Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Zinc deficiency as a codeterminant for airway epithelial barrier dysfunction in an ex vivo model of COPD
Authors Roscioli E, Jersmann HPA, Lester S, Badiei A, Fon A, Zalewski P, Hodge S
Received 20 August 2017
Accepted for publication 16 October 2017
Published 5 December 2017 Volume 2017:12 Pages 3503—3510
DOI https://doi.org/10.2147/COPD.S149589
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
Eugene Roscioli,1,2 Hubertus PA Jersmann,1,2 Susan Lester,2,3 Arash Badiei,1,2 Andrew Fon,1,2 Peter Zalewski,2,4 Sandra Hodge1,2
1Department of Thoracic Medicine, Royal Adelaide Hospital, 2Department of Medicine, The University of Adelaide, Adelaide, 3Department of Rheumatology, 4Cardiology Unit, The Queen Elizabeth Hospital, Woodville, SA, Australia
Abstract: There is now convincing evidence that the airway epithelium drives the pathogenesis of COPD. A major aspect of this is the disease-related reduction in barrier function that is potentiated by dysregulation of tight junction (TJ) protein complexes. However, a significant number of studies using in vitro smoke exposure models have not observed alterations in barrier permeability. We have previously shown that zinc (Zn) is an influential cytoprotective factor for the airway epithelium, and its depletion by cigarette smoke produces disease-related modifications consistent with inflammatory changes in COPD. We hypothesized that Zn deficiency is a significant co-stimulus with cigarette smoke extract (CSE) for potentiating the leaky barrier phenotype exhibited in COPD. We employed an ex vivo model of differentiated human airway epithelium exposed to Zn depletion and CSE to determine the contribution of Zn in maintaining normal epithelial permeability. Western blot analysis demonstrated a significant downregulation of the TJ proteins such as ZO-1 (–1.93-fold, P<0.05) and Claudin-1 (–3.37-fold, P<0.01) with the combination exposure. Assessment of barrier function via paracellular ionic conductance and tracer permeability also showed that Zn depletion was an important factor, which potentiated an increase in epithelial permeability (P<0.001 for both) compared to Zn depletion or CSE exposures in isolation. Visual inspection of the epithelium using transmission electron microscopy revealed a marked reduction in junction complexes between the adjacent airway epithelial cells treated with a combination of Zn depletion and CSE. These observations identify Zn deficiency as a significant codeterminant with CSE as a factor leading to an increase in airway epithelial permeability. Hence, as Zn dyshomeostasis has been reported in the airway epithelium exposed to chronic cigarette smoke and inflammation, targeting these phenomena may represent a promising strategy to ameliorate the leaky barrier phenotype that is synonymous with COPD.
Keywords: COPD, airway epithelium, barrier function, cigarette smoke, zinc
Introduction
The complexes that couple and enable signal transfer between airway epithelial cells (AECs), which include tight junctions (TJs), adherens junctions (AJs), and gap junctions (GJs), are essential to maintain and coordinate the mucociliary escalator and selective permeability imparted by the epithelial barrier. It is widely established that during COPD and cigarette smoke exposure, the epithelium is in a state of injury and aberrant repair that is synonymous with the dysregulation of TJ complexes and a leaky barrier phenotype that contributes to exacerbations, disease progression, and airway remodeling.1–3 However, a surprising number of studies that employed in vitro models of cigarette smoke exposure did not find alterations in epithelial permeability or modulation of the expression of TJ proteins such as the Occludins, Zonula Occludens (ZOs), and Claudins.1,4–8 A reason for this may relate to the challenges of modeling a chronic and heterogeneous disease such as cigarette smoke-induced COPD, where important contributing factors that promote epithelial permeability in vivo are not always captured by short-term in vitro systems.
One such factor is the metal ion zinc (Zn). Apart from its essential requirement for proper cellular and protein function, Zn is highly concentrated in the luminal margins of AECs, where it has a number of pro-survival and anti-inflammatory roles.9,10 Further, we have shown dyshomeostasis of Zn regulatory factors in a chronic cigarette smoke-exposed mouse model, where a significant reduction in free Zn within the epithelial compartment, inflammation, and dysregulation of pro-survival activities was noted.11 While at least one report has described a role for Zn in supporting AJs in the context of acute lung injury,12 the greatest understanding has come from studies examining TJ disassembly and aberrant epithelial permeability during Zn deficiency in the context of inflammatory bowel diseases such as Crohn’s and ulcerative colitis.13
In this study, we employed an ex vivo model that demonstrates that Zn deficiency is an influential co-stimulus with cigarette smoke extract (CSE) for potentiating the leaky barrier phenotype exhibited in COPD.
Subjects and methods
COPD-related disease model of human AECs differentiated at an air–liquid interface (ALI)
Ethical approval was obtained for the collection of bronchial cells from the Human Ethics Committee of the Royal Adelaide Hospital, and the experiments were conducted with the understanding and the written consent of each participant. All the participants were nonsmokers recruited from our volunteer database (N=11; four females; median age 46±16.5 years [SD]), who have no history of respiratory or allergic diseases and had normal spirometry. Bronchial brushing was used to collect basal progenitor AECs, which were subsequently propagated at an ALI and exposed to inflammatory cytokines, Zn-depleted conditions, and 10% CSE as previously reported.11,14,15 The timing, and placement (afforded by an apical and basal reservoir of the transwell system) of each exposure, was configured to approximate the fragile epithelium caused by inflammation, cigarette smoke, and Zn-depleted conditions. For the combination exposure, differentiated AEC ALI cultures were treated with Zn-depleted media in the basal chamber for the assay period (64 h), TNFα and IFNγ (both 5 ng/mL) were added to the basal chamber after 24 h (40 h duration), and finally 10% CSE was applied to the apical chamber after 40 h (24 h duration). The placement of the CSE exposure in the apical reservoir (vs both apical and basal) was used to moderate broad cytotoxic effects, because smoke exposure is an environmental toxin that interfaces with the innate protection imparted by the mucus-producing goblet cells and the cilium. Routine microscopic observation showed that the unstimulated epithelial cells were indistinguishable from those subject to the disease-related exposures (including the preservation of motile cilia) in this model, providing evidence that the treatment responses were due to biochemical effects vs excessive and protracted cytotoxicity. To ensure each culture formed an epithelial layer that closely approximates the situation in vivo, cultures included in the study exhibited full mucociliary differentiation (Figure S1), and the formation of cohesive cellular connections between adjacent cells as evidence by transepithelial electrical resistance (TEER) measures (using the EVOM2; World Precision Instruments, Sarasota, FL, USA) that exceed 500 Ω·cm2.
Western blot analysis
Protein was isolated from differentiated AECs within transwells in situ, and Western blot analysis was performed as previously described.16 Blots were probed using antibodies directed to Claudin-1, Occludin-1, ZO-1 (all; Thermo Fisher Scientific, Waltham, MA, USA), and β-actin (Sigma-Aldrich Co., St Louis, MO, USA). Densitometry of histogram analyses was performed using Multi Gauge software (V3.1; Fujifilm, Tokyo, Japan). Density scores were analyzed by a gamma (log link) mixed-model regression to allow for correlated treatment responses within each culture. Results were normalized to both β-actin and the biological control and expressed as relative abundance. The statistical analysis was performed using R statistical software (release 3.2.3) for N=6 cultures.
Measurement of epithelial electrical impedance
Electrical impedance (to restrict ionic conductance) imparted by differentiated epithelial cultures was measured at t=0 h and at the end of the treatment period (64 h) to determine the integrity of TJs as a result of the exposures. Culture plates containing transwell ALI cultures were allowed to acclimatize for 30 min on a 37°C heating platform (Leica Microsystems, Wetzlar, Germany) within a biosafety cabinet, before reading electrical impedance using the EVOM2 Ohm meter (World Precision Instruments). Raw resistance values were converted to Ω·cm2 by subtracting the resistance of a blank transwell insert and factoring for the surface area of the membrane support (0.33 cm2). Results were double normalized using the control exposure and the t=0 min time interval value, analyzed using a Gamma regression model, with robust standard errors adjusted for the number of replicate cultures (N=6), and expressed as relative ionic conductance. Statistical analysis was performed using Stata statistical software (release 14; StataCorp LP, College Station, TX, USA).
Assessment of epithelial paracellular permeability
Paracellular permeability was assessed in the transwell ALI culture system using a fluorescent tracer assay method. Sodium fluorescein (NaFl; Sigma-Aldrich Co.) was suspended in growth media (0.5 mg/mL) and applied to the apical reservoir after the treatment period. The concentration of NaFl that permeated across the epithelial layer and into the basal reservoir was determined using fluorimetry (FLUOstar Optima; BMG Labtech, Mornington, VIC, Australia) after 1, 6 and 24 h time intervals. Log-transformed data were analyzed using a linear regression model with robust standard errors adjusted for the number of replicates (N=3). Results were normalized to the control exposure at each time point and expressed as relative paracellular permeability. Statistical analysis was performed using Stata statistical software.
Transmission electron microscopy (TEM)
Ex vivo cultures of human AEC were fixed in situ in transwells with 4% paraformaldehyde/1.25% glutaraldehyde (both from ProSciTech, Kirwan, QLD, Australia) in PBS/4% sucrose (pH 7.2) for 24 h, and then postfixed in 2% aqueous osmium tetroxide (Electron Microscopy Sciences, Hatfield, PA, USA). The fixed samples were then dehydrated in increasing concentrations of ethanol and infiltrated with Procure/Araldite epoxy resin (ProSciTech). Samples were allowed to polymerize overnight at 70°C, then sectioned in a Leica UC6 Ultramicrotome using a diamond blade (Leica Microsystems). Sections were stained with uranyl acetate and lead citrate (Sigma-Aldrich Co.) and examined in an FEI Tecnai G2 Spirit Transmission Electron Microscope (FEI, Hillsboro, OR, USA). Experienced TEM operators assisted in identifying subcellular structures during image acquisition.
Results
Exposure to Zn depletion and CSE causes a downregulation of TJ proteins
The essential TJ proteins such as ZO-1 (connects Claudin and Occludin to the actin cytoskeleton) and Claudin-1 (links adjacent cells through another Claudin-1 protein) were significantly downregulated by the combination exposure in differentiated AECs (−1.93-fold, P=0.046, 95% CI [0.93, 0.27] and −3.37-fold, P=0.01, 95% CI [0.61, 0.14], respectively; N=6; Figure 1). While both the CSE and Zn depletion exposures contributed to this effect, the dual exposure was an important factor for the negative regulation of these TJ proteins in this model. However, the relative influence of the Zn depletion exposure may have concealed a more significant effect imparted by the inclusion of the CSE treatment in the combination exposure. Occludin-1 (links adjacent cells through another Occludin-1 molecule) demonstrated a similar downregulation, but was unaffected by the CSE, and therefore, its reduction was not significant for the combination exposure. Overall, this pattern suggests that there is differential regulation of ZO-1/Claudin-1 vs Occludin-1 expression in the presence of CSE, but a similar tendency for their downregulation in conditions where the cell is deprived of Zn. Given this result, we examined the impact these treatments have on epithelial barrier function.
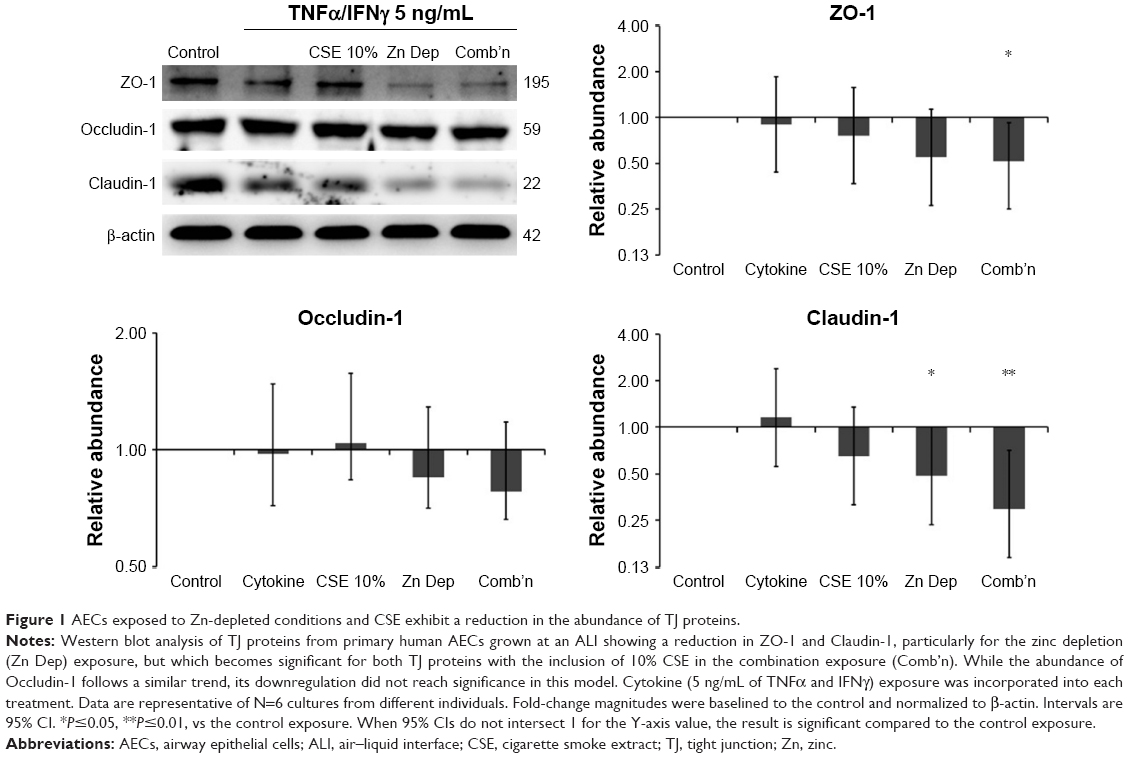 | Figure 1 AECs exposed to Zn-depleted conditions and CSE exhibit a reduction in the abundance of TJ proteins. |
Differentiated AECs exhibit a leaky phenotype when treated with Zn depletion and CSE
Barrier function was assessed to ascertain whether the exposures produced the leaky epithelium phenotype. TEER was used to measure the integrity of TJs by measuring their capacity to impede electric current applied across the epithelial layer. While the CSE and Zn depletion treatments produced a moderate reduction in TEER (ie, increased ionic conductance; 1.45-fold, 95% CI [1.16, 1.79] and 2.2-fold, 95% CI [1.89, 2.56], respectively, both P<0.001; N=6), the combination exposure elicited a comparatively large decline in electrical impedance (4.39-fold, P=0.001, 95% CI [2.94, 6.25], N=6; Figure 2A). Comparative analysis showed that the increase in ionic conductance elicited by Zn depletion was significantly higher than that produced by 10% CSE (Zn depletion/10% CSE ratio =0.66, 95% CI [0.52, 0.82], P<0.001), while the combination exposure was more influential than Zn depletion alone (combination/Zn depletion ratio =0.51, 95% CI [0.35, 0.73], P<0.001).
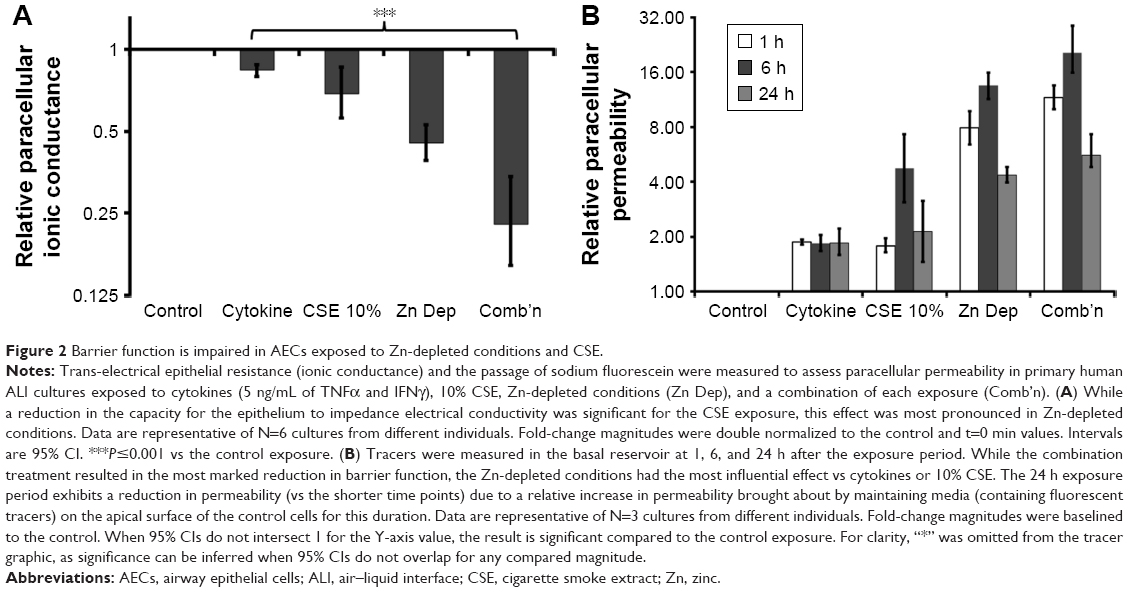 | Figure 2 Barrier function is impaired in AECs exposed to Zn-depleted conditions and CSE. |
The passage of fluorescent tracers across the epithelial layer was then quantified to directly assess the permeability of the paracellular pathway. For each time point, the exposures incorporating Zn depletion most deleteriously affected epithelial barrier function, with tracer permeability peaking for the combination exposure at the 6 h time interval (20.46-fold, 95% CI [13.77, 28.24], P=0.0009; N=3; Figure 2B). Indeed, a comparative analysis that integrated the results for each time point showed that Zn depletion potentiated an increase in epithelial permeability greater than the effect of 10% CSE (average Zn depletion/10% CSE ratio =3.9, 95% CI [3.1, 4.9], P=0.003), and that the addition of 10% CSE for the combination exposure was more effective than Zn depletion in isolation (average combination/Zn depletion ratio =1.41, 95% CI [1.02, 1.97], P=0.044). It should also be noted that the effect of maintaining the tracer dye on the epithelial layer over the course of the 24 h time point caused the control cells to become leaky. Hence, as the experimental exposure values were normalized in relation to the control, this produced an artifactual reduction in permeability for the 24 h interval relative to the shorter (1 h) and 6 h assay intervals.
As expected, the reduction in epithelial barrier function largely corresponded to the observations made for the downregulation of TJ protein expression observed for ZO-1 and Claudin-1. However, greater magnitudes of change (vs the control) were observed in the context of the TEER and tracer permeability assays. As other factors also contributed to the maintenance of the epithelial barrier that could be effected by the Zn depletion and CSE exposures, we next used TEM to provide a visual assessment of other junction complexes, such as the AJs and GJs, that are shared by adjacent AECs.
ALI cultures exposed to Zn depletion and CSE exhibit a marked reduction in junction complexes
TEM was used to ascertain whether a reduction in cell-binding complexes was elicited by the combination exposure. Primary AEC propagated at an ALI until mucociliary differentiation was achieved exhibited frequent junctional complexes between the adjacent cells in the epithelial layer (Figures 3 and S1). In contrast, similar examination of cell-to-cell margins for AECs treated with the combination exposures revealed few discernible junction complexes, and those that were observed exhibited a marked reduction in electron density compared to the control cells (Figure 3, right and lower panels). A reduction in electron density, consistent with the downregulation of Claudin-1 and ZO-1 proteins, supported the findings of an increase in ionic conductance and tracer paracellular permeability observed during the functional assays for epithelial permeability.
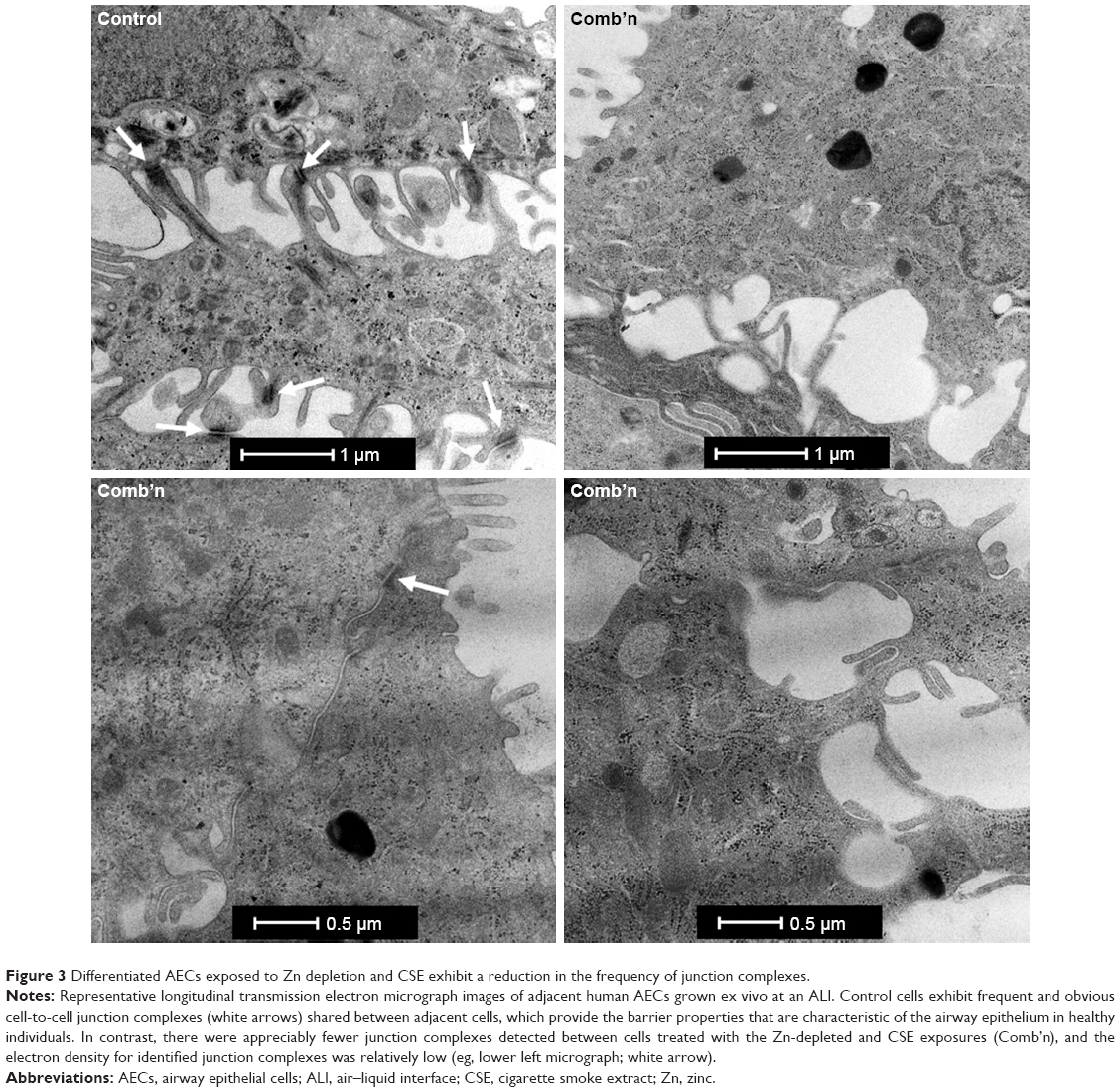 | Figure 3 Differentiated AECs exposed to Zn depletion and CSE exhibit a reduction in the frequency of junction complexes. |
Discussion
We reported that a combination of CSE and Zn depletion downregulates TJ protein expression (Figure 1), with a corresponding increase in ionic and molecular paracellular permeability (Figure 2A and B, respectively) and a reduction in the frequency of junction complexes (Figure 3). These new findings are consistent with our previous observations that chronic cigarette smoke exposure depletes Zn from the airway epithelium.11 Compared to inconsistent reports using in vitro models of cigarette smoke exposure, there is strong evidence that Zn deficiency potentiates paracellular permeability in models of inflammatory gastrointestinal disease via the downregulation of ZO-1, Occludins, and Claudins,17–19 and that Zn supplementation ameliorates this effect.20–22 Similarly, in this study, we found that Zn depletion was an influential exposure leading to TJ dysfunction, and this effect was amplified by the incorporation of CSE. TEM showed that the other junctional complexes such as AJs may also be disrupted by the combination exposure (Figure 3). This is consistent with evidence that AJs also govern paracellular permeability via interactions to TJs through ZO-1–cytoskeleton linkages, which promote the formation of TJs, and it has been shown that disruption of AJs precedes a reduction in TJ protein expression.12,23–25
Given that disease-related Zn insufficiency is associated with a leaky barrier phenotype in a range of epithelial compartments such as the intestine,13 kidneys,26 and airways,12 its appropriate regulation may be a common requisite for the maintenance of epithelial barriers. For COPD, the chronic inflammation and cigarette smoke exposures alone are factors that can disrupt Zn homeostasis,11,27 and in doing so, they likely potentiate a more considerable, and potentially longer-lasting reduction in barrier function than either exposure in isolation.
Many in vitro models of COPD rely on secondary cell lines and commercially derived frozen cells. Our findings suggest that the more physiologically relevant AECs from participant-derived isolates are required for accurate investigations of barrier function. There is little doubt that mucus and cilia serve to protect the epithelium from apical exposures such as cigarette smoke. In addition, their formation is a reliable indicator that a continuous and functional epithelial barrier has been established. In this study, every effort has been made to ensure that mucociliary differentiation was complete in cultures from different donors (Figure S1). Indeed, the ex vivo model employed in this study can be advanced by replacing CSE with a smoke aerosol systems to more closely approximate the in vivo situation, and by comparing outcomes in AECs derived from control vs COPD participants. Our future investigations will follow these next steps, with a view to determining the significance of using AECs that were previously subject to COPD-related exposures in vivo before being entered into the ex vivo culture model, and whether the reintroduction of Zn elicits a restoration in barrier function in the context of cigarette smoke exposure or COPD status.
Conclusion
We show that Zn deficiency is an influential codeterminant that drives epithelial barrier dysfunction in an ex vivo model of CSE exposure. Targeting this phenomenon may be a previously unidentified and critical avenue toward ameliorating epithelial dysfunction that is synonymous with COPD.
Acknowledgments
We are grateful for the expertise and support of the University of Adelaide Microscopy Suite, the clinical staff and donors of the Royal Adelaide Hospital Thoracic Unit, and Mr Josh Woenig who provided technical support for the tracer assay. We acknowledge the funding provided by the National Health and Medical Research Council (APP1099040), the Thoracic Society of Australia and New Zealand, AstraZeneca Pty Ltd, and the Royal Adelaide Hospital Research Fund.
Disclosure
The authors report no conflicts of interest in this work.
References
Gamez AS, Gras D, Petit A, et al. Supplementing defect in club cell secretory protein attenuates airway inflammation in COPD. Chest. 2015;147(6):1467–1476. | ||
Ganesan S, Sajjan US. Repair and remodeling of airway epithelium after injury in chronic obstructive pulmonary disease. Curr Respir Care Rep. 2013;2(3). | ||
Shaykhiev R, Otaki F, Bonsu P, et al. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci. 2011;68(5):877–892. | ||
Brekman A, Walters MS, Tilley AE, Crystal RG. FOXJ1 prevents cilia growth inhibition by cigarette smoke in human airway epithelium in vitro. Am J Respir Cell Mol Biol. 2014;51(5):688–700. | ||
Glader P, Moller S, Lilja J, Wieslander E, Lofdahl CG, von Wachenfeldt K. Cigarette smoke extract modulates respiratory defence mechanisms through effects on T-cells and airway epithelial cells. Respir Med. 2006;100(5):818–827. | ||
Iskandar AR, Xiang Y, Frentzel S, et al. Impact assessment of cigarette smoke exposure on organotypic bronchial epithelial tissue cultures: a comparison of mono-culture and coculture model containing fibroblasts. Toxicol Sci. 2015;147(1):207–221. | ||
Schamberger AC, Staab-Weijnitz CA, Mise-Racek N, Eickelberg O. Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci Rep. 2015;5:8163. | ||
Unwalla HJ, Ivonnet P, Dennis JS, Conner GE, Salathe M. Transforming growth factor-beta1 and cigarette smoke inhibit the ability of beta2-agonists to enhance epithelial permeability. Am J Respir Cell Mol Biol. 2015;52(1):65–74. | ||
Roscioli E, Hamon R, Lester S, Murgia C, Grant J, Zalewski P. Zinc-rich inhibitor of apoptosis proteins (IAPs) as regulatory factors in the epithelium of normal and inflamed airways. Biometals. 2013;26(2):205–227. | ||
Zalewski PD, Truong-Tran AQ, Grosser D, Jayaram L, Murgia C, Ruffin RE. Zinc metabolism in airway epithelium and airway inflammation: basic mechanisms and clinical targets. A review. Pharmacol Ther. 2005;105(2):127–149. | ||
Roscioli E, Tran HB, Jersmann H, et al. The uncoupling of autophagy and zinc homeostasis in airway epithelial cells as a fundamental contributor to COPD. Am J Physiol Lung Cell Mol Physiol. 2017;313(3):L453–L465. | ||
Bao S, Knoell DL. Zinc modulates cytokine-induced lung epithelial cell barrier permeability. Am J Physiol Lung Cell Mol Physiol. 2006;291(6):L1132–L1141. | ||
Skrovanek S, DiGuilio K, Bailey R, et al. Zinc and gastrointestinal disease. World J Gastrointest Pathophysiol. 2014;5(4):496–513. | ||
Su Y, Han W, Giraldo C, Li YD, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol. 1998;19(5):819–825. | ||
Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, Reynolds PN. Smoking alters alveolar macrophage recognition and phagocytic ability. Am J Respir Cell Mol Biol. 2007;37(6):748–755. | ||
Hodge G, Roscioli E, Jersmann H, et al. Steroid resistance in COPD is associated with impaired molecular chaperone Hsp90 expression by pro-inflammatory lymphocytes. Respir Res. 2016;17(1):135. | ||
Finamore A, Massimi M, Conti Devirgiliis L, Mengheri E. Zinc deficiency induces membrane barrier damage and increases neutrophil transmigration in Caco-2 cells. J Nutr. 2008;138(9):1664–1670. | ||
Miyoshi Y, Tanabe S, Suzuki T. Cellular zinc is required for intestinal epithelial barrier maintenance via the regulation of claudin-3 and occludin expression. Am J Physiol Gastrointest Liver Physiol. 2016;311(1):G105–G116. | ||
Ranaldi G, Ferruzza S, Canali R, et al. Intracellular zinc is required for intestinal cell survival signals triggered by the inflammatory cytokine TNFalpha. J Nutr Biochem. 2013;24(6):967–976. | ||
Mercado J, Valenzano MC, Jeffers C, et al. Enhancement of tight junctional barrier function by micronutrients: compound-specific effects on permeability and claudin composition. PLoS One. 2013;8(11):e78775. | ||
Shao Y, Wolf PG, Guo S, Guo Y, Gaskins HR, Zhang B. Zinc enhances intestinal epithelial barrier function through the PI3K/AKT/mTOR signaling pathway in Caco-2 cells. J Nutr Biochem. 2017;43:18–26. | ||
Zhu C, Lv H, Chen Z, et al. Dietary zinc oxide modulates antioxidant capacity, small intestine development, and jejunal gene expression in weaned piglets. Biol Trace Elem Res. 2017;175(2):331–338. | ||
Taddei A, Giampietro C, Conti A, et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10(8):923–934. | ||
Ganesan S, Comstock AT, Sajjan US. Barrier function of airway tract epithelium. Tissue Barriers. 2013;1(4):e24997. | ||
Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778(3):660–669. | ||
Wang X, Valenzano MC, Mercado JM, Zurbach EP, Flounders CJ, Mullin JM. Zinc enhancement of LLC-PK(1) renal epithelial barrier function. Clin Nutr. 2014;33(2):280–286. | ||
Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc Natl Acad Sci U S A. 2005;102(19):6843–6848. |
Supplementary material
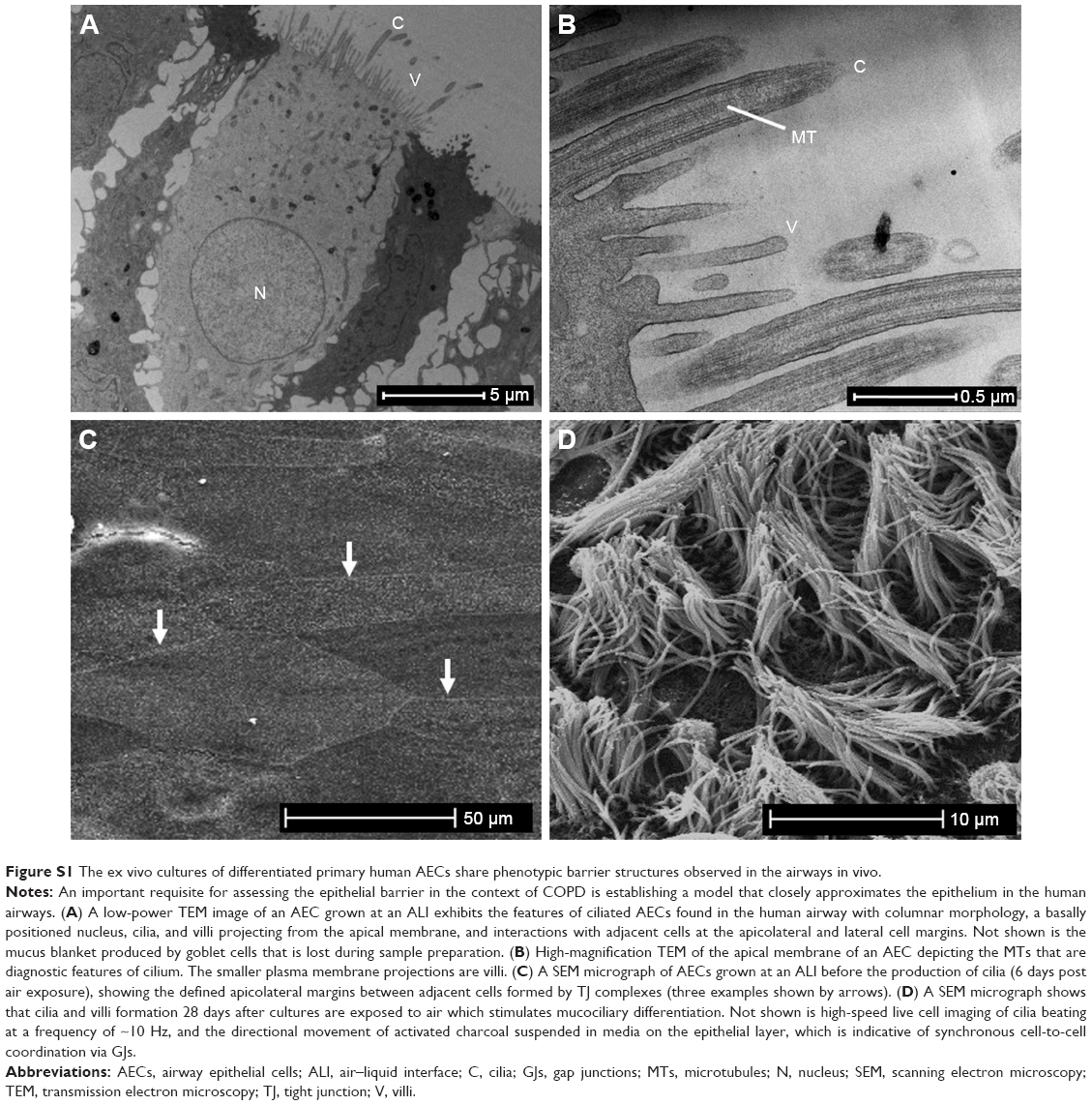 | Figure S1 The ex vivo cultures of differentiated primary human AECs share phenotypic barrier structures observed in the airways in vivo. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.