Back to Journals » Drug Design, Development and Therapy » Volume 19
YK-4-279 Induces Osteosarcoma Cell Cycle Arrest, DNA Damage Response, and Apoptosis by Regulating the MAPK Cascade
Authors Wang W, Zhang Y ![]() , Jia X
, Jia X ![]() , Han L, Xin M
, Han L, Xin M ![]()
Received 27 May 2025
Accepted for publication 22 December 2025
Published 26 December 2025 Volume 2025:19 Pages 11749—11766
DOI https://doi.org/10.2147/DDDT.S542983
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Leonidas Panos
Weifeng Wang,1– 3 Yuli Zhang,4,5 Xiuqin Jia,6 Liren Han,1 Ming Xin7
1Department of Orthopedic Surgery, Beijing Jishuitan Hospital Liaocheng Hospital, Liaocheng, Shandong, People’s Republic of China; 2Department of Orthopedic Surgery, Liaocheng People’s Hospital, Liaocheng, Shandong, People’s Republic of China; 3School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, People’s Republic of China; 4Department of Dermatology, Liaocheng People’s Hospital, Liaocheng, Shandong, People’s Republic of China; 5Department of Dermatology, The Second Hospital of Shandong University, Jinan, Shandong, People’s Republic of China; 6The Key Laboratory of Clinical Pharmacology, Liaocheng People’s Hospital, Liaocheng, Shandong, People’s Republic of China; 7The Key Laboratory of Molecular Pharmacology, Liaocheng People’s Hospital, Liaocheng, Shandong, People’s Republic of China
Correspondence: Ming Xin, The Key Laboratory of Molecular Pharmacology, Liaocheng People’s Hospital, Liaocheng, Shandong, People’s Republic of China, Email [email protected] Liren Han, Department of Orthopedic Surgery, Beijing Jishuitan Hospital Liaocheng Hospital, Liaocheng, Shandong, People’s Republic of China, Email [email protected]
Background: YK-4-279, a promising anticancer agent, has demonstrated therapeutic potential against various tumors. Osteosarcoma (OS), an aggressive bone cancer primarily affecting adolescents, lacks effective treatment options. Investigating YK-4-279’s mechanisms in OS is critical for evaluating its clinical utility.
Methods: Using in vitro models, we examined YK-4-279’s effects on OS cell viability, proliferation, apoptosis, cell cycle progression, and DNA damage. We also assessed its impact on MAPK signaling pathway activation. To clarify the pathway’s role, we combined YK-4-279 treatment with a P38 inhibitor.
Results: YK-4-279 markedly suppressed OS cell viability and proliferation, triggered G2/M phase arrest, and enhanced apoptosis and DNA damage. Furthermore, it activated the MAPK pathway, elevating phosphorylation of ERK1/2, JNK, and P38 MAPK. Co-treatment with a P38 inhibitor partially reversed these effects, confirming MAPK’s involvement in YK-4-279’s antitumor action.
Conclusion: YK-4-279 inhibits OS cell growth, induces DNA damage and cell cycle arrest, and promotes apoptosis via MAPK pathway activation. These findings highlight its strong therapeutic potential for OS treatment.
Keywords: YK-4-279, osteosarcoma, MAPK signaling pathway, cell cycle, DNA damage, apoptosis
Introduction
Osteosarcoma (OS) is a prevalent and highly malignant bone tumor, with a global incidence of approximately 3 per million, representing 11.7% of all primary bone tumors.1 Osteosarcoma primarily affects children and adolescents, with its onset typically occurring in the distal femur, tibia, proximal humerus, and other long bone epiphyses.2 OS is highly invasive and associated with a poor prognosis, with approximately 80–90% of patients already exhibiting micrometastases at the time of diagnosis.3 Although the 5-year survival rate can reach approximately 70% following comprehensive treatment with chemotherapy, surgery, and radiotherapy, it drops to around 20% for patients with recurrent or metastatic OS.4 This imposes significant psychological pressure and economic burden on both the patient’s family and society, while the treatment of OS has reached a bottleneck in recent years.5 Therefore, there is an urgent need to explore novel therapeutic approaches and strategies to provide patients with improved treatment options and enhance their prognosis.
DNA damage, cell cycle arrest, and induction of apoptosis are promising strategies to selectively eliminate cancer cells.6 These mechanisms also represent important targets for the development of novel antitumor drugs, by interacting with DNA through specific drugs, cell division is inhibited, leading to the induction of cell death.7 The DNA damage response initiates with the efficient recognition of damage by sensor proteins (ATM, CHK), which activate other molecules in the DNA repair pathway (p21, CDK).8–12 Concurrently, the DNA damage response halts cell cycle progression, leading to cell cycle arrest.13 Severe unrepaired DNA damage subsequently activates cell death pathways, such as apoptosis.14 Apoptosis is a programmed cell death mechanism in multicellular organisms that plays a crucial role in eliminating unwanted cells in response to cellular development, stress, and various stimuli, including irreparable DNA damage. Evasion of apoptosis promotes cancer development.15
Currently, promising emerging treatment strategies include targeted therapy16 and nanotherapy.17 In the study of malignant tumors, it has been observed that tumor cells frequently overexpress specific signature molecules, which may be tumor proto-oncogene products or involved in normal signaling pathways. These signature molecules are targeted to bind specifically to them, thereby blocking their activity and inhibiting tumor growth or inducing tumor cell death.18 The precision of this treatment method is more robust and specific than that of chemotherapy drugs, with relatively fewer toxic side effects. Numerous targeted drugs have been approved for use in first- and second-line treatments of various malignant tumors, achieving favorable responses.19–22 Molecular targeted therapy can undoubtedly be considered a novel approach for the treatment of malignant tumors.
The E26 transformation-specific (ETS) family consists of 28 transcription factors. The activation of ETS is regulated by several intracellular signaling pathways, including MAPK kinases (Erk, p38, and JNK), and disruption of its activity typically leads to aberrant oncogenic changes involved in tumorigenesis and progression,23 including Ewing’s sarcoma (ES),24–26 brain tumors,27 hematologic malignancies,28,29 and prostate cancer.30,31 Among these, ETS variant 1 (ETV1) plays a crucial role in the metastatic progression of both Ewing’s sarcoma and OS.32,33 YK-4-279 is a small molecule antagonist of ETV1 that has demonstrated promising anti-tumor activity in Ewing’s sarcoma (ES),34 prostate cancer,35 and neuroblastoma.36 YK-4-279 binds to EWS-FLI1 and blocks its interaction with DHX9, leading to growth arrest and apoptosis in Ewing’s sarcoma (ES) cells. YK-4-279 inhibits the function of ETS factors by disrupting protein-protein interactions between various ETS proteins and their binding partners.37 This disruption of protein interactions, rather than inhibiting a relatively ubiquitous enzyme, is believed to offer a more specific treatment for cancer cells.38 However, studies on the mechanism of action of YK-4-279 in OS remain limited, thus, this study aimed to investigate the effects of YK-4-279 on OS cell viability and proliferation, DNA damage, cell cycle, apoptosis, and the MAPK signaling pathway, which could provide a new foundation for the treatment and prognosis of OS.
Materials and Methods
Reagents and Antibodies
YK-4-279 (Cat. No. Y10002; CAS: 1037184–44-3) was procured from Beijing Psaitong Biotechnology Co., Ltd. This compound has the following molecular formula: C17H13Cl2NO4, with a molecular weight of 366.20 g/mol. Its physicochemical properties include a density of 1.5 ± 0.1 g/cm3, a boiling point of 608.9 ± 55.0 °C at 760 mmHg, a melting point of 149–151 °C, and a flash point of 322.1 ± 31.5 °C. For experimental use, a 50 mM stock solution was prepared in dimethyl sulfoxide (DMSO; Solarbio, Cat. No. D8371), aliquoted, and stored at −80 °C to maintain compound stability. The following reagents were used in the experiments: CCK-8 (C0039, Beyotime), Crystalline Violet Staining Solution (0.1%) (G1063, Solarbio), DNA Content Quantification Assay (Cell Cycle) (CA1510, Solarbio), Annexin V-FITC Apoptosis Detection Kit (BD PharmingenTM, 556547), Hoechst 33342/PI (Beyotime, P0137), SB203580 (Psaitong Bio, S10108).
The following antibodies were used in this study: Phospho-ATM (Ser1981) (4526s, 1:1000), γ-H2AX (Ser139)(9718s, 1:1000), Phospho-p44/42 MAPK (Erk1/2)(Thr202/Tyr204)(4370s, 1:1000), Phospho-SAPK/JNK (Thr183/Tyr185) (9255s, 1:1000), β-actin (Ms)(3700s, 1:1000), GAPDH(Rb)(5147s, 1:1000), purchased from Cell Signaling Technology (Danvers, MA, USA). Additional antibodies included ATM (ET1606-20, 1:1000), Chk2 (ET1610-52, 1:1000), p27 (ET1608-61, 1:1000), Cyclin B1 (ET1608-27, 1:1000), Cyclin D1 (ET1601-31, 1:1000), purchased from HUABIO (China). PARP1 (13371-1-AP, 1:500), Caspase 8(13423-1-AP, 1:500), ERK1/2 (16443-1-AP, 1:1000), JNK (66210-1-Ig, 1:2000), Phospho-CHEK2 (Thr68)(29012-1-AP, 1:1000), p21 (10355-1-AP, 1:1000), p38 MAPK (14064-1-AP, 1:1000), Phospho-p38 MAPK (Thr180/Tyr182) (28796-1-AP, 1:1000), purchased fromProteintech(China). Goat Anti-Rabbit IgG H&L (HRP)(ab205718, Rb: 1:20000), Goat Anti-Mouse IgG H&L (HRP) (ab205719, Ms: 1:10000), Goat Anti-Rabbit IgG H&L (Alexa Fluor® 488) (ab150077, 1:500), Goat Anti-Mouse IgG H&L (Alexa Fluor® 594)(ab150116, 1:500), were procured from Abcam (Cambridge, UK).
Cells and Cell Culture
HOS (ZQ0952) and MG63 (ZQ0403) were kindly provided Shanghai Zhong Qiao Xin Zhou Biotechnology Co., ltd. All cell lines were authenticated by Short Tandem Repeat (STR) profiling to confirm their identity. Cells were cultured in Minimum Essential Medium (MEM) with Non-Essential Amino Acids (NEAA)(Pricella, PM150410), supplemented with 10% fetal bovine serum (FBS)(Abwbio, AB-FBS0500) and 1% Penicillin-Streptomycin (P1400, Solarbio). Cultures were maintained at 37 °C in a humidified incubator with 5% CO2, and the medium was refreshed every 2 to 3 days.
Cell Viability Assay
The effect of YK-4-279 on OS cell viability was assessed using the CCK-8 assay. HOS and MG63 cells in the logarithmic growth phase were collected by centrifugation, counted, and inoculated into 96-well plates, with 1×104 cells/100 μL of cell suspension added to each well. Six replicate wells were set for each concentration. The cells were incubated for 24 hours. YK-4-279 was diluted according to the following concentration gradient: 8×10−5, 6×10−5, 4×10−5, 2×10−5, 1×10−6, 1×10−7, and 1×10−8 mol/L, and added to the corresponding wells. The cells were then incubated for 24 or 48 hours before performing the CCK-8 assay. Two hours prior to the assay, CCK-8 reagent was added to each well, followed by incubation at 37°C in a light-protected incubator. Absorbance (OD) was measured at 450 nm, and the cell survival rate was calculated using the following formula: OD value of experimental wells - OD value of blank control wells) / (OD value of control wells - OD value of blank control wells). The IC50 of YK-4-279 was subsequently calculated.
Cell Proliferation Assay
The effect of YK-4-279 on OS cell proliferation was assessed using Crystalline Violet Staining Solution in a cell colony formation assay. HOS and MG63 cells, in the logarithmic growth phase, were seeded in 6-well plates at a density of 1000 cells per well and cultured overnight to allow adherence. YK-4-279 was then added to the plates at concentrations of 0, 1, and 5 µM, and the culture medium was replaced every 2–3 days. After 14 days of culture, the medium was removed, and the cells were washed twice with ice-cold PBS. The cells were then fixed in 4% paraformaldehyde (PFA) for 30 minutes, after which the fixative was removed. The cells were washed once with ice-cold PBS, stained with 0.1% Crystalline Violet Staining Solution (G1063, Solarbio) for 10 minutes, and subsequently washed with PBS before being air-dried. Colony formation was observed, images were captured, and the number of colonies formed was quantified.
Cell Cycle Analysis
Flow cytometry with PI staining was employed to assess the effect of YK-4-279 on the OS cell cycle. HOS and MG63 cells, in the logarithmic growth phase, were seeded in 6-well plates and treated with YK-4-279 at concentrations of 0, 5, and 10 µM for 24 hours. The cells from each group were collected in flow cytometry tubes, washed once with PBS, and centrifuged. The cell concentration was then adjusted to 106 cells/mL, and 1 mL of single-cell suspension was prepared.
To fix the cells, 500 µL of 70% ice-cold ethanol was added, and the cells were incubated at 4°C overnight. After fixation, the cells were washed with PBS before staining. 100 µL of RNase A solution was added to the cell pellet, the cells were resuspended, and incubated in a water bath at 37°C for 30 minutes. 400 µL of PI staining solution was added and mixed. The cells were incubated at 4°C for 30 minutes, protected from light, and subsequently analyzed by flow cytometry.
Immunofluorescence Analysis
Phosphorylation of histone H2AX was employed to label DNA double-strand breaks (DSBs) and evaluate the effect of YK-4-279 on DNA damage in OS cells. HOS and MG63 cells (5 × 105 cells/mL) were seeded onto coverslips in 24-well plates and treated with YK-4-279 (5 µM) for 24 hours. After treatment, the HOS and MG63 cells were washed with PBS and fixed in 4% paraformaldehyde for 30 minutes. The cell membrane was then permeabilized with 0.1% Triton-X 100 (Beyotime, P0096) for 30 minutes, followed by blocking with 5% BSA at 4°C for 1 hour. Cells were incubated overnight at 4°C with primary antibodies: rabbit γ-H2AX (Ser139, 9718S, 1:100, green) and mouse α-Tubulin (66031-1-Ig, 1:100, red). After washing away excess primary antibodies with PBS, the HOS and MG63 cells were incubated with fluorescently labeled secondary antibodies: Goat anti-Rabbit IgG H&L (Alexa Fluor® 488, ab150077, 1:500) and Goat anti-Mouse IgG H&L (Alexa Fluor® 594, ab150116, 1:500) for 1 hour at room temperature. The HOS and MG63 cells were then incubated for 1 hour at room temperature. Finally, the nuclei were stained and counterstained with an antifluorescence quenching solution containing DAPI (Beyotime, P0131). Fluorescence microscopy was employed to observe and analyze the expression of fluorescent signals in individual cells.
Hoechst33342/PI Analysis
To quantitatively evaluate whether YK-4-279 induces OS cell death, a Hoechst 33342/PI double-staining assay (Beyotime, P0137) was employed. The HOS and MG63 cells were cultured in a suspension of 100,000 cells/mL per well on coverslips in 24-well plates and treated with YK-4-279 at a concentration of 5 µM for 24 hours. Hoechst 33342 and PI stains were then added, and the HOS and MG63 cells were incubated at 4°C for 15 minutes. The HOS and MG63 cells were then washed with PBS. Co-localization of Hoechst 33342 (blue) and PI (red) indicates cell death. The images were captured and analyzed by fluorescence microscopy.
Cell Apoptosis Analysis
The effect of YK-4-279 on OS cell apoptosis was further assessed using flow cytometry. Apoptosis was detected using the Annexin V-FITC Apoptosis Detection Kit (BD Pharmingen, 556547). HOS and MG63 cells in the logarithmic growth phase were collected, counted, and diluted into a cell suspension of 1 × 106 cells/mL. The cells were then seeded into 6-well plates and treated with YK-4-279 at concentrations of 0, 5, and 10 µM for 24 hours. Adherent cells were dissociated with 0.25% trypsin (without EDTA), and suspended cells were collected by centrifugation for 5–10 minutes at room temperature. After washing the cells with ice-cold PBS, the cells were resuspended in 300 μL of 1× binding buffer. Subsequently, 5 μL of Annexin V-FITC and 5 μL of PI were added, gently mixed, and incubated for 15 minutes at room temperature, protected from light. Prior to analysis, 200 μL of 1× binding buffer was added, and flow cytometric analysis was performed using a CytoFLEX SRT flow cytometer (BECKMAN COULTER, BF17071).
Western Blot Analysis
Healthy HOS and MG63 cells were washed with PBS, and adherent cells were collected and lysed using RIPA buffer (Beyotime, P0013) containing protease and phosphatase inhibitors. The cells were lysed on ice for 30 minutes to extract the protein. Protein concentration was quantified using the BCA assay kit (Beyotime, P0010). A 30 µg protein sample was separated by 10% SDS-PAGE and transferred to a PVDF membrane after electrophoresis.
The membrane was blocked with 5% non-fat dry milk (BD Difco™ Skim Milk, 232100). The blocking solution was prepared by mixing 0.5 g of non-fat dry milk with 10 mL of 1x TBST (0.1% Tween 20) and incubated at room temperature on a shaking platform for 1 hour.
Following blocking, the membrane was incubated overnight at 4°C with the following primary antibodies: γ-H2AX (Ser139) (9718s, Rb, 1:1000), ATM (ET1606-20, Rb, 1:1000), Phospho-ATM (Ser1981) (4526s, Ms, 1:1000), Chk2 (ET1610-52, Rb, 1:1000), Phospho-CHEK2 (Thr68) (29012-1-AP, Rb, 1:1000), p21 (10355-1-AP, Rb, 1:1000), p27 (ET1608-61, Rb, 1:1000), Cyclin B1 (ET1608-27, Rb, 1:1000), Cyclin D1 (ET1601-31, Rb, 1:1000), PARP1 (13371-1-AP, Rb, 1:500), Caspase 8 (13423-1-AP, Rb, 1:500), ERK1/2 (16443-1-AP, Rb, 1:1000), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (4370s, Rb, 1:1000), JNK (66210-1-Ig, Ms, 1:2000), Phospho-SAPK/JNK (Thr183/Tyr185) (9255s, Ms, 1:1000), p38 MAPK (14064-1-AP, Rb, 1:1000), Phospho-p38 MAPK (Thr180/Tyr182) (28796-1-AP, Rb, 1:1000), β-actin (3700s, Ms, 1:1000), and GAPDH (5147s, Rb, 1:1000).
After primary antibody incubation, the membrane was washed with 1x TBST and incubated with HRP-labeled secondary antibodies at room temperature for 1 hour. The secondary antibodies used were Goat Anti-Rabbit IgG H&L (HRP) (ab205718, Rb, 1:20000) and Goat Anti-Mouse IgG H&L (HRP) (ab205719, Ms, 1:10000). The membrane was washed three times with 1x TBST.
Finally, the blot was developed and analyzed using an ECL chemiluminescence detection system. The relative expression level of the target protein was calculated as the ratio of the target protein to the internal control protein.
Animal Study
Female BALB/c-nu nude mice (SPF grade, approximately 15 g, 5–6 weeks old; Certificate No. 37072610100546123) were supplied by Jinan Pengyue Experimental Animal Breeding Co., Ltd. (License No. SCXK [Lu] 20220006). All mice were housed under standard specific pathogen-free (SPF) barrier conditions, maintained at 22 ± 2°C, 50–60% humidity, and a 12-hour light/dark cycle, with free access to food and water. After one week of acclimatization, the mice (n = 12) were subcutaneously inoculated in the right axilla with 1 × 107 MG63 cells suspended in 200 µL of a 1:1 mixture of Matrigel and PBS. The mice were randomly divided into two groups: a control group (administered PBS) and a YK-4-279 treatment group (30 mg/kg/day). All agents were delivered via intraperitoneal (i.p.) injection, and the mice were closely monitored throughout the dosing period. Tumor size was measured every three days, and tumor volume was calculated using the formula: Volume = L × S2 × 0.5, where L represents the longest diameter and S the shortest diameter. After 28 days, all mice were anesthetized, euthanized, and the tumors were excised and weighed. Collected samples were preserved for further analysis.
Statistical Analysis
Each experiment was conducted at least five times, and the results are expressed as the mean ± standard deviation (SD). Statistical significance between the two groups was determined using a paired two-tailed Student’s t-test. Comparisons between multiple groups were analyzed using one-way ANOVA with post hoc multiple comparisons (Dunnett’s test) performed in GraphPad Prism 9.0 software, if applicable. A p-value of < 0.05 was considered statistically significant.
Results
YK-4-279 Significantly Inhibits the Growth of OS Cells
The cytotoxicity of YK-4-279 in OS HOS and MG63 cell lines was assessed using the CCK-8 assay. After 24 and 48 hours of treatment, the viability of HOS and MG63 cells was significantly reduced in the presence of YK-4-279 concentrations of 80, 60, 40, 20, 1, 0.1, and 0.01 μM. The effect was dose-dependent. The half-maximal inhibitory concentration (IC50) values were calculated to be 2.41 ± 0.94 μM for HOS cells at 24 hours and 9.69 ±3.96 μM at 48 hours, while MG63 cells had IC50 values of 3.01 ± 0.72 μM at 24 hours and 0.92 ±0.32 μM at 48 hours (Figure 1A and B).
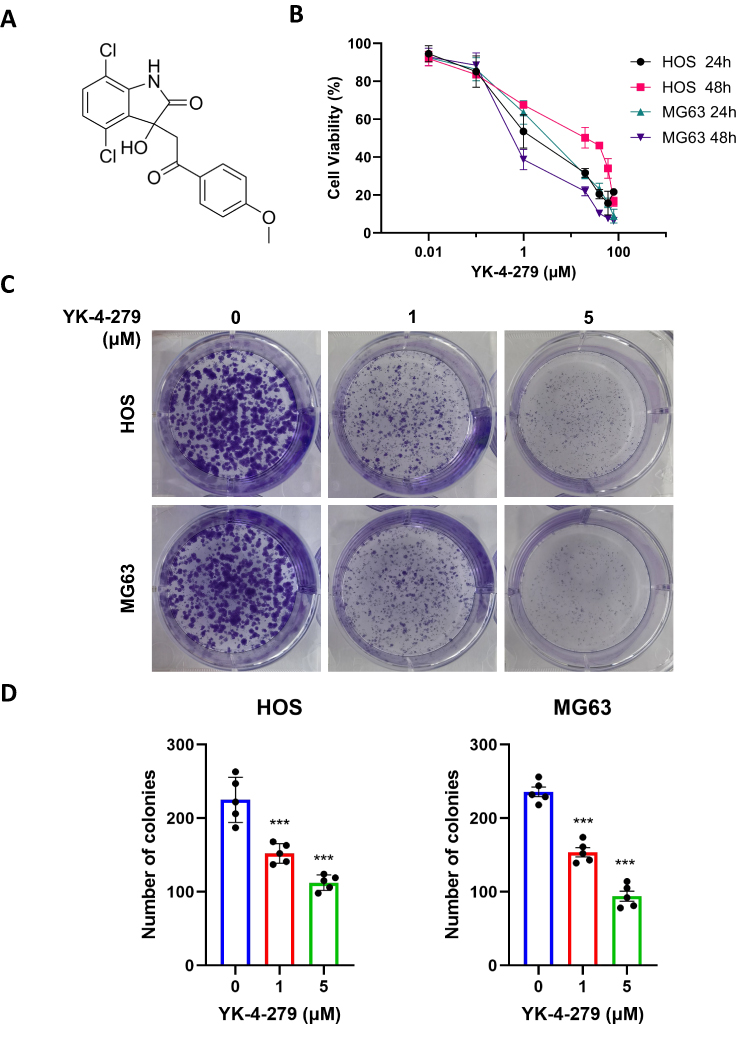 |
Figure 1 YK-4-279 significantly inhibits the growth of OS cells. (A) Chemical structure of YK-4-279 (CAS No.: 1,037,184–44-3). (B) HOS and MG63 cells were exposed to increasing concentrations of YK-4-279 for 24 and 48 hours, followed by evaluation of cell viability and IC50 values using the CCK-8 assay. (C) The effect of YK-4-279 (0, 1, 5 µM) on the clonogenic ability of HOS and MG63 cells was assessed using a colony formation assay. Colonies were stained with 0.1% crystalline violet staining solution and subsequently counted. (D) Clone formation was statistically analyzed. Experiments were performed with five biological replicates per treatment, and results are presented as mean ± SD. ***P < 0.001. |
To assess the cytotoxic effects of YK-4-279 on normal cells, osteoblast MC3T3-E1 cells and rat renal podocytes were exposed to various concentrations of YK-4-279 (80, 60, 40, 20, 1, 0.1, and 0.01 μM). The calculated IC50 values were 10.63 ± 4.95 μM for MC3T3-E1 cells and 14.47 ± 6.94 μM for rat renal podocytes, both measured over a 24 hours period. These results indicate that YK-4-279 exerts a lesser cytotoxic effect on normal cells compared to its effects on cancer cells (Figure S1).
The effect of YK-4-279 on cell proliferation in HOS and MG63 cells was then evaluated using a colony formation assay. HOS and MG63 cells were treated with 0, 1, and 5 µM concentrations of YK-4-279 for 24 hours, and the colony areas were significantly reduced at increasing concentrations of YK-4-279. Taken together, these results show that YK-4-279 significantly reduced the viability and proliferation of HOS and MG63 cells (Figure 1C and D).
YK-4-279 Induces Cell Cycle Arrest at the G2/M Phase in OS Cells
Cell cycle arrest contributes to the inhibition of cell growth.39 We assessed the mechanism by which YK-4-279 induces G2/M cell cycle arrest using flow cytometry. As shown in Figure 2A and B, treatment of HOS and MG63 cells with 0, 5, and 10 μM YK-4-279 for 24 hours resulted in a significant decrease in the percentage of cells in the G0/G1 phase compared with the control group, and a significant increase in the percentage of cells in the G2/M phase. The percentage of cells in the S phase decreased in treated HOS and MG63 cells compared to control cells. These results indicated that YK-4-279 induced cell cycle arrest in the G2/M phase in both HOS and MG63 cells.
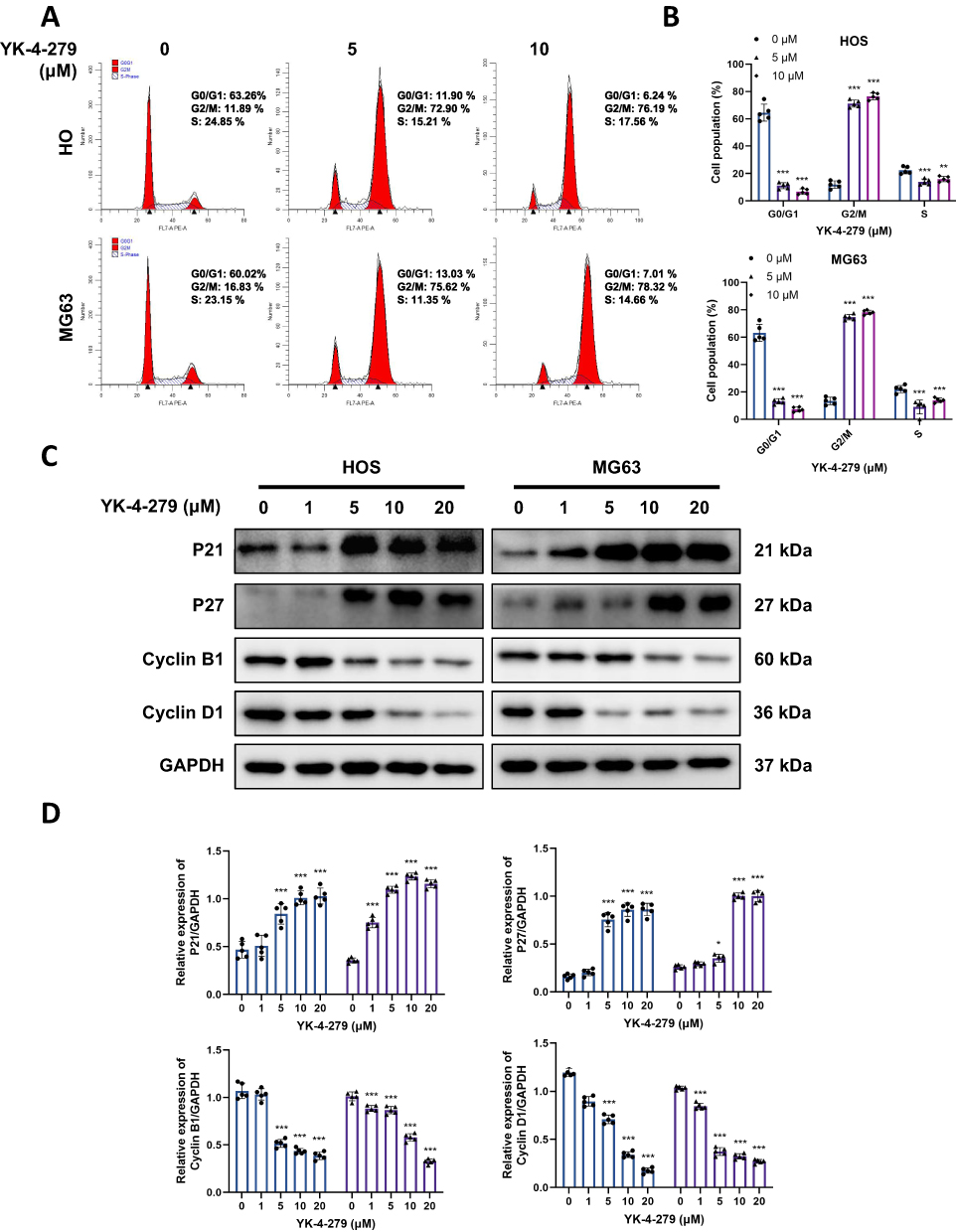 |
Figure 2 YK-4-279 induces cell cycle arrest at the G2/M phase in OS cells. HOS and MG63 cells were treated with YK-4-279 (0, 1, 5, 10, 20 µM in Western blot, 0, 5 µM in flow cytometry) for 24 h. (A) The average percentage of cells in G0/G1, S, and G2/M phases was determined and calculated by flow cytometry in HOS and MG63 cells. (B) Quantification of cell cycle distribution in HOS and MG63 cells was performed. (C) The protein expression levels of P21, P27, Cyclin B1, and Cyclin D1 were analyzed by Western blot in HOS and MG63 cells, with GAPDH serving as the loading control. (D) Quantitative analysis of the protein expression levels of P21, P27, Cyclin B1, and Cyclin D1 in HOS and MG63 cells was performed (●: HOS; ▲: MG63). Experiments were performed with five biological replicates per treatment, and results are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. |
Cyclin-dependent kinase (CDK) inhibitors p21 and p27 are well-known proteins that arrest cell cycle progression in response to various stimuli. Cyclin B1 and cyclin D1 promote the G2/M phase transition. We further tested whether YK-4-279 regulated the expression of p21, p27, cyclin B1, and cyclin D1 in HOS and MG63 cells by Western blot. The findings indicated that YK-4-279 treatment considerably increased p21 and p27 expression, while cyclin B1 and cyclin D1 expression was inhibited upon YK-4-279 treatment (Figure 2C and D). These findings suggest that YK-4-279 may inhibit OS cell proliferation by blocking the cell cycle at the G2/M phase.
YK-4-279 Triggered a Significant DNA Damage by DNA Damage Sensor Kinases in OS Cells
γ-H2AX is a sensitive marker of DSBs.40 To investigate whether the cytotoxic effects of YK-4-279 are correlated with DNA damage, we first analyzed the effects of YK-4-279 on DSBs by assessing the expression of γ-H2AX. After exposure to YK-4-279 for 24 hours, we assessed the protein level of γ-H2AX by Western blot. The results showed that YK-4-279 increased the expression of γ-H2AX (Figure 3A and B). We then used immunofluorescence to detect γ-H2AX expression. The immunofluorescence staining showed that YK-4-279 increased γ-H2AX formation (green) in the nucleus (Figure 3C and D), consistent with the Western blot results. The results indicated that YK-4-279 upregulated γ-H2AX expression in a concentration-dependent manner.
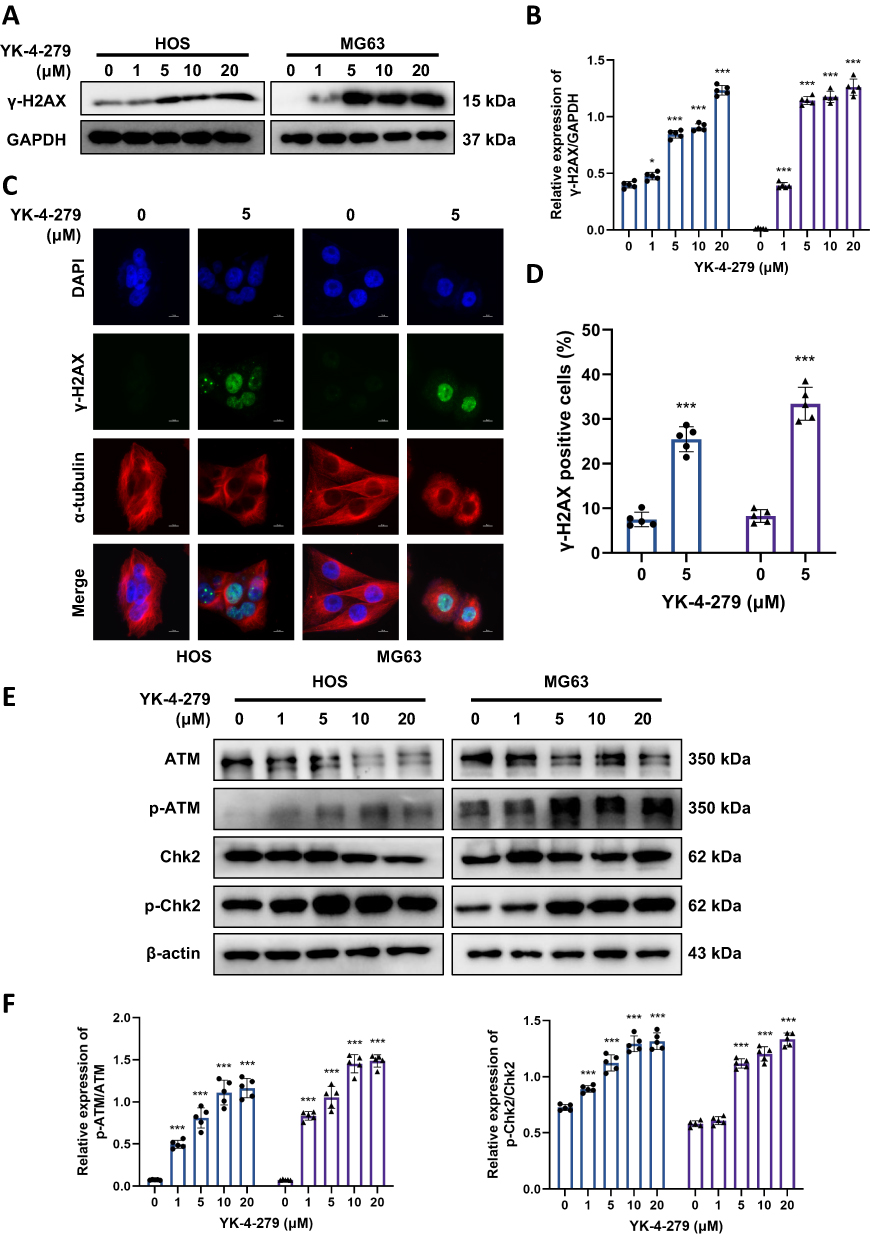 |
Figure 3 YK-4-279 triggered a significant DNA damage by DNA damage sensor kinases in OS cells. HOS and MG63 cells were treated with YK-4-279 (0, 1, 5, 10, 20 µM in Western blot, 0, 5 µM in immunofluorescence) for 24 h. (A) The protein expression levels of γ-H2AX in HOS and MG63 cells were analyzed by Western blot, with GAPDH serving as the loading control. (B) Quantitative analysis of the protein expression levels of γ-H2AX in HOS and MG63 cells was performed. (C) The localization of γ-H2AX expression was assessed by immunofluorescence in HOS and MG63 cells following treatment with YK-4-279. Scale bar = 10 µm. (D) Quantification of γ-H2AX fluorescence intensity per nucleus in HOS and MG63 cells was performed. (E) The protein expression levels of ATM, p-ATM, Chk2, and p-Chk2 were analyzed by Western blot, with β-actin serving as the loading control. (F) Quantitative analysis of the protein expression levels of ATM, p-ATM, Chk2, and p-Chk2 in HOS and MG63 cells was performed (●: HOS; ▲: MG63). Experiments were performed with five biological replicates per treatment, and results are presented as mean ± SD. *P < 0.05, ***P < 0.001. |
To further elucidate the mechanisms underlying YK-4-279-mediated DNA damage, we examined the phosphorylation of ATM and Chk2 following YK-4-279 exposure in both HOS and MG63 cells. Western blot results showed that YK-4-279 increased the phosphorylation of ATM and Chk2 in a concentration-dependent manner (Figure 3E and F). These results suggest that YK-4-279 can induce DNA damage, thereby inhibiting OS cell viability and proliferation.
YK-4-279 Triggers Apoptosis in OS Cells
When severe DNA damage exceeds the cell’s ability to repair, it may lead to cell death.41 To determine whether YK-4-279 could induce OS cell death, we assessed it using the Hoechst 33342/PI double staining kit. After 24 hours of treatment with YK-4-279 (5 µM), a red fluorescent reaction was observed in the nuclei of HOS and MG63 cells. This showed that YK-4-279 significantly induced cell death in HOS and MG63 cells (Figure 4A and B). To further determine whether apoptosis occurred under YK-4-279 exposure, we performed Annexin V-FITC/PI staining by flow cytometry. The results showed that the percentage of apoptotic cells increased gradually in HOS and MG63 cells upon exposure to 5 μM or 10 μM YK-4-279. These results showed that YK-4-279 could induce apoptosis in HOS and MG63 cells (Figure 4C and D). Next, we also used Western blotting to assess the activation of the apoptosis-related proteins PARP1 and Caspase 8. The results showed that the levels of cleaved PARP1 and cleaved Caspase 8 increased in a concentration-dependent manner (Figure 4E and F). These results suggest that YK-4-279 confers cytotoxicity through apoptosis in HOS and MG63 cells.
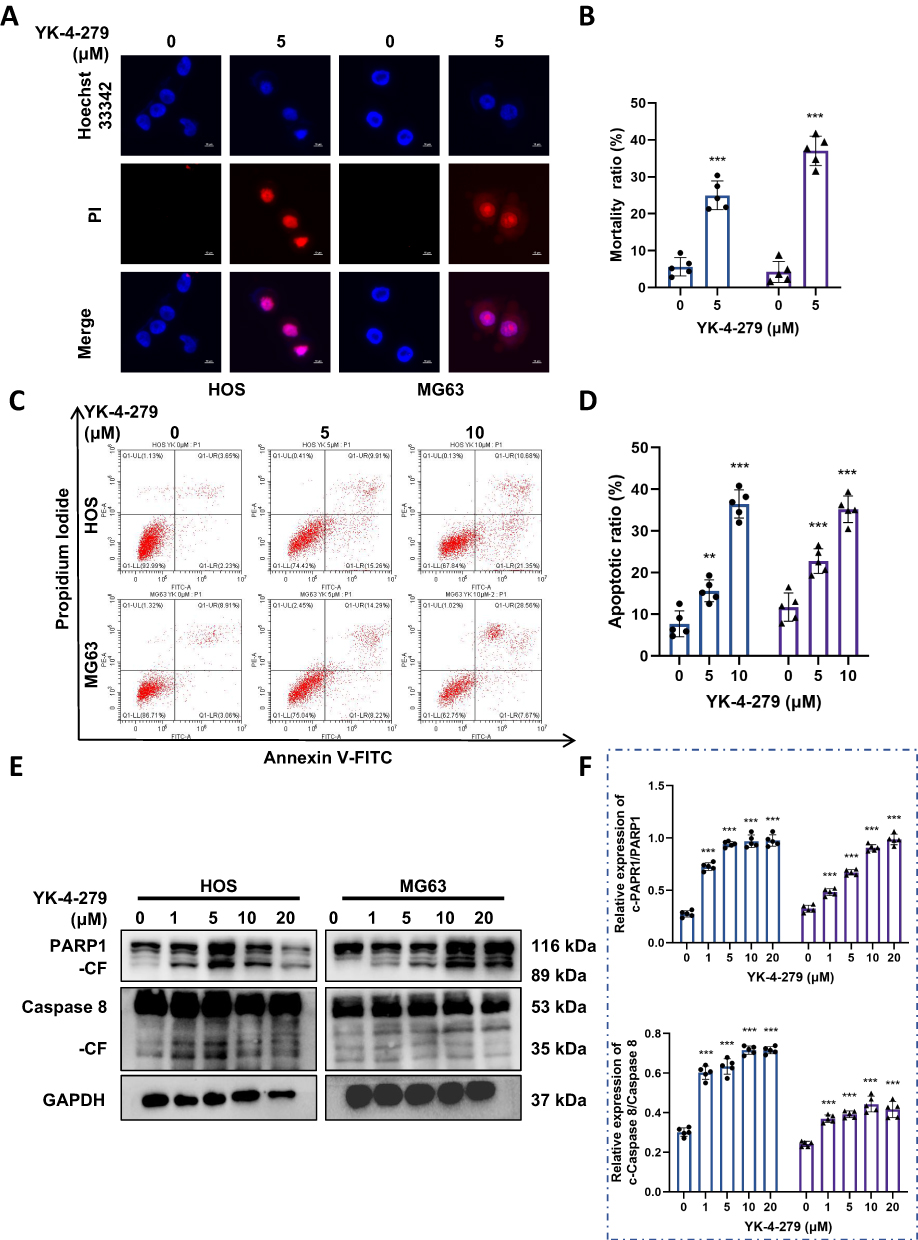 |
Figure 4 YK-4-279 triggers apoptosis in OS cells. HOS and MG63 cells were treated with YK-4-279 (0, 5 µM in immunofluorescence, 0, 5, 10 µM in flow cytometry, 0, 1, 5, 10, 20 µM in Western blot) for 24 h. (A) Cell death was assessed using Hoechst 33342/PI staining in HOS and MG63 cells, with scale bar = 10 µm. (B) Quantification of cell death marker-positive cells in HOS and MG63 cells was performed. (C) Apoptosis in HOS and MG63 cells was assessed using Annexin V-FITC/PI staining followed by flow cytometry analysis. (D) Quantification of the apoptosis rate in HOS and MG63 cells was performed. (E) The protein expression levels of PARP1 and caspase 8 were analyzed by Western blot in HOS and MG63 cells, with GAPDH serving as the loading control. (F) Quantitative analysis of the protein expression levels of PARP1 and caspase 8 in HOS and MG63 cells was performed (●: HOS; ▲: MG63). Experiments were performed with five biological replicates per treatment, and results are presented as mean ± SD. **P < 0.01, ***P < 0.001. |
YK-4-279 Induces Activation of MAPK Pathways in OS Cells
After confirming that YK-4-279 induces DNA damage, G2/M phase arrest, and apoptosis in osteosarcoma cells, we sought to identify the key signaling pathway potentially coordinating these diverse phenotypic responses. Given the critical role of the MAPK pathway (particularly the stress-responsive p38 and JNK subfamilies) in integrating DNA damage signals and transmitting them to cell cycle checkpoints and apoptotic outcomes,42,43 we hypothesized that this pathway might serve as the core mediator of YK-4-279’s effects. To validate this hypothesis, we first assessed whether YK-4-279 affected the MAPK pathway in HOS and MG63 cells by detecting the phosphorylation levels of ERK1/2, JNK, and p38 MAPK. Western blot analysis revealed that YK-4-279 treatment activated the phosphorylation of ERK1/2, JNK, and p38 MAPK in both cell lines in a dose-dependent manner (Figure 5A and B). This preliminary finding demonstrates the rapid activation of the MAPK pathway following drug exposure, providing direct evidence for subsequent functional studies. These results indicate that YK-4-279 activates the MAPK signaling pathway in osteosarcoma cells.
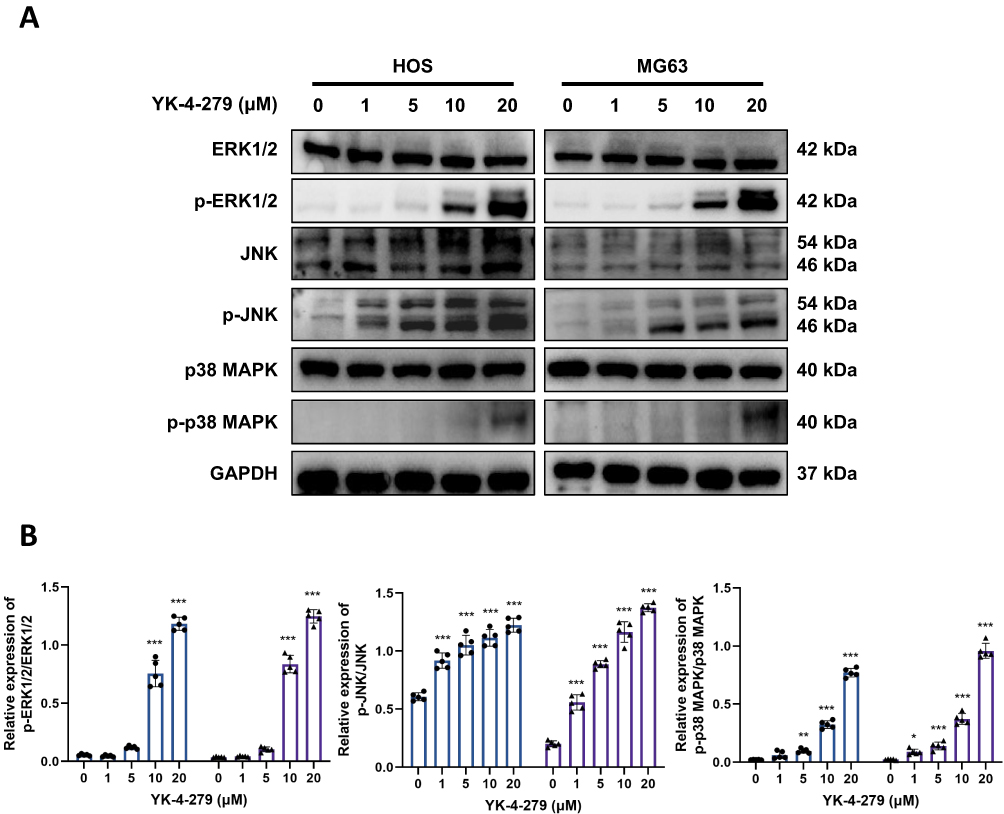 |
Figure 5 YK-4-279 induces activation of MAPK pathways in OS cells. (A and B) The expressions of ERK1/2, JNK, and p38 MAPK, along with their phosphorylation levels, were measured following treatment with various concentrations (0, 1, 5, 10, and 20 µM) of YK-4-279 for 24 hours in HOS and MG63 cells using Western blot analysis. Data were then analyzed (●: HOS; ▲: MG63). Experiments were performed with five biological replicates per treatment, and results are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. |
Inhibiting MAPK Pathways Attenuates YK-4-279 Cytotoxic Activity on OS Cells
To further investigate whether YK-4-279 inhibits cell viability, induces DNA damage, and triggers apoptosis through activation of the MAPK signaling pathway, we used SB203580 (p38 MAPK inhibitor) with or without YK-4-279 treatment in HOS and MG63 cells.
First, we assessed DNA damage in HOS and MG63 cells using fluorescent staining. As shown, the intensity of γ-H2AX fluorescence was significantly diminished in the SB203580 combined with YK-4-279 group compared with the YK-4-279 alone group (Figure 6A and B). These results suggest that YK-4-279 induces DNA damage through activation of the MAPK signaling pathway.
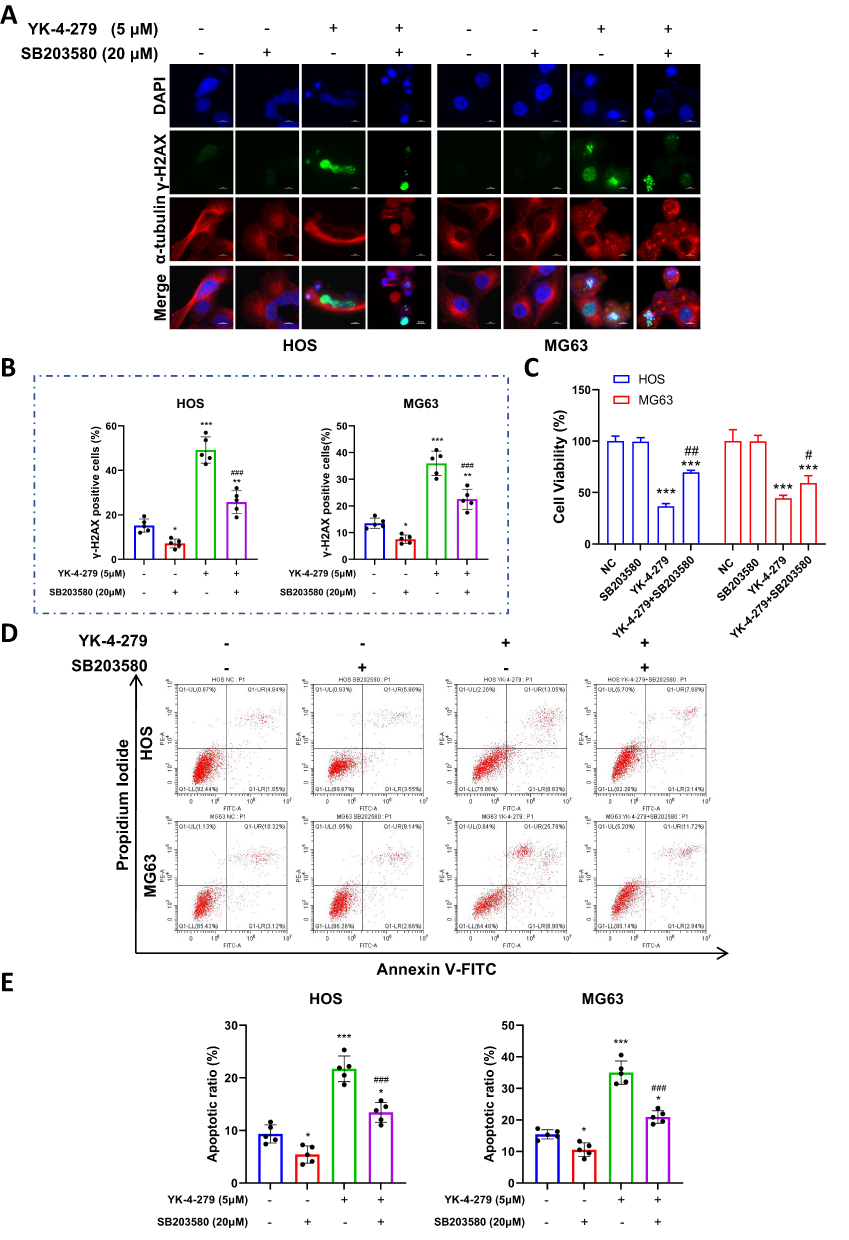 |
Figure 6 Inhibiting MAPK pathways attenuates YK-4-279 cytotoxic activity on OS cells. HOS and MG63 cells were treated with YK-4-279 and SB203580 (p38 MAPK inhibitor) for 24 hours. (A and B) Changes in γ-H2AX fluorescence foci following drug treatment were assessed by immunofluorescence in HOS and MG63 cells, and the data were analyzed. (C) Cell viability changes following drug treatment were assessed using the CCK-8 assay in HOS and MG63 cells, and the data were analyzed. (D and E) Changes in apoptosis in HOS and MG63 cells following drug treatment were assessed using Annexin V-FITC/PI staining followed by flow cytometry, and the data were analyzed. Experiments were performed with five biological replicates per treatment, and results are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001: drug treatment group vs control. # < 0.05, ## P < 0.01, ###P < 0.001: co-treatment group vs YK-4-279. |
A CCK-8 assay was then performed to assess changes in cell viability. YK-4-279 significantly reduced cell viability in HOS and MG63 cells, but in the YK-4-279 co-administered with SB203580 group, cell viability in HOS and MG63 cells increased (Figure 6C). These results suggest that YK-4-279 inhibits cell viability through activation of the MAPK signaling pathway.
Next, we assessed changes in apoptosis in HOS and MG63 cells using flow cytometry. The results showed that in HOS cells, the apoptosis rate increased in the YK-4-279-exposed group, while it decreased in the SB203580 combined with YK-4-279 group. In MG63 cells, the apoptosis rate increased in the YK-4-279-exposed group, while it decreased in the SB203580 combined with YK-4-279 group (Figure 6D and E).
Finally, we used Western blot analysis to detect the expression of p-p38 MAPK, PARP1, and γ-H2AX proteins. The findings showed that the combination of SB203580 and YK-4-279 significantly decreased the protein levels of p-p38 MAPK, cleaved PARP1 (CF-PARP1), and γ-H2AX in HOS and MG63 cells compared to YK-4-279 alone (Figure 7A and B).
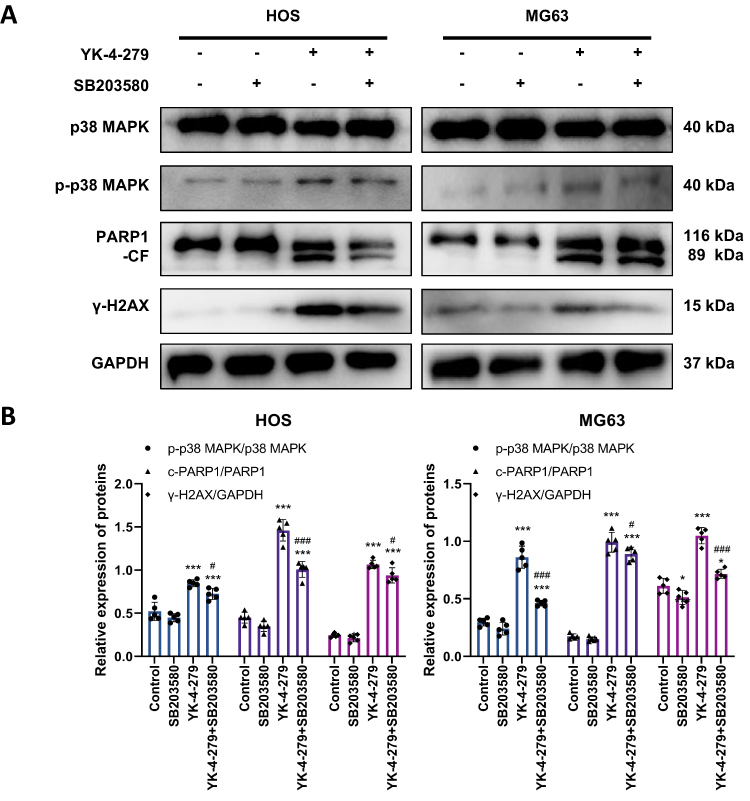 |
Figure 7 Inhibition MAPK pathway attenuates the effects of YK-4-279 on apoptosis and DNA damage proteins in OS cells. HOS and MG63 cells were treated with YK-4-279 and SB203580 (p38 MAPK inhibitor) for 24 hours. (A) Protein expression levels of p38 MAPK, p-p38 MAPK, PARP1, and γ-H2AX were analyzed by Western blot in HOS and MG63 cells after drug treatment. GAPDH was used as the internal control. (B) Quantitative analysis of the protein expression levels of p38 MAPK, p-p38 MAPK, PARP1, and γ-H2AX in HOS and MG63 cells was performed. Experiments were performed with five biological replicates per treatment, and results are presented as mean ± SD. *P < 0.05, ***P < 0.001: drug treatment group vs control. # < 0.05, ### P < 0.001: co-treatment group vs YK-4-279. |
Altogether, these findings indicated that YK-4-279 inhibited cell viability, induced DNA damage, and triggered apoptosis through activation of the MAPK signaling pathway.
YK-4-279 Inhibits Tumor Growth in Nude Mice
To evaluate the in vivo effect of YK-4-279 on osteosarcoma, MG63 cells were subcutaneously injected into nude mice, which were then treated daily with either control (PBS) or YK-4-279 at a specified dose (30 mg/kg/day). Tumor size was measured every three days to generate the tumor growth curve (Figure S2A). Notably, the administration of YK-4-279 was well-tolerated, with no observed side effects such as poor mental state or body weight loss (Figure S2B). After 28 days, the mice were euthanized, and tumors were collected and weighed (Figure S2C and D). Compared with the control group, YK-4-279 treatment significantly slowed tumor growth. Western blot analysis revealed that YK-4-279 treatment activated the expression of γ-H2AX and induced the cleavage of PARP1, along with an increase in p-P38 MAPK levels (Figure S2E and F). Additionally, serum biochemical analysis of the mice showed no significant differences in liver and kidney function indicators between the two groups (Figure S2G). These results indicate that YK-4-279 not only effectively inhibits osteosarcoma growth but also exhibits no significant toxicity at the therapeutic dose.
Discussion
Our results showed that YK-4-279 significantly inhibited OS cell viability and proliferation, induced DNA damage, blocked cell cycle progression, and triggered apoptosis. In addition, YK-4-279 significantly activated the MAPK signaling pathway in OS cells, and may inhibit cell viability and proliferation, induce DNA damage, arrest cell cycle progression, and trigger apoptosis through MAPK pathway activation. This study provides both an experimental basis and a theoretical rationale for the use of the ETV1 inhibitor YK-4-279 in future clinical applications, offering new perspectives for anti-OS therapy. Targeted inhibition of ETS activity by YK-4-279 has the potential to offer a promising therapeutic modality for OS.
Tumors arise from cells that have lost control of their proliferation, making it crucial to inhibit the abnormal proliferation of tumor cells. YK-4-279 can inhibit prostate cancer growth and metastasis in a mouse xenograft model.44 YK-4-279 inhibits the activity of lymphoma cell lines, and its antiproliferative effects are dose-dependent.35 In melanoma studies, YK-4-279 was shown to significantly reduce the growth and viability of three different melanoma cell lines, It has a significant impact on blocking or delaying tumor progression.45 Our results showed that YK-4-279 significantly reduced OS cell viability and inhibited cell proliferation, indicating its therapeutic potential for OS.
OS is a disease characterized by abnormal cell cycle regulation, closely linked to disruptions in its molecular mechanisms. Cell cycle blockade is tightly regulated by specific cyclins and CDK complexes.46 Cyclin-dependent kinase interacting proteins, such as p21 and p27, act as tumor suppressors by inhibiting CDK/cyclin complexes, thereby restricting the cell cycle and blocking DNA replication.47,48 Cyclin D1, a key target in cancer therapy, influences the cell cycle and cell growth.49 Cyclin B plays a crucial role in activating mitosis during the G2/M phase.50 YK-4-279 rapidly induces G2/M phase blockade in ES cells, reduces Cyclin B1 abundance, decreases microtubule-associated protein production, and ultimately leads to growth arrest and apoptosis.51 Previous studies have shown that methyl protodioscin induces apoptosis in human osteosarcoma cells through caspase-dependent and MAPK signaling pathways.52 Xie DM’s research indicates that Tanshinone II A enhances the sensitivity of osteosarcoma cells to chemotherapeutic drugs by activating the p38/MAPK pathway. This activation leads to the upregulation of cleaved caspase-3 and the pro-apoptotic gene Bax, while downregulating the expression of caspase-3 and the anti-apoptotic gene Bcl-2.53 Our data showed that YK-4-279 blocked the OS cell cycle at the G2/M phase, significantly activated p21 and p27, and inhibited the expression levels of Cyclin B1 and Cyclin D1. These findings suggest that YK-4-279 may inhibit OS cell proliferation by blocking the cell cycle at the G2/M phase.
Anti-cancer drugs inhibit the proliferation of cancer cells by inducing DNA damage through targeting growth signaling molecules involved in cell replication and the division of rapidly proliferating cells.54,55 DNA damage activates the upstream DNA damage response kinase, ataxia telangiectasia mutated (ATM), and the downstream checkpoint kinase CHK2. ATM then induces the phosphorylation of the histone variant H2A histone family member X (H2AX), leading to the formation of γ-H2AX.56 We observed γ-H2AX foci formation and elevated γ-H2AX protein expression in OS cells after treatment with YK-4-279, indicating early signs of DSBs in OS cells. When DNA is damaged, the ATM-CHK2 pathway is activated, triggering cell cycle checkpoints, apoptosis, or DNA repair, among other responses. Our results show that YK-4-279 induces the phosphorylation of ATM and CHK2 in OS cells. These results suggest that YK-4-279 can induce DNA damage, thereby inhibiting OS cell viability and proliferation.
It is well known that inhibition of cell proliferation is closely linked to apoptosis, which plays a crucial role in anti-tumor mechanisms. Therefore, apoptosis has become a key indicator for assessing anticancer efficacy.57 Apoptosis is an irreversible cascade process driven by the activation of pro-apoptotic proteins, specifically the cysteine protease family (caspases).58 Caspase 8 plays a crucial role in death receptor-mediated apoptosis.59,60 Cleavage of PARP1 is commonly considered a marker of apoptosis. YK-4-279 is a potent mitotic inhibitor that induces apoptosis in various neuroblastoma cell lines with different oncogenic drivers.36 In a xenograft mouse model, YK-4-279 induced apoptosis and slowed tumor growth in ES cell lines by inhibiting EWS-FLI1 activity.61 The combination of low-dose docetaxel with YK-4-279 enhanced apoptosis induction, reduced motility, and decreased invasiveness in prostate cancer cells.62 In this study, we found that YK-4-279 significantly increased the rate of apoptosis in OS cells and promoted elevated levels of PARP1 and Caspase 8 cleavage products, consistent with previous reports. These results suggest that YK-4-279 may have therapeutic potential in the treatment of OS by promoting apoptosis in OS cells, effectively inhibiting the growth and progression of OS.
The Mitogen-Activated Protein Kinase (MAPK) signaling pathway plays a crucial role in the induction of apoptosis by anticancer drugs. Key members of the MAPK family, including ERK, JNK, and P38, regulate cell differentiation, proliferation, apoptosis, and survival.63 Since several ETS factors are direct targets of the MAPK signaling pathway.64,65 Studying the activation of this pathway by YK-4-279 is crucial for the treatment of OS. Our experimental results show that YK-4-279 activates ERK1/2, JNK, and P38 MAPK, significantly increasing the expression levels of their phosphorylated forms. Studies have shown that activation of JNK and P38 MAPK can induce apoptosis through G2/M phase arrest,66,67 which aligns with our results.
To further verify whether YK-4-279 exerts its inhibitory effects on OS through the MAPK signaling pathway, we co-treated cells with YK-4-279 and a P38 MAPK inhibitor to observe whether the inhibitor could reverse YK-4-279’s effects. The results showed that the P38 inhibitor partially neutralized the effects of YK-4-279, reducing apoptosis in OS cells, partially restoring DNA integrity, and attenuating the activation of the MAPK signaling pathway induced by YK-4-279. These findings suggest that YK-4-279 may inhibit OS cell viability and proliferation by activating the MAPK signaling pathway, inducing DNA damage and cell cycle arrest, and promoting apoptosis, thereby exerting its anti-OS activity.
YK-4-279 has demonstrated anticancer effects across a variety of tumor types, highlighting its potential as a promising therapeutic approach. Our in vitro experiments in OS cells show that YK-4-279 holds significant potential for the future treatment of OS. In summary, YK-4-279 inhibited OS cell viability and proliferation, induced cell cycle arrest, promoted apoptosis, and suppressed OS cell growth and progression. The antitumor activity of YK-4-279 in OS may be mediated through the activation of the MAPK signaling pathway. YK-4-279 shows strong potential as a therapeutic agent for OS treatment.
Furthermore, this study demonstrated that YK-4-279 significantly suppressed tumor growth in an MG63 osteosarcoma xenograft model. The underlying mechanism may be associated with the activation of the DNA damage response (as evidenced by upregulated γ-H2AX expression), the apoptotic pathway (indicated by PARP1 cleavage), and the p38 MAPK stress signaling pathway. Notably, at the effective dose (30 mg/kg/day), the treatment did not induce significant changes in body weight or abnormalities in hepatic and renal function parameters, indicating that YK-4-279 possesses both potent in vivo antitumor efficacy and a favorable safety profile.
This study has limitations, and future research will expand in multiple directions. First, we plan to include a broader variety of osteosarcoma cell lines to better simulate the tumor microenvironment and comprehensively evaluate the antitumor activity and mechanisms of action of YK-4-279. Additionally, we will refine our animal experiments to assess the efficacy and safety of YK-4-279 in vivo. Furthermore, pharmacokinetic and pharmacodynamic studies are essential for determining the optimal dosing regimen and evaluating the drug’s stability in circulation. We may also explore structural modifications to YK-4-279 to enhance solubility, reduce potential toxicity, and maintain target specificity. Additionally, we will investigate potential combination therapy regimens with traditional chemotherapy or immunotherapy to potentially enhance its antitumor efficacy and overcome resistance mechanisms. This approach aims to advance the preclinical optimization of YK-4-279 and establish a solid foundation for its clinical translation.
Conclusion
This study investigates the anticancer effects and underlying mechanisms of YK-4-279 in OS. The results of our in vitro experiments revealed that YK-4-279 effectively inhibits OS cell viability and proliferation, induces G2/M phase cell cycle arrest, promotes apoptosis and induces DNA damage. We further observed that YK-4-279 activates the MAPK signaling pathway, including key proteins such as ERK1/2, JNK, and P38 MAPK. Phosphorylation of these proteins was significantly increased upon treatment with YK-4-279. Additionally, co-treatment with a P38 MAPK inhibitor partially reversed the effects of YK-4-279, highlighting the crucial role of P38 MAPK in mediating its antitumor effects. In conclusion, YK-4-279 exerts its antitumor activity in OS through the activation of the MAPK signaling pathway, leading to inhibition of cell viability, cell cycle arrest, DNA damage, and apoptosis. These findings suggest that YK-4-279 holds great promise as a therapeutic agent for the treatment of osteosarcoma.
Abbreviations
OS, Osteosarcoma; ETS, The E26 transformation-specific; ETV1, ETS variant 1; CDK, Cyclin-dependent kinase; DSBs, DNA double-strand breaks; ATM, ataxia telangiectasia mutated; MAPK, Mitogen-Activated Protein Kinase.
Ethics Statement
All animal experiments and the use of cell lines in this study were approved by the Medical Ethics Committee of Liaocheng People’s Hospital (Approval No. 2022063) and were conducted in strict accordance with the NIH guidelines for animal care and use and the principles of the Declaration of Helsinki. The HOS (ZQ0952) and MG63 (ZQ0403) cell lines were purchased by Dr. Zhang Lu from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd., and were authenticated by short tandem repeat (STR) profiling to confirm their identity and exclude cross-contamination.
Funding
This study was funded by the Natural Science Foundation of Shandong Province (No. ZR2023QH034).
Disclosure
The authors declare that there are no conflicts of interest in this work.
References
1. Moukengue B, Lallier M, Marchandet L, et al. Origin and therapies of osteosarcoma. Cancers. 2022;14(14):3503. doi:10.3390/cancers14143503
2. Shoaib Z, Fan TM, Irudayaraj JMK. Osteosarcoma mechanobiology and therapeutic targets. Br J Pharmacol. 2022;179(2):201–217. doi:10.1111/bph.15713
3. Li J, Gu A, Tang N, et al. Patient-derived xenograft models in pan-cancer: from bench to clinic. Interdiscip Med. 2025;3(5):e20250016. doi:10.1002/INMD.20250016
4. Zhao J, Dean DC, Hornicek FJ, Yu X, Duan Z. Emerging next-generation sequencing-based discoveries for targeted osteosarcoma therapy. Cancer Lett. 2020;474:158–167. doi:10.1016/j.canlet.2020.01.020
5. Chen R, Wang G, Zheng Y, Hua Y, Cai Z. Drug resistance-related microRNAs in osteosarcoma: translating basic evidence into therapeutic strategies. J Cell Mol Med. 2019;23(4):2280–2292. doi:10.1111/jcmm.14064
6. Barbosa FAR, Siminski T, Canto RFS, et al. Novel pyrimidinic selenourea induces DNA damage, cell cycle arrest, and apoptosis in human breast carcinoma. Eur J Med Chem. 2018;155:503–515. doi:10.1016/j.ejmech.2018.06.026
7. Hurley LH. DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer. 2002;2(3):188–200. doi:10.1038/nrc749
8. Huang RX, Zhou PK. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Trans Targeted Ther. 2020;5(1):60. doi:10.1038/s41392-020-0150-x
9. Li Z, Pearlman AH, Hsieh P. DNA mismatch repair and the DNA damage response. DNA Repair. 2016;38:94–101. doi:10.1016/j.dnarep.2015.11.019
10. Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59(4):928–942. doi:10.1016/j.ijrobp.2004.03.005
11. He G, Siddik ZH, Huang Z, et al. Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene. 2005;24(18):2929–2943. doi:10.1038/sj.onc.1208474
12. Wang JYJ. Cell death response to DNA damage. Yale J Biol Med. 2019;92(4):771–779.
13. Ishikawa K, Ishii H, Saito T. DNA damage-dependent cell cycle checkpoints and genomic stability. DNA Cell Biol. 2006;25(7):406–411. doi:10.1089/dna.2006.25.406
14. Angius G, Tomao S, Stati V, Vici P, Bianco V, Tomao F. Prexasertib, a checkpoint kinase inhibitor: from preclinical data to clinical development. Cancer Chemother Pharmacol. 2020;85(1):9–20. doi:10.1007/s00280-019-03950-y
15. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
16. Huang M, Gong G, Deng Y, et al. Crosstalk between cancer cells and the nervous system. Med Adv. 2023;1(3):173–189. doi:10.1002/med4.27
17. Ma P, Wang G, Men K, et al. Advances in clinical application of nanoparticle-based therapy for cancer treatment: a systematic review. Nano TransMed. 2024;3:100036. doi:10.1016/j.ntm.2024.100036
18. Zinn S, Vazquez-Lombardi R, Zimmermann C, Sapra P, Jermutus L, Christ D. Advances in antibody-based therapy in oncology. Nat Cancer. 2023;4(2):165–180. doi:10.1038/s43018-023-00516-z
19. Zhou C, Solomon B, Loong HH, et al. First-line selpercatinib or chemotherapy and pembrolizumab in RET fusion-positive NSCLC. N Engl J Med. 2023;389(20):1839–1850. doi:10.1056/NEJMoa2309457
20. Tilly H, Morschhauser F, Sehn LH, et al. Polatuzumab vedotin in previously untreated diffuse large B-cell lymphoma. N Engl J Med. 2022;386(4):351–363. doi:10.1056/NEJMoa2115304
21. Qin S, Chan SL, Gu S, et al. Camrelizumab plus rivoceranib versus sorafenib as first-line therapy for unresectable hepatocellular carcinoma (CARES-310): a randomised, open-label, international Phase 3 study. Lancet. 2023;402(10408):1133–1146. doi:10.1016/S0140-6736(23)00961-3
22. Zhang Y, Zheng WH, Zhou SH, et al. Molecular genetics, therapeutics and RET inhibitor resistance for medullary thyroid carcinoma and future perspectives. Cell Commun Signal. 2024;22(1):460. doi:10.1186/s12964-024-01837-x
23. Wang Y, Huang Z, Sun M, Huang W, Xia L. ETS transcription factors: multifaceted players from cancer progression to tumor immunity. Biochimica Et Biophysica Acta-Rev Cancer. 2023;1878(3):188872. doi:10.1016/j.bbcan.2023.188872
24. Saulnier O, Guedri-Idjouadiene K, Aynaud -M-M, et al. ERG transcription factors have a splicing regulatory function involving RBFOX2 that is altered in the EWS-FLI1 oncogenic fusion. Nucleic Acids Res. 2021;49(9):5038–5056. doi:10.1093/nar/gkab305
25. Lin L, Huang M, Shi X, et al. Super-enhancer-associated MEIS1 promotes transcriptional dysregulation in Ewing sarcoma in co-operation with EWS-FLI1. Nucleic Acids Res. 2019;47(3):1255–1267. doi:10.1093/nar/gky1207
26. Boone MA, Taslim C, Crow JC, et al. Identification of a novel FUS/ETV4 fusion and comparative analysis with other Ewing sarcoma fusion proteins. Mol Cancer Res. 2021;19(11):1795–1801. doi:10.1158/1541-7786.MCR-21-0354
27. Liu Z, Ren Z, Zhang C, et al. ELK3: a new molecular marker for the diagnosis and prognosis of glioma. Front Oncol. 2021;11:608748. doi:10.3389/fonc.2021.608748
28. Mehtonen J, Teppo S, Lahnalampi M, et al. Single cell characterization of B-lymphoid differentiation and leukemic cell states during chemotherapy in ETV6-RUNX1-positive pediatric leukemia identifies drug-targetable transcription factor activities. Genome Med. 2020;12(1):99. doi:10.1186/s13073-020-00799-2
29. Ben-David Y, Gajendran B, Sample KM, Zacksenhaus E. Current insights into the role of Fli-1 in hematopoiesis and malignant transformation. CMLS-Cellular Mol Life Sci. 2022;79(3):163. doi:10.1007/s00018-022-04160-1
30. Hwang SY, Park S, Jo H, et al. Interrupting specific hydrogen bonds between ELF3 and MED23 as an alternative drug resistance-free strategy for HER2- overexpressing cancers. J Adv Res. 2023;47:173–187. doi:10.1016/j.jare.2022.08.003
31. Ma Y, Xu B, Yu J, et al. Fli-1 activation through targeted promoter activity regulation using a novel 3’, 5’-diprenylated chalcone inhibits growth and metastasis of prostate cancer cells. Int J Mol Sci. 2020;21(6):2216. doi:10.3390/ijms21062216
32. Eid W, Abdel-Rehim W. Genome-wide analysis of ETV1 targets: insights into the role of ETV1 in tumor progression. J Cell Biochem. 2019;120(6):8983–8991. doi:10.1002/jcb.28169
33. Wu SP, Cooper BT, Bu F, et al. DNA methylation-based classifier for accurate molecular diagnosis of bone sarcomas. JCO Precis Oncol. 2017;2017. doi:10.1200/PO.17.00031
34. Erkizan HV, Kong Y, Merchant M, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat Med. 2009;15(7):750–756. doi:10.1038/nm.1983
35. Rahim S, Beauchamp EM, Kong Y, Brown ML, Toretsky JA, Üren A. YK-4-279 inhibits ERG and ETV1 mediated prostate cancer cell invasion. PLoS One. 2011;6(4):e19343. doi:10.1371/journal.pone.0019343
36. Kollareddy M, Sherrard A, Park JH, et al. The small molecule inhibitor YK-4-279 disrupts mitotic progression of neuroblastoma cells, overcomes drug resistance and synergizes with inhibitors of mitosis. Cancer Lett. 2017;403:74–85. doi:10.1016/j.canlet.2017.05.027
37. Huang L, Zhai Y, Fajardo CD, et al. YK-4-279 attenuates progression of pre-existing pigmented lesions to nodular melanoma in a mouse model. Cancers. 2021;14(1):143. doi:10.3390/cancers14010143
38. Spriano F, Chung EYL, Gaudio E, et al. The ETS inhibitors YK-4-279 and TK-216 are novel antilymphoma agents. Clin Cancer Res. 2019;25(16):5167–5176. doi:10.1158/1078-0432.CCR-18-2718
39. Liu Z, Zhu Q, Song E, Song Y. Polybrominated diphenyl ethers quinone exhibits neurotoxicity by inducing DNA damage, cell cycle arrest, apoptosis and p53-driven adaptive response in microglia BV2 cells. Toxicology. 2021;457:152807. doi:10.1016/j.tox.2021.152807
40. Saito Y, Zhou H, Kobayashi J. Corrigendum: chromatin modification and NBS1: their relationship in DNA double-strand break repair [Genes Genet. Syst. (2015) 90, p. 195-208]. Genes Genet Syst. 2016;91(4):241. doi:10.1266/ggs.91.241
41. Zhang YY, Feng PP, Wang HF, et al. Licochalcone B induces DNA damage, cell cycle arrest, apoptosis, and enhances TRAIL sensitivity in hepatocellular carcinoma cells. Chem Biol Interact. 2022;365:110076. doi:10.1016/j.cbi.2022.110076
42. Moruno-Manchón JF, Pérez-Jiménez E, Knecht E. Glucose induces autophagy under starvation conditions by a p38 MAPK-dependent pathway. Biochem J. 2013;449(2):497–506. doi:10.1042/BJ20121122
43. Notte A, Ninane N, Arnould T, Michiels C. Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast cancer cells: role of autophagy and JNK activation. Cell Death Dis. 2013;4(5):e638. doi:10.1038/cddis.2013.167
44. Rahim S, Minas T, Hong SH, et al. A small molecule inhibitor of ETV1, YK-4-279, prevents prostate cancer growth and metastasis in a mouse xenograft model. PLoS One. 2014;9(12):e114260. doi:10.1371/journal.pone.0114260
45. Huang L, Zhai Y, La J, et al. Targeting Pan-ETS factors inhibits melanoma progression. Cancer Res. 2021;81(8):2071–2085. doi:10.1158/0008-5472.CAN-19-1668
46. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–166. doi:10.1038/nrc2602
47. Torres-Martinez Z, Delgado Y, Ferrer-Acosta Y, et al. Key genes and drug delivery systems to improve the efficiency of chemotherapy. Cancer Drug Resistance. 2021;4:163–191. doi:10.20517/cdr.2020.64
48. Shamloo B, Usluer S. p21 in cancer research. Cancers. 2019;11(8):1178. doi:10.3390/cancers11081178
49. Zheng J, Li Q, Wang W, et al. Apoptosis-related protein-1 acts as a tumor suppressor in cholangiocarcinoma cells by inducing cell cycle arrest via downregulation of cyclin-dependent kinase subunits. Oncol Rep. 2016;35(2):809–816. doi:10.3892/or.2015.4422
50. Fischer M, Müller GA. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit Rev Biochem Mol Biol. 2017;52(6):638–662. doi:10.1080/10409238.2017.1360836
51. Zöllner SK, Selvanathan SP, Graham GT, et al. Inhibition of the oncogenic fusion protein EWS-FLI1 causes G2-M cell cycle arrest and enhanced vincristine sensitivity in Ewing’s sarcoma. Sci Signaling. 2017;10(499). doi:10.1126/scisignal.aam8429
52. Tseng SC, Shen TS, Wu CC, et al. Methyl protodioscin induces apoptosis in human osteosarcoma cells by caspase-dependent and MAPK signaling pathways. J Agric Food Chem. 2017;65(13):2670–2676. doi:10.1021/acs.jafc.6b04800
53. Xie DM, Li ZY, Ren BK, et al. Tanshinone II A facilitates chemosensitivity of osteosarcoma cells to cisplatin via activation of p38 MAPK pathway. Chin J Integr Med. 2025;31(4):326–335. doi:10.1007/s11655-024-4118-5
54. Haider T, Tiwari R, Vyas SP, Soni V. Molecular determinants as therapeutic targets in cancer chemotherapy: an update. Pharmacol Ther. 2019;200:85–109. doi:10.1016/j.pharmthera.2019.04.011
55. Sadiq Z, Varghese E, Büsselberg D. Cisplatin’s dual-effect on the circadian clock triggers proliferation and apoptosis. Neurobiol Sleep Circadian Rhythms. 2020;9:100054. doi:10.1016/j.nbscr.2020.100054
56. Wang L, Chen J, Li Q, et al. Cigarette smoke extract induces malignant transformation and DNA damage via c-MET phosphorylation in human bronchial epithelial cells. Ecotoxicol Environ Saf. 2024;283:116985. doi:10.1016/j.ecoenv.2024.116985
57. Jiang G, Liu J, Ren B, et al. Anti-tumor and chemosensitization effects of cryptotanshinone extracted from salvia miltiorrhiza bge. on ovarian cancer cells in vitro. J Ethnopharmacol. 2017;205:33–40. doi:10.1016/j.jep.2017.04.026
58. Bai L, Wang S. Targeting apoptosis pathways for new cancer therapeutics. Ann Rev Med. 2014;65(1):139–155. doi:10.1146/annurev-med-010713-141310
59. Jiang C, Fang X, Jiang Y, et al. TNF-α induces vascular endothelial cells apoptosis through overexpressing pregnancy induced noncoding RNA in Kawasaki disease model. Int J Biochem Biotechnol. 2016;72:118–124. doi:10.1016/j.biocel.2016.01.011
60. Wu R, Shen D, Sohun H, et al. miR‑186, a serum microRNA, induces endothelial cell apoptosis by targeting SMAD6 in Kawasaki disease. IntJ Mol Med. 2018;41(4):1899–1908. doi:10.3892/ijmm.2018.3397
61. Minas TZ, Han J, Javaheri T, et al. Correction: YK-4-279 effectively antagonizes EWS-FLI1 induced leukemia in a transgenic mouse model. Oncotarget. 2024;15(1):143. doi:10.18632/oncotarget.28524
62. Yu L, Wu X, Chen M, et al. The effects and mechanism of YK-4-279 in combination with docetaxel on prostate cancer. Int J Med Sci. 2017;14(4):356–366. doi:10.7150/ijms.18382
63. Nussinov R, Tsai CJ, Jang H, Korcsmáros T, Csermely P. Oncogenic KRAS signaling and YAP1/β-catenin: similar cell cycle control in tumor initiation. Semin Cell Dev Biol. 2016;58:79–85. doi:10.1016/j.semcdb.2016.04.001
64. Charlot C, Dubois-Pot H, Serchov T, Tourrette Y, Wasylyk B. A review of post-translational modifications and subcellular localization of Ets transcription factors: possible connection with cancer and involvement in the hypoxic response. Methods Mol Biol. 2010;647:3–30. doi:10.1007/978-1-60761-738-9_1
65. Hollenhorst PC, Ferris MW, Hull MA, Chae H, Kim S, Graves BJ. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011;25(20):2147–2157. doi:10.1101/gad.17546311
66. Liu W, Ning R, Chen RN, et al. Aspafilioside B induces G2/M cell cycle arrest and apoptosis by up-regulating H-Ras and N-Ras via ERK and p38 MAPK signaling pathways in human hepatoma HepG2 cells. Mol Carcinogenesis. 2016;55(5):440–457. doi:10.1002/mc.22293
67. Thongsom S, Suginta W, Lee KJ, Choe H, Talabnin C. Piperlongumine induces G2/M phase arrest and apoptosis in cholangiocarcinoma cells through the ROS-JNK-ERK signaling pathway. Apoptosis. 2017;22(11):1473–1484. doi:10.1007/s10495-017-1422-y
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.