Back to Journals » Drug Design, Development and Therapy » Volume 10
Xyloketal-derived small molecules show protective effect by decreasing mutant Huntingtin protein aggregates in Caenorhabditis elegans model of Huntington’s disease
Authors Zeng Y, Guo W, Xu G, Wang Q, Feng L, Long S, Liang F, Huang Y, Lu X, Li S, Zhou J, Burgunder J, Pang J, Pei Z ![]()
Received 18 August 2015
Accepted for publication 4 February 2016
Published 13 April 2016 Volume 2016:10 Pages 1443—1451
DOI https://doi.org/10.2147/DDDT.S94666
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Yixuan Zeng,1,2,* Wenyuan Guo,1,* Guangqing Xu,3 Qinmei Wang,4 Luyang Feng,1,2 Simei Long,1 Fengyin Liang,1 Yi Huang,1 Xilin Lu,1 Shichang Li,5 Jiebin Zhou,5 Jean-Marc Burgunder,6 Jiyan Pang,5 Zhong Pei1,2
1Department of Neurology, National Key Clinical Department and Key Discipline of Neurology, Guangdong Key Laboratory for Diagnosis and Treatment of Major Neurological Disease, The First Affiliated Hospital, Sun Yat-sen University, 2Guangzhou Center, Chinese Huntington’s Disease Network, 3Department of Rehabilitation, The First Affiliated Hospital, 4Key laboratory on Assisted Circulation, Ministry of Health, Department of Cardiovascular Medicine of the First Affiliated Hospital, 5School of Chemistry and Chemical Engineering, Sun Yat-sen University, Guangzhou, Guangdong, People’s Republic of China; 6Swiss Huntington’s Disease Center, Department of Neurology, University of Bern, Bern, Switzerland
*These authors contributed equally to this work
Abstract: Huntington’s disease is an autosomal-dominant neurodegenerative disorder, with chorea as the most prominent manifestation. The disease is caused by abnormal expansion of CAG codon repeats in the IT15 gene, which leads to the expression of a glutamine-rich protein named mutant Huntingtin (Htt). Because of its devastating disease burden and lack of valid treatment, development of more effective therapeutics for Huntington’s disease is urgently required. Xyloketal B, a natural product from mangrove fungus, has shown protective effects against toxicity in other neurodegenerative disease models such as Parkinson’s and Alzheimer’s diseases. To identify potential neuroprotective molecules for Huntington’s disease, six derivatives of xyloketal B were screened in a Caenorhabditis elegans Huntington’s disease model; all six compounds showed a protective effect. Molecular docking studies indicated that compound 1 could bind to residues GLN369 and GLN393 of the mutant Htt protein, forming a stable trimeric complex that can prevent the formation of mutant Htt aggregates. Taken together, we conclude that xyloketal derivatives could be novel drug candidates for treating Huntington’s disease. Molecular target analysis is a good method to simulate the interaction between proteins and drug compounds. Further, protective candidate drugs could be designed in future using the guidance of molecular docking results.
Keywords: Huntington’s disease, mutant Huntingtin, xyloketal derivatives, Caenorhabditis elegans, protein misfolding, molecular target
Introduction
Huntington’s disease (HD) is a devastating, rare, dominantly inherited brain disorder with typical symptoms of choreatic movements, neuropsychiatric disturbance, and cognitive impairment.1 It is caused by an elongated polyglutamine (polyQ)-encoding CAG (cytosine-adenine-guanine) repeat in the IT15 gene.2,3 There is still no effective treatment or cure for HD. Pathologically, it is characterized by the presence of protein inclusions containing mutant Huntingtin (mHtt) and progressive neuronal loss in the brain.2,4 Mutations, extensions of the polyglutamate portion of the protein, alter the stability and native structure of Htt protein, leaving it prone to aggregation; accumulation of these protein aggregates disrupts normal cellular function, eventually leading to neurodegeneration,5 whereas inhibition of protein aggregation has been shown to reduce neuronal death in experimental studies.6 Some specific proteins can also interact with mHtt to change the protein structure. For example, the interaction of protein Rhes with mHtt might improve mouse behavior in HD mouse model.7 Glyceradehyde-3-phosphate dehydrogenase could reduce the aggregation of long polyQ repeats and cytotoxicity in the cell and in a fly model.8 Thus, inhibition of pathogenic protein aggregation may have potent clinical applications to ameliorate HD.
The marine environment often provides a sink for the discovery of many novel, natural, and active compounds. Xyloketal B, an extract from marine mangrove fungi,9 has exhibited potent neuroprotection in different models of neurological diseases, including Parkinson’s disease and epilepsy.10–13 Xyloketal B has unique bicyclic acetal moieties fused to its aromatic core structure (Figure 1), which can be easily modified to improve and expand its activity.5,14 Given that some xyloketal derivatives have the ability to hydrogen bond, xyloketal may have the potential to bind to mHtt proteins and disrupt the process of Htt aggregation, thereby attenuating the pathological progression of HD.
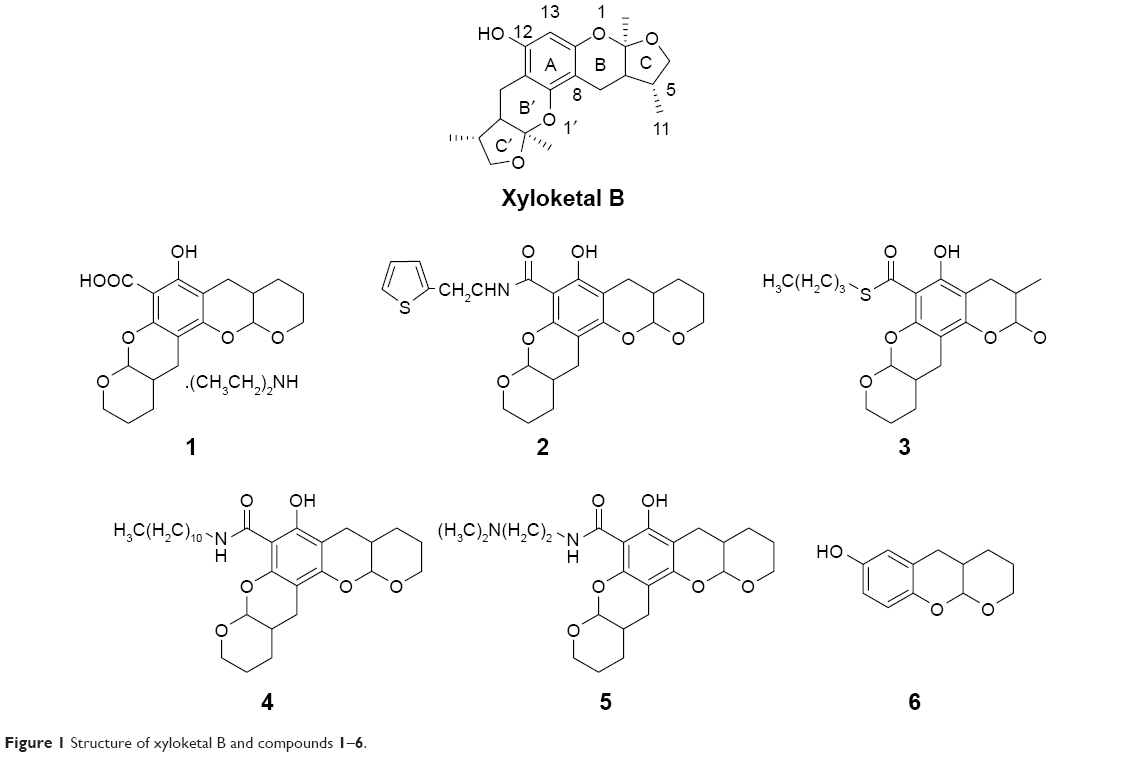 | Figure 1 Structure of xyloketal B and compounds 1–6. |
Caenorhabditis elegans is a small soil nematode that has a very short life span.15,16 C. elegans has emerged as an ideal model for the study of neurodegenerative diseases because of the high degree of conservation in molecular and cellular pathways between these worms and mammals.17 Additionally, the transparent body and expression of fluorescent tags such as green fluorescent protein (GFP) in transgenic C. elegans allow simultaneous investigation of both cellular and behavioral phenotypes. Several C. elegans HD models have been developed recently, which recapitulate key features of HD pathogenesis.18–21 For example, expression of different-length polyQ variants in C. elegans can induce length-dependent aggregation and toxicity.22–24
In the present study, we generated a C. elegans model expressing polyQ fused with GFP in muscle cells. We then used the model to examine the anti-mHtt toxin properties of six xyloketal derivatives and investigated the underlying mechanisms of action by conducting molecular docking studies.
Materials and methods
Chemicals and reagents
Xyloketal derivatives 1–6 (Figure 1, compounds 1–6) were synthesized and purified by the School of Chemistry and Chemical Engineering, Sun Yat-sen University, People’s Republic of China. The purity of xyloketal B and its derivatives was >99% as examined using high performance liquid chromatography and nuclear magnetic resonance spectroscopy. All compounds were dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C until use.
Culture conditions and generation of C. elegans model
C. elegans strains N2 (wild type) and CB4088 were purchased from CGC (Caenorhabditis Genetic Center, Minneapolis, MN, USA). All worms were handled using standard methods unless otherwise stated.25 They were cultivated on nematode growth medium (NGM) plates in a 20°C incubator and fed on living Escherichia coli strain OP50. The E. coli OP50 cells were precultured overnight at 37°C after spreading on the surface of NGM plates.
Plasmid construction for expressing polyQ fused with GFP in C. elegans muscle cells was performed essentially as described previously.26 The target polyQ fragments were obtained from plasmid pPcDNA3.1/Myc-HisA, which encoded the N-terminal 63 amino acids of the huntingtin gene plus a normal or expanded polyQ repeat (Htt-N63-16Q or Htt-N63-148Q). The polyQ-encoding sequence was then ligated into vector pPD95.86 between the XhoI and BamHI sites. GFP was cloned by polymerase chain reaction and inserted into pPD95.86 between sites KpnI and SacI, creating myo-3::Q16-GFP and myo-3::Q148-GFP for expression in the muscle cells.
Transgenic worms were generated by microinjection of engineered plasmid into the gonad of C. elegans N2. To allow integration of the polyQ-GFP construct, worms were subjected to UV exposure. They were then mated with CB4088 worms to decrease the UV-induced random damage to the whole genome sequence.27 The HD transgenic line was selected based on fluorescence in muscle cells under optical imaging.
Bioactivity assays
Age-matched worm specimens were used for all bioactivity assays. Adult worms were lysed by hypochlorite treatment to collect synchronized eggs. After 24 hours, the hatched L1 larvae were transferred to fresh NGM plates supplemented with DMSO or the indicated compound, which had been previously dissolved in E. coli OP50 and were present on the plates at a final concentration of 100 μM. As the concentration of DMSO was not more than 1 μL per 1,000 μL, the DMSO showed no bioactivity. All plates were cultured in a 20°C incubator until the animals were 4 days old.
In the heat-shock life span assay, the synchronized, treated worms were incubated at 37°C. At least 50 animals were randomly selected after they were treated by each compounds. Their viability and mortality rate were counted every hour. Death was defined as a halt of food intake or nonresponsiveness to gentle mechanical touch.28 In bending assays, at least 30 randomly selected animals from each group were used to examine the body bending frequency. Bending was determined in M9 buffer as a change in the direction of the bend in the midbody until the animal returned to its original position. The number of bends in 20 seconds was counted for each assay.29 All bioactivity assays were repeated at least three times and the mean values were assessed.
Imaging and mHtt aggregate quantification
The number of mHtt aggregates per worm was counted in live animals using confocal microscopy (Leica SP5II) equipped with epifluorescence optics at 20× magnification. The aggregates were defined as discontinuous structures in the form of dots or string-like fibrils. The procedure for worm preparation was the same as for heat-shock/bending assays. Age-synchronized, day 4 worms were loaded onto fresh agar slides which were prepared 2 hours before and treated with 20 μM NaN3.
Molecular docking
Molecular docking was performed using Surflex-Dock (SYBYL®8.1 molecular modeling software Tripos, St. Louis, MO, USA). The crystal structure of Htt36Q3H-EX1-C1 (Protein Data Bank code 4FE8), a trimer of MBP-Htt36Q3H-EX1, was retrieved from the PDB (http://www.pdb.org/). As the critical point for understanding mHtt toxicity, the mHtt polyQ region was extracted for molecular docking study.30 Ligands were sketched and minimized using Powell optimization in the presence of the Tripos force field with a convergence criterion of 0.001 kcal/mol Å and then assigned with Gasteiger–Hückel charges. Automatic docking was employed. Other parameters were established by default in the software.
Western blot
In order to detect the level of mHtt expression in worms’ lysates, animals weighing ~50 mg (all in L4 stage) were selected from the control group and drug-treated group and washed with water for three times in order to remove the OP50 cells around C. elegans body. Craked the worms in radio immunoprecipitation assay (containing 1 mM phenylmethanesulfonyl fluoride) and treated with low temperature ultrasound pyrolysis processing method. And then, the protein was boiled for 10 minutes in sodium dodecyl sulfate (SDS) loading buffer. After electrophoresis on 8% SDS gel, proteins were electroblotted onto a polyvinylidene difluoride (PVDF) membrane under semidry conditions and subjected to Western detection. Primary antibody was the polyclonal anti-GFP antibody at a dilution of 1:3,000 (NB600-308; Novus Biologicals, Littleton, CO, USA). At the same time, 1:4,000 dilution of mouse monoclonal anti-β actin was used as control. Secondary antibody was a HRP-linked anti-rabbit lgG antibody at a dilution of 1:5,000 (#7074; Cell Signaling Technology, Beverly, MA, USA). All the antibodies were diluted in TBST/2% milk powder. All the pictures were captured by ChemiDoc Imaging System (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Statistics
All the data were analyzed using SPSS Statistics 18 software. Survival data were analyzed by the Kaplan–Meier method. One-way analysis of variance (ANOVA) was followed by a Tukey’s post hoc test to evaluate the effects of different compounds on mean survival time in the heat-shock life span assay and bending frequency. Probability values of P<0.05 were considered significant.
Results and discussion
mHtt protein aggregation in C. elegans inhibits development, motility, and survival
In the C. elegans model, we established that polyQ-GFP proteins tagged with GFP could be observed directly in muscle cells under a fluorescence microscope. The animals expressing the normal polyQ repeat, Q16, showed smooth green lines in both the ventral and the dorsal muscle cells, while Q148-expressing nematodes showed discontinuous foci in muscle cells throughout their life span.31 The foci were mHtt aggregations, presenting as dots or even fibril-like strings (Figure 2A and B). In contrast to the relatively normal presentation of Q16-expressing worms, Q148-expressing worms were much shorter at the same stage of development (Figure 2A). They also had a shorter life span and weakened motility. The life span of Q148 worms decreased under heat stress, and total mortality was observed after heat treatment for 7 hours, whereas 30% of Q16 worms remained alive at that time (Figure 2C). Furthermore, Q148 worms also had a defect in bending, which can be reduced up to 70% in frequency in contrast to Q16 worms (Figure 2D).
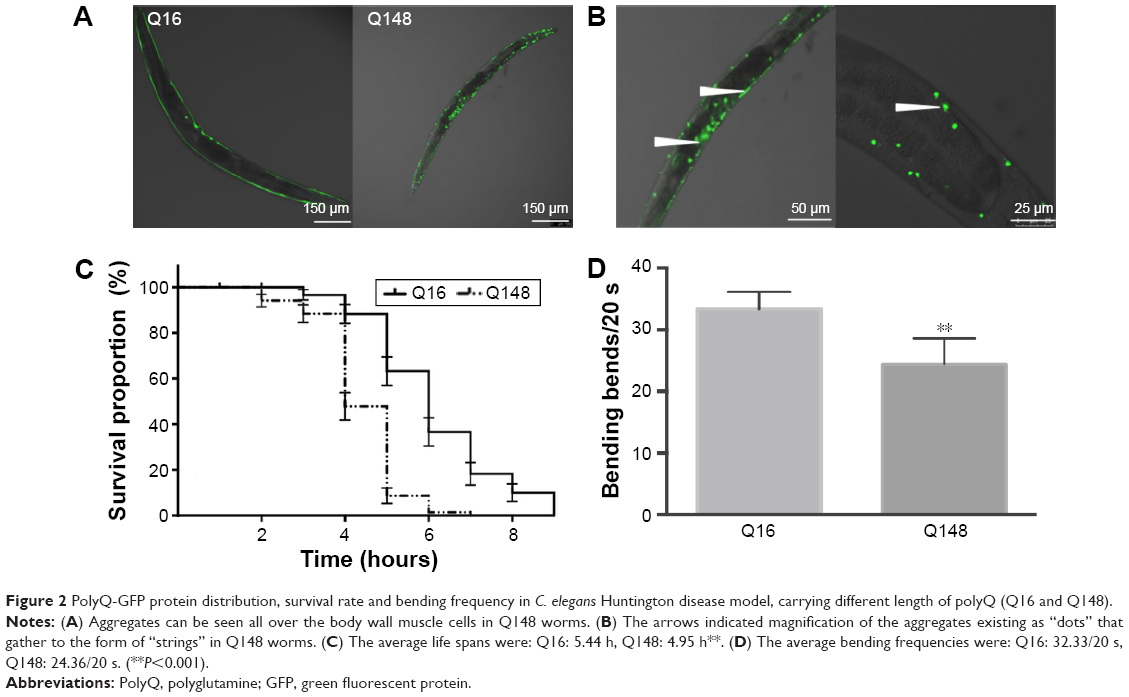 | Figure 2 PolyQ-GFP protein distribution, survival rate and bending frequency in C. elegans Huntington disease model, carrying different length of polyQ (Q16 and Q148). |
These findings are in accordance with previously reported length-dependent toxicity of polyQ repeats. As lengthy polyQ tracts are more likely to cause binding between mHtt monomers, aggregates would accumulate, leading to the loss of original protein function and the gain of aggregated mHtt toxin;32,33 such behavior results in the deficits exhibited in the Q148 C. elegans model.
Xyloketal derivatives attenuate Q148-mediated defects in C. elegans
As expected, Q148 worms displayed a clear phenotype, providing a suitable model to examine the potential anti-mHtt toxin properties of xyloketals. All the Q148 model worms exposed to xyloketal compounds 1–6 showed marked improvement in the bioactivity assays; all six derivatives prolonged the life span under stress and improved motility (Figure 3A and B).
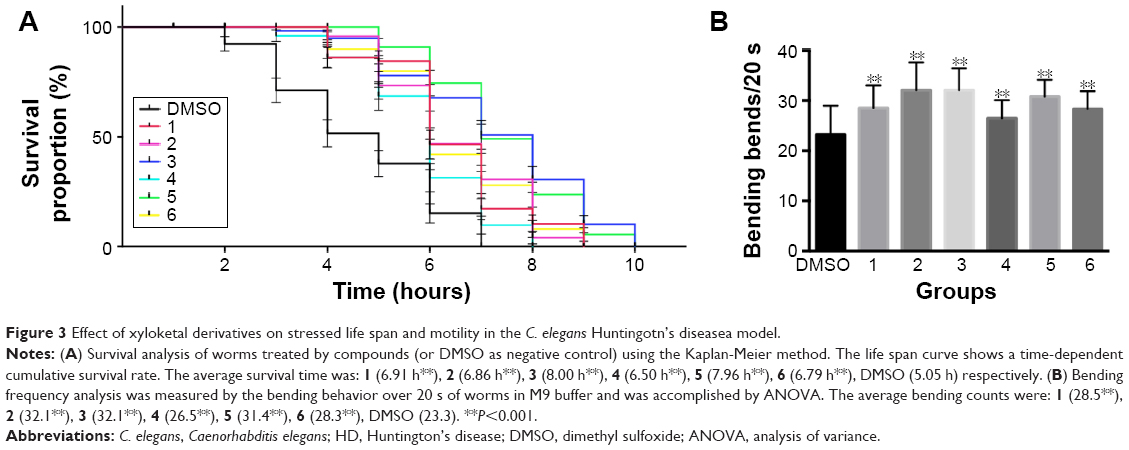 | Figure 3 Effect of xyloketal derivatives on stressed life span and motility in the C. elegans Huntingotn’s diseasea model. |
Notably, compound 1 reduced mortality in the first hour by 36% and improved motor function and growth development. Compared with the DMSO-treated worms (negative control), worms treated with compound 1 showed a 14% increase in the bending frequency. Also, no distinct shortening of body size was observed. Compounds 2 and 3 were found to have an extraordinary effect on the impaired motility of Q148; worms treated with these compounds showed nearly 38% increase in the bending frequency. Mean survival time was extended by nearly 3 hours on treatment with compound 3 relative to DMSO treatment. Compounds 4, 5, and 6 could effectively mitigate the PolyQ-mediated phenotype. Compound 5 showed the most significant effect among these three, with 50% and 35% improvements in heat-shock life span and bending assays, respectively, relative to the DMSO treatment. Overall, xyloketal derivatives 1–6 demonstrated a clear protective effect in the C. elegans model of HD, in both the heat-shock life span and bending assays.
Xyloketal derivatives 1, 4, 5, and 6 prevent mHtt aggregate formation
As previously reported, polyQ pathogenesis indicates that cellular dysfunction is often accompanied by aggregation of mHtt.34 Consistent with this, the formation of visible polyQ aggregate dots correlated with muscle dysfunction in transgenic worms bearing Q148. To explore whether xyloketals exert their protective effect by inhibiting mHtt aggregation, we examined the aggregate foci in Q148 worms after treatment.
The number of foci in body muscle cells was assessed in late-L4-stage worms. The protective effect of compounds 1, 4, 5, and 6 was accompanied by a reduction of aggregation in muscle cells. After 60 hours of treatment, the numbers of green foci located along the body wall cells decreased and fibril-like strings significantly reduced. The intensity of foci also weakened after treatment (Figure 4A). All these compounds diminished the aggregate foci to varying degrees, with 1 being the most effective. Statistical analysis revealed that compound 1 could decrease the aggregation by 20%, while compounds 4, 5, and 6 showed an average reduction of 10% (Figure 4B). However, compounds 2 and 3 showed no significant effect in the aggregation assay, which could suggest a different mechanism for their protective effects. Overexpression of mHtt has been known to change the chaperone activity and the global proteostasis network of C. elegans, and xyloketals might have protective properties mediated through these pathways.35 Therefore, it is possible that compounds 2 and 3 protected the muscle cells by alerting the proteostasis network either singularly or in parallel with other molecules to clear aggregates. Western blotting showed that the formation of mHtt aggregates was affected by treatment with different compounds (Figure 4C).
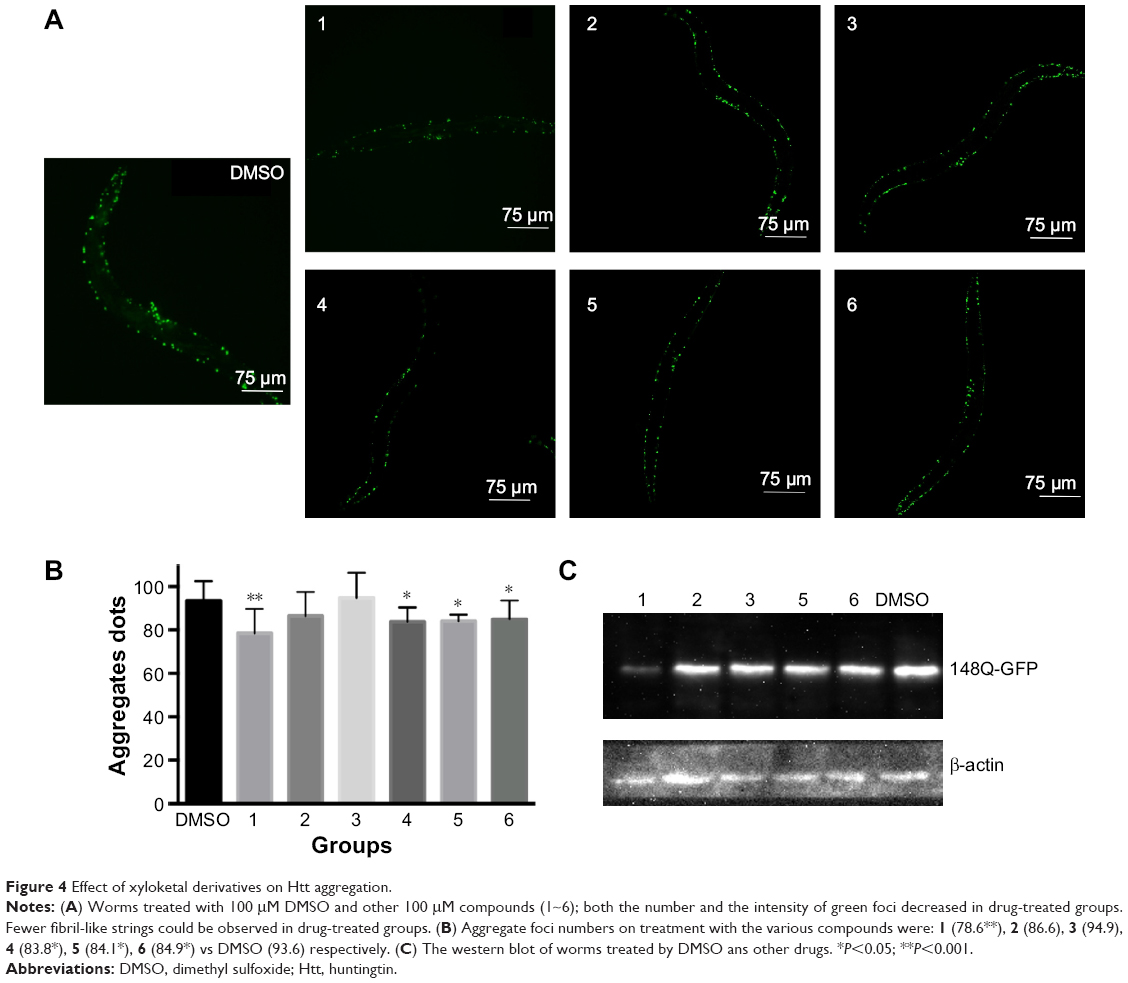 | Figure 4 Effect of xyloketal derivatives on Htt aggregation. |
For compounds 1, 4, 5, and 6, the decrease in mHtt aggregation in worm muscle cells was in accordance with the functional recovery these compounds promoted in the bioactivity assays, indicating that disruption of mHtt aggregation may be the major neuroprotective mechanism for these xyloketals. Based on our results, we hypothesized that the screened compounds might act as inhibitors of Htt aggregation. This theory was tested in silico.
Xyloketal derivatives target the GLN site of Htt protein
HD is an autosomal-dominant neurodegenerative disorder caused by a polyQ expansion (>35Q) in the first exon (EX1) of Htt protein. mHtt protein is thought to adopt one or more toxic conformations that are involved in pathogenic interactions in cells. However, the structure of mHtt is not known. To investigate the mechanism of interaction of xyloketals with mHtt, we simulated contact between various compounds and Htt in silico. Since the polyQ region of mHtt is critical for mHtt toxicity, but no structure is available, Htt36Q3H-EX1 trimers in the crystal X1 (PDB code 4EF8) were used to conduct the molecular docking analyses as previously reported.36 The structures of MBP and 3A linker were removed for clarity. Molecules were sketched according to the procedure described in the “Materials and methods” section. Automatic docking was employed. Other parameters were established by default in the software. In in silico analysis, the most potent compound, 1, was able to form hydrogen bonds with GLN396 using two benzopyran oxygens and a carboxyl oxygen (bond distances: 1.91, 2.05, and 2.58 Å, respectively). There was also a hydrogen bond between a hydroxyl group and the carboxyl oxygen from GLN393 (2.24 Å) (Figure 5A and B). As a result, mHtt protein and compound 1 may link together to form a stable trimeric complex (Figure 5C and D), which may attenuate further protein aggregation by preventing further interactions between β-strands. Pathologically, polyQ protein is thought to adopt one or more conformations that are involved in pathogenic interactions in cells.37–39 Compound 1 may abolish these conformations by interacting with GLN393 and GLN396 of the mHtt protein.
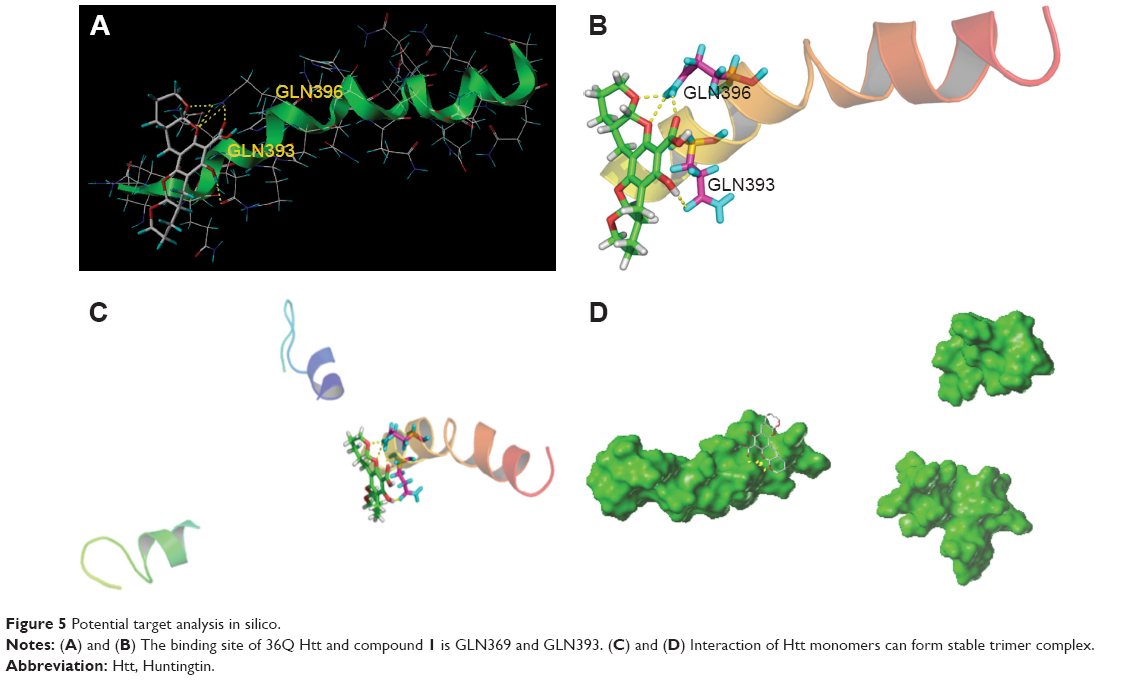 | Figure 5 Potential target analysis in silico. |
Docking scores for compounds 1–6 bound to mHtt showed varying capabilities in polyQ protein clearance (Table 1); greater docking scores indicate better clearance. The analysis results were in good agreement with functional recovery in the bioactivity assays described earlier, except for compound 6, probably because its smaller structure allows easy access into a cavity in the mHtt protein, leading to a distinct binding mechanism. Regardless of the specific mechanisms involved, our evidence supports the hypothesis that these compounds act as inhibitors of mHtt aggregation.
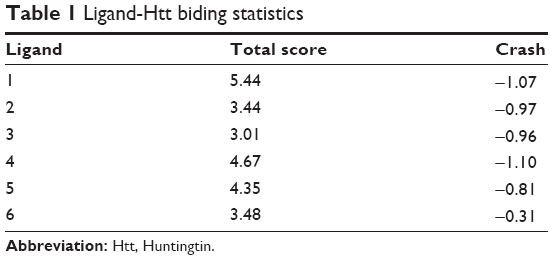 | Table 1 Ligand-Htt biding statistics |
Consideration of the docking target analyzed earlier (GLN393 and GLN396) might suggest possible mechanisms for the antiaggregation properties of the xyloketal derivatives in the misfolding of mHtt. The glutamine residues of mHtt can form polar zipper structures,40,41 which are part of β-strands, and can be linked together by hydrogen bonding between main-chain and side-chain amides.42,43 As the number of polyQ residues increases, these expanded sequences will show more affinity for each other and hold together in successive turns to form β-helix tubes.44,45 As the trimer complex of xyloketal-mHtt is formed, the secondary and tertiary structure of the mHtt protein may change. Formation of this complex could prevent self-propagation of mHtt aggregates via the polyQ sequences by influencing hydrogen bonding. As a result, the affinity for self-assembled formations of polyQ would decrease, which in turn might weaken the stability of the β-helix tubes. Consequently, the basic structure of mHtt aggregates will be interrupted by this process.46
Conclusion
HD is a monogenic disease. The abnormal extension of CAG nucleotide sequences in the IT15 gene seems to be the only key determinant. Disease severity and onset age is strongly associated with CAG triplet repeat number. Larger Htt protein, with abundant glutamine (Q), is more likely to form toxic aggregates. In the HD C. elegans model, all the tested xyloketal B derivative compounds were found to have a protective effect, which was mainly related to the attenuation of Htt aggregate formation. By simulating mHtt protein and compound interaction in silico, we demonstrated that the compounds could specifically bind to GLN residues in mHtt to prevent the proteins from forming bigger aggregates and therefore restrain further degeneration. Preventing aggregation of mutant proteins may be an attractive treatment strategy. As a novel drug candidate, xyloketal could be a promising therapeutic tool for both HD and other neurodegenerative diseases involving misfolded proteins.
In light of these findings, we may consider docked conformations of compounds with proteins to identify compounds with similar activities for future drug discovery. This strategy can significantly benefit the analysis of the potential therapeutic efficiency of drugs. By performing docking studies beforehand, we may guide new synthesis of compounds that target specific protein-binding sites. The same strategy can be applied to other neurodegenerative diseases with similar pathology of amyloidosis, such as Parkinson’s, Alzheimer’s, and prion diseases.
Acknowledgments
Thanks for the support of Prof Wenzhen Duan (Division of Neurobiology, Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA Baltimore). This study was supported by grants from the National Key Clinical Department, National Key Discipline, Guangdong Key Laboratory for Diagnosis and Treatment of Major Neurological Disease, 2014B030301035. The National Natural Science Foundation of China (No 813712555; No 21172271); National Science and Technology support program (No 2015BAIO7B01); Guandong Science and Technology project (No 2013B051000018, 2014A030304018, 2014B040404053, 2015B050501003).
Disclosure
The authors report no conflicts of interest in this work.
References
A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–983. | ||
Hoffner G, Island ML, Djian P. Purification of neuronal inclusions of patients with Huntington’s disease reveals a broad range of N-terminal fragments of expanded huntingtin and insoluble polymers. J Neurochem. 2005;95:125–136. | ||
Schilling G, Sharp AH, Loev SJ, et al. Expression of the Huntington’s disease (IT15) protein product in HD patients. Hum Mol Genet. 1995;4:1365–1371. | ||
Hoffner G, Djian P. Monomeric, oligomeric and polymeric proteins in huntington disease and other diseases of polyglutamine expansion. Brain Sci. 2014;4:91–122. | ||
Qin ZH, Gu ZL. Huntingtin processing in pathogenesis of Huntington disease. Acta Pharmacol Sin. 2004;25(10):1243–1249. | ||
Chánez-Cárdenas ME, Vázquez-Contreras E. The aggregation of Huntingtin and α-Synuclein. J Biophys. 2012;2012:606172. | ||
Swarnkar S, Chen Y, Pryor WM, Shahani N, Page DT, Subramaniam S. Ectopic expression of the striatal-enrich GTPase Rhes elicits cerebellar degeneration and an ataxia phenotype in Huntinton’s diseae. Neurobiol Dis. 2015;82:66–77. | ||
Lazarev VF, Benken KA, Semenyuk PI, et al. GAPDH binders as potential drugs for the therapy of polyglutamine disease, design of a new screening assay. FEBS Lett. 2015;589(5):581–587. | ||
Lin Y, Wu X, Feng S, et al. Five unique compounds: xyloketals from mangrove fungus Xylaria sp. from the South China Sea coast. J Org Chem. 2001;66:6252–6256. | ||
Long SM, Liang FY, Wu Q, et al. Identification of marine neuroactive molecules in behaviour-based screens in the larval zebrafish. Mar Drugs. 2014;12:3307–3322. | ||
Li S, Shen C, Guo W, et al. Synthesis and neuroprotective action of xyloketal derivatives in Parkinson’s disease models. Mar Drugs. 2013;11:5159–5189. | ||
Xiao AJ, Chen W, Xu B, et al. Sun. Marine compound xyloketal B reduces neonatal hypoxic-ischemic brain injury. Mar Drugs. 2015;13:29–47. | ||
Su J, Chang C, Xiang Q, et al. Xyloketal B, a marine compound, acts on a network of molecular proteins and regulates the activity and expression of rat cytochrome P450 3a: a bioinformatic and animal study. Drug Des Devel Ther. 2014;8:2555–2602. | ||
Pettigrew JD, Cadieux JA, So SS, Wilson PD. Phenylboronic acid mediated triple condensation reactions of phloroglucinol and unsaturated carbonyl compounds. Org Lett. 2005;7:467–470. | ||
Li J, Le W. Modeling neurodegenerative diseases in Caenorhabditis elegans. Exp Neurol. 2013;250:94–103. | ||
Parker JA, Holbert S, Lambert E, Abderrahmane S, Neri C. Genetic and pharmacological suppression of polyglutamine-dependent neuronal dysfunction in Caenorhabditis elegans. J Mol Neurosci. 2004;23:61–68. | ||
Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. | ||
Gama Sosa MA, De Gasperi R, Elder GA. Modeling human neurodegenerative diseases in transgenic systems. Hum Genet. 2012;131:535–563. | ||
Faber PW, Alter JR, MacDonald ME, Hart AC. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc Natl Acad Sci U S A. 1999;96:179–184. | ||
Holbert S, Denghien I, Kiechle T, et al. The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: neuropathologic and genetic evidence for a role in Huntington’s disease pathogenesis. Proc Natl Acad Sci U S A. 2001;98:1811–1816. | ||
Gohil VM, Offner N, Walker JA, et al. Meclizine is neuroprotective in models of Huntington’s disease. Hum Mol Gen. 2011;20: 294–300. | ||
Steffan JS, Thompson LM. Targeting aggregation in the development of therapeutics for the treatment of Huntington’s disease and other polyglutamine repeat diseases. Expert Opin Ther Targets. 2003;7:201–213. | ||
Voisine C, Varma H, Walker N, Bates EA, Stockwell BR, Hart AC. Identification of potential therapeutic drugs for Huntington’s disease using Caenorhabditis elegans. PloS One. 2007;2:e504. | ||
Wang H, Lim PJ, Yin C, Rieckher M, Vogel BE, Monteiro MJ. Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington’s disease by ubiquilin. Hum Mol Gen. 2006;15:1025–1041. | ||
Waterston RH, Epstein HF, Brenner S. Paramyosin of Caenorhabditis elegans. J Mol Biol. 1974;90:285–290. | ||
Satyal SH, Schmidt E, Kitagawa K, et al. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2000;97:5750–5755. | ||
Mariol MC, Walter L, Bellemin S, Gieseler K. A Rapid Protocol for integrating extrachromosomal arrays with high transmission rate into the C. elegans genome. J Vis Exp. 2013;82:50773. | ||
Stiernagle T, Ceenorhabditis Genetic Center. Maintenance of C. elegans. Worm book. 2006:1–11. (PMID: 18050451 in pubmed). | ||
Wang X, Yang C, Chai J, Shi Y, Xue D. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science. 2002; 298:1587–1592. | ||
Kahlem P, Green H, Djian P. Transglutaminase action imitates Huntington’s disease: selective polymerization of Huntingtin containing expanded polyglutamine. Mol Cell. 1998;1:595–601. | ||
Iuchi S, Hoffner G, Verbeke P, Djian P, Green H. Oligomeric and polymeric aggregates formed by proteins containing expanded polyglutamine. Proc Natl Acad Sci U S A. 2003;100:2409–2414. | ||
Trottier Y, Devys D, Imbert G, et al. Cellular localization of the Huntington’s disease protein and discrimination of the normal and mutated form. Nat Genet. 1995;10:104–110. | ||
Andre W, Sandt C, Dumas P, Djian P, Hoffner G. Structure of inclusions of Huntington’s disease brain revealed by synchrotron infrared microspectroscopy: polymorphism and relevance to cytotoxicity. Anal Chem. 2013;85:3765–3773. | ||
Becher MW, Kotzuk JA, Sharp AH, et al. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis. 1998;4:387–397. | ||
Liang V, Ullrich M, Lam H, et al. Altered proteostasis in aging and heat shock response in C. elegans revealed by analysis of the global and de novo synthesized proteome. Cell Mol Life Sci. 2013;71:3339–3361. | ||
Kim M. Beta conformation of polyglutamine track revealed by a crystal structure of Huntingtin N-terminal region with insertion of three histidine residues. Prion. 2013;7:221–228. | ||
Davranche A, Aviolat H, Zeder-Lutz G, et al. Huntingtin affinity for partners is not changed by polyglutamine length: aggregation itself triggers aberrant interactions. Hum Mol Gen. 2011;20:2795–2806. | ||
Gao T, Collins RE, Horton JR, et al. The ankyrin repeat domain of Huntingtin interacting protein 14 contains a surface aromatic cage, a potential site for methyl-lysine binding. Proteins. 2009;76:772–777. | ||
Gupta S, Misra G, Pant MC, Seth PK. Prediction of a new surface binding pocket and evaluation of inhibitors against huntingtin interacting protein 14: an insight using docking studies. J Mol Model. 2011;17:3047–3056. | ||
Perutz MF, Staden R, Moens L, De Baere I. Polar zippers. Curr Biol. 1993;3:249–253. | ||
De Baere I, Liu L, Moens L, et al. Polar zipper sequence in the high-affinity hemoglobin of Ascaris suum: amino acid sequence and structural interpretation. Proc Natl Acad Sci U S A. 1992;89:4638–4642. | ||
Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natil Acad Sci U S A. 1994;91:5355–5358. | ||
Mangiarini L, Sathasivam K, Seller M, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. | ||
Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I. Secondary structure of Huntingtin amino-terminal region. Structure. 2009;17:1205–1212. | ||
Darnell G, Orgel JP, Pahl R, Meredith SC. Flanking polyproline sequences inhibit beta-sheet structure in polyglutamine segments by inducing PPII-like helix structure. J Mol Biol. 2007;374:688–704. | ||
Nakano M, Watanabe H, Starikov EB, Rothstein SM, Tanaka S. Mutation effects on structural stability of polyglutamine peptides by molecular dynamics simulation. Interdiscip Sci. 2009;1:21–29. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.