Back to Journals » International Journal of Nanomedicine » Volume 15
Zinc Oxide Nanoparticles Induce Ferroptotic Neuronal Cell Death in vitro and in vivo
Authors Qin X, Tang Q, Jiang X, Zhang J ![]() , Wang B
, Wang B ![]() , Liu X, Zhang Y, Zou Z
, Liu X, Zhang Y, Zou Z ![]() , Chen C
, Chen C ![]()
Received 18 February 2020
Accepted for publication 10 June 2020
Published 27 July 2020 Volume 2020:15 Pages 5299—5315
DOI https://doi.org/10.2147/IJN.S250367
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Xia Qin,1,* Qianghu Tang,2,* Xuejun Jiang,3,* Jun Zhang,4 Bin Wang,4 Xuemei Liu,2 Yandan Zhang,4 Zhen Zou,4,5 Chengzhi Chen2,5
1Department of Pharmacy, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, People’s Republic of China; 2Department of Occupational and Environmental Health, School of Public Health and Management, Chongqing Medical University, Chongqing 400016, People’s Republic of China; 3Center of Experimental Teaching for Public Health, Experimental Teaching and Management Center, Chongqing Medical University, Chongqing 400016, People’s Republic of China; 4Institute of Life Sciences, Chongqing Medical University, Chongqing 400016, People’s Republic of China; 5Dongsheng Lung-Brain Disease Joint Lab, Chongqing Medical University, Chongqing 400016, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhen Zou; Chengzhi Chen Email [email protected]; [email protected]
Purpose: Zinc oxide nanoparticles (ZnONPs) are one of the most important nanomaterials that are widely used in the food, cosmetic and medical industries. Humans are often exposed to ZnONPs via inhalation, and they may reach the brain where neurotoxic effects could occur via systemic distribution. However, the mechanisms underlying how ZnONPs produce neurotoxic effects in the brain remain unclear. In this study, we aimed to investigate the novel mechanism involved in ZnONPs-induced neurotoxicity.
Methods and Results: We demonstrated for the first time that pulmonary exposure to ZnONPs by intratracheal instillation could trigger ferroptosis, a new form of cell death, in the neuronal cells of mouse cerebral cortex. A similar phenomenon was also observed in cultured neuron-like PC-12 cell line. By using a specific inhibitor of ferroptosis ferrostatin-1 (Fer-1), our results showed that inhibition of ferroptosis by Fer-1 could significantly alleviate the ZnONPs-induced neuronal cell death both in vivo and in vitro. Mechanistic investigation revealed that ZnONPs selectively activated the JNK pathway and thus resulted in the ferroptotic phenotypes, JNK inhibitor SP600125 could reverse lipid peroxidation upregulation and ferroptotic cell death induced by ZnONPs in PC-12 cells.
Conclusion: Taken together, this study not only demonstrates that pulmonary exposure of ZnONPs can induce JNK-involved ferroptotic cell death in mouse cortex and PC-12 cells, but also provides a clue that inhibition of ferroptosis by specific agents or drugs may serve as a feasible approach for reducing the untreatable neurotoxicity induced by ZnONPs.
Keywords: zinc oxide nanoparticles, ferroptosis, neuron, cerebral cortex
Introduction
Ferroptosis is a novel non-apoptotic and iron-dependent form of cell death.1 The hallmarks of ferroptosis are defined as the inactivation of phospholipid hydroperoxidase glutathione peroxidase 4 (GPX4), the enhancement of iron and the accumulation of lipid oxidation.2 Previous investigations have demonstrated that ferroptosis can be triggered by natural stimuli, synthetic agents and environmental toxicants in many types of cells.2–4 Intriguingly, the studies also reported that the engineered nanoparticles could cause ferroptotic cell death in mouse xenograft tumors, and thus suppressed the growth of tumors.5,6 These findings suggest that nanoparticles are capable of inducing ferroptosis in animals and/or cultured cells.
Zinc oxide nanoparticles (ZnONPs) are one of the most important nanomaterials that are widely used in the food, cosmetic and medical industries.7 Increasing evidence is available regarding the toxicological effects of ZnONPs in animals, particularly in the respiratory,8 cardiovascular9 and neuronal systems.10 These toxicological effects have been identified mainly through oxidative stress, inflammation and even cell death.8,11,12 ZnONPs can induce several forms of cell death in cultured cells or animals, including apoptosis,13 necrosis14 and autophagy.8,15,16 Importantly, ZnONPs recently have been found to interact with iron within the cells and significantly elevate the generation of peroxidation products (such as malondialdehyde, MDA),17,18 both of which are highly linked to the hallmarks of ferroptosis. Therefore, it is reasonable to speculate that ferroptosis may be involved in the process of cell death induced by ZnONPs. However, to date, whether ZnONPs can trigger ferroptotic cell death in vivo and/or in vitro remains unknown.
ZnONPs can distribute in various tissues of mice, such as liver, lung and kidney, etc.19 Accumulation of ZnONPs in these tissues may further contribute to the adverse effects by induction of oxidative stress, inflammatory responses and cytotoxicity.8 After systemic distribution, ZnONPs are also capable of reaching the brain via blood–brain barrier penetration or translocation along the olfactory nerve pathway.20 Consequently, ZnONPs have been demonstrated to cause spatial learning and memory deficits of animals by altering the synaptic plasticity in the hippocampus.21 In cultured cells, ZnONPs also induce the strongest cytotoxicity via induction of DNA damage and cell apoptosis in many types of neuronal cell lines, such as neural stem cells, Neuro-2a cells, SH-SY5Y cells and primary cultured astrocytes, etc.22–25
In this study, using both C57BL/6J mice and cultured PC-12 cells, we aimed to test the hypothesis that ferroptosis was involved in ZnONPs-induced neuronal cell death in vivo and in vitro. To verify the results obtained from animals and cells, a specific inhibitor of ferroptosis was used. The data showed that inhibition of ferroptosis could remarkably ameliorate the cytotoxicity caused by ZnONPs. These findings will provide a clue that inhibition of ferroptosis by specific agents or drugs may serve as a feasible approach for intervention and/or prevention of ZnONPs-induced neurotoxicity.
Materials and Methods
Chemicals and Reagents
ZnONPs, <50 nm particle size, was purchased from Sigma-Aldrich (St. Louis, MO, USA). MDA assay kit, adenosine triphosphate (ATP) assay kit and tissue iron assay kit were obtained from Nanjing Jiancheng Institute of Bioengineering (Jiangsu, China); anti-glutathione peroxidase 4 (GPX4), anti-voltage-dependent anion channel 3 (VDAC3), anti-cystine/glutamate antiporter (SLC7A11), anti-neuronal nuclei antigen (NeuN), anti-microtubule-associated protein 2 (MAP2), anti-Ki67 and anti-GAPDH antibodies were all obtained from Bioss Biological Technology Co. Ltd. (Beijing, China); β-actin antibody was obtained from ABclonal Biotechnology (Woburn, MA, USA); p-JNK (4668), p-ERK1/2 (4370) and p-p38 (4511) antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Cy3 AffiniPure Goat anti-Rabbit IgG (H+L) was from EarthOx Life Sciences (San Francisco, CA, USA); 4.6-diamidino-2-phenylindole (DAPI), phenylmethanesulfonyl fluoride (PMSF) and bicinchoninic acid assay kit were all obtained from Beyotime Institute of Biotechnology (Shanghai, China); fetal bovine serum (FBS) was from Ausbian (Australia); Dulbecco’s modified Eagle’s medium (DMEM) was obtained from Gibco Life Technologies (Grand Island, NY, USA); CCK-8 assay kit was obtained from Dojindo Laboratories (Shanghai, China); ferrostain-1 (Fer-1) was purchased from APExBIO (Houston, TX, USA); zinc-specific indicator FluoZin-3, AM was provided by Molecular Probes Inc., Invitrogen (Eugene, OR, USA); deferoxamine (DFO) was obtained from Shandong Topscience Biotech Co., Ltd. (Shandong, China). Diethylenetriaminepentaacetic acid (DTPA) was obtained from Sigma-Aldrich (St. Louis, MO, USA). SP600125 (S1460) was purchased from Selleckchem (Houston, TX, USA).
Animal Husbandry
Healthy male specific pathogen-free C57BL/6J mice, aged 8 weeks and weighing 21–25 g, were purchased from Experimental Animal Center of Chongqing Medical University [Chongqing, China, license numbers: SCXK (Yu) 2012–001]. The mice were housed in propylene cages fitted with filter covers at a controlled ambient temperature (23 ± 1 °C) and relative humidity (55 ± 10%). The animals were maintained under standard laboratory conditions (12-hour light/dark cycle) and were given ad-libitum access to food and tap water. This study was approved by Institutional Animal Care and Use Committee of Chongqing Medical University. All procedures were conducted following the guidelines contained in the guide for the care and use of laboratory animals. All efforts were made to ensure minimal suffering to the animals.
ZnONPs Characterization and Preparation of Exposure Stock
The nature of particles and level of agglomeration were determined by transmission electron microscopy (TEM, Hitachi-7500, Hitachi, Ltd., Tokyo, Japan). Field emission scanning electron microscopy (Hitachi-SU8010, Hitachi, Ltd., Tokyo, Japan) with energy dispersive spectroscopy (Oxford X-MAN50, Oxford Instruments, UK) was used to detect the chemical elemental composition of ZnONPs. Size and zeta potential of ZnONPs were detected by dynamic light scattering and phase analysis light scattering (Zetasizer Nano-ZS, ModelZEN3600, Malvern Instruments Ltd., Malvern, UK). The obtained TEM image, chemical composition, Z-average, PDI and Zeta potential of ZnONPs were all shown in our previous work.8 ZnONPs were suspended in 2% heat-inactivated sibling mouse serum in MilliQ water. The solution of ZnONPs was freshly prepared as quickly as possible and sonicated with an ultrasonic cleaner (SB-5200DT, Ningbo Scientz Biotechnology Co., Ltd., China) for 20 min in an ice water bath before use.26,27
ZnONPs Intratracheal Instillation
The mice were randomly divided into four groups: control, low-dose group, medium-dose group and high-dose group. After one-week adaption, the animals were intratracheally instilled with a single dose of ZnONPs for 3 μg (low), 6 μg (medium) and 12 μg (high) per animal, and the vehicle control animals received 0.9% sterile physiological saline for instillation. The doses of ZnONPs used in the current study were calculated according to the Chinese occupational exposure limit of ZnONPs; detailed information about the process of calculation was described in our previous work.8 Moreover, these concentrations of ZnONPs were also similar to the previous studies.28,29
The animals were instilled intratracheally once as described previously by Jacobsen et al.28 In brief, after anesthesia, the mice were placed on a 40-degree slope and the trachea was intubated by an insyte catheter with a shortened needle. A total volume of 50 μL ZnONPs suspension or sterile physiological saline was instilled followed by 150 μL air by a syringe. Subsequently, the mice were held with head up and gently transferred on a 37 °C heating plate until they recovered from anesthesia. After 1, 3 and 7 days of ZnONPs administration, three mice from each group were sacrificed and the cerebral cortex tissues were quickly collected for morphological observation. At day 3 after ZnONPs exposure, eight animals from each group were used for the further designed experiments. For the rescue experiments in vivo, 100 μL Fer-1 (10 mg/kg) or vehicle was injected intraperitoneally 1 h before ZnONPs (12 μg/mouse) instillation, six mice per treatment were used. All samples were collected 3 days after ZnONPs instillation for the further indicated analysis in the rescue experiments.
Hematoxylin–Eosin Staining
The pathological changes of cerebral cortex tissues tissue were evaluated by hematoxylin–eosin (H&E) staining according to protocols described previously.4 Briefly, the tissue-containing slides were deparaffinized in xylene and dehydrated with ethanol. After staining with hematoxylin at room temperature, the sections were rinsed with distilled water and then stained with eosin. Subsequently, the sections were dehydrated by ethanol, cleared in xylene, and finally mounted with neutral balsam.
Immunofluorescence Assay
Immunofluorescence assay was conducted according to the protocols described previously.4 Briefly, the tissue-containing slides were rinsed and incubated in the blocking solution containing serum. The sections were incubated with the primary anti-NeuN (1:500), anti-MAP2 (1:500) and anti-Ki67 (1:500) antibodies at 4 °C overnight. Next day, the Cy3-conjugated secondary antibody (1:500) was incubated at room temperature followed by staining with DAPI. The sections were then observed under a fluorescent microscope (Olympus, IX53, Tokyo, Japan). The average fluorescence intensities were obtained from Image-Pro Plus software (version 6.0, Media Cybernetics, USA).
Transmission Electron Microscopy
The ultrastructure of mitochondria was observed by transmission electron microscopy as described previously.4 Briefly, the cerebral cortex tissues were rapidly collected by heart perfusion-fixed technique with 4% paraformaldehyde and 2.5% (w/v) glutaraldehyde buffer. Afterwards, the tissues were dissected and cut into 1 mm3 in size. The tissues were then fixed in glutaraldehyde and 1.0% osmic acid, and dehydrated by ethanol. After embedding, the tissue-containing slides were coated with celloidin membrane and stained with uranyl acetate and lead citrate. The slides were observed under transmission electron microscope (Hitachi-7500, Hitachi, Ltd., Tokyo, Japan).
Measurement of ATP, MDA and Iron Contents
The contents of ATP, MDA and iron were determined by commercial available kits according to the procedures of manufacturers.4 Briefly, the MDA contents were measured at a wavelength of 532 nm, the ATP contents were detected at a wavelength of 636 nm, and the levels of iron were tested at a wavelength of 520 nm by an ultraviolet spectrophotometer (UV-1750, Shimazu, Co., Ltd., Japan) and micro-plate reader (ELX808, Bio-Tek Instruments, Winooski, VT, USA), respectively.
Inductively Coupled Plasma–Mass Spectrometry (ICP-MS)
The determination of zinc ion concentration in the cerebral cortex tissue was performed as described previously.8 In brief, the tissues were rapidly dissected and homogenized after the indicated treatment. The homogenate and digestion solution were then added to a Teflon digestion tank (Sineo Microwave Chemistry Technology Co., Ltd., Shanghai, China), followed by heating on an electronic temperature control heating plate (EHC-1, Sineo Microwave Chemistry Technology Co., Ltd., Shanghai, China) until the solution was colorless and dry. After mixing, the concentration of zinc ion was detected by a PerkinElmer NexION™ 300X ICP-MS (Norwalk, CT, USA).
Western Blot Analysis
Western blot analysis was carried out according to procedures as previously described.30 Briefly, the total proteins from tissues or cells were extracted by radioimmunoprecipitation assay buffer containing PMSF and protease inhibitors. The protein concentration was determined by bicinchoninic acid method. Subsequently, the sample's protein was denatured and subjected to sodium dodecyl sulfate polyacrylamide gels. After transferring the protein to polyvinylidencedifluoride (PVDF) membrane, the membrane was blocked in 5% non-fat milk and incubated with primary antibodies (SLC7A11, 1:500; VDAC3, 1:500; GPX4, 1:500, p-JNK, 1:1000; p-p38, 1:1000; p-ERK1/2, 1:1000) overnight at 4 °C. The membrane was incubated with secondary antibodies at room temperature for 1 h. The bands were visualized by enhanced chemiluminescence reagents with a Molecular Imager Gel Doc XR System (Bio-Rad, Hercules, CA, USA) and analyzed by Image J software (NIH, Bethesda, MD, USA).
Quantitative PCR Assay
Quantitative PCR assay was performed according to the protocols described by Tang et al.4 Briefly, total RNA was extracted by TRizol reagent (KeyGen Biotech, Nanjing, China) according to the manufacturer’s instruction. cDNA was then synthesized with PrimeScript™ RT Master Mix (Perfect Real Time) obtained from TAKARA Co., Ltd. (Tokyo, Japan). Quantitative RT-PCR was conducted by SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) on CFX Connect™ Real-Time System (Bio-Rad, Hercules, CA, USA). The specific primers were synthesized by Sangon Biotechnology Co., Ltd. (Shanghai, China) and are shown in Supplementary Table 1. PCR reactions were carried out under the conditions: 95 °C for 3 min, followed by amplification in 40 cycles of 95 °C for 1030 s at 55 °C for annealing, using the CFX Connect™ Real-Time System (Bio-Rad, Hercules, CA, USA).
Cell Culture
The PC-12 Adh (ATCC CRL-1721.1TM) cell line was obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were cultured in high-glucose DMEM supplemented with 10% (v/v) FBS, 100 units/mL penicillin and 100 µg/mL streptomycin. Cells were maintained in 25-cm2 culture flask in a humidified incubator at 37 °C supplied with 5% CO2.
Measurement of Cell Viability
Cell viability was determined by using the CCK-8 assay according to protocols described previously.30 Briefly, cells were seeded into 96-well plates and then treated with designed concentration of DTPA, DFO, Fer-1, SP600125 or/and ZnONPs for 24 h. Subsequently, the cells were incubated with a CCK-8 solution at 37 °C in 5% CO2 for 4 h in the dark. The absorbance was measured at a wavelength of 490 nm by a micro-plate reader (ELX808, Bio-Tek Instruments, VT, USA). Relative cell viability was calculated and adjusted as the percentage of untreated cells.
Measurement of Intracellular Zinc Concentration
Zinc concentration in the cell was detected by FluoZin-3, AM probe using fluorescence-activated cell sorting analysis (FACS). FACS was carried out according to the protocols described previously.31 Briefly, cells were seeded into six-well plates and treated with designed concentration of DTPA and/or ZnONPs. The probe was added into the cells at the final concentration of 500 nM and incubated in the dark for 30 min. Subsequently, cells were washed by the complete medium followed by an additional incubation for 30 min in the dark. FACS analysis were performed on Becton Dickinson (BD) Influx Cell Sorter (BD Biosciences, San Jose, CA, USA) and the results were analyzed by BD FACS Software (San Jose, CA, USA).
Statistical Analysis
All experimental results were reported as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) followed by Tukey’s honestly significant different post-hoc test was applied. If the original data were in the face of heterogeneity of variance, a non-parametric Kruskal–Wallis test was used. Statistical analysis was performed using GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA), and statistical significance was set at P< 0.05.
Results
Pulmonary Exposure to ZnONPs Caused the Reduced Number of Neurons in Mouse Cerebral Cortex
After intratracheally administration with low- (3 μg/animal), medium- (6 μg/animal) or high- (12 μg/animal) dose ZnONPs, the cerebral cortex tissues of mice were collected on days 1, 3, and 7. H&E staining was used to observe morphological changes of cerebral cortex. The results illustrated that treatment of ZnONPs reduced the number of neuronal cells, and caused the aberrant shape, shrunken or fractured nucleus of cells in the cerebral cortex. Moreover, these pathological changes last for 7 days. These results indicated that intratracheal administration of ZnONPs led to the loss of neuronal cells in the cerebral cortex (Figure 1). In our previous work, we demonstrated that ZnONPs-induced lung damage might diminish gradually to the normal level with time on post-exposure days 7.8 Based on these findings, the time point at day 3 after ZnONPs exposure was chosen for the following designed experiments.
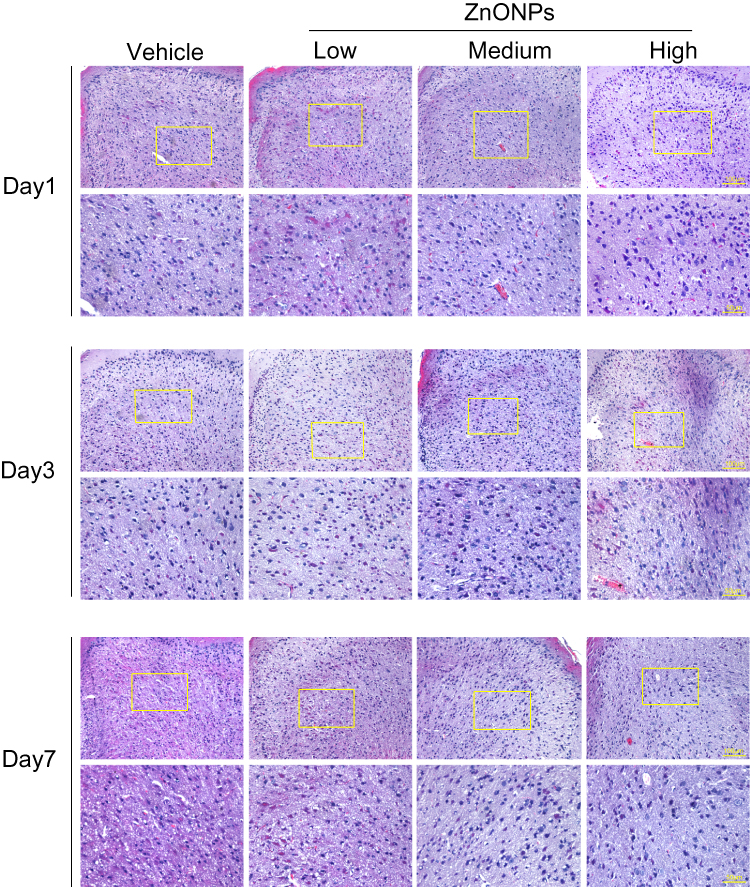 |
Figure 1 Pulmonary exposure to ZnONPs caused the pathological changes in mouse cerebral cortex. After a single treatment of 3, 6, 12 μg/animal ZnONPs via tracheal instillation, the cerebral cortex tissues were collected at post-exposure days 1, 3 and 7. H&E staining was used to observe the morphological changes in the cerebral cortex (n= 3). Scale bar = 100 μm in original figures or 50 μm in zoom-in figures. |
To verify the loss of neuronal cells induced by ZnONPs in the H&E staining, the specific neuronal protein marker, NeuN, was determined by immunofluorescence assay. The data showed that the fluorescent intensities were remarkably reduced after ZnONPs treatment as compared with the controls, suggesting that ZnONPs gradually decreased number of NeuN-positive neurons with increasing concentrations (Figure 2A). Because the reduction of NeuN labeling in the brain may be affected by depletion of the protein or loss of its antigenicity,32 expression of another neuronal marker, MAP2, was assessed. Results demonstrated that the average fluorescence intensities of MAP2-labeled cells also sharply declined in a dose-dependent manner in ZnONPs-treated mice (Figure 2B). Ki67 is a marker of cell proliferation, the initial step in the process leading to neurogenesis in the adult brain.33 To investigate the effect of ZnONPs on the adult neurogenesis, we further tested the expression of Ki67 in the cerebral cortex. Our results showed that ZnONPs caused a decrease in the number of Ki67-positive neurons (Figure 2C), indicating that the neurogenesis might be affected by ZnONPs treatment. Taken together, these findings suggest that pulmonary inhaled ZnONPs can translocate from lung to brain via systemic translocation and then cause significant reduction on the number of neurons in the cerebral cortex of mice.
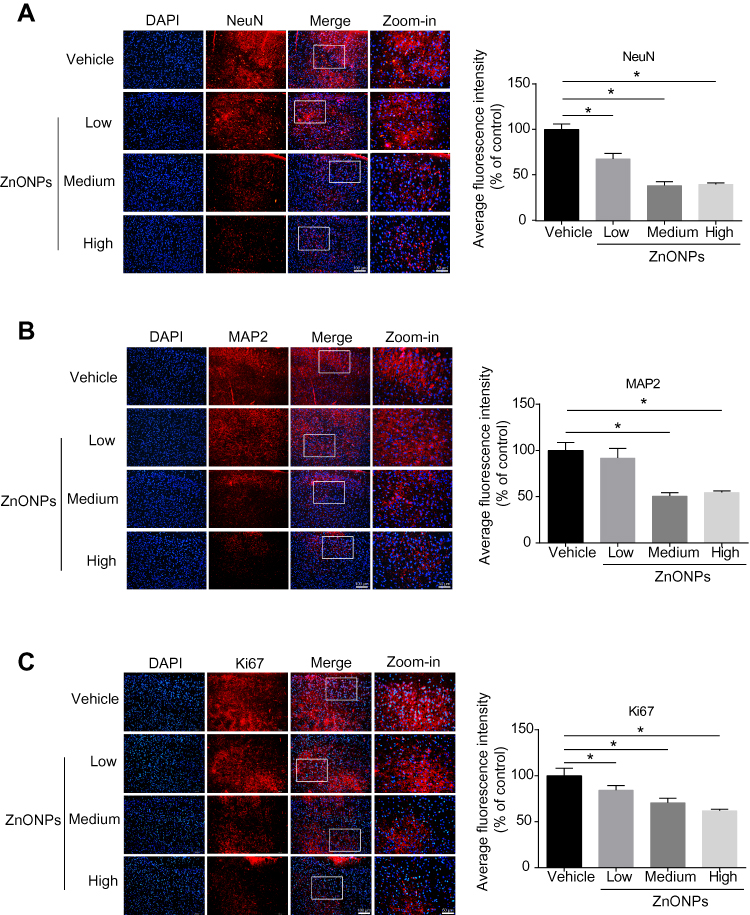 |
Figure 2 Pulmonary exposure to ZnONPs caused the reduced number of neurons in mouse cerebral cortex. After a single treatment of 3, 6, 12 μg/animal ZnONPs, cerebral cortex tissues were collected for immunofluorescence assay at post-exposure day 3. (A–C) Representative images obtained from immunofluorescence reflecting NeuN, MAP2 and Ki67 expressions in mouse cortex tissue. Average fluorescence intensities of NeuN, MAP2 and Ki67 were analyzed by Image-Pro Plus image analysis. Scale bar = 100 μm in original figures or 50 μm in zoom-in figures. Data were derived from at least three independent experiments and were reported as mean ± SD. *Denoted P< 0.05, compared with the vehicle control. |
Pulmonary Exposure to ZnONPs Resulted in Ferroptosis-Related Mitochondrial Oxidative Damage in Mouse Cerebral Cortex
The process of ferroptosis is typically characterized by mitochondrial dysfunction and lipid peroxidation derived from iron excess.3 Therefore, to test whether ZnONPs exposure triggers ferroptotic cell death in mouse cerebral cortex, ultra-morphological changes of mitochondria were observed by TEM. Our results showed that mitochondrial cristae were significantly reduced or vanished with severe vacuolar lesions, and the density of the mitochondrial membrane was partially condensed (Figure 3A). As shown in Figures 3B and C, the data illustrated the ATP contents were sharply reduced while the levels of iron within the tissues were significantly enhanced in response to ZnONPs exposure. Given MDA is a typical and sensitive indicator for lipid peroxidation, its contents were further detected after different doses of ZnONPs treatment. We ascertained that ZnONPs were capable of increasing the accumulation of lipid peroxidation products (Figure 3D). The protein expressions of GPX4 and SCL7A11, both of which were key regulators and used for reflecting ferroptosis, showed significant decrease of their expressions as compared with vehicle controls (Figures 3E and F). On the contrary, the opposite trends were observed in the protein expressions of VDAC3, which was found to be involved in the induction of ferroptosis (Figures 3E and F). These findings together indicate that pulmonary ZnONP exposure can result in iron accumulation and cause ferroptosis-related mitochondrial oxidative damage in the mouse cerebral cortex.
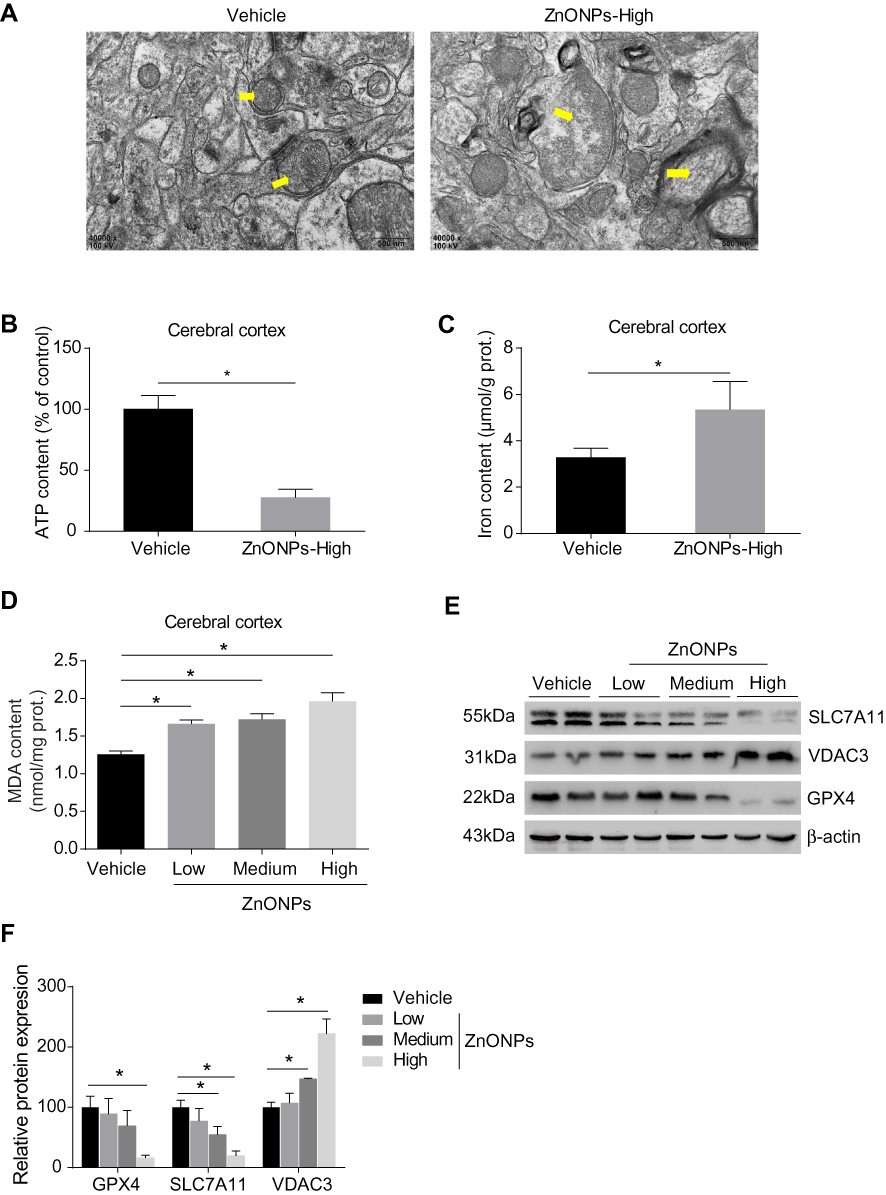 |
Figure 3 Pulmonary exposure to ZnONPs resulted in ferroptosis-related mitochondrial oxidative damage in mouse cerebral cortex. After treatment of ZnONPs, cerebral cortex tissues were collected for designed experiments at post-exposure day 3. (A) The ultrastructure changes on the mitochondria were observed by TEM. The yellow arrows showed the representative mitochondria. Scale bar=500 nm. (B–D) The total ATP, iron, and MDA contents in the cerebral cortex of mouse were determined by commercial kits. (E) Representative Western blot reflecting ferroptosis-related protein levels in mouse cerebral cortex tissues. (F) The protein expression of GPX4, VDAC3 and SLC7A11 obtained from at least three independent experiments. β-actin was served as loading control. Data were derived from at least three independent experiments and were reported as mean ± SD. *Denoted P< 0.05, compared with the vehicle control. |
Pulmonary Exposure to ZnONPs Increased the Level of Zinc in the Mouse Cerebral Cortex
Because of their small size, inhaled ZnONPs have been found to penetrate the blood–air barrier and the blood–brain barrier, thus translocating to the brain by systemic circulation via blood.10,19,21 Indeed, in this study, by using ICP-MS analysis, our results observed that the level of zinc in the cerebral cortex was significantly increased in a dose-dependent manner, although a low concentration of ZnONPs did not cause a statistical change of the level of zinc in mouse cerebral cortex (Figure 4A). Metallothioneins (MTs) are small, cysteine-rich proteins that can bind zinc and act as a zinc reservoir. The metallothionein 2 (Mt2) isoform is predominantly found in the brain. At physiological conditions, zinc is terminally bound to the cysteine thiols of Mt2, making bead-like structures, while the expression of Mt2 can be elevated in response to an increased level of zinc.34,35 To further verify the results obtained from ICP-MS analysis, we detected the mRNA expression of Mt2 after ZnONPs treatment. Our results showed that the mRNA expression of Mt2 was significantly enhanced in ZnONPs-treated mice in a related dose-dependent manner (Figure 4B). These findings indicate that pulmonary ZnONPs exposure increased the level of zinc in the cerebral cortex.
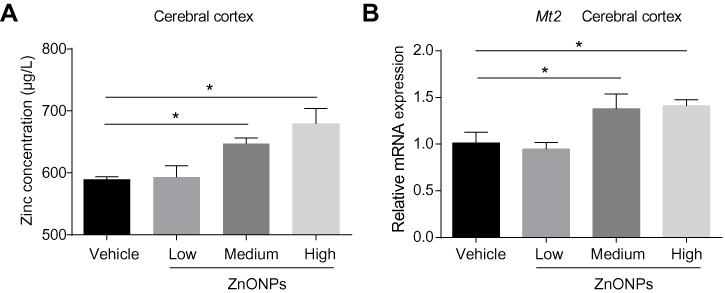 |
Figure 4 Pulmonary exposure to ZnONPs increased the level of zinc in the mouse cerebral cortex. After treated of animals with ZnONPs, cerebral cortex tissues were collected at post-exposure day 3. (A) The level of zinc was measured by using ICP-MS. (B) The mRNA expression of Mt2 was determined by qPCR assay. Data were derived from at least three independent experiments and were reported as mean ± SD. *Denoted P< 0.05, compared with the vehicle control. |
ZnONPs Reduced Cell Viability via Increasing Concentrations of Zinc in PC-12 Cells
Because the enhancement of zinc was observed in the cerebral cortex, we further determined that the role of zinc in ZnONPs induced a reduced number of neuronal cells by using an in vitro culture system. First, we performed a CCK-8 assay to obtain the dose-related cytotoxic effects of ZnONPs on PC-12 cells. Our data showed 30 μg/mL ZnONPs treatment for 24 h significantly reduced cell viability (Figure 5A). Based on these results, this dose was chosen for the further designed experiments. Second, DTPA (200 μM), a potent metal chelator to exchange zinc for a metal of greater binding capacity, was applied to test the role of zinc in the regulation of neuron-like cell viability. Our results demonstrated that DTPA co-treatment with ZnONPs did not cause any cytotoxic effects in PC-12 cells, although DTPA itself could trigger inhibition of cell viability (Figure 5B). On the one hand, these results suggest a reduction of zinc concentration may lead to a significant decrease on cell viability; on the other hand, the obtained data also indicate that chelating with zinc by DTPA can attenuate the cytotoxicity of ZnONPs. As illustrated in Figure 5C, single ZnONPs exposure remarkably elevated the transcriptional Mt2 mRNA level compare d with untreated cells, whereas this enhancement of Mt2 mRNA expression could be relieved by co-treatment with DTPA. A similar phenomenon was observed on the intracellular zinc concentration in PC-12 cells (Figure 5D). These findings indicate that ZnONPs reduce cell viability by increasing zinc concentration within PC-12 cells.
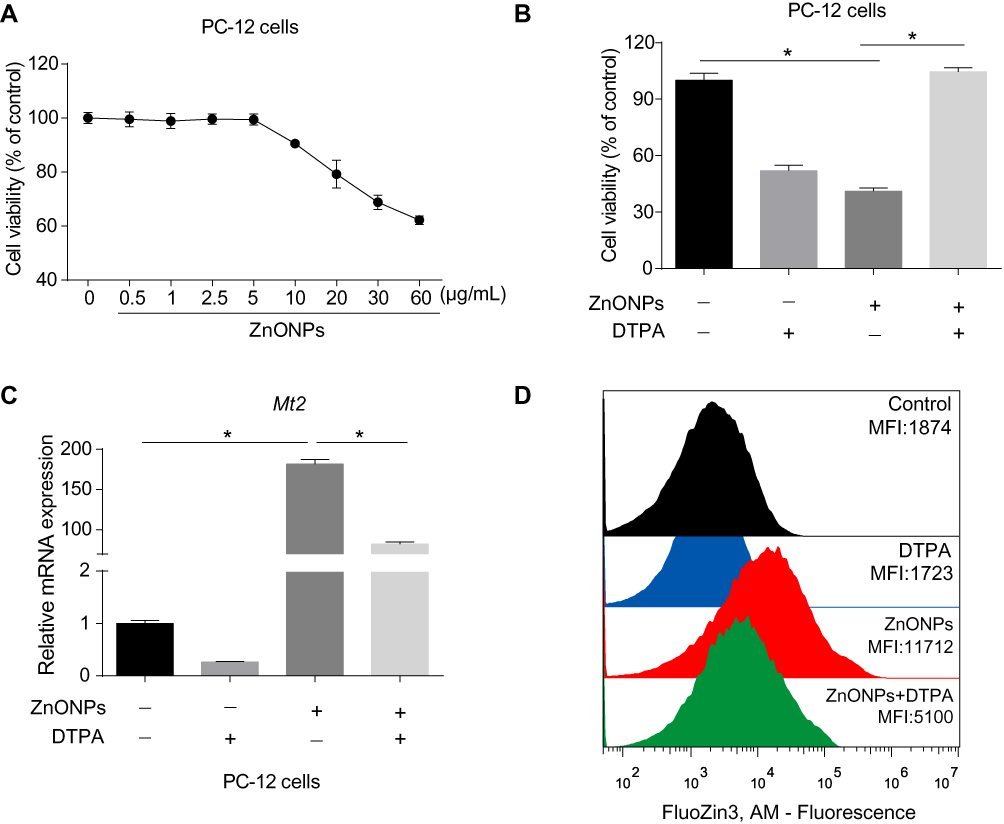 |
Figure 5 ZnONPs reduced cell viability via increasing concentrations of zinc in PC-12 cells. Cells were treated with various concentrations of ZnONPs ranged from 0 to 60 μg/mL for 24 h. (A) Cell viability was detected by CCK-8 assay. (B) The effect of zinc chelator DTPA in regulation of PC-12 cell viability in absence and presence of ZnONPs. (C) Effects of DTPA on regulation of Mt2 mRNA expression in absence and presence of ZnONPs treatment. Zinc concentration in the cell was detected by FluoZin-3, AM probe using fluorescence-activated cell sorting analysis, and the effects of DTPA on regulation of intracellular zinc concentration are shown in (D). Data were derived from at least three independent experiments and were reported as mean ± SD. *Denoted P< 0.05. |
ZnONPs Triggered Ferroptotic Cell Death in Cultured PC-12 Cells
Pulmonary exposure to ZnONPs was capable of inducing ferroptosis-related mitochondrial oxidative damage in mouse cerebral cortex. To further verify the involvement of ferroptosis in ZnONPs-triggered cell death, a selective inhibitor of ferroptosis, Fer-1 (2 μM), was used in the cultured PC-12 cells. Our data found the reduction of cell viability induced by ZnONPs was significantly relieved when co-treated with Fer-1 (Figure 6A). As the ferroptotic cell death typically occurred via the production of excess intracellular iron, the iron chelator deferoxamine (DFO, 100 μM) was used to test if ZnONPs-triggered cell death was initiated in an iron-dependent manner. Indeed, our results illustrated that, after co-treatment with DFO for 24 h, the cell viability could return to near normal levels, suggesting ZnONPs-induced cell death could be rescued by chelating iron (Figure 6B). These findings were confirmed by detection of ferroptosis-related protein expressions. As depicted in Figures 6C and D, the protein expression levels of GPX4 and SCL7A11 were markedly decreased by ZnONPs in a dose-dependent manner. Importantly, the reduced protein expressions of these two proteins could be ameliorated by co-treatment of DFO or Fer-1 (Figures 6E and F). A similar trend was also observed in the determination of MDA, a classic lipid peroxidation product, showing that Fer-1 co-treatment significantly reduced the enhanced content of MDA induced by ZnONPs (Figures 6G and H). These findings together suggest ferroptosis is involved in ZnONP-triggered cell death in PC-12 cells.
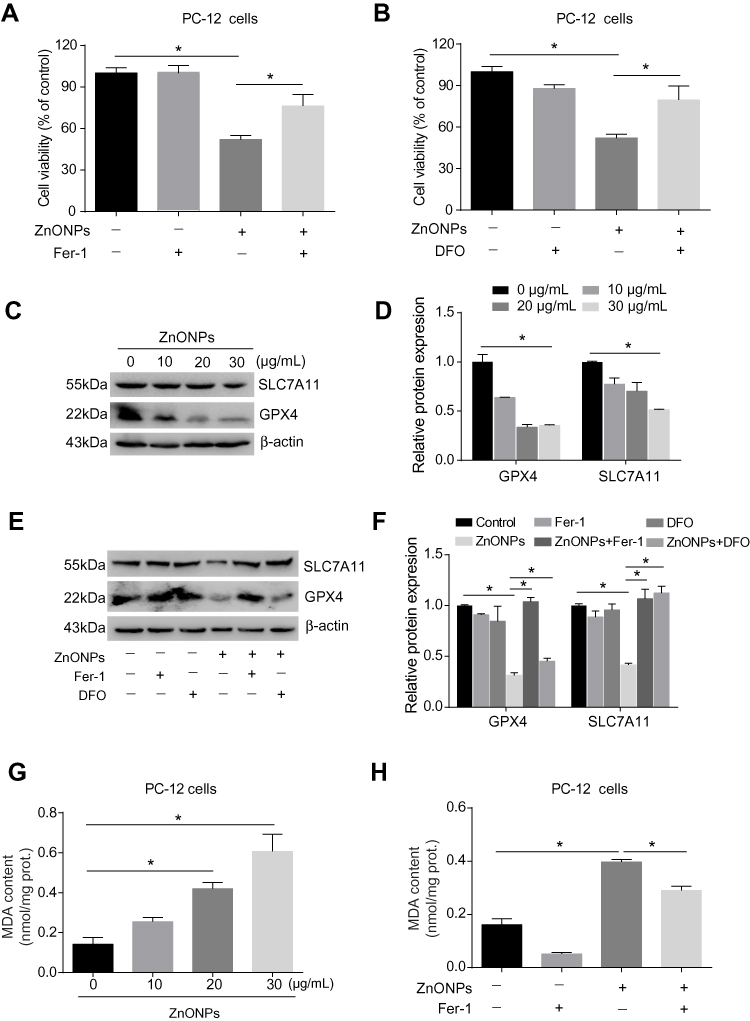 |
Figure 6 ZnONPs triggered ferroptotic cell death in cultured PC-12 cells. Cells were treated with 2 μM Fer-1 and 100 μM DFO in absence and presence of ZnONPs. The cell viability was evaluated by using CCK-8 assay, and the results are shown in (A) and (B), respectively. The protein expressions of ferroptosis-related markers were assessed by Western blot analysis. (C) Representative Western blot reflecting GPX4 and SCL7A11 protein levels in PC-12 cells. β-actin was served as loading control. (D) Relative protein expression of GPX4 and SLC7A11 obtained from at least three independent experiments. (E,F) Effects of Fer-1 and DFO on the protein expressions of ferroptosis-related markers, both of which were detected by Western blot. (G) Effects of ZnONPs on the contents of MDA. (H) Effects of Fer-1 on the content of MDA in absence and presence of ZnONPs. Data were derived from at least three independent experiments and were reported as mean ± SD. *Denoted P< 0.05. |
Fer-1 Inhibited ZnONPs-Induced Ferroptotic Cell Death in Mouse Cerebral Cortex
To further verify the results obtained in PC-12 cells, we pre-treated mice with Fer-1 (10 mg/kg) by intraperitoneal injection 1 h before ZnONPs administration. The method of pre-treatment of Fer-1 had been reported to exhibit better antagonistic effects against ferroptosis than either post-treatment or co-treatment.36 In this study, similar protective effects of Fer-1 against ZnONPs-induced ferroptotic cell death were also observed. Our results showed that pre-treatment of Fer-1 significantly elevated the protein expressions of GPX4 and SLC7A11 in ZnONPs-treated mice (Figure s 7A and B). Similarly, the enhanced MDA contents in ZnONPs-treated mice were significantly reduced by Fer-1 (Figure 7C). However, we did not find any changes in the mRNA expressions of Mt1, Mt2, Mt3 and Mt4 in the ZnONP-exposed mice pre-treated with Fer-1 when compared with single ZnONPs treated mice (Figure 7D). These results indicate that Fer-1 could inhibit ferroptosis in the cerebral cortex but might not influence the release of zinc ions.
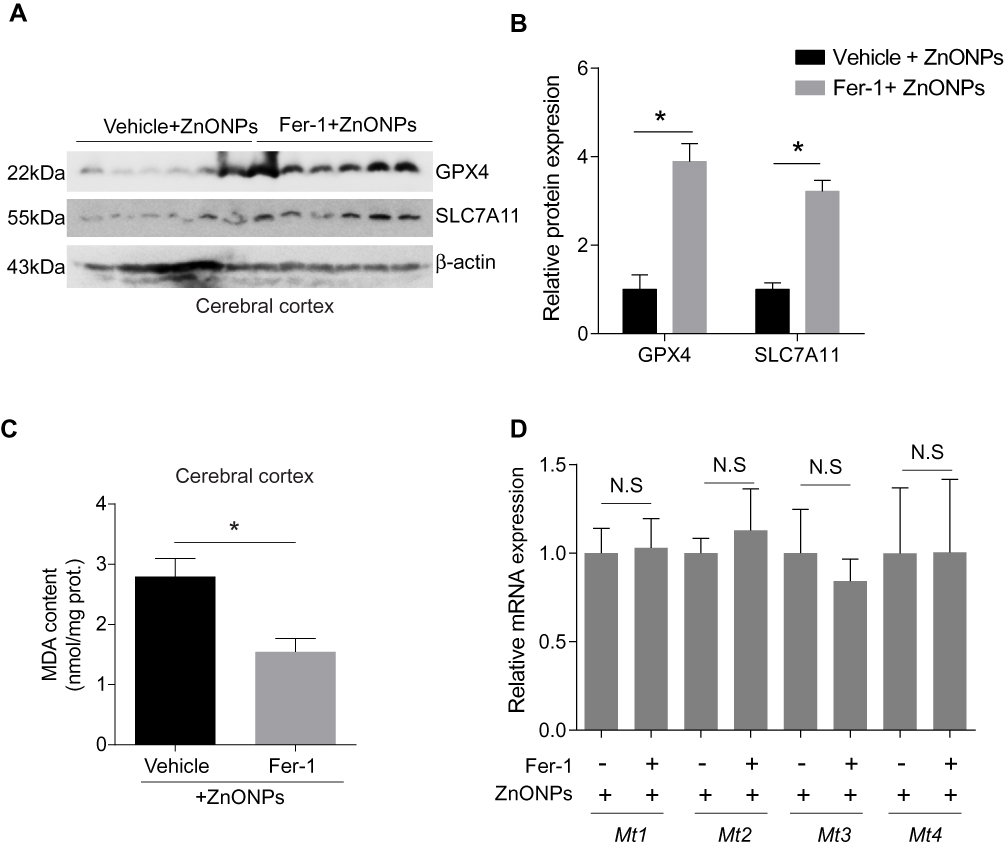 |
Figure 7 Fer-1 inhibited ZnONPs-induced ferroptotic cell death in mouse cerebral cortex. The animals were pre-treated with 10 mg/kg Fer-1 by intraperitoneal injection 30 min before ZnONPs administration (n= 6). The protein expressions of GPX4 and SCL7A11 were detected by Western blot. (A) Representative Western blot reflecting ferroptosis-related protein levels in mouse cerebral cortex tissues. (B) Relative protein expression of GPX4 and SLC7A11 obtained from at least three independent experiments. β-actin was served as loading control. (C) The MDA contents were measured by using commercial kits. (D) The mRNA expression of Mt1, Mt2, Mt3 and Mt4 were assessed by qPCR assay. Data were derived from at least three independent experiments and were reported as mean ± SD. *Denoted P< 0.05. |
Activation of c-Jun NH2-Terminal Kinases (JNK) Pathway Involved in ZnONPs-Induced Ferroptotic Cell Death
Activation of the mitogen-activated protein kinase (MAPK) pathway has been revealed to contribute to ferroptotic cell death.3 The MAPK pathway is mediated by extracellular signal-regulated kinase (ERK), JNK, and p38 protein kinases.37 After treatment with ZnONPs, we found the protein expressions of p-p38 did not show any significant changes in cerebral cortex tissues as compared with vehicle controls (Figure 8A). The protein expressions of p-ERK1/2 and p-JNK were both enhanced in ZnONPs-treated mice, but the expression of p-JNK showed greater enhancements than that of p-ERK1/2 (Figure 8A). Consistently, ZnONPs remarkably increased the level of p-JNK in a dose-dependent manner in cultured PC-12 cells (Figure 8B). To further verify the role of activation of JNK involved in ZnONPs-triggered ferroptosis, the specific inhibitor of p-JNK, SP600125 (10 μM), was used. Our results illustrated that inhibition of the JNK pathway not only significantly reduced the increased contents of MDA (Figure 8C), but also markedly reversed cell death in ZnONPs-treated PC-12 cells (Figure 8D). These findings suggest that activation of the JNK pathway may be involved in ZnONPs-induced ferroptotic cell death.
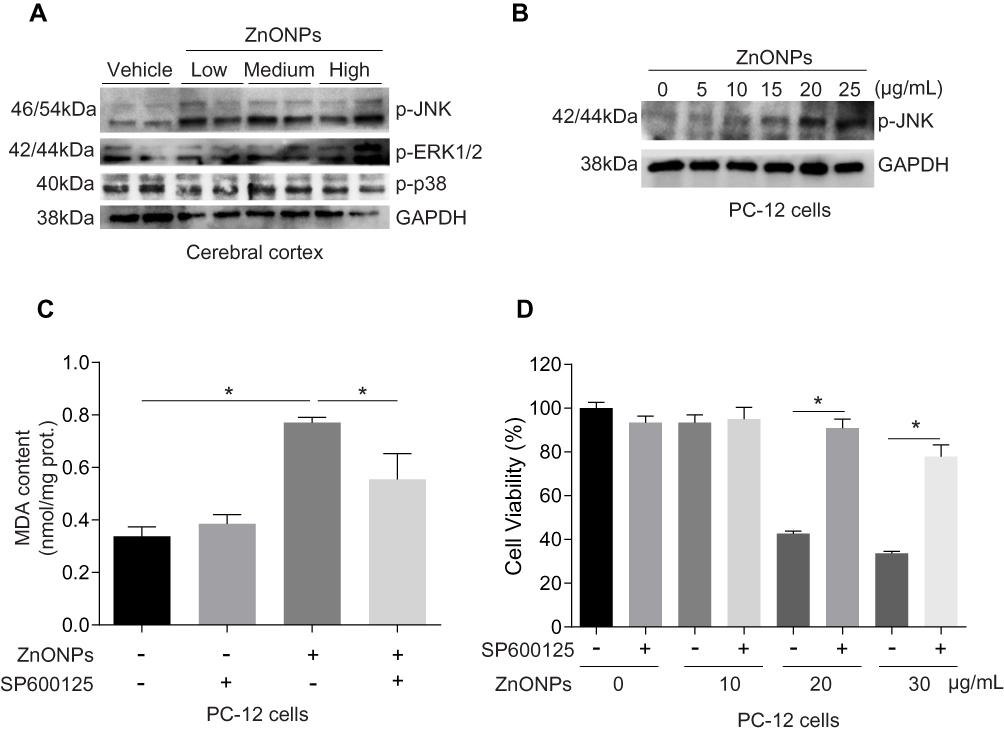 |
Figure 8 Activation of JNK pathway involved in ZnONPs-induced ferroptotic cell death. After treatment of mice with ZnONPs, cerebral cortex tissues were collected at post-exposure day 3. The protein expressions of p-JNK, p-ERK1/2 and p-p38 were assessed by Western blot. (A) Representative Western blot reflecting p-JNK, p-ERK1/2 and p-p38 protein levels in mouse cerebral cortex tissues. PC-12 cells were treated with different concentrations of ZnONPs (0, 5, 10, 20 and 25 μg/mL) and the protein expression of p-JNK was determined. (B) Representative Western blot reflecting p-JNK protein levels. GAPDH was served as loading control. SP600125 was a specific inhibitor of p-JNK. After treatment of SP600125 (10 μM) with or without ZnONPs, the MDA contents and cell viability were measured. (C) Effects of SP600125 on the contents of MDA in absence and presence of ZnONPs. (D) Effects of SP600125 on the cell viability in absence and presence of ZnONPs. Data were derived from at least three independent experiments and were reported as mean ± SD. *Denoted P< 0.05. |
Discussion
Inhalation is the main and typical route of entry to human for ZnONPs exposure in workplaces. This occurs mostly by releasing zinc oxide fumes during industrial activities, such as thermal cutting, welding of steel and melting, etc.38 Such exposure of ZnONPs via airway may result in metal fume fever in workers. The systemic inflammatory effects of ZnONPs inhalation further cause permanent pathological changes in different organs, including the brain.38–40 In the present study, to ensure consistent amounts of inhaled nanoparticles per animal, a single intratracheal instillation of ZnONPs was used. Because the bioavailability of metal oxide nanoparticles is often related to the ability of nanoparticles to deliver soluble metal ions to adjacent tissues,41 the increased levels of zinc ion and Mt2 expression observed in the mouse cerebral cortex tissues may suggest administration of ZnONPs by this way spray directly into the airway tract of animals and facilitate their absorption and distribution. Intriguingly, we also found that the pathological changes in cortex tissues at day 7 were not more serious than that at day 3 after ZnONPs treatment. Similar trends were also found in lung tissues,8 and this manifestation may be due to metabolism or excretion of nanoparticles via urine or feces with increasing time.
Growing evidence has demonstrated that ZnONPs are capable of reaching the brain because of their extremely small size.10,21,42 There are two explanations for the accumulation of inhaled ZnONPs in the brain. First, ZnONPs that are small enough can reach the lower airways and gain access to the air–blood barrier, which is the most permeable barrier in the body. ZnONPs may translocate across the air–blood barrier permitting systemic access to extra-pulmonary organs.43 They are also able to penetrate the blood–brain barrier directly by transcytosis and consequently produce neuronal injury.44 Second, inhaled ZnONPs reach the brain indirectly via an olfactory bulb–brain translocation pathway.42,45 In this study, we found administration of ZnONPs by intratracheal instillation caused the pathological changes in the cerebral cortex tissues, mainly manifested by a reduced number of neuronal cells. These results partially indicate that pulmonary exposed ZnONPs can aggregate in the brain via the above two ways to reach the central nervous system where neurotoxic effects could develop. In addition, it is now well-documented that small-sized nanoparticles (less than 50 nm) produce greater neuronal injury than larger ones.42 A previous report illustrated that nanoparticles with approximate size of 50–60 nm were able to disrupt the blood–brain barrier of an in vivo system, therefore leading to the brain pathology.46 Similarly, we observed pathological changes in the brain when mice were treated with ZnONPs under 50 nm particle size. Some studies have reported the opposite results. For instance, Deng et al. revealed no size-dependent cytotoxic effects of ZnONPs on neural stem cells after a 24 h short-term exposure.23 This discrepancy implies in vitro studies may lead to results that do not correspond to circumstances occurring in a living organism. Considering this potential discrepancy between in vivo and in vitro, in this study, we used both animals and cell models to test whether ZnONPs triggered ferroptosis in neuronal cells.
Fer-1 is a potent and selective inhibitor of ferroptosis that effectively slows the accumulation of lipid hydroperoxides and thereby inhibits cell death.47 Indeed, in this study, Fer-1 obviously inhibited the phenotype of ferroptosis induced by ZnONPs in both cultured PC-12 cells and animals. It is noteworthy that we did not find any significant alterations on the mRNA expressions of Mt1, Mt2, Mt3, Mt4, although zinc ion chelation by DTPA evidently ameliorated the cytotoxicity of ZnONPs. On the one hand, these results signify the protective effects of Fer-1 against ferroptosis are not associated with the level of zinc ion within the cells. On the other hand, it is believed that the cytotoxicity of ZnONPs may occur as a result of release of zinc ions in the biological micro-environment. Similar to other metal nanoparticles, zinc ions play a critical role in ZnONPs cytotoxicity, despite the effects of the intact nanoparticle form which cannot be excluded.48–51 Intriguingly, some epidemiological investigations have revealed that iron and zinc may interact in the body and thereby decrease the efficacy of iron supplementation.52,53 Moreover, zinc also has an inhibitory effect on iron absorption.54 These results together suggest that the elevated concentration of zinc ions in the brain may be a risk factor for the initiation of iron-dependent cell death.
Both iron metabolism and lipid peroxidation are recognized as central mediators in the initiation and execution of ferroptosis.3 However, cellular sensitivity to ferroptosis is also modulated by several other pathways, such as production of coenzyme Q10, upregulation of endoplasmic reticulum stress and activation of MAPKs pathways, etc.55,56 Among these pathways, MAPK cascades are the vital signaling pathway that regulate diverse cellular programs, including cell proliferation, differentiation, development and cell death.57 In mammals, there are more than a dozen MAPK enzymes, and the best-known conventional enzymes are ERK, p38 and JNK.57 The role of MAPKs in the regulation of ferroptosis has been revealed for several years. Blocking the ERK pathway inhibited ferroptosis in Ras-mutated cancer cells, but JNK and p38 appeared to be important in leukemia cells.55,58 Inhibition of JNK phosphorylation significantly decreased the cytotoxicity induced by ferroptosis inducers in leukemia cells.58 These results indicate that MAPKs may serve in a cell type-specific way in the process of ferroptosis. In this study, we observed a similar phenomenon in the cultured PC-12 cells, which originated from a pheochromocytoma of the rat adrenal medulla with neuronal phenotype. Our results showed ZnONPs markedly enhanced the level of p-JNK expression, but they did not activate the p-ERK and p-p38 pathway significantly. Inhibition of p-JNK by its specific agent not only relieved the overproduced lipid peroxidation product, but also reduced the cytotoxicity of ZnONPs. These findings confirm the notion that the MAPK pathway is tightly linked to the initiation of ferroptosis, but the activation of specific MAPK pathways may vary depending on the cell types, environmental stimuli or drugs.
There are several limitations regarding this study that should be considered. First, the toxicity of ZnONPs may depend on the duration of exposure time, the size of nanoparticle and the dose used, etc.10 In this study, we did not treat mice with ZnONPs for a long time based on the following two reasons. One was that the beneficial effects of a ferroptosis-specific inhibitor might not last for a long time because of drug metabolism. The other was because the toxicity of ZnONPs had been found to recover as time went by.8 Second, Fer-1 usually acts as a lipid ROS scavenger, and the inhibited effects of Fer-1 against ferroptosis may be affected by the other potential mechanisms, such as anti-oxidative effects. Third, although ferroptosis is distinct from canonical apoptosis, autophagy and necroptosis, we did not exclude whether airway exposure to ZnONPs initiated the other forms of cell death. Also, we even did not know which form of cell death dominates the cytotoxicity of ZnONPs for the neurons. Do these forms of cell death all or partially exist in response to ZnONPs exposure? Is ferroptosis independent of other known cell death pathways? These questions and related mechanisms remain unsolved and need further investigation.
In summary, in this study, we demonstrated for the first time that pulmonary exposure to ZnONPs triggered ferroptosis in the neuronal cells of mouse cerebral cortex. Similar phenomena were also observed in cultured PC-12 cells. Importantly, our results showed that inhibition of ferroptosis could alleviate ZnONPs-induced neuronal cell death both in vivo and in vitro. The molecular findings from this study indicated that ZnONPs selectively activated the JNK pathway and therefore resulted in ferroptotic phenotypes (Figure 9). These results will provide the novel evidence that inhibition of ferroptosis by specific agents or drugs may be the potential strategy for reducing untreatable neurotoxicity induced by ZnONPs airway exposure.
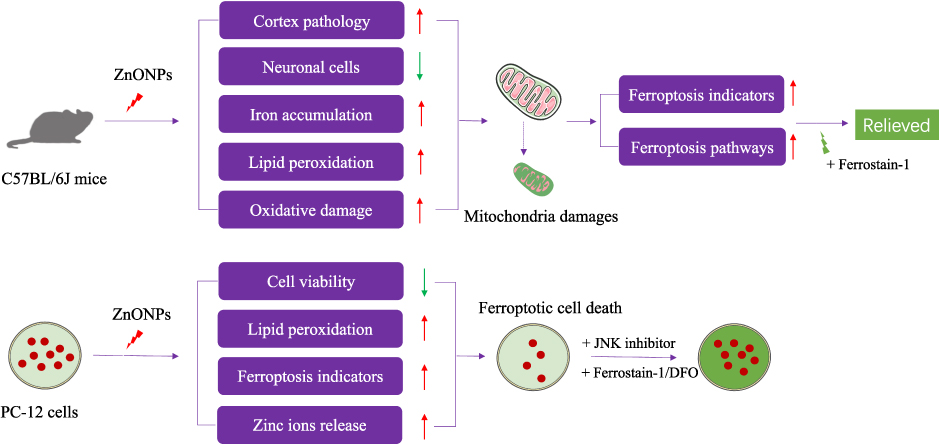 |
Figure 9 The experimental design and main results of this study are summarized. |
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no competing interests.
References
1. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
2. Dixon SJ, Stockwell BR. The hallmarks of ferroptosis. Annual Rev Cancer Biol. 2019;3(1):35–54. doi:10.1146/annurev-cancerbio-030518-055844
3. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26(3):165–176. doi:10.1016/j.tcb.2015.10.014
4. Tang Q, Bai L, Zou Z, et al. Ferroptosis is newly characterized form of neuronal cell death in response to arsenite exposure. Neurotoxicology. 2018;67:27–36. doi:10.1016/j.neuro.2018.04.012
5. Kim SE, Zhang L, Ma K, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11(11):977. doi:10.1038/nnano.2016.164
6. Shen Z, Liu T, Li Y, et al. Fenton-reaction-acceleratable magnetic nanoparticles for ferroptosis therapy of orthotopic brain tumors. ACS Nano. 2018;12(11):11355–11365. doi:10.1021/acsnano.8b06201
7. Srivastava V, Gusain D, Sharma YC. Synthesis, characterization and application of zinc oxide nanoparticles (n-ZnO). Ceramics Int. 2013;39(8):9803–9808. doi:10.1016/j.ceramint.2013.04.110
8. Jiang X, Tang Q, Zhang J, et al. Autophagy-dependent release of zinc ions is critical for acute lung injury triggered by zinc oxide nanoparticles. Nanotoxicology. 2018;1–24.
9. Cao Y, Gong Y, Liao W, et al. A review of cardiovascular toxicity of TiO 2, ZnO and Ag nanoparticles (NPs). Bio Metals. 2018;31(4):457–476. doi:10.1007/s10534-018-0113-7
10. Singh S. Zinc oxide nanoparticles impacts: cytotoxicity, genotoxicity, developmental toxicity, and neurotoxicity. Toxicol Mech Methods. 2019;29(4):300–311. doi:10.1080/15376516.2018.1553221
11. Arakha M, Roy J, Nayak PS, Mallick B, Jha S. Zinc oxide nanoparticle energy band gap reduction triggers the oxidative stress resulting into autophagy-mediated apoptotic cell death. Free Radic Biol Med. 2017;110:42–53. doi:10.1016/j.freeradbiomed.2017.05.015
12. Huang K-L, Lee Y-H, Chen H-I, Liao H-S, Chiang B-L, Cheng T-J. Zinc oxide nanoparticles induce eosinophilic airway inflammation in mice. J Hazard Mater. 2015;297:304–312. doi:10.1016/j.jhazmat.2015.05.023
13. Kim D-Y, Kim J-H, Lee J-C, et al. Zinc oxide nanoparticles exhibit both cyclooxygenase-and lipoxygenase-mediated apoptosis in human bone marrow-derived mesenchymal stem cells. Toxicol Res. 2019;35(1):83–91. doi:10.5487/TR.2019.35.1.083
14. Wilhelmi V, Fischer U, Weighardt H, et al. Zinc oxide nanoparticles induce necrosis and apoptosis in macrophages in a p47phox-and Nrf2-independent manner. PLoS One. 2013;8(6):e65704. doi:10.1371/journal.pone.0065704
15. Zhang J, Qin X, Wang B, et al. Zinc oxide nanoparticles harness autophagy to induce cell death in lung epithelial cells. Cell Death Dis. 2017;8(7):e2954e2954.
16. Wang B, Zhang J, Chen C, et al. The size of zinc oxide nanoparticles controls its toxicity through impairing autophagic flux in A549 lung epithelial cells. Toxicol Letters. 2018;285:51–59.
17. Wilson MR, Foucaud L, Barlow PG, et al. Nanoparticle interactions with zinc and iron: implications for toxicology and inflammation. Toxicol Appl Pharmacol. 2007;225(1):80–89. doi:10.1016/j.taap.2007.07.012
18. Ali D, Alarifi S, Kumar S, Ahamed M, Siddiqui MA. Oxidative stress and genotoxic effect of zinc oxide nanoparticles in freshwater snail Lymnaea luteola L. Aquatic Toxicol. 2012;124:83–90. doi:10.1016/j.aquatox.2012.07.012
19. Choi S-J, Choy J-H. Biokinetics of zinc oxide nanoparticles: toxicokinetics, biological fates, and protein interaction. Int J Nanomedicine. 2014;9(Suppl 2):261.
20. Kao -Y-Y, Cheng T-J, Yang D-M, Wang C-T, Chiung Y-M, Liu P-S. Demonstration of an olfactory bulb–brain translocation pathway for ZnO nanoparticles in rodent cells in vitro and in vivo. J Mol Neurosci. 2012;48(2):464–471. doi:10.1007/s12031-012-9756-y
21. Han D, Tian Y, Zhang T, Ren G, Yang Z. Nano-zinc oxide damages spatial cognition capability via over-enhanced long-term potentiation in hippocampus of Wistar rats. Int J Nanomedicine. 2011;6:1453. doi:10.2147/IJN.S25646
22. Valdiglesias V, Costa C, Kiliç G, et al. Neuronal cytotoxicity and genotoxicity induced by zinc oxide nanoparticles. Environ Int. 2013;55:92–100. doi:10.1016/j.envint.2013.02.013
23. Deng X, Luan Q, Chen W, et al. Nanosized zinc oxide particles induce neural stem cell apoptosis. Nanotechnology. 2009;20(11):115101. doi:10.1088/0957-4484/20/11/115101
24. Jeng HA, Swanson J. Toxicity of metal oxide nanoparticles in mammalian cells. J Environ Sci Health Part A. 2006;41(12):2699–2711. doi:10.1080/10934520600966177
25. Wang J, Deng X, Zhang F, Chen D, Ding W. ZnO nanoparticle-induced oxidative stress triggers apoptosis by activating JNK signaling pathway in cultured primary astrocytes. Nanoscale Res Lett. 2014;9(1):117. doi:10.1186/1556-276X-9-117
26. Gosens I, Kermanizadeh A, Jacobsen NR, et al. Comparative hazard identification by a single dose lung exposure of zinc oxide and silver nanomaterials in mice. Plos One. 2015;10:5.
27. Lee H, Park K. In vitro cytotoxicity of zinc oxide nanoparticles in cultured statens seruminstitut rabbit cornea cells. Toxicol Res. 2019;35(3):287.
28. Jacobsen NR, Stoeger T, Van Den Brûle S, et al. Acute and subacute pulmonary toxicity and mortality in mice after intratracheal instillation of ZnO nanoparticles in three laboratories. Food Chem Toxicol. 2015;85:84–95. doi:10.1016/j.fct.2015.08.008
29. Pan C-H, Chuang K-J, Chen J-K, et al. Characterization of pulmonary protein profiles in response to zinc oxide nanoparticles in mice: A 24-hour and 28-day follow-up study. Int J Nanomed. 2015;10:4705.
30. Bai L, Tang Q, Zou Z, et al. m6A demethylase FTO regulates dopaminergic neurotransmission deficits caused by arsenite. Toxicol Sci. 2018;165(2):431–446. doi:10.1093/toxsci/kfy172
31. Zhang J, Qin X, Wang B, et al. Zinc oxide nanoparticles harness autophagy to induce cell death in lung epithelial cells. Cell Death Dis. 2017;8(7):e2954. doi:10.1038/cddis.2017.337
32. Ünal-Çevik I, Kılınç M, Gürsoy-Özdemir Y, Gurer G, Dalkara T. Loss of NeuN immunoreactivity after cerebral ischemia does not indicate neuronal cell loss: a cautionary note. Brain Res. 2004;1015(12):169–174. doi:10.1016/j.brainres.2004.04.032
33. Kee N, Sivalingam S, Boonstra R, Wojtowicz J. The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods. 2002;115(1):97–105. doi:10.1016/S0165-0270(02)00007-9
34. Baltaci AK, Yuce K, Mogulkoc R. Zinc metabolism and metallothioneins. Biol Trace Elem Res. 2018;183(1):22–31. doi:10.1007/s12011-017-1119-7
35. Jayawardena DP, Heinemann IU, Stillman MJ. Zinc binds non-cooperatively to human liver metallothionein 2a at physiological pH. Biochem Biophys Res Commun. 2017;493(1):650–653. doi:10.1016/j.bbrc.2017.08.137
36. Li Q, Han X, Lan X, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight. 2017;2:7. doi:10.1172/jci.insight.90777
37. Soares-Silva M, Diniz FF, Gomes GN, Bahia D. The mitogen-activated protein kinase (MAPK) pathway: role in immune evasion by trypanosomatids. Front Microbiol. 2016;7:183. doi:10.3389/fmicb.2016.00183
38. Monsé C, Hagemeyer O, Raulf M, et al. Concentration-dependent systemic response after inhalation of nano-sized zinc oxide particles in human volunteers. Particle Fibre Toxicol. 2018;15(1):8.
39. Rossner JP, Vrbova K, Strapacova S, et al. Inhalation of ZnO nanoparticles: splice junction expression and alternative splicing in mice. Toxicol Sci. 2019;168(1):190–200.
40. Adamcakova-Dodd A, Stebounova LV, Kim JS, et al. Toxicity assessment of zinc oxide nanoparticles using sub-acute and sub-chronic murine inhalation models. Particle Fibre Toxicol. 2014;11(1):15.
41. Xia T, Kovochich M, Liong M, et al. Comparison of the mechanism of toxicity of zinc oxide and cerium oxide nanoparticles based on dissolution and oxidative stress properties. ACS nano 2. 2008;2(10):2121–2134.
42. Nicolosi A, Cardoit L, Pasquereau P, et al. Acute exposure to zinc oxide nanoparticles critically disrupts operation of the respiratory neural network in neonatal rat. Neurotoxicology. 2018;67:150–160.
43. Poh TY, NAtBM A, Mac Aogáin M, et al. Inhaled nanomaterials and the respiratory microbiome: clinical, immunological and toxicological perspectives. Particle Fibre Toxicol. 2018;15(1):46.
44. Simko M, Mattsson M-O. Interactions between nanosized materials and the brain. Current Med Chem. 2014;21(37):4200–4214.
45. Kao -Y-Y, Cheng T-J, Yang D-M, Wang C-T, Chiung Y-M, Liu P-S. Demonstration of an olfactory bulb–brain translocation pathway for ZnO nanoparticles in rodent cells in vitro and in vivo. J Mol Neurosci. 2012;48(2):464–471.
46. Shim KH, Hulme J, Maeng EH, Kim M-K, An S. Analysis of zinc oxide nanoparticles binding proteins in rat blood and brain homogenate. Int J Nanomed. 2014;9(Suppl 2):217.
47. Zilka O, Shah R, Li B, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Central Sci. 2017;3(3):232–243.
48. Liu J, Feng X, Wei L, Chen L, Song B, Shao L. The toxicology of ion-shedding zinc oxide nanoparticles. Critical Rev Toxicol. 2016;46(4):348–384.
49. Kao -Y-Y, Chen Y-C, Cheng T-J, Chiung Y-M, Liu P-SJT-S. Zinc oxide nanoparticles interfere with zinc ion homeostasis to cause cytotoxicity. Toxicol Sci. 2012;125(2):462–472.
50. Babu Maddinedi S, Mandal BK, Patil SH, et al. Diastase induced green synthesis of bilayered reduced graphene oxide and its decoration with gold nanoparticles. J Photochem Photobio B Biol. 2017;166:252–258.
51. Maddinedi SB, Mandal BK, Anna K. Pharmacology. Environment friendly approach for size controllable synthesis of biocompatible silver nanoparticles using diastase. Environ Toxicol Pharmacol. 2017;49:131–136.
52. Nguyen P, Grajeda R, Melgar P, et al. Effect of zinc on efficacy of iron supplementation in improving iron and zinc status in women. J Nut Metab. 2012;2012.
53. Donangelo CM, Woodhouse LR, King SM, Viteri FE, King J. Supplemental zinc lowers measures of iron status in young women with low iron reserves. J Nut. 2002;132(7):1860–1864.
54. Olivares M, Pizarro F, Gaitán D, Ruz M. Acute inhibition of iron absorption by zinc. Nut Res. 2007;27(5):279–282.
55. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379.
56. Stockwell BR, Angeli JPF, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285.
57. Cargnello M, Roux PPJMMBR. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75(1):50–83.
58. Yu Y, Xie Y, Cao L, et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2015;2(4):e1054549.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.