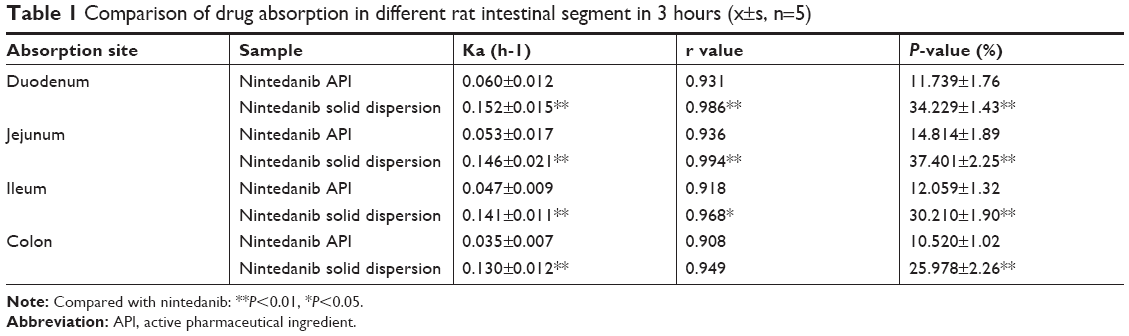 | Table 1 Comparison of drug absorption in different rat intestinal segment in 3 hours (x±s, n=5) |
According to Figure 5, the absorption of the nintedanib solid dispersion group in each intestinal segment was significantly higher than that of the nintedanib solution group (P<0.01), indicating that the preparation of nintedanib as a solid dispersion can effectively increase the absorption rate of the drug in each intestinal segment, which is conducive to improving the oral absorption of drugs. Because of the high dispersion and larger specific surface area of the nintedanib solid dispersion, the drug is easier to dissolve and release in vitro. Therefore, after oral administration, the solid dispersion will dissolve in the intestinal segment and be absorbed by the gastrointestinal tract in the form of a solution. However, the solubility of the API is poor, and it is still in the state of insoluble powder, which is difficult to absorb.
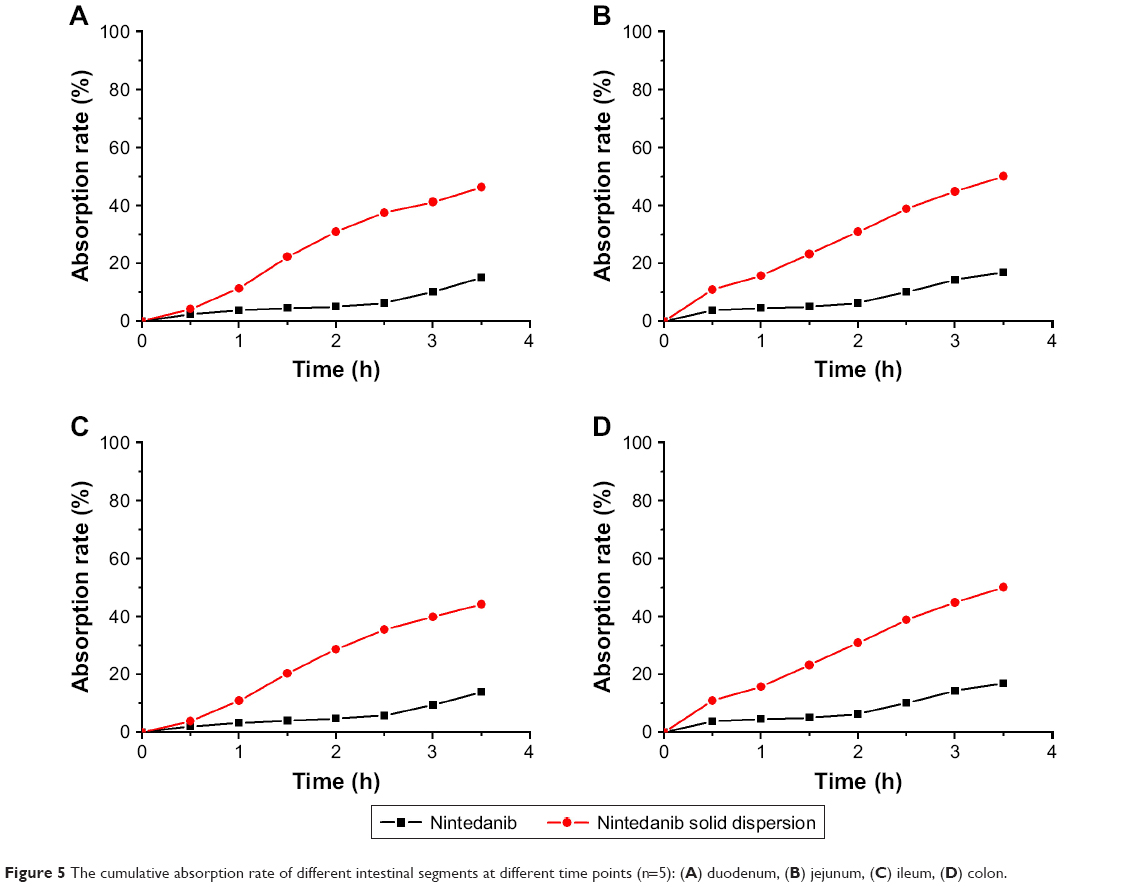 | Figure 5 The cumulative absorption rate of different intestinal segments at different time points (n=5): (A) duodenum, (B) jejunum, (C) ileum, (D) colon. |
Influence of HPMC viscosity on drug release from the sustained release capsule
The effects of the HPMC viscosity on nintedanib release from the sustained release capsules are shown in Figure 6A. The results showed that the HPMC viscosity had a significant effect on the release of nintedanib. As can be seen from dissolution curve, the nintedanib release rate slowed down as the HPMC viscosity increased, because the high viscosity of HPMC hydrated quickly, resulting in a larger viscosity of the gel layer, nintedanib went through the gel layer slowly, thus delaying the release of nintedanib. In this study, HPMC K100M was chosen as the most suitable material for the preparation of the nintedanib sustained release preparations.
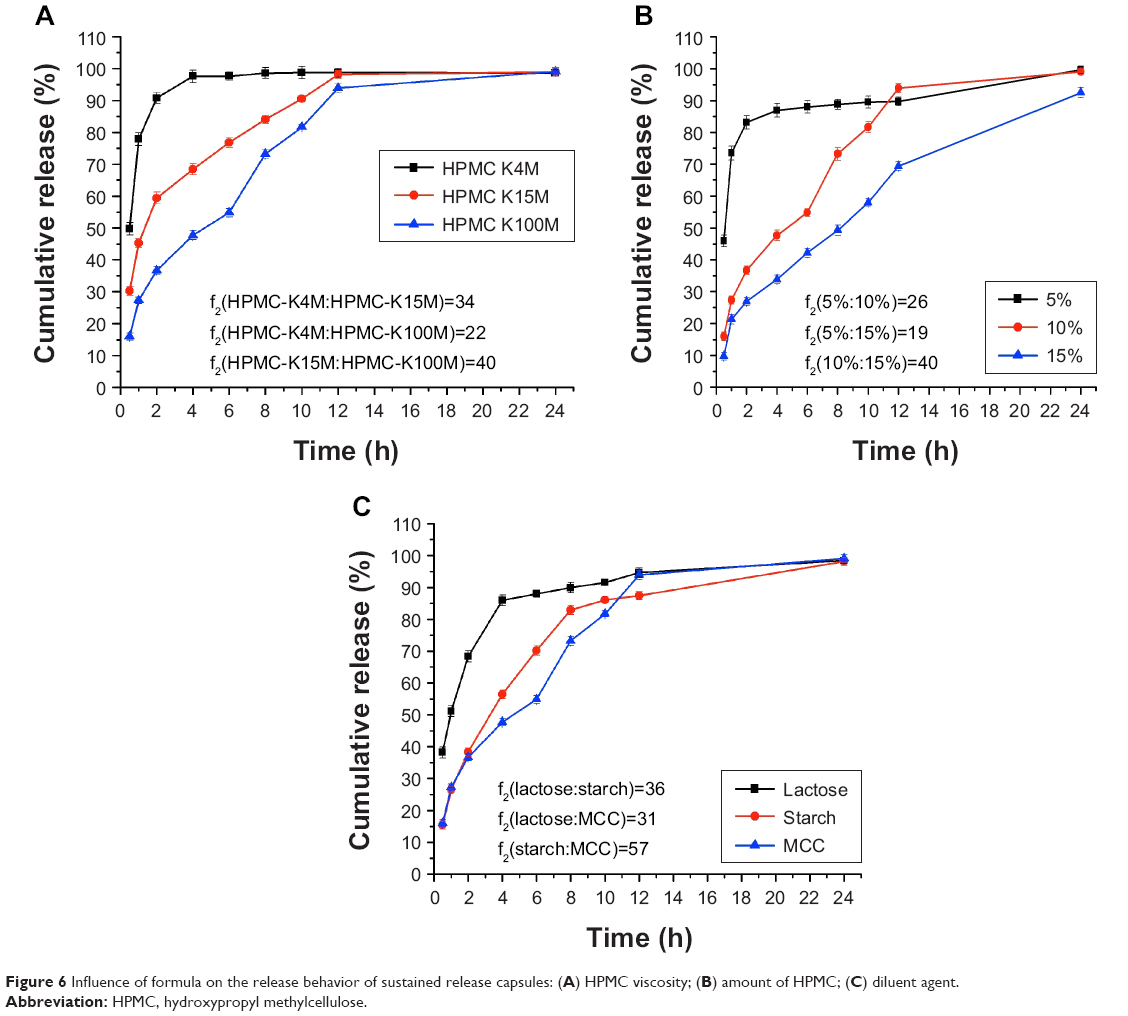 | Figure 6 Influence of formula on the release behavior of sustained release capsules: (A) HPMC viscosity; (B) amount of HPMC; (C) diluent agent. |
Influence of the amount of HPMC on the release behavior of sustained release capsules
The effects of different amounts of HPMC on nintedanib release from the sustained release capsules are shown in Figure 6B. The results showed that the effect of HPMC amount on the release of nintedanib sustained release capsules was significantly obvious.
When the amount of HPMC was 5%, the drug released at 2 hours was more than 80%, and the drug released was much greater than that released from 10% or 15% of the HPMC amount at the same time point. The release rate was the slowest at 15% HPMC, mainly because the gel layer became thicker and the diffusion speed slowed with the increase of the HPMC amount, the cumulative release rate of the drug at 12 hours was only 69%, and the drug could not be released in the form of a steady state. Therefore, the dosage of 10% was selected as the final amount of the sustained release material.
Influence of diluent agent on the release behavior of sustained release capsules
The effects of different diluents on nintedanib release from the sustained release capsules are shown in Figure 6C. The experimental results showed that the sustained release effect of lactose was weaker than that of starch and MCC. The dissolution rate from the sustained release capsule reached more than 80% at 4 hours when lactose was used as a filler. Because lactose is a water-soluble excipient, lactose dissolves during dissolution, and pores form in the gel layer of the sustained release tablet, which promotes the early release of nintedanib. The sustained release capsule with MCC as a filler had obviously a sustained release effect in the later period than that with the starch as the filling agent, so MCC was chosen as the filler.
The in vitro drug release results of the three batches of the optimized nintedanib sustained release capsules are shown in Figure 7. The results showed that the cumulative release rates of nintedanib sustained release capsules were about 37.17%, 55.45%, 94.61%, and 99.42% at 2, 6, 12, and 24 hours, respectively. The results showed an obviously sustained release in the release process and complete release at the end, also with good reproducibility among the three batches.
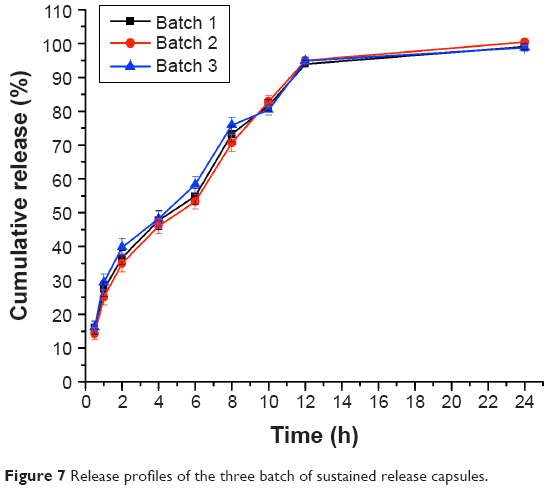 | Figure 7 Release profiles of the three batch of sustained release capsules. |
Fitting of drug release model
In previous studies, a variety of model systems have been established, such as the Higuchi model,42,43 Ritger–Peppas model,44–46 Hixson–Crowell model, diffusion-dissolution model, diffusion-relaxation model, and Weibull distribution.47–49 In this study, the release of nintedanib sustained release capsules in pH 6.8 buffer containing 0.5% Tween 80 was fitted according to the model formula in Table 2. The results of the fitting are shown in Table 3.
 | Table 2 Release model equations |
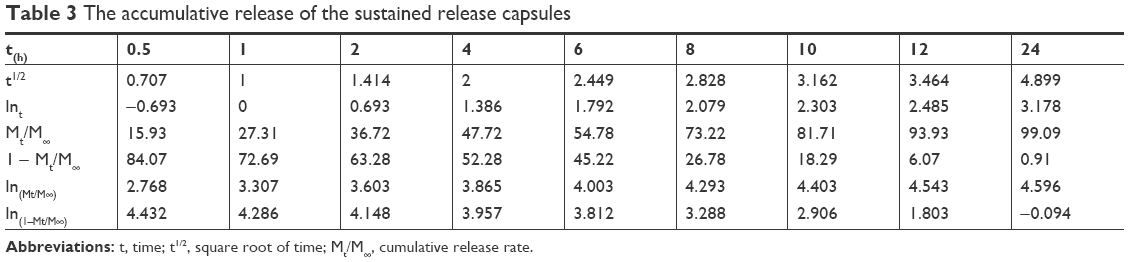 | Table 3 The accumulative release of the sustained release capsules |
The correlation coefficient R2 was calculated by the various release models (Table 4), in the order: Ritger–Peppas model > first-order model > Hixson–Crowell model > Higuchi model > zero-order model > Baker–Lonsdale model, where the Ritger–Peppas model fit the drug release best.
 | Table 4 The release pattern of sustained release capsules |
Assay of nintedanib concentration in plasma samples and validation of the analysis methods
According to Figure 8, the result showed that no interfering peaks were detected at the retention times of piperine (22.3 minutes) and nintedanib (8.7 minutes). A linear correlation (r2=0.9981) between the ratio of peak area and the nintedanib concentration was obtained at the range of 200–6,000 ng/mL. The limit of quantitation was 4.0 ng. The precision of the method was evaluated at concentrations of 200, 1,500, and 6,000 ng/mL. The coefficient of variation of intra- and inter-day precision was 0.33%–1.52% and 0.45%–1.73% at all concentrations, respectively. No decrease in the content of quality control samples was observed in the freezer or auto-sampler.
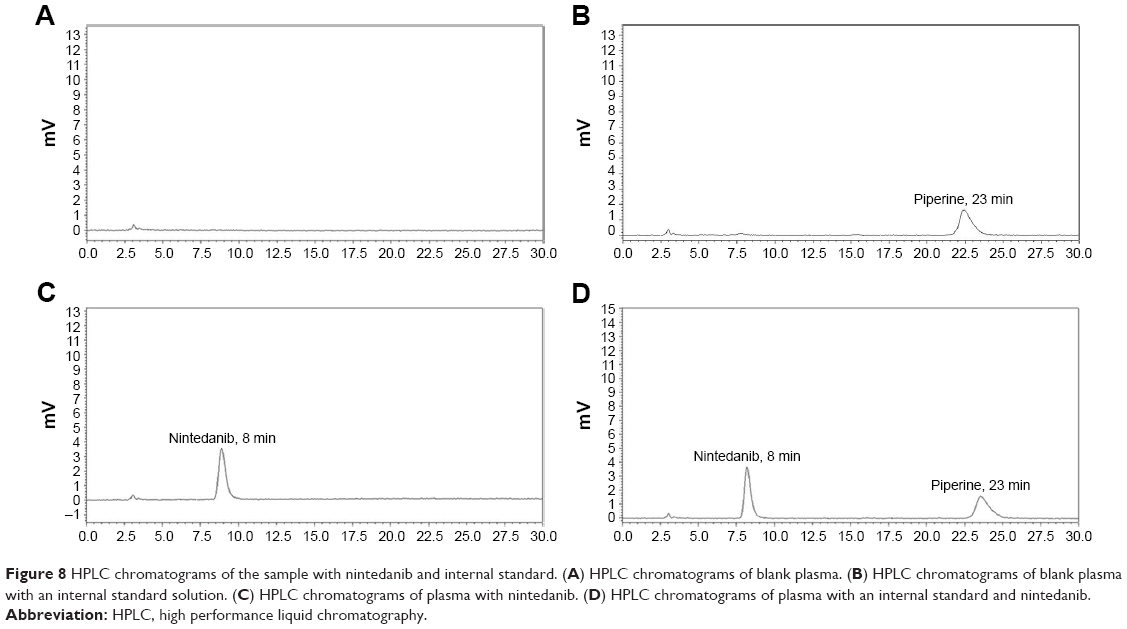 | Figure 8 HPLC chromatograms of the sample with nintedanib and internal standard. (A) HPLC chromatograms of blank plasma. (B) HPLC chromatograms of blank plasma with an internal standard solution. (C) HPLC chromatograms of plasma with nintedanib. (D) HPLC chromatograms of plasma with an internal standard and nintedanib. |
In vivo study
Figure 9 shows a comparison of the plasma concentration–time profiles of nintedanib after oral administration of nintedanib (50 mg/kg) of each formulation to 18 SD rats. Table 5 summarizes the pharmacokinetic parameters. Furthermore, we calculated the relative bioavailability of the test preparations, obtained using the following equation:
|
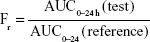
 | Figure 9 Mean plasma drug concentration–time profile of nintedanib capsule, nintedanib solid dispersion, and nintedanib sustained release capsule (n=6). |
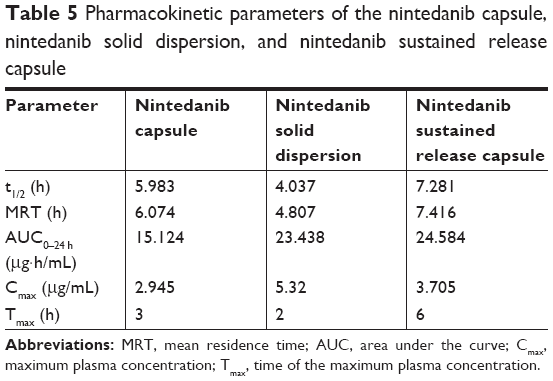 | Table 5 Pharmacokinetic parameters of the nintedanib capsule, nintedanib solid dispersion, and nintedanib sustained release capsule |
The results in vivo showed that the Tmax of the nintedanib soft capsule, solid dispersion, and sustained release capsule were 3, 2, and 6 hours, respectively, the Cmax was 2.945, 5.32, and 3.75 μg/mL, respectively, and the AUC0–24 was 15.124, 23.438, and 24.584 μg·h/mL, respectively. The average bioavailability of nintedanib sustained release capsules relative to the soft capsules was 162.55%, and the average relative bioavailability of the nintedanib sustained-release capsules relative to the solid dispersions was 104.89%. It proved that the solid dispersion increased nintedanib solubility, thus increased the bioavailability of nintedanib in vivo as compared with the soft capsules, and the nintedanib sustained-release capsules was bioequivalent with the solid dispersions. In this study, nintedanib was prepared as a solid dispersion, and the drug was highly dispersed in the soluble carrier PVP. Because of its high dispersion and larger specific surface area, the drug was easier to dissolve and release in vitro. Maybe it is because this formulation method can induce super-saturated drug dissolution and with that enhance absorption.50 Therefore, after oral administration, the solid dispersion will dissolve in the intestinal segment and be absorbed by the gastrointestinal tract in the form of a solution, thus increasing the bioavailability to 162.55%, improving the efficacy and reducing the side effects in vivo. However, the solubility of the nintedanib is poor, and it is still in the state of an insoluble powder, which is difficult to absorb. Therefore, the bioavailability was improved compared with nintedanib. Meanwhile, in order to reduce the side effects of patients, reduce the number of medications, and maintain a relatively constant and effective blood concentration level, the study further prepared a sustained release capsule by using a solid dispersion as API. When the drug is released from the sustained-release capsule, it is still in the form of a solid dispersion, resulting in a bioequivalence (104%) with the solid dispersion. However, the nintedanib sustained release capsule sustained the drug release by drug diffusing from the gel skeleton material. The in vivo sustained release also results in vivo sustained absorption. Therefore, the nintedanib solid dispersion has a higher Cmax than that of the nintedanib sustained release capsule.
The in vivo drug absorption and in vitro drug release correlation of nintedanib sustained release capsules were also evaluated in Table 6. The linear relationship between in vitro dissolution and in vivo absorption fraction was as follows: Y=1.274X−25.165, R=0.9628. The results showed that the in vivo absorption of nintedanib sustained release capsules had a good correlation with the in vitro drug release in the release medium of pH 6.8 PBS containing 0.5% Tween 80. The result showed that in the first few hours, the in vivo absorption had a significant hysteresis compared with in vitro release. As the drug is substantially released completely and the absorption site is saturated, the in vivo absorption ratio increases to substantially equivalent or slightly faster than the in vitro release rate. Maybe it is because the nintedanib solid dispersion sustained release capsules are orally administered, but the drugs need to be released, dissolved, absorbed, and further delivered into the blood circulation.
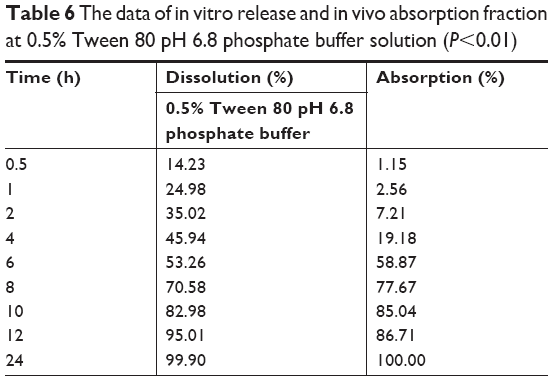 | Table 6 The data of in vitro release and in vivo absorption fraction at 0.5% Tween 80 pH 6.8 phosphate buffer solution (P<0.01) |
Conclusion
In this article, first, nintedanib solid dispersions were prepared by an optimized electrospraying technology to increase its solubility, and the prepared solid dispersion had a particle size of 340 nm, with uniform size distribution and a smooth surface. The presence of nintedanib was at an amorphous state in the nintedanib solid dispersion. Drug release of the solid dispersion in the dissolution medium was about 60%, within 2 hours, and the cumulative dissolution rate reached 100%. Second, nintedanib solid dispersions were used as API to prepare the sustained release capsules using HPMC as a sustained release matrix material. The content of nintedanib was between 98% and 102%, and the release rate of the sustained release capsules was 35.17%, 54.78%, 70.58%, and 93.93%, respectively, in the dissolution medium of 2, 6, 8, and 12 hours, indicating a better controlled release profile than solid dispersion itself. The release of the nintedanib sustained release capsules accords with the Ritger–Peppas model, which suggests diffusion and dissolution at the same time. In vivo drug absorption studies indicated that the preparation of nintedanib as a solid dispersion can effectively increase the absorption rate of the drug in each intestinal segment which is conducive to improving the oral absorption of drugs.
Finally, we investigated the blood concentration of the nintedanib soft capsule, nintedanib solid dispersions, nintedanib sustained release capsules in SD rats in vivo by HPLC. The relative bioavailability of nintedanib sustained release capsules was 162.55% as compared to the nintedanib soft capsule and 104.89% as compared to the nintedanib solid dispersion. The results showed that the test dosage form significantly improved the bioavailability compared to the reference dosage form and rendered a promising sustained release effect in vivo.
Acknowledgments
This study was sponsored by The Research Foundation for Advanced Scholars of Jiangsu University (No 11JDG122), China Postdoctoral Science Foundation (2017M610309), Training Project of Jiangsu University for Young Key Teachers (5521290003), and the Department of Education of Guangdong Province (No 2016KCXTD005, 2017KSYS010).
Disclosure
The authors report no conflicts of interest in this work.
References
Parhizkar M, Reardon PJT, Knowles JC, et al. Performance of novel high throughput multi electrospray systems for forming of polymeric micro/nanoparticles. Mater Des. 2017;126(1):73–84. | ||
Nath SD, Son S, Sadiasa A, Min YK, Lee BT. Preparation and characterization of PLGA microspheres by the electrospraying method for delivering simvastatin for bone regeneration. Int J Pharm. 2013;443(1–2):87–94. | ||
Dormer NH, Gupta V, Scurto AM, Berkland CJ, Detamore MS. Effect of different sintering methods on bioactivity and release of proteins from PLGA microspheres. Mater Sci Eng C Mater Biol Appl. 2013;33(7):4343–4351. | ||
Jadhav A, Wang LJ, Lawrence C, Padhye R. Study of electrospraying characteristics of polymer solution coating on textile substrate. Adv Mater. 2011;332:710–715. | ||
Papadopoulou SK, Tsioptsias C, Pavlou A, Kaderides K, Sotiriou S, Panayiotou C. Superhydrophobic surfaces from hydrophobic or hydrophilic polymers via nanophase separation or electrospinning/electrospraying. Colloids Surf A Physicochem Eng Asp. 2011;387(1–3):71–78. | ||
Wang M, Yu JH, Hsieh AJ, Rutledge GC. Effect of tethering chemistry of cationic surfactants on clay exfoliation, electrospinning and diameter of PMMA/clay nanocomposite fibers. Polymer. 2010;51(26):6295–6302. | ||
Widiyandari H, Hogan CJ, Yun KM, Iskandar F, Biswas P, Okuyama K. Production of narrow-size-distribution polymer-pigment-nanoparticle composites via electrohydrodynamic atomization. Macromol Mater Eng. 2007;292(4):495–502. | ||
Bohr A, Kristensen J, Stride E, Dyas M, Edirisinghe M. Preparation of microspheres containing low solubility drug compound by electrohydrodynamic spraying. Int J Pharm. 2011;412(1–2):59–67. | ||
Nath SD, Son S, Sadiasa A, Min YK, Lee BT. Preparation and characterization of PLGA microspheres by the electrospraying method for delivering simvastatin for bone regeneration. Int J Pharm. 2013;443(1–2):87–94. | ||
Li X, Li H, Huang K, et al. Durable superamphiphobic nano-silica/epoxy composite coating via coaxial electrospraying method. Appl Surf Sci. 2018;436:283–292. | ||
Greiner A, Wendorff JH. Electrospinning: a fascinating method for the preparation of ultrathin fibers. Angew Chem Int Ed Engl. 2007;46(30):5670–5703. | ||
Nath SD, Son S, Sadiasa A, Min YK, Lee BT. Preparation and characterization of PLGA microspheres by the electrospraying method for delivering simvastatin for bone regeneration. Int J Pharm. 2013;443(1–2):87–94. | ||
Zhang CX, Wang P. Fabrication of polymer microspheres and microparticles by electrospraying. Mate Rev. 2013;27(17):95–97. | ||
Xue LW. The research on the preparation of biodegradable phosphazene/polyester drug-loaded microparticles by electrohydrodynamic atomization and the formation mechanism of different microparticles morphologies. Beijing: Beijing University of Chemical Technology; 2010. | ||
Zhou YY, Lw X, Mao JF, Cai Q, Jin RG. Preparation of ion of polyphosphazene microparticles via electrospray. Acta Polymerica Sinica. 2010;1:125–130. | ||
Borchardt J. S2K guideline on diagnosis and treatment of idiopathic pulmonary fibrosis. Pneumologie. 2013;67(6):356. | ||
Wuyts WA, Kolb M, Stowasser S, Stansen W, Huggins JT, Raghu G. First data on efficacy and safety of nintedanib in patients with idiopathic pulmonary fibrosis and forced vital capacity of ≤50% of predicted value. Lung. 2016;194(5):739–743. | ||
King CS, Nathan SD. POINT: should all patients with idiopathic pulmonary fibrosis, even those with more than moderate impairment, be treated with nintedanib or pirfenidone? Yes. Chest. 2016;150(2):273–275. | ||
Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–1087. | ||
Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther. 2014;349(2):209–220. | ||
Hostettler KE, Zhong J, Papakonstantinou E, et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res. 2014;15(1):157. | ||
Antoniu SA, Kolb MR. Intedanib, a triple kinase inhibitor of VEGFR, FGFR and PDGFR for the treatment of cancer and idiopathic pulmonary fibrosis. IDrugs. 2010;13(5):332–345. | ||
Luca R, Bois RMD, Ganesh R. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. New Engl Med. 2014;370(22):2071–2082. | ||
Taniguchi H, Xu Z, Azuma A, et al. Subgroup analysis of Asian patients in the INPULSIS® trials of nintedanib in idiopathic pulmonary fibrosis. Respirology. 2016;21(8):1425–1430. | ||
Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. | ||
Guan J, Liu Q, Zhang X, et al. Alginate as a potential diphase solid dispersion carrier with enhanced drug dissolution and improved storage stability. Eur J Pharm Sci. 2018;114:346–355. | ||
Deshmane S, Deshmane S, Shelke S, Biyani K. Enhancement of solubility and bioavailability of ambrisentan by solid dispersion using Daucus carota as a drug carrier: formulation, characterization, in vitro, and in vivo study. Drug Dev Ind Pharm. 2018;44(6):1001–1011. | ||
Han G, Yan Lq S, Wang CQ, Liu L, Zhao Y. Preparation of emodin solid dispersion and determination of its dissolution. China Tradit and Herb Drug. 2001;42(3):487–490. | ||
Yu DG, Branford-White C, White K, Li XL, Zhu LM. Dissolution improvement of electrospun nanofiber-based solid dispersions for acetaminophen. AAPS PharmSciTech. 2010;11(2):809–817. | ||
Yue PF, Zheng Q, Zhu GH, Wu ZF, Hu PY, Yang M. “Total amount” release kinetics evaluation of traditional Chinese medicine sustained-release preparations based on material rough set theory. Yao Xue Xue Bao. 2010;45(11):1354–1360. | ||
Zhang M, Wang W, Cui Y, Zhou N, Shen J. Near-infrared light-mediated photodynamic/photothermal therapy nanoplatform by the assembly of Fe3O4 carbon dots with graphitic black phosphorus quantum dots. Int J Nanomedicine. 2018;13:2803–2819. | ||
Zanela da Silva Marques T, Santos-Oliveira R, Betzler de Oliveira de Siqueira L, et al. Development and characterization of a nanoemulsion containing propranolol for topical delivery. Int J Nanomedicine. 2018;13:2827–2837. | ||
Liu HF, Shi SS, Sun CS, Zhao X, Shen S, He H. Investigation of the pharmacokinetics of a novel sustained release metformin hydrochloride suspension with ion exchange resin as carriers in beagle dogs. Afr J Pharm Pharmacol. 2012;6(7):502–504. | ||
Greiner A, Wendorff JH. Electrospinning: a fascinating method for the preparation of ultrathin fibers. Angew Chem Int Ed Engl. 2007;46(30):5670–5703. | ||
Liu HF, X JM, Chen BG. Formulation and in vitro evaluation of lysozyme-loaded PLGA microspheres fabricated by one-step electrospraying method. Lat Am J Pharm. 2016;35(2):274–283. | ||
Nath SD, Son S, Sadiasa A, Min YK, Lee BT. Preparation and characterization of PLGA microspheres by the electrospraying method for delivering simvastatin for bone regeneration. Int J Pharm. 2013;443(1–2):87–94. | ||
Zhang CX, Wang P. Fabrication of polymer Microspheres and Microparticles by Electrospraying. Mater Rev. 2013;27(17):95–97. | ||
Dahlgren D, Roos C, Lundqvist A, et al. Effect of absorption-modifying excipients, hypotonicity, and enteric neural activity in an in vivo model for small intestinal transport. Int J Pharm. 2018;549(1–2):239–248. | ||
Li X, Uehara S, Sawangrat K, et al. Erratum to ‘Improvement of intestinal absorption of curcumin by cyclodextrins and the mechanisms underlying absorption enhancement’. 2018, 535, 340–349. Int J Pharm. 2018;541(1–2):261. | ||
Purnachand D, Veerareddy A, Ramadevi B. Development and validation of a simple and sensitive stability indicating RP-HPLC assay method for determination of Nintedanib and stress degradation studies. J Chem Pharm Res. 2015;78:774–782. | ||
Hartlllan RPA, Brunner DJ, Camelot DMA. Electrohydynamic atomi-zation in the cone-jet modeling of the liquid cone and jet. J Aerosol Sci. 1999;30(7):823–849. | ||
Higuchi T. Mechanism of sustained-action medication, theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52:1145–1149. | ||
Higuchi WI. Analysis of data on the medicament release from ointments. J Pharm Sci. 1962;51:802–804. | ||
Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15(1):25–35. | ||
Sinclair GW, Peppas NA. Analysis of non-fickian transport in polymers using simplified exponential expressions. J Memb Sci. 1984;17(3):329–331. | ||
Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Controlled Release. 1987;5(1):37–42. | ||
Siepmann J, Siepmann F. Mathematical modeling of drug dissolution. Int J Pharm. 2013;453(1):12–24. | ||
Rahman MM, Hasan S, Alam MA. Formulation and evaluation of Ranolazine sustained release matrix tablets using Eudragit and HPMC. J Pharmaceut Biomed. 2011;2(1):172–177. | ||
Rahman MM, Khalipha ABR, Azad MAK, et al. Effect of natural and synthetic polymer on release of ketotifen fumarate matrix tablets:a sustained release dosage form. IJPSR. 2013;4(4):1401–1408. | ||
Sawicki E, Schellens JH, Beijnen JH, Nuijen B. Inventory of oral anticancer agents: Pharmaceutical formulation aspects with focus on the solid dispersion technique. Cancer Treat Rev. 2016;50:247–263. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.