")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Xyloketal B, a marine compound, acts on a network of molecular proteins and regulates the activity and expression of rat cytochrome P450 3a: a bioinformatic and animal study
Authors Su J, Chang C, Xiang Q , Zhou Z, Luo R, Yang L, He Z, Yang H, Li J, Bei Y, Xu J, Zhang M, Zhang Q, Su Z, Huang Y, Pang J, Zhou S
Received 30 August 2014
Accepted for publication 22 September 2014
Published 12 December 2014 Volume 2014:8 Pages 2555—2602
DOI https://doi.org/10.2147/DDDT.S73476
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Junhui Su,1–3,* Cui Chang,3,* Qi Xiang,1,2 Zhi-Wei Zhou,4 Rong Luo,5 Lun Yang,6 Zhi-Xu He,7 Hongtu Yang,2,3 Jianan Li,1 Yu Bei,1 Jinmei Xu,1,2 Minjing Zhang,1 Qihao Zhang,1 Zhijian Su,1 Yadong Huang,1 Jiyan Pang,5 Shu-Feng Zhou4,7
1Institute of Biomedicine and Guangdong Provincial Key Laboratory of Bioengineering Medicine, 2Department of Pharmacy, Jinan University, Guangzhou, 3The People’s Hospital of Shenzhen City, Shenzhen, People’s Republic of China; 4Department of Pharmaceutical Science, College of Pharmacy, University of South Florida, Tampa, FL, USA; 5School of Chemistry and Chemical Engineering, Sun Yat-Sen University, Guangzhou, 6Bio-X Institutes, Key Laboratory for the Genetics of Development and Neuropsychiatric Disorders (Ministry of Education), Shanghai Jiao Tong University, Shanghai, 7Guizhou Provincial Key Laboratory for Regenerative Medicine, Stem Cell and Tissue Engineering Research Center and Sino-US Joint Laboratory for Medical Sciences, Guiyang Medical University, Guiyang, People’s Republic of China
*These two authors contributed equally to this work
Abstract: Natural compounds are becoming popular for the treatment of illnesses and health promotion, but the mechanisms of action and safety profiles are often unknown. Xyloketal B (XKB) is a novel marine compound isolated from the mangrove fungus Xylaria sp., with potent antioxidative, neuroprotective, and cardioprotective effects. However, its molecular targets and effects on drug-metabolizing enzymes are unknown. This study aimed to investigate the potential molecular targets of XKB using bioinformatic approaches and to examine the effect of XKB on the expression and activity of rat cytochrome P450 3a (Cyp3a) subfamily members using midazolam as a model probe. DDI-CPI, a server that can predict drug–drug interactions via the chemical–protein interactome, was employed to predict the targets of XKB, and the Database for Annotation, Visualization and Integrated Discovery (DAVID) was used to analyze the pathways of the predicted targets of XKB. Homology modeling was performed using the Discovery Studio program 3.1. The activity and expression of rat hepatic Cyp3a were examined after the rats were treated with XKB at 7 and 14 mg/kg for 8 consecutive days. Rat plasma concentrations of midazolam and its metabolite 1'-OH-midazolam were determined using a validated high-performance liquid chromatographic method. Bioinformatic analysis showed that there were over 324 functional proteins and 61 related signaling pathways that were potentially regulated by XKB. A molecular docking study showed that XKB bound to the active site of human cytochrome P450 3A4 and rat Cyp3a2 homology model via the formation of hydrogen bonds. The in vivo study showed that oral administration of XKB at 14 mg/kg to rats for 8 days significantly increased the area under the plasma concentration-time curve (AUC) of midazolam, with a concomitant decrease in the plasma clearance and AUC ratio of 1'-OH-midazolam over midazolam. Further, oral administration of 14 mg/kg XKB for 8 days markedly reduced the activity and expression of hepatic Cyp3a in rats. Taken together, the results show that XKB could regulate networks of molecular proteins and related signaling pathways and that XKB downregulated hepatic Cyp3a in rats. XKB might cause drug interactions through modulation of the activity and expression of Cyp3a members. More studies are warranted to confirm the mechanisms of action of XKB and to investigate the underlying mechanism for the regulating effect of XKB on Cyp3a subfamily members.
Keywords: Xyloketal B, molecular target, cytochrome P450 3A, DDI-CPI tool, DAVID, midazolam, pharmacokinetics, rat, bioinformatics
Introduction
There is an increasing prevalence of natural products, including marine products, that are used to improve body function and manage various ailments due to their diverse pharmacological activity,1 although there is limited or sparse clinical evidence for their applications. In particular, there is a lack of data on molecular targets, mechanisms of action, pharmacokinetics (PKs, mainly focused on absorption, distribution, metabolism and excretion [ADME]), and toxicology for most natural products.2,3 On the other hand, many of the natural products have the ability to regulate important drug-metabolizing enzymes and drug transporters, such as the cytochrome P450 enzymes (CYPs) and P-glycoprotein. This has raised a safety concern regarding the use of natural products due to their modulatory effect on the activity and expression of drug-metabolizing enzymes and drug transporters, resulting in potentially harmful drug interactions and eventually adverse drug reactions.4
Human CYPs are a superfamily consisting of 57 functional genes that oxidize over 95% of the drugs in clinical use.5,6 The human CYP3A subfamily contains four members, with CYP3A4 being the most abundant enzyme in the liver and intestine.5 Importantly, CYP3A4/5 is predisposed to induction and inhibition when exposed to a number of endogenous and exogenous factors, probably resulting in an altered PK profile of the victim drug and adverse drug reactions, in particular, for those drugs with a narrow therapeutic index, such as digoxin, warfarin, and carboplatin.7,8 The rat Cyp3a subfamily contains Cyp3a1, 3a2, 3a9, and 3a62. Both rat Cyp3a1 and 3a2 have ≥72% identity in amino acid sequence to human CYP3A4 and share a lot of substrate specificity and inhibitor selectivity with CYP3A4.9,10
Cardiovascular disease (CVD), such as coronary heart disease and stroke, is the leading cause of death throughout the world. About 17.3 million people died from CVD in 2008, accounting for 30% of all global deaths.11,12 Of these, about 7.3 million deaths resulted from coronary heart disease and 6.2 million deaths were due to stroke.11,12 The incidence of CVD is increasing, exposing individuals, family, and society to a great burden. The pathogenesis of CVD is complicated, and oxidative stress has been proposed to be a causal factor in the pathogenesis.13,14 Reducing oxidative stress has been reported to be a promising strategy for the treatment of CVD.13 A number of natural products show potent antioxidative effects, and coadministration of natural products with cardiovascular drugs has gained increasing popularity because of these effects, although there is a lack of systematic preclinical and clinical data on their PKs, molecular targets, mechanisms of action, and toxicology.15,16
Marine organisms are an attractive source for drug development. Numerous compounds have been isolated from marine organisms, with a wide range of pharmacological activity, including antioxidative, anti-inflammatory, and anticancer effects.17–18 Xyloketal B (XKB, Figure 1) is a novel marine compound isolated from the mangrove fungus Xylaria sp., and has potent antioxidative, neuroprotective, and cardioprotective effects.19,20 We have found that XKB has a relaxing effect on blood vessels and protects endothelial cells against oxidative injury induced by oxidized low-density lipoprotein.10 It directly scavenged the free radical-generating compound 1,1-diphenyl-2-picrylhydrazyl and protected PC12 cells against oxygen–glucose deprivation injury.9 Further, it has a strong inhibitory effect on the L-calcium channel.21 The potent antioxidative effect of XKB suggests that it is a promising agent in the treatment of oxidative stress-associated conditions, such as CVD. To establish the dose-response and dose-toxicity relationships of XKB, we investigated its metabolism and PKs in rats. In vivo, the plasma PKs of XKB followed a two-compartment model with a distribution phase and an elimination phase.22 XKB showed a short elimination half-life (22–27 minutes), suggesting that it was rapidly distributed in the body and also rapidly eliminated. However, the molecular targets in response to XKB treatment and the effect of XKB on regulation of drug-metabolizing enzymes are unknown.
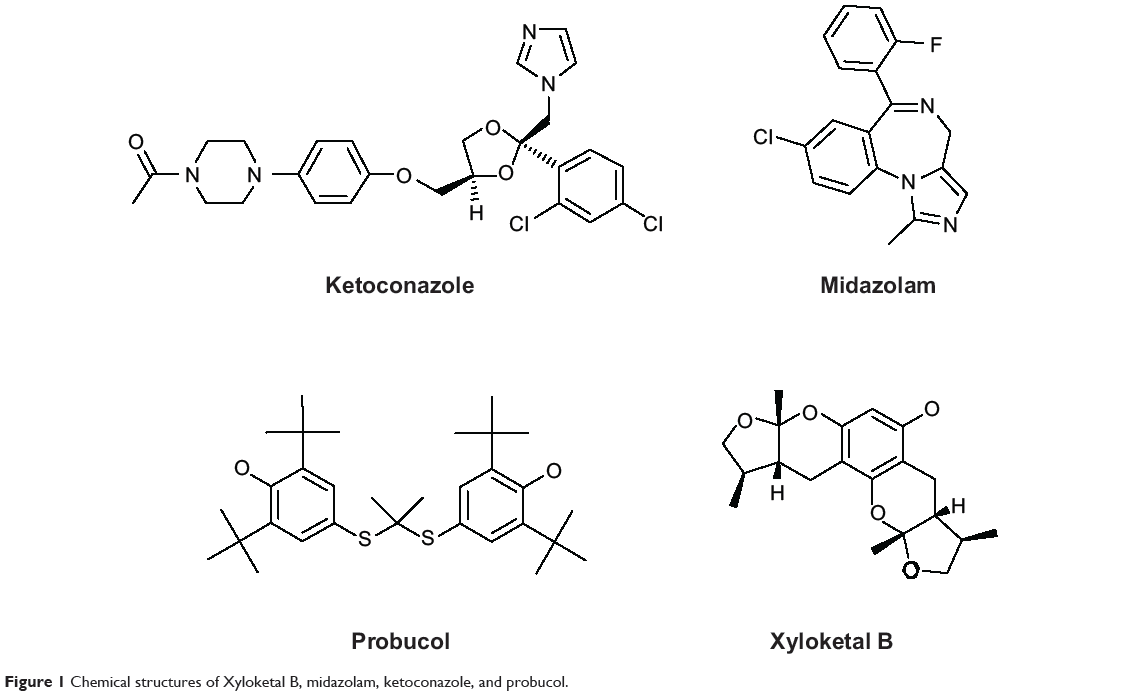
|
Figure 1 Chemical structures of Xyloketal B, midazolam, ketoconazole, and probucol. |
In the present study, in order to investigate the molecular targets of XKB, we predicted these targets using a bioinformatic platform, analyzed the interactions between XKB and rat Cyp3a2 using a homology model followed by molecular docking, and examined the effect of oral XKB on the activity and expression of hepatic Cyp3a in rats using midazolam as the model substrate.
Materials and methods
Prediction of interactome of XKB and pathway analysis by molecular docking and bioinformatic approach
The interactome of XKB was predicted using the DDI-CPI tool, a web-based server that can predict drug–drug interactions via the chemical–protein interactome.23,24 Protein targets were obtained from a third-party protein structure database named PDBbind.25 There is a total of 1,780 Protein Data Bank (PDB) entries for human proteins available in PDBbind, and a total of 301 nonredundant PDBs corresponding to 353 ligand binding pockets were identified, 86% of which have resolutions of less than 2.5å. The docking boxes for each of the pockets were defined by expanding the circumscribed cube of the pocket with a margin of 8å in six directions (up, down, front, back, left, and right). For preparation of XKB, the two-dimensional structure of the XKB was downloaded from PubChem. The hydrogen and Gasteiger charge were added and the file format was transformed into mol2 using Vega ZZ. The docking program AutoDock 4.2 was used to dock the XKB molecule into all 353 pockets, generating a score vector of 353 dimensions. Z’-scores were then calculated using methodologies described elsewhere.23,26,27 Herein, an empirical threshold of −0.6 for the Z’-score was set to indicate that the binding of XKB to this target was likely to be true.
Pathway analysis by the Database for Annotation, Visualization and Integrated Discovery
Following target prediction, the Database for Annotation, Visualization and Integrated Discovery version 6.7 (DAVID, http://david.abcc.ncifcrf.gov/) was used to provide a biological functional interpretation of the potential targets of XKB derived from DDI-CPI. The protein IDs of these targets from UniProtKB, National Center for Biotechnology Information, and other sources were converted into gene lists by using the Gene ID conversion tool in DAVID. DAVID adds biological function annotation including gene ontology, pathway, protein–protein interactions, functional groups of genes (ie, clustering), and disease associations derived from main public data sources. The genes of interest were visualized using BioCarta and Kyoto Encyclopedia of Genes and Genomes pathway maps. The highest classification stringency was selected for functional annotation clustering. Enrichment scores and Fisher’s Exact test P-values (and the corresponding false discovery rate) were then calculated to identify which functional-related gene groups are significantly enriched in the target list. These significant enriched gene groups could provide clues on how XKB interacts with molecular targets in a systematic way.
Molecular docking of human CYP3A4 and homology modeling of rat Cyp3a2
We employed the Discovery Studio program 3.1 designed by Accelrys Inc (San Diego, CA, USA) to dock XKB, midazolam, ketoconazole, and probucol into the active site of human CYP3A4 and rat Cyp3a2 homology model as previously described,28 but with some modifications. Due to the lack of availability of a crystal structure for rat hepatic Cyp3a2, we built a homology model based on the reported human CYP3A4 crystal structure (PDB ID 4K9W; http://www.rcsb.org/pdb/). For the establishment of a homology model of rat hepatic Cyp3a2, we first retrieved the rat Cyp3a2 sequence from the National Center for Biotechnology Information (NCBI Reference Sequence NP_695224.2; http://www.ncbi.nlm.nih.gov/protein/). We aligned the rat Cyp3a2 sequence based on the template of human CYP3A4. Following the establishment of this homology model, both the human CYP3A4 and rat Cyp3a2 homology models were cleaned, modified, and prepared. For the preparation for XKB, midazolam, ketoconazole, and probucol, the duplicate chemical structures were deleted and ionization change, tautomer or isomer generation, Lipinski filter, and three-dimensional generator were all set true. After ligand preparation, XKB, midazolam, ketoconazole, and probucol were docked into the active sites of the human CYP3A4 and rat Cyp3a2 homology model. Electrostatic energy and van der Waals forces were considered during the docking process. A harmonic potential with a force constant of 300 kcal/mol was applied outside the grid boundary.28
Prediction of ADME and toxicity of XKB, midazolam, ketoconazole, and probucol
Using the Discovery Studio program 3.1 designed by Accelrys Inc, the ADME and toxicity (ADMET) profiles of XKB, midazolam, ketoconazole, and probucol were predicted. Briefly, the chemical structures of XKB, midazolam, ketoconazole, and probucol were retrieved from the PubChem database (Figure 1). All the structures were introduced into the Discovery Studio program and prepared. During preparation of these structures, the duplicate chemical structures were deleted and the ionization change, tautomer or isomer generation, Lipinski filter, and three-dimensional generator were all set true. Following preparation of the structures, XKB, midazolam, ketoconazole, and probucol were subject to ADMET evaluation using the ADMET Predictor module. Parameters including solubility, absorption, permeability across the blood–brain barrier, interactions with CYP2D6, hepatotoxicity, and plasma protein binding were predicted. There are four different ADMET absorption levels, ranging from 0, 1, 2, to 3, indicating good, moderate, low, and very low absorption, respectively. The ADMET aqueous solubility level is classified as extremely low (0), no, very low, but possible (1), yes, low (2), yes, good (3), yes, optimal (4), no, too soluble (5), and warning, molecules with one or more unknown AlogP98 types (6). ADMET blood–brain barrier permeability has six different levels, including very high (0), high (1), medium (2), low (3), undefined (4), and warning, molecules with one or more unknown AlogP calculation (5). There are two predicted classes of ADMET CYP2D6 ligand, ie, noninhibitor (0) and inhibitor (1). ADMET hepatotoxicity is categorized into nontoxic (0) and toxic (1) effects. There are three different ADMET plasma protein binding levels, ie, binding <90% (0), binding >90% (1), and binding >95% (2).
Chemicals and reagents
XKB (purity >99%) was synthesized at Sun Yat-Sen University, Guangzhou, People’s Republic of China. Nicotinamide adenine dinucleotide phosphate (NADPH) and midazolam were purchased from Sigma-Aldrich (St Louis, MO, USA). Ketoconazole was obtained from Nanjing Pharmaceutical Co (Baijingyu, People’s Republic of China, catalog number 65277-42-1). 1′-Hydroxy midazolam (1′-OH-MDZ) was purchased from Cayman Chemical Co (Ann Arbor, MI, USA). Midazolam injections were sourced from Pharmaceutical Group Co Ltd Jiangsu Nhwa (Jiangsu, People’s Republic of China). Diazepam injections were obtained from Amino Acid Co Ltd Tianjin Jin Yao (Tianjin, People’s Republic of China, catalog number 439-14-5). A P450-Glo™ assay kit was purchased from Promega (Madison, WI, USA). Clarity Western enhanced chemiluminescence (ECL) substrate was obtained from Bio-Rad Laboratories (Hercules, CA, USA). Primary antibody against rat Cyp3a2 was purchased from Merck Millipore (Darmstadt, Germany). GADPH and anti-rabbit immunoglobulin G horseradish peroxidase-linked antibody were purchased from Cell Signaling Technology (Boston, MA, USA). High-performance liquid chromatography (HPLC)-grade acetonitrile and ammonium acetate were obtained from Merck (Darmstadt, Germany). Ultrapure water was prepared in our laboratory using an Arium 611VF system (Sartorius Corporation, NY, USA).
Animals
Healthy male Sprague-Dawley rats weighing 250–280 g and aged 2–3 months were purchased from Guangdong Medical Laboratory Animal Center, Guangzhou, People’s Republic of China. The rats were housed in an accredited animal housing facility under controlled temperature (22°C±2°C) and on a 12-hour dark/light cycle. All animals were allowed to acclimatize for 5–7 days prior to the experiments. All experiments were conducted according to the guidelines for animal care and use of the People’s Republic of China and were approved by the animal ethics committee of the Chinese Academy of Medical Science (Beijing, People’s Republic of China).
Drug administration
The animals were randomly divided into five groups (n=8) and treated intraperitoneally once daily with saline (blank control), soybean oil (vehicle), ketoconazole (75 mg/kg, positive control), or XKB (7 and 14 mg/kg) for 8 consecutive days. All animals had free access to standard laboratory food and water. In order to reduce the intestinal contents, animals were fasted overnight before termination of the study. Midazolam 20 mg/kg was administered orally 12 hours after the last dose of XKB. Blood samples were collected from the post ocular vein 0, 5, 15, 30, 45, 60, 90, 120, 180, 240, 300, and 360 minutes after administration of midazolam. All blood samples were immediately centrifuged at 1,000× g for 10 minutes to obtain the plasma and stored at −20°C until further analysis. The animals were euthanized by cervical dislocation after blood collection, and liver samples were collected for further assessment of activity and expression of Cyp3a.
Preparation of rat hepatic microsomes
Rat liver microsomes were prepared as previously described.29 Briefly, liver tissues were collected and washed with ice-cold KCl buffer (0.2 M, pH 7.4) until the tissues became pale or whitish, and then blotted dry and weighed. All subsequent steps were performed at 4°C. The liver samples were homogenized with three volumes of 0.05 M Tris/KCl buffer at pH 7.4 using a tissue homogenizer. The liver homogenates were then centrifuged at 9,000× g for 30 minutes. The supernatant was collected and centrifuged further at 105,000× g for 60 minutes. The supernatant was thereafter decanted and the microsomal pellets were resuspended in homogenization buffer and centrifuged again at 105,000× g for 60 minutes to remove the hemoglobin. Rat liver microsomes were resuspended in 0.1 M sodium phosphate buffer (0.8 mM ethylenediamine tetraacetic acid, 1 mM dithiothreitol, and 20% glycerol; pH 7.4) and stored at −80°C until analysis. The protein concentration of each microsomal sample was determined using the bicinchoninic acid protein assay (Pierce Chemicals, Rockford, IL, USA) with bovine serum albumin as the standard.
Determination of midazolam and its metabolite 1′-OH-midazolam by HPLC
The plasma concentrations of midazolam and its metabolites 1′-OH-MDZ were measured by HPLC as previously described,30 with slight modifications. The HPLC method was validated prior to determination of the plasma PKs of midazolam as per the International Conference on Harmonization guidelines on validation of analytical procedures, including the specificity (ability to unequivocally determine the analyte), linearity (response curve of the analyte), lower limit of quantification (concentration of the analyte which has a signal to noise ratio of 3:1), precision and accuracy (results of repeatability), recovery (satisfactory accuracy and precision), and stability (degradation of analyte).2,4 A 100 μL aliquot of plasma was diluted with 400 μL of 0.1 M NaOH, and diazepam 8 μg/mL was added as an internal standard. The mixture was extracted with 2.5 mL of dichloromethane and pentane (at a ratio of 3:7), then vortexed for 20 seconds. The upper organic phase was transferred into a clean glass tube and the residual liquid was extracted again. The upper organic phase mixture was evaporated to dryness. The dried residue was dissolved in 100 μL of mobile phase and centrifuged at 12,000× g for 10 minutes, and the supernatant was subjected to HPLC analysis and detected using an ultraviolet detector at 254 nm. An HPLC system with a Shim-Pack 250 mm ×4.6 mm reverse phase column packed with 5 μm VP-ODS C18 (Shimadzu Corporation, Tokyo, Japan) was used. The column temperature was set at 40°C. The mobile phase was 10 mM ammonium acetate buffer (pH 4.5) and acetonitrile (63:37, v/v) at a flow rate of 1.0 mL per minute. All samples were analyzed within the validated stability timeframe.
Pharmacokinetic analysis
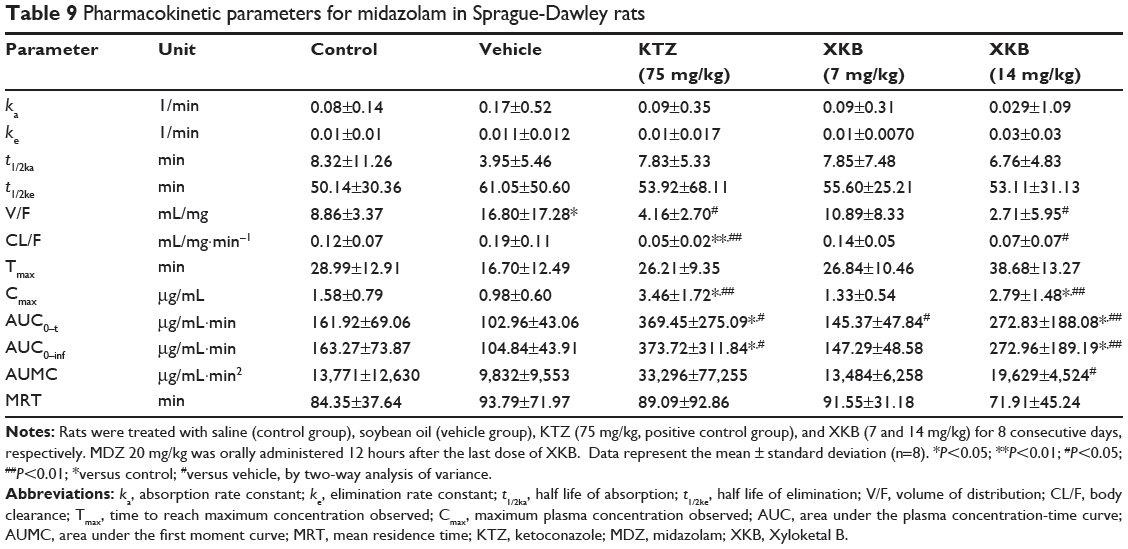
Following determination of their plasma concentrations, the PK parameters of midazolam and 1′-OH-MDZ were calculated by single-compartmental analysis using the PKSlover program (China Pharmaceutical University, Nanjing, People’s Republic of China),31,32 which is a freely available menu-driven add-in program for Microsoft Excel written in Visual Basic for Applications for PK data analysis. The PK parameters calculated were the absorption rate constant (ka), elimination rate constant (ke), half-life of absorption (t1/2ka), half-life of elimination (t1/2ke), volume of distribution (V/F), body clearance (CL/F), maximum plasma concentration observed (Cmax), time to reach Cmax (Tmax), area under the plasma concentration-time curve (AUC0–t), AUC0–inf, area under the first moment curve (AUMC), and mean residence time. The t1/2ke was calculated using the formula t1/2ke =0.693/ke. The AUC was calculated using the trapezoidal rule. CL was calculated by the formula CL/F = dose/AUC. The V/F following oral administration was calculated using the V/F = CL/F/ke equation.
Measurement of rat hepatic Cyp3a activity
The effect of XKB on rat hepatic Cyp3a activity was examined using the luminescent assay (P450-Glo) according to the manufacturer’s instructions. Briefly, rat liver homogenates were prepared as described previously.29 The reaction mixture (25 μL) was added to a 96-well microtiter plate containing microsomal protein (1 μg), Luciferin-IPA substrate (16 μM), and 200 mM potassium phosphate buffer solution (pH 7.4), and mixed gently. The plate was preincubated at 37°C for 10 minutes. The reaction was initiated by adding 25 μL NADPH and the mixture was incubated at 37°C for 10 minutes. Next, 50 μL of reconstituted Luciferin detection reagent was added to all wells. The reaction mixture was mixed on a plate shaker for 10 seconds. The plate was then incubated at 37°C for 20 minutes. Cyp3a activity was examined by luminescence reading. The relative Cyp3a activity was expressed as fold change over the control.
Western blotting analysis
Western blotting was used to investigate the regulating effect of XKB on expression of Cyp3a in the rat liver. Briefly, rat liver microsomes (10 mg) were denatured at 95°C for 10 minutes then centrifuged at 9,000× g for 3 minutes at 4°C. Protein samples were resolved in 12% sodium dodecyl sulfate-polyacrylamide gel and then transferred onto polyvinylidene difluoride membranes. The membranes were blocked for one hour with 5% nonfat milk in Tris-buffered saline/0.1% Tween 20 buffer, followed by incubation overnight with their respective primary antibodies at 4°C. The membranes were then washed with Tris-buffered saline/0.1% Tween 20 buffer and incubated with secondary antibody for one hour at 37°C. Signals were detected using the Clarity Western ECL substrate and Bio-Rad imaging system. Quantification was performed using the ImageJ Analyzer 4.0 software Media (Cybernetics, Bethesda, MD, USA). The relative expression level of Cyp3a2 in rat liver microsomes was expressed as the ratio of the intensity of Cyp3a2 blots to that of GAPDH.
Data analysis
The data are expressed as the mean ± standard deviation. The PK parameters were compared using one-way analysis of variance, followed by Tukey’s multiple comparison. A value of P<0.05 was considered to be statistically significant.
Results
XKB likely interacts with a number of important functional proteins
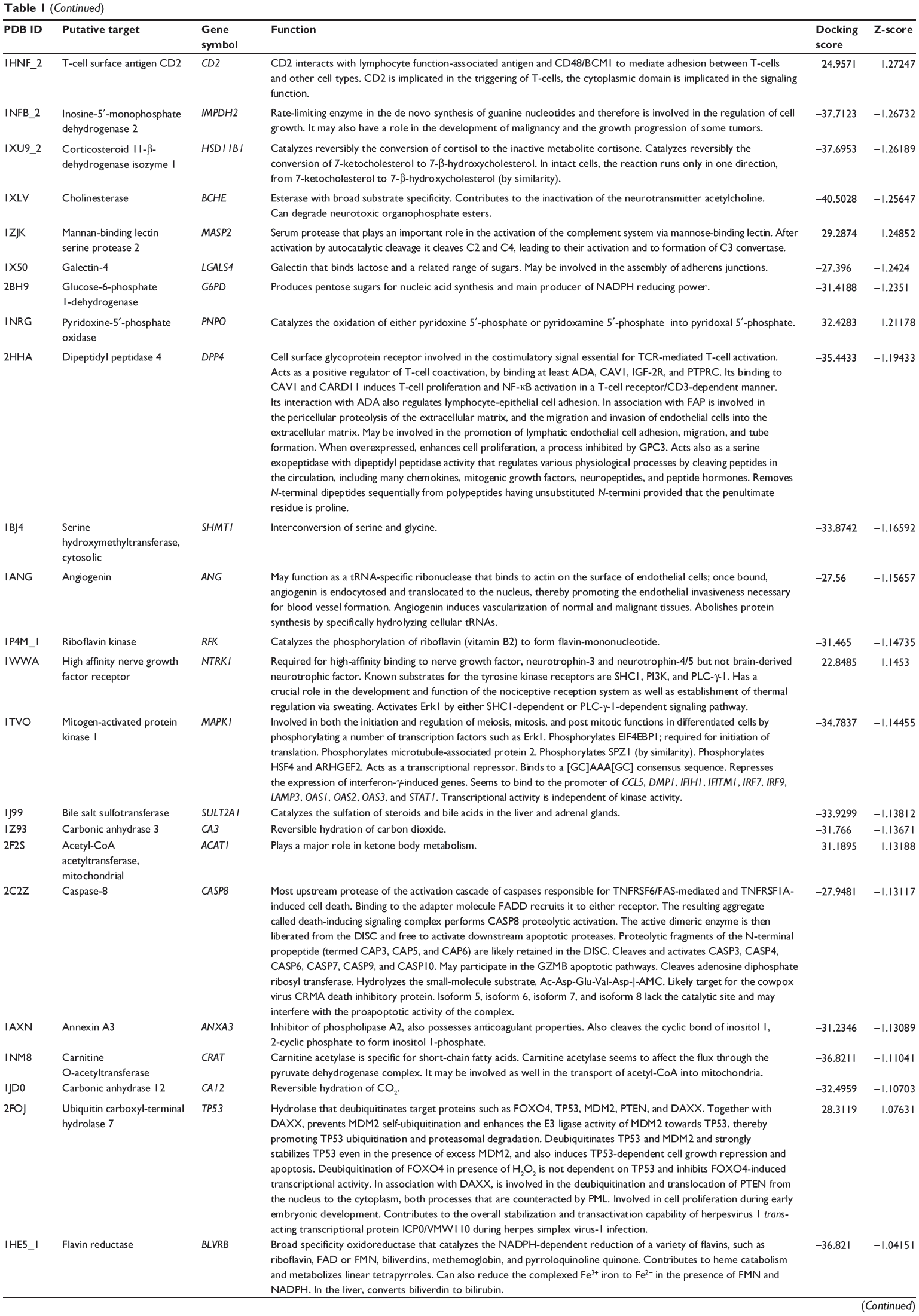
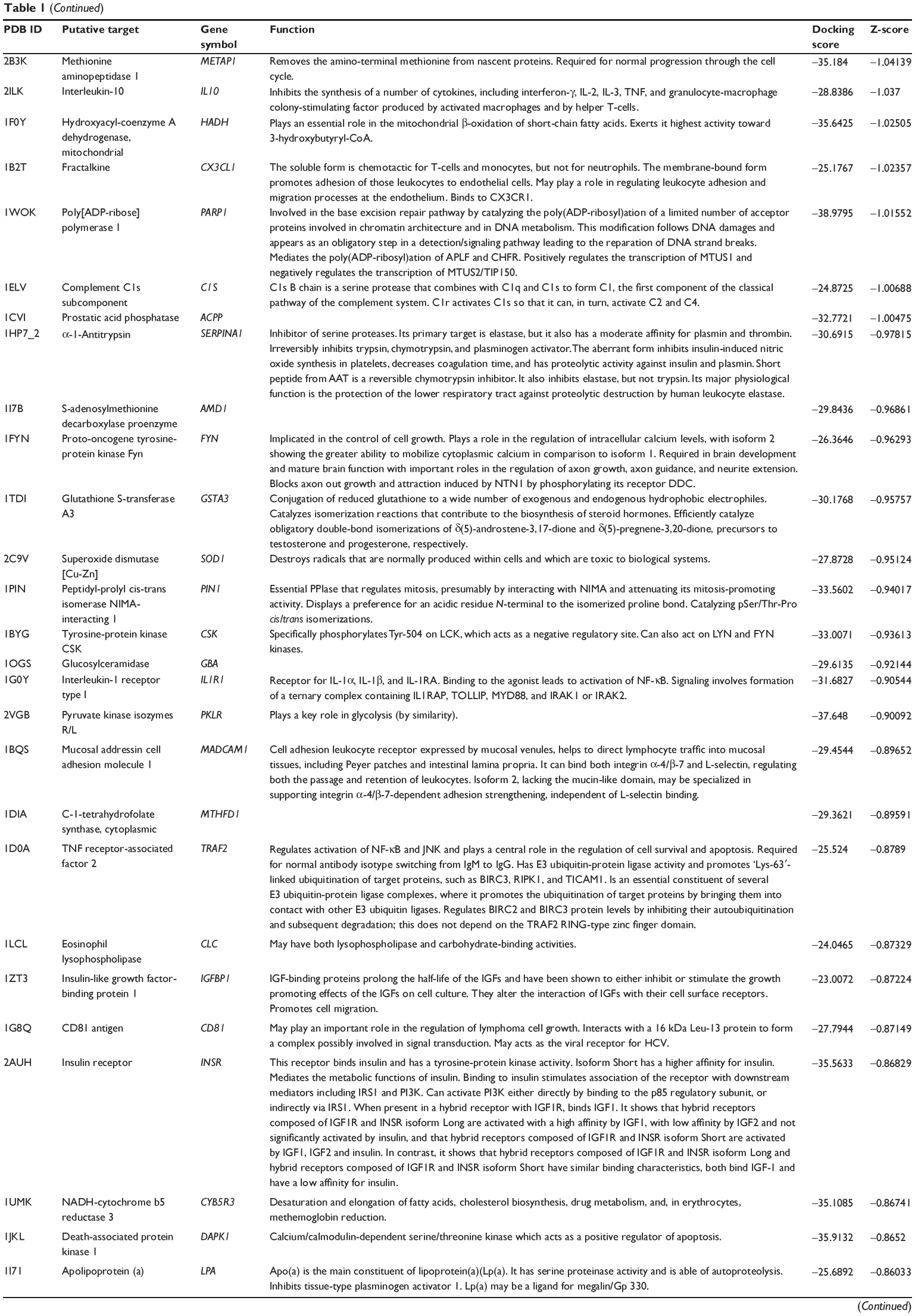
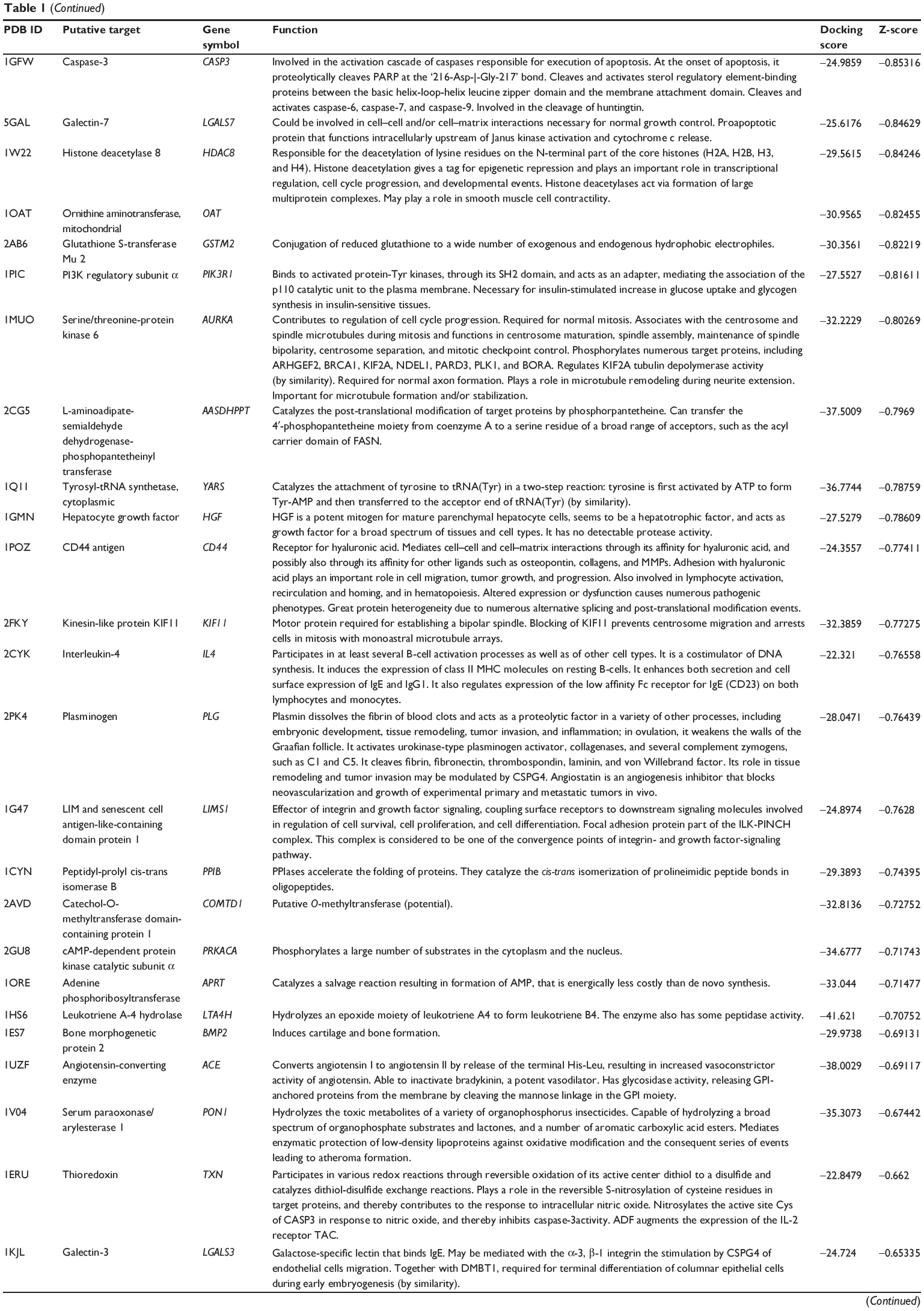
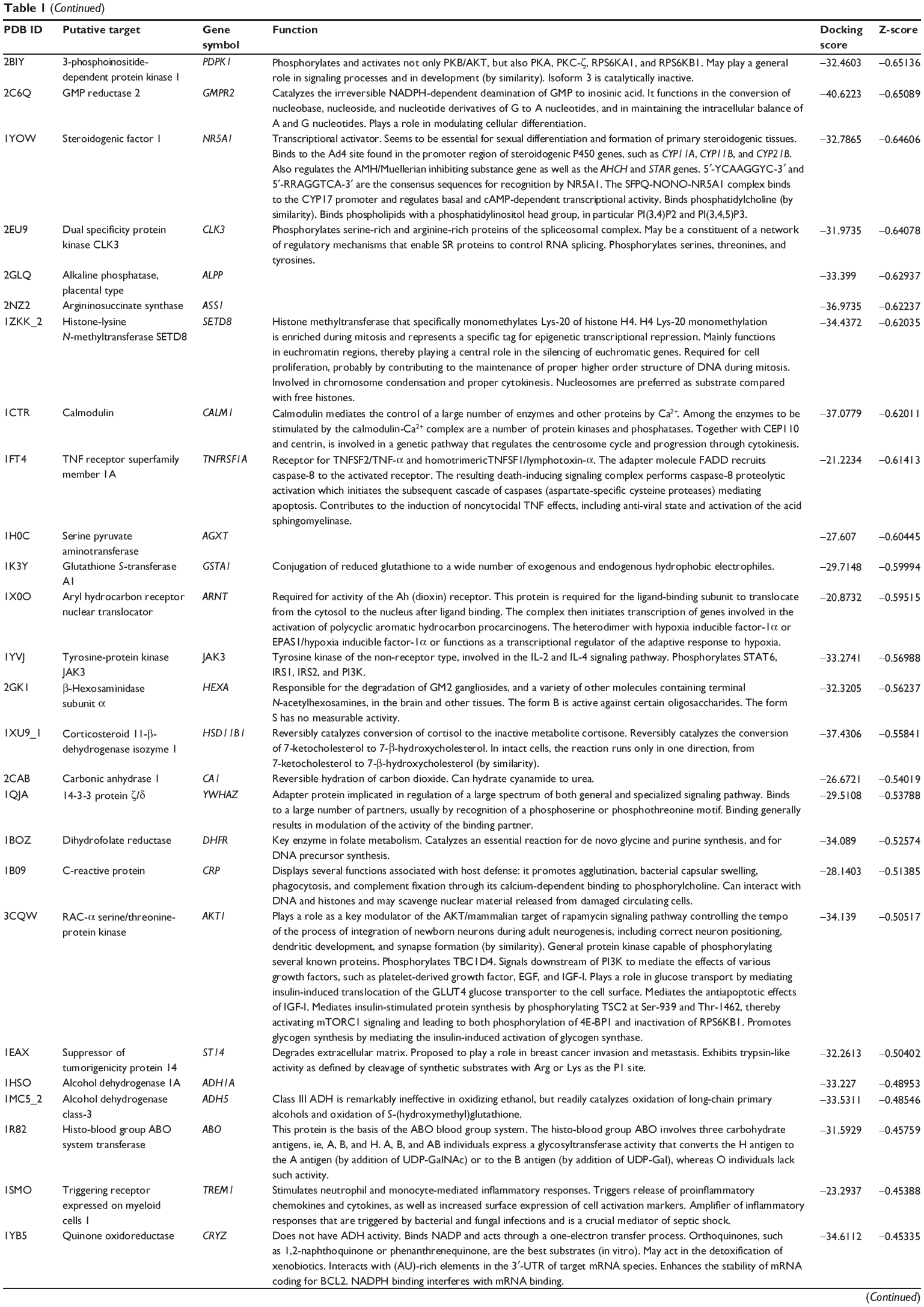
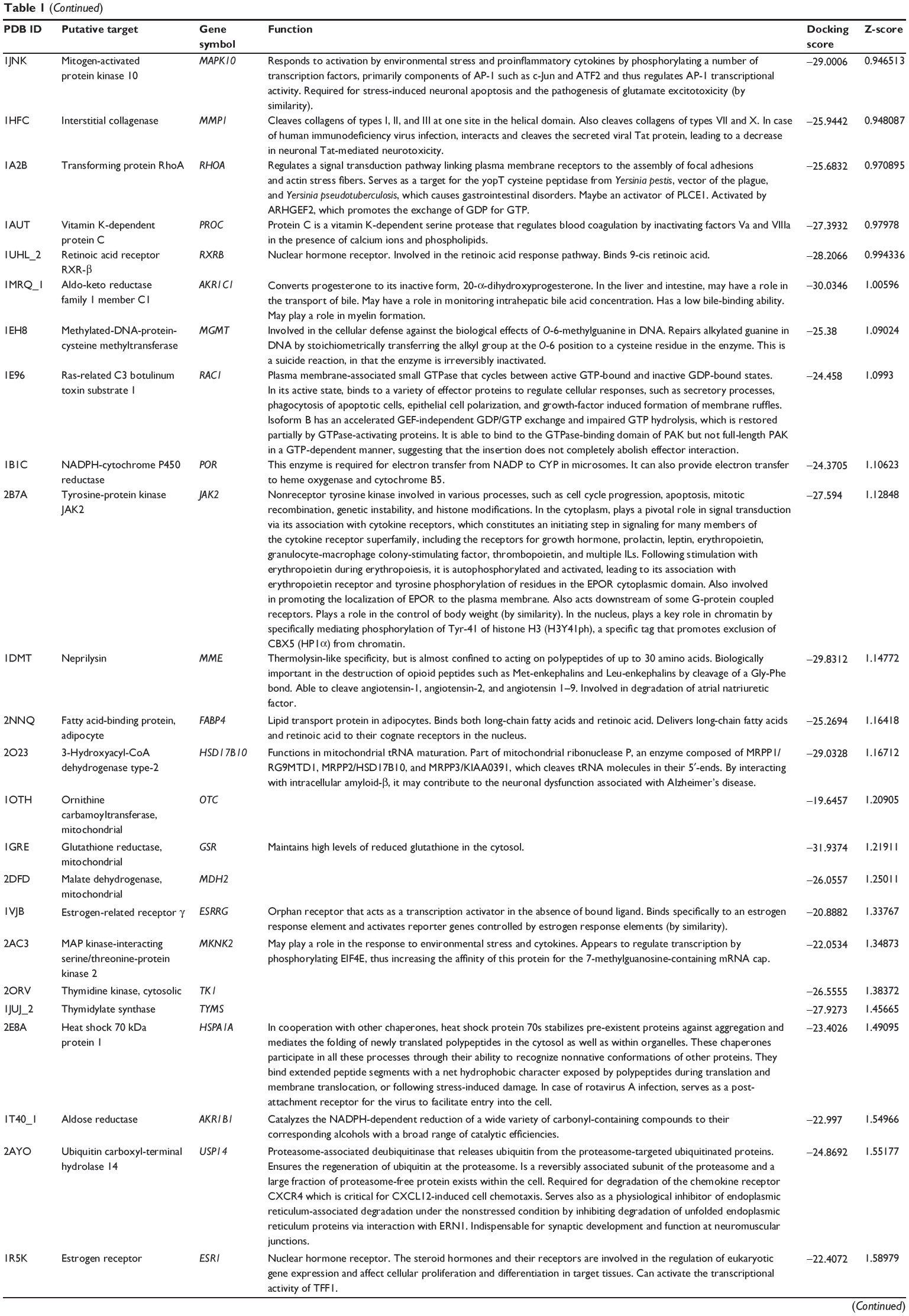
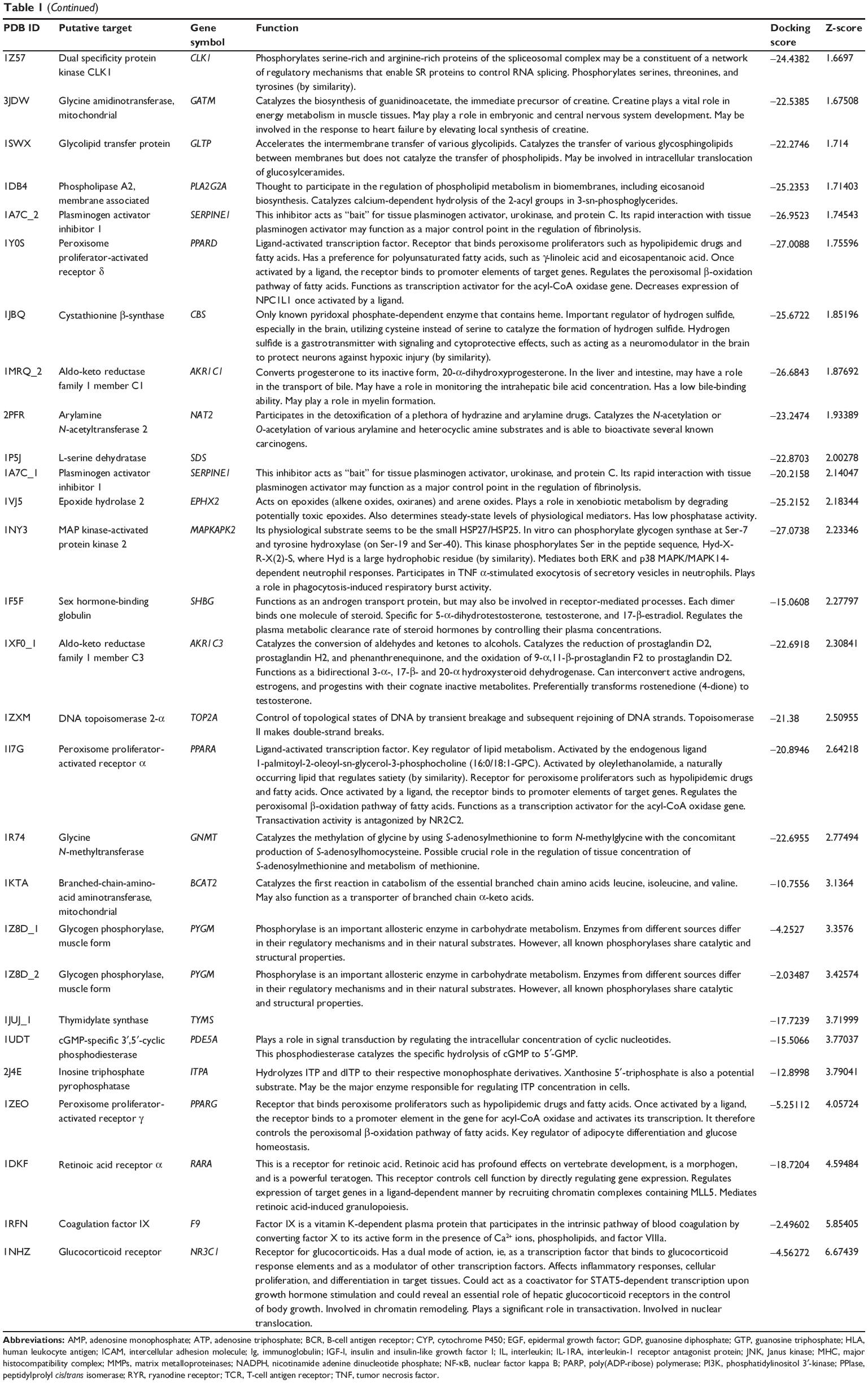
First, we predicted the molecular targets of XKB using our web-based DDI-CPI tool. There were 324 proteins that possibly interacted with XKB (Table 1), including those involved in redox homeostasis (eg, TXNRD1, PRDX2, TXN, RAC2, and GSTZ1), xenobiotic metabolism (eg, NR1I2, POR, ACAT1, CYB5R3, DPEP1, ADH7, and MTAP), cell proliferation (eg, LIMS1, SETD8, CLIC4, PIM1, and CHEK1), apoptosis (eg, CASP1, CASP3, CASP7, CASP8, PARP1, TRAF2, TNFRSF1A, and MAPK1), lipid metabolism (eg, FABP4, PPARA, and PPARD), signal transduction (eg, AKT1, CSNK2A1, CSNK1G2, IGF1R, GSK3B, RHOA, JAK2, JAK3, PDE5A, and RGS17), and immune and inflammatory response (eg, LILRB1, LCK, CD2, DPP4, IL1R1, CD81, and PDPK1).
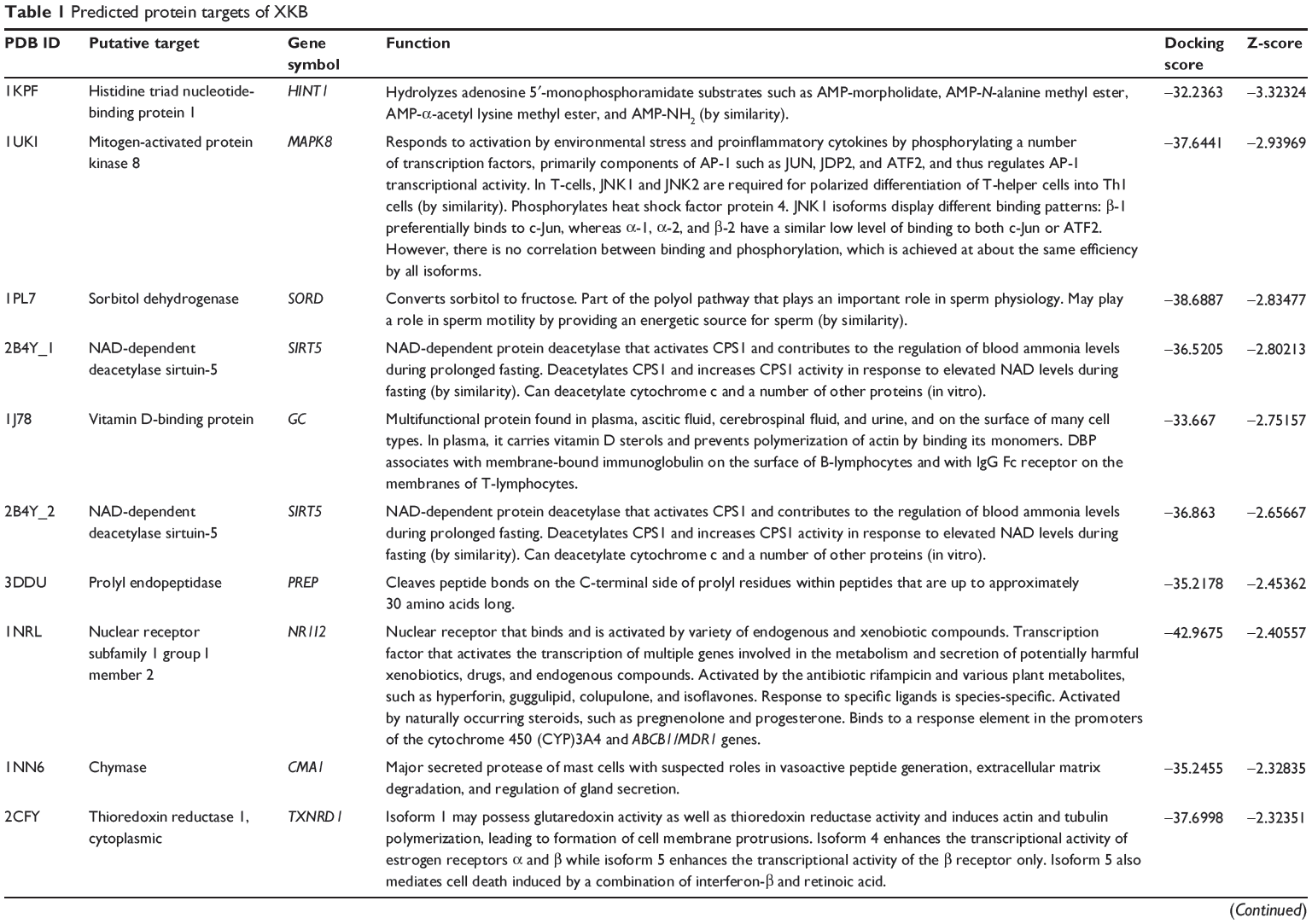 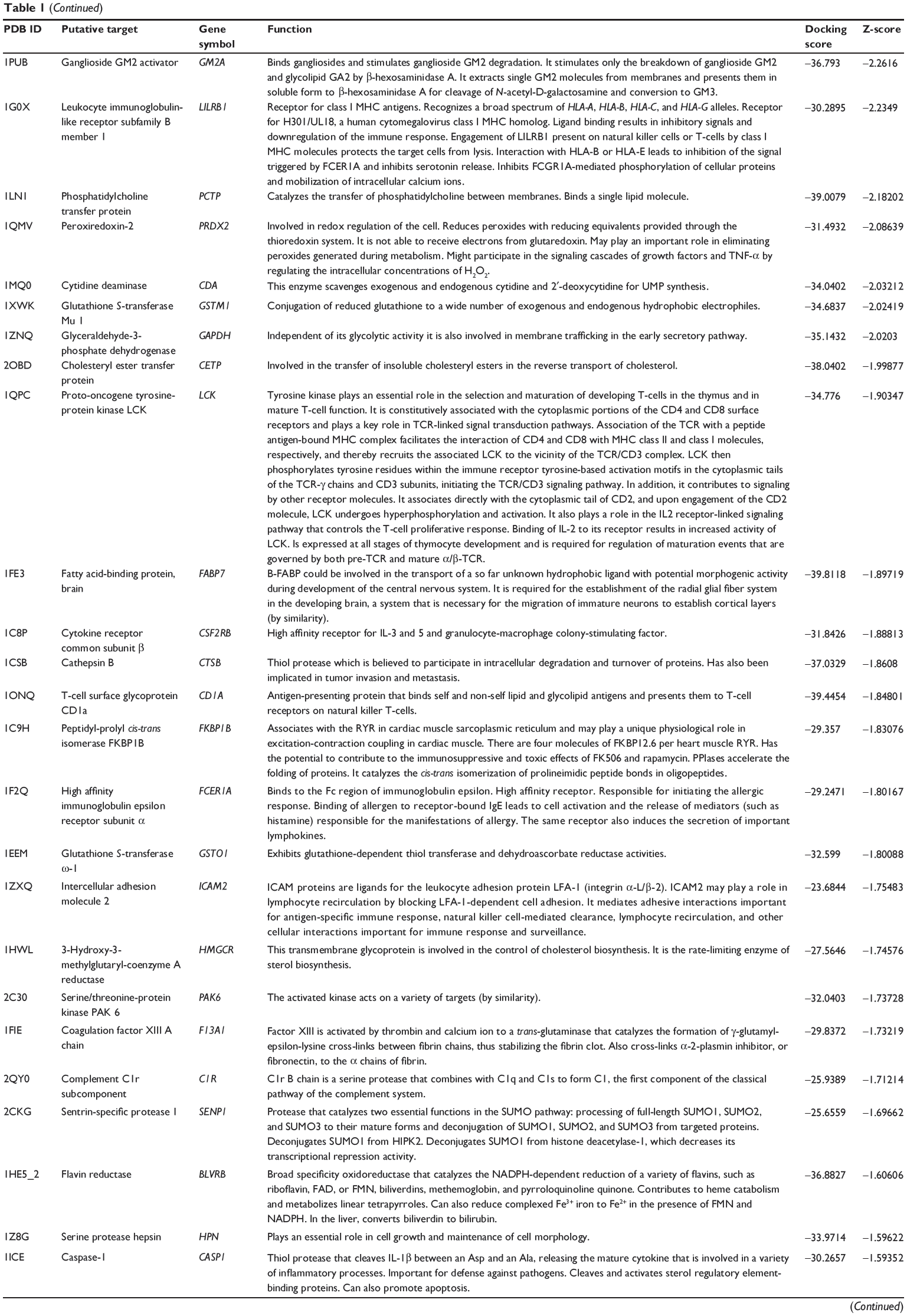 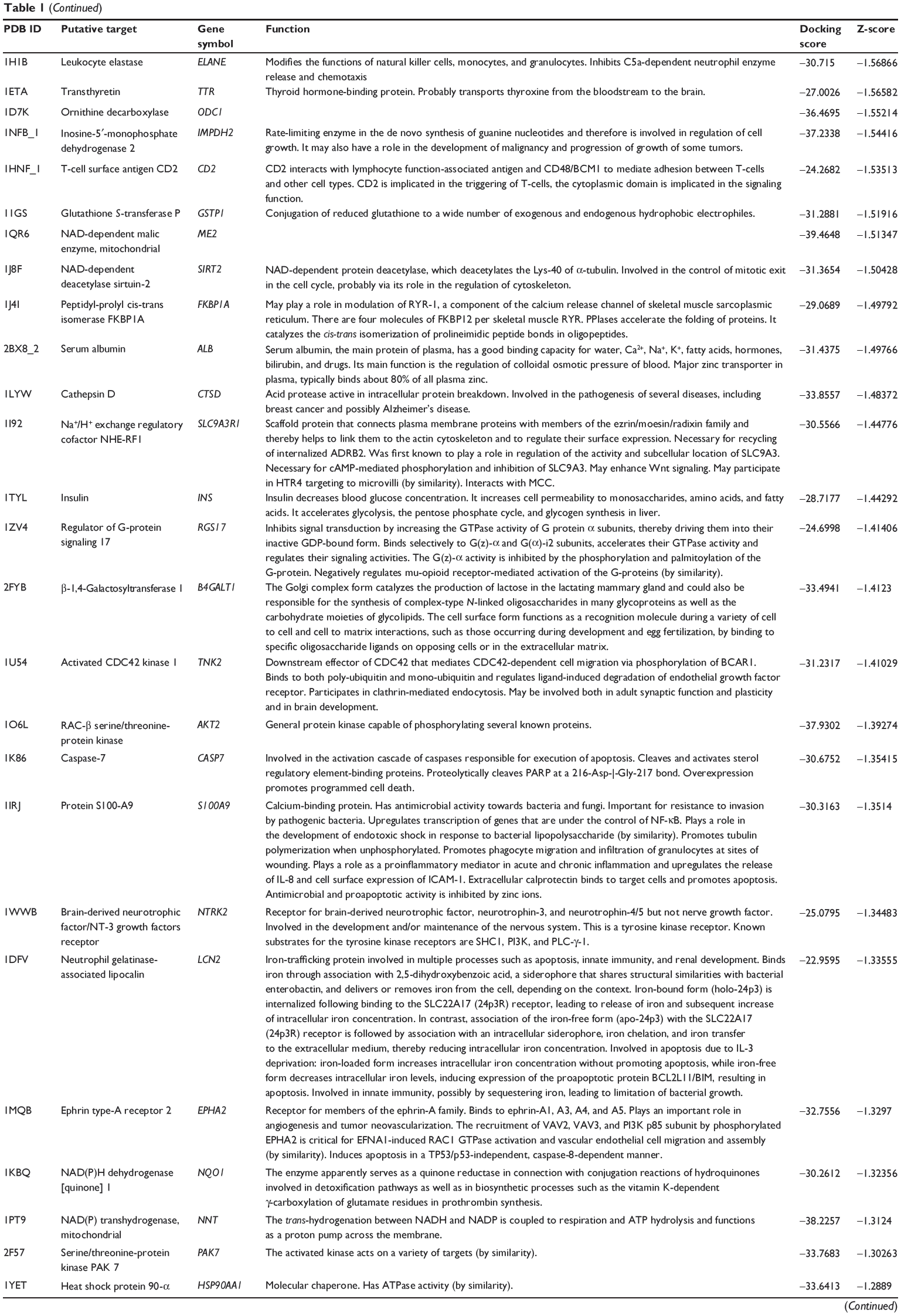     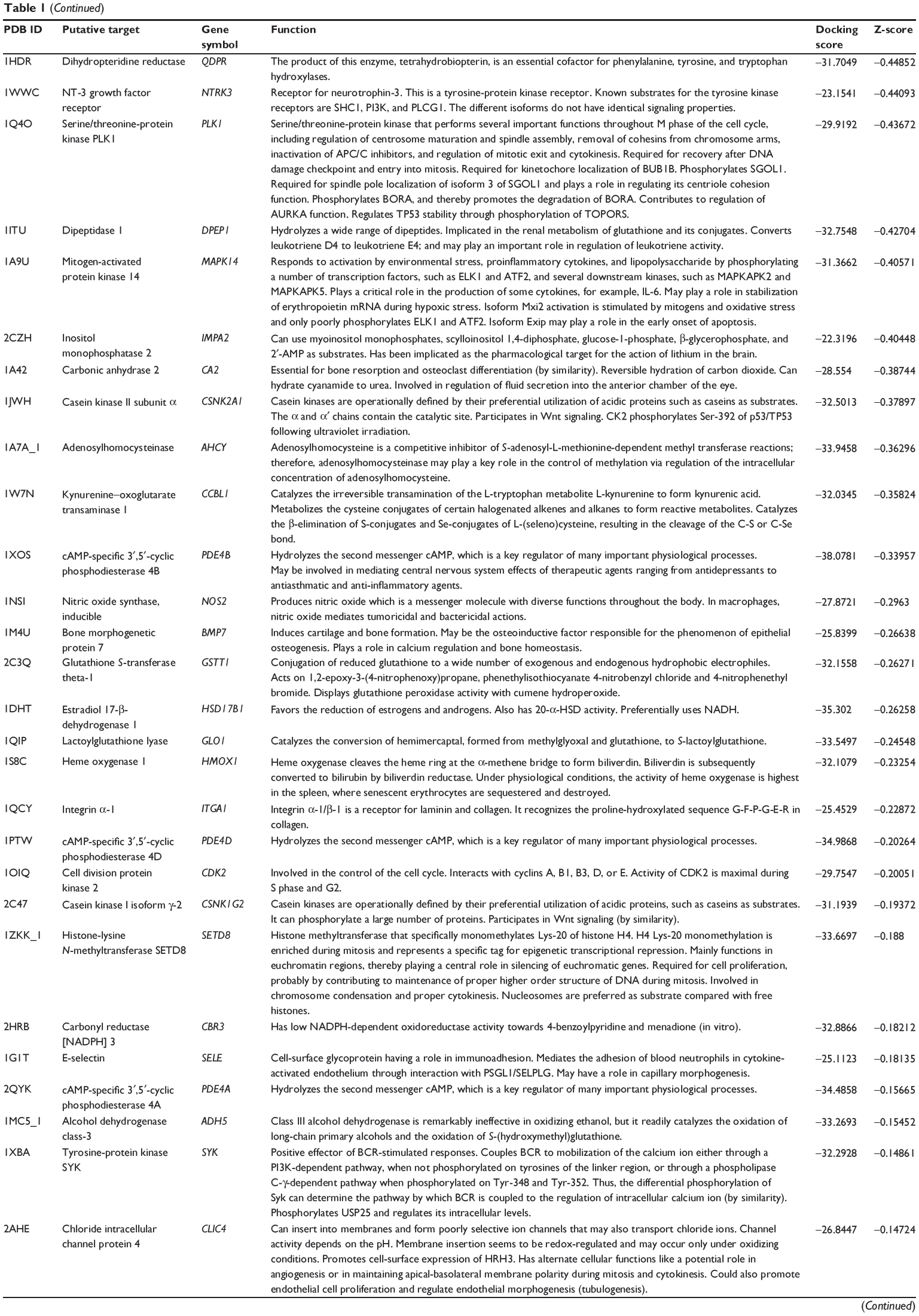 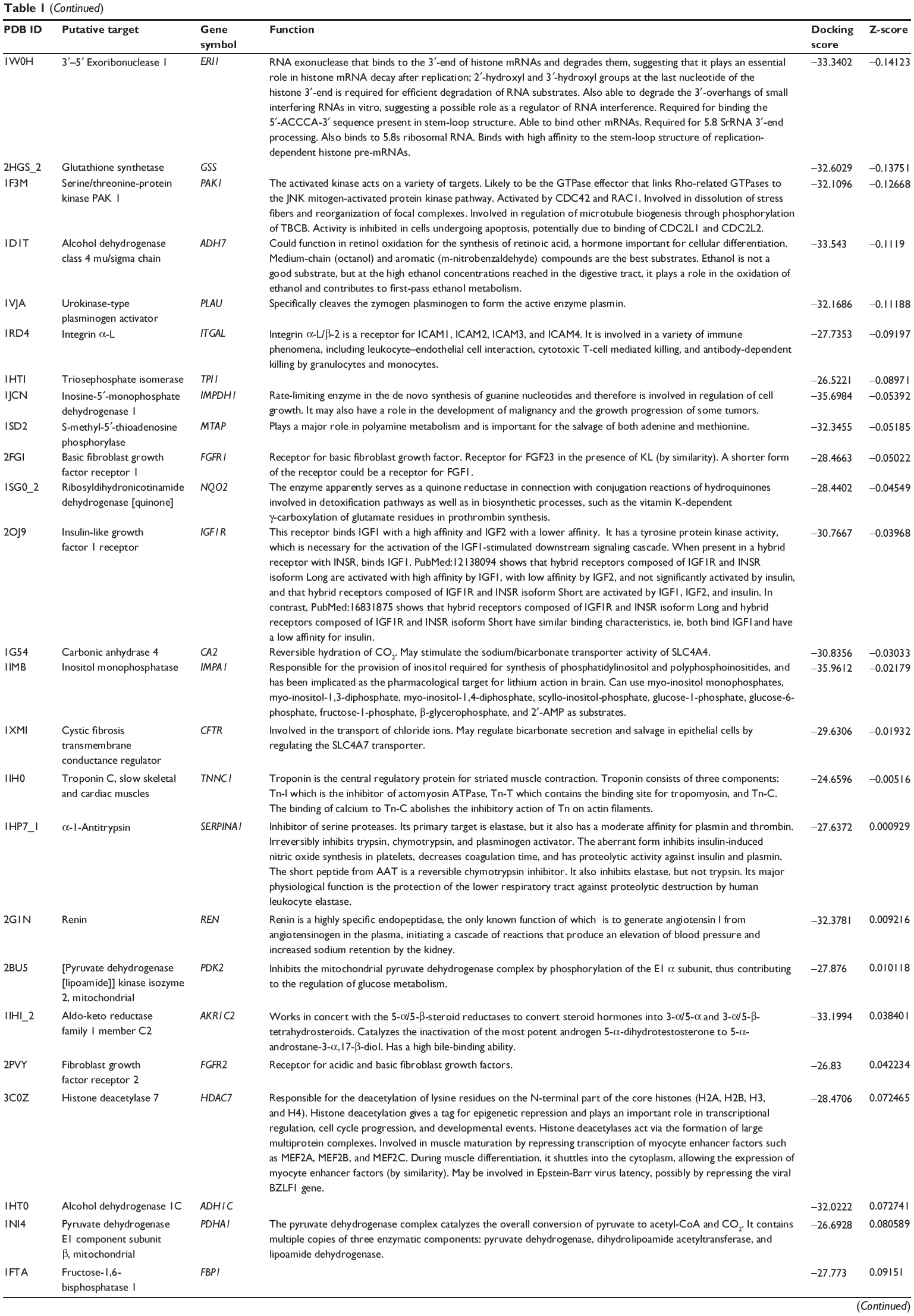 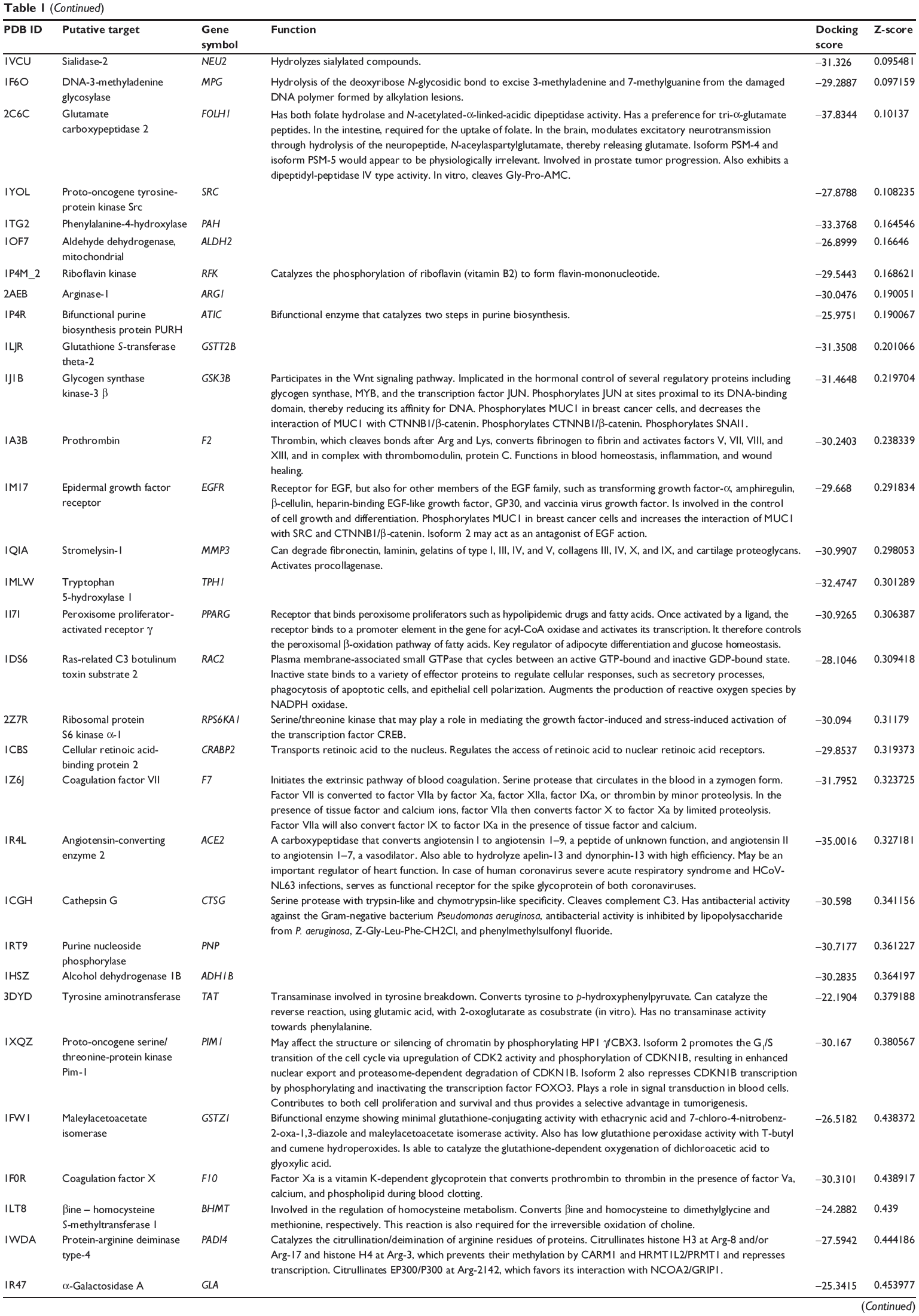 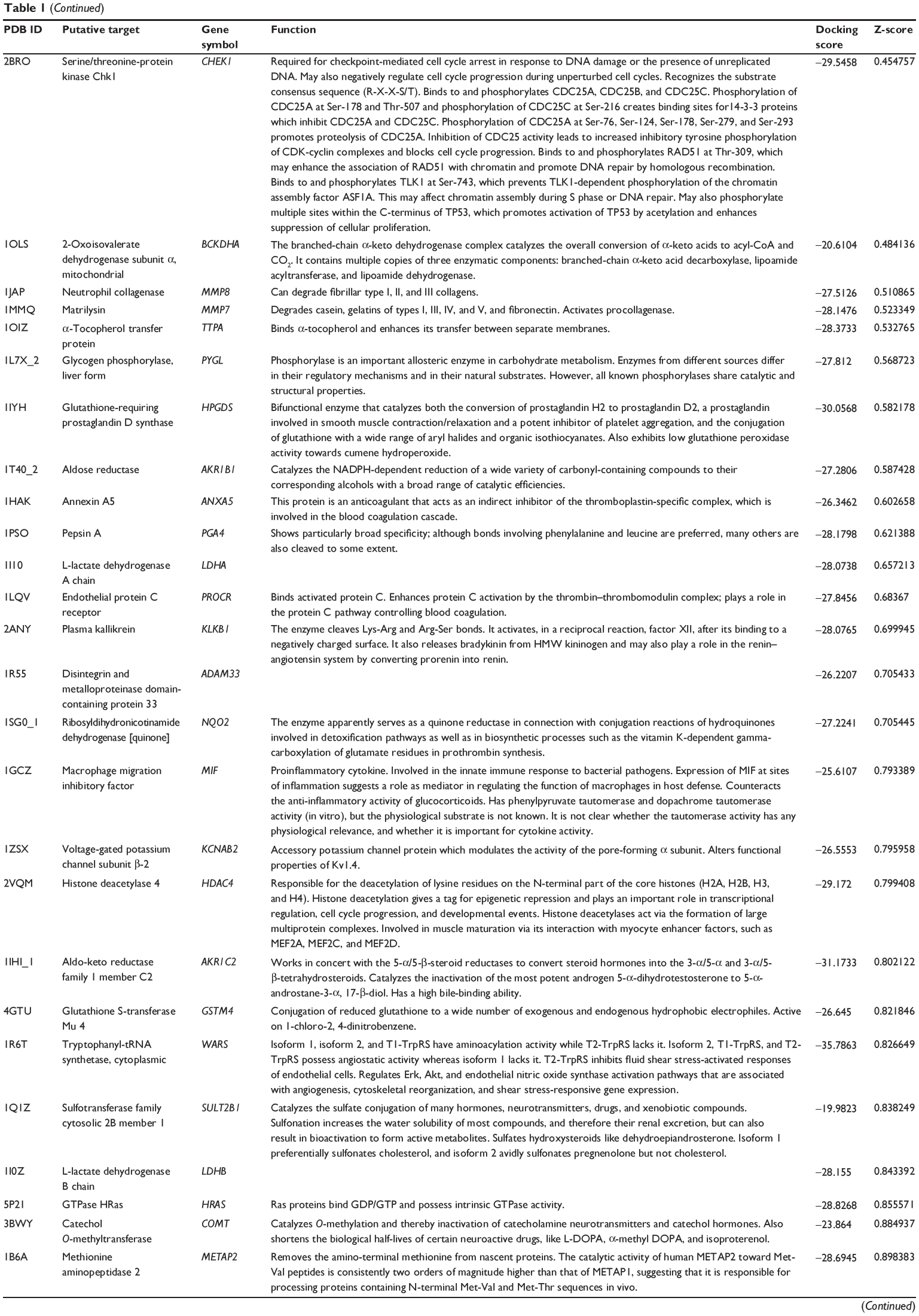  
|
Table 1 Predicted protein targets of XKB |
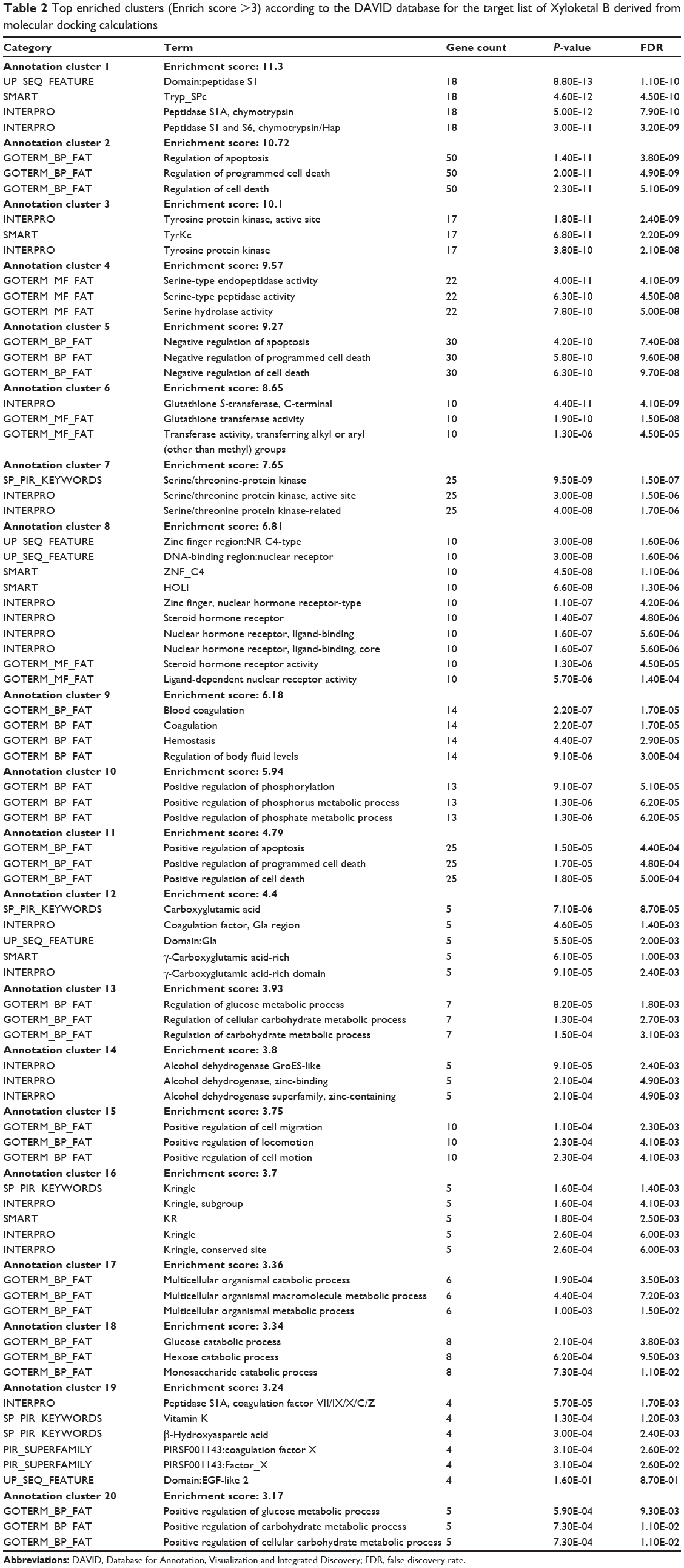
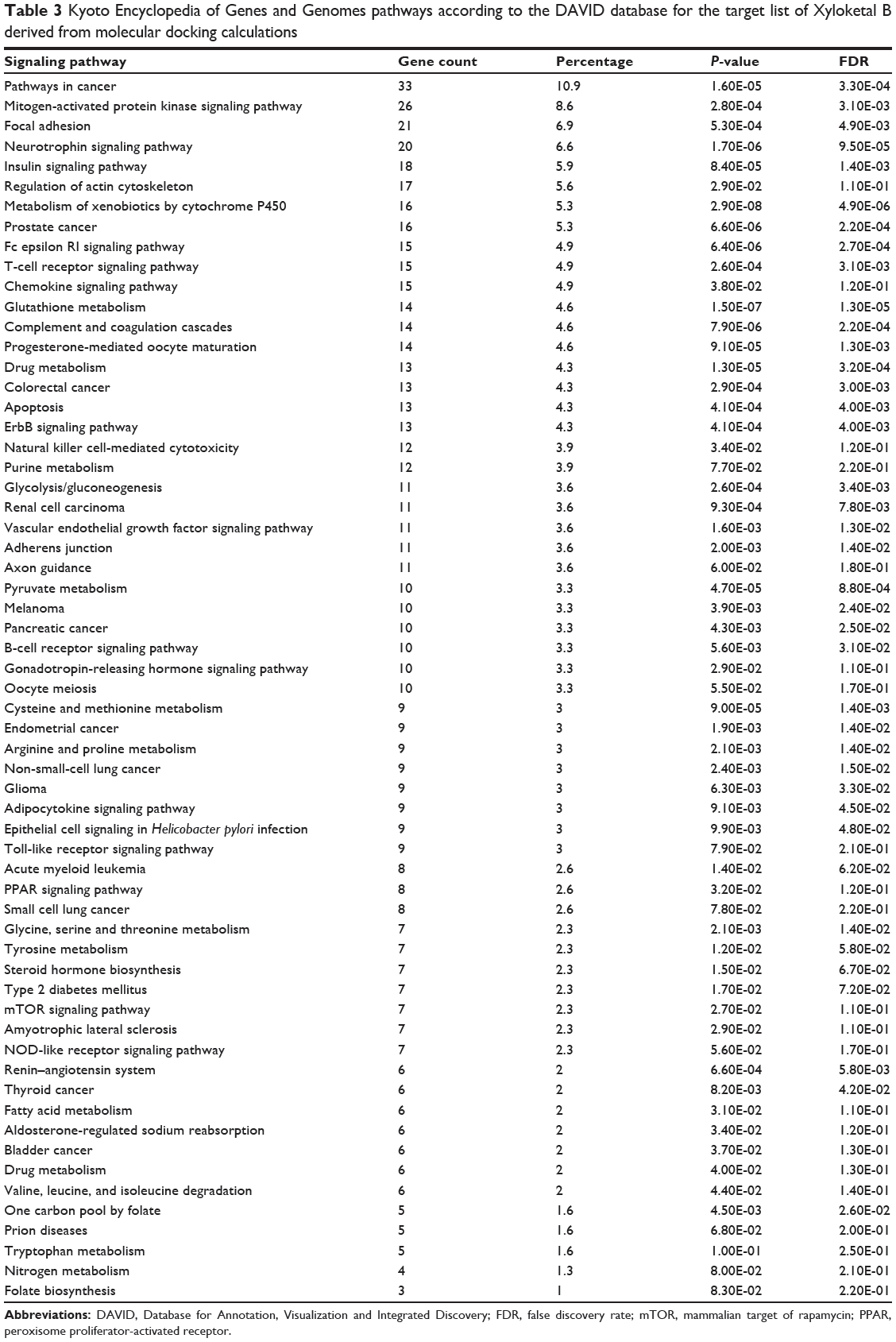
As shown in Table 2, DAVID analysis identified 20 functional clusters that were significantly enriched (enrichment score >3) in the target list derived from molecular docking calculations, including energy metabolism, signal transduction, vascular regulation, and carbohydrate metabolism. Further, 61 Kyoto Encyclopedia of Genes and Genomes pathways were found to be significantly enriched in the target list (Table 3). In particular, the pathways for metabolism of xenobiotics by CYPs and other enzymes were predicted to be modulated by XKB by DAVID analysis.
|
Table 2 Top enriched clusters (Enrich score >3) according to the DAVID database for the target list of Xyloketal B derived from molecular docking calculations |
|
Table 3 Kyoto Encyclopedia of Genes and Genomes pathways according to the DAVID database for the target list of Xyloketal B derived from molecular docking calculations |
In addition, DAVID analysis revealed overlapped molecular targets between XKB and probucol (Table 4), including CD1A, FKBP1A, PCTP, ME2, G6PD, FKBP1B, FABP7, CETP, PREP, PAK6, SIRT5, PRDX2, S100A9, SIRT5, F13A1, GM2A, CMA1, SORD, IMPDH2, GSTP1, CDA, CTSB, and GSTO1. Probucol is an antihyperlipidemic drug originally developed for the treatment of coronary artery disease but withdrawn from the US market in 1995.33 The targets shared by both XKB and probucol suggest that XKB may be a promising agent for lowering of blood lipids.
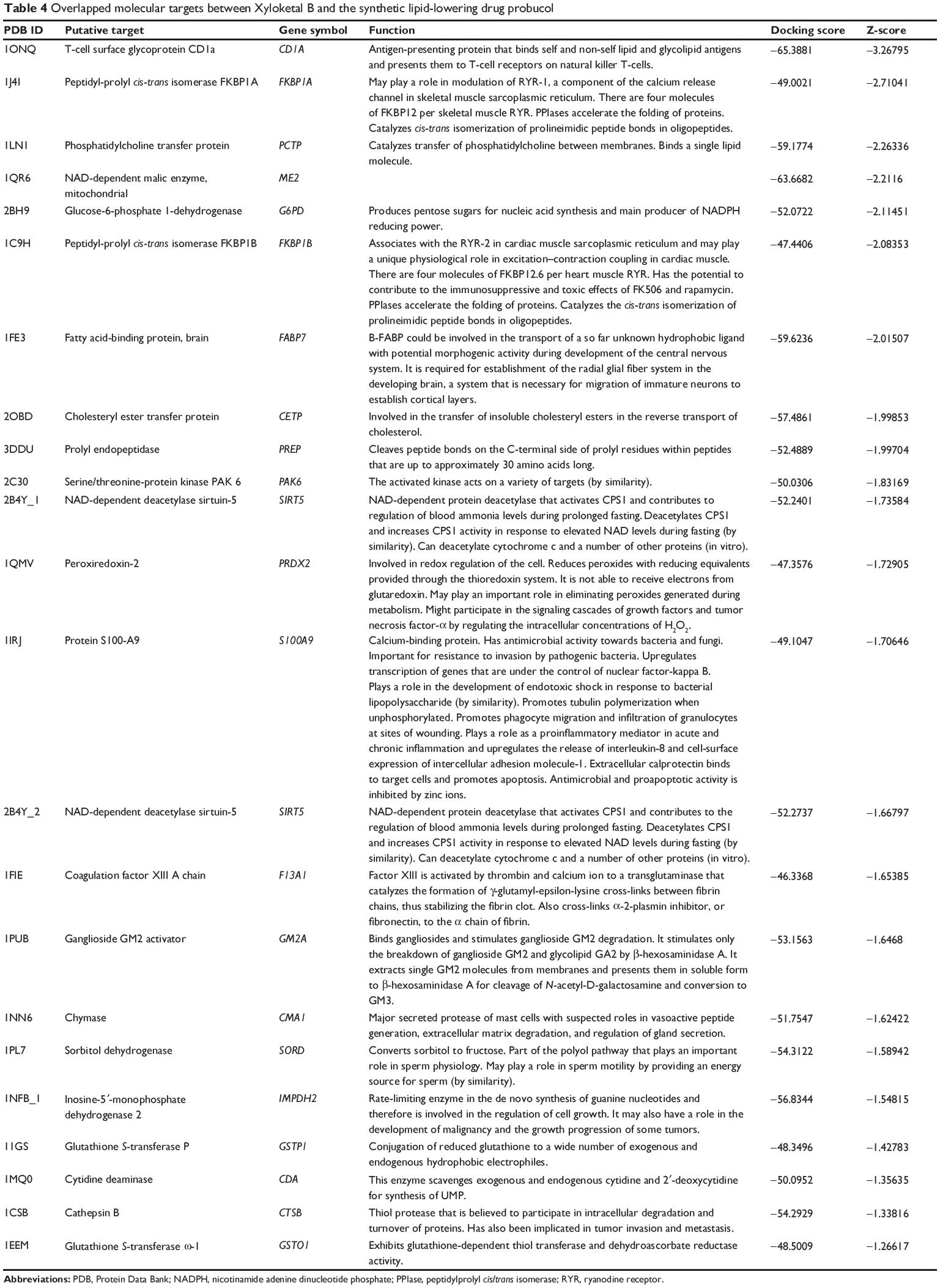
|
Table 4 Overlapped molecular targets between Xyloketal B and the synthetic lipid-lowering drug probucol |
Validation of predicted molecular targets of XKB by published data
Given that we had predicted a number of molecular targets and related signaling pathways possibly regulated by XKB, we performed a validation study using published data after an extensive search via PubMed (from inception to August 2014). There were four published papers reporting the molecular targets and mechanisms of action of XKB.34–37 XKB reduced oxidative stress via induction of heme oxygenase-1 and suppression of NADPH oxidase activity involving the PI3K/Akt/Nrf2 signaling pathways in human umbilical vein endothelial cells.37 In consistency with previous studies,34–37 our previous study also showed that XKB decreased the activity of NADPH oxidase and the expression of gp91phox and p47phox and that XKB increased the expression of Bcl-2 in human umbilical vein endothelial cells with injury induced by oxidized low-density lipoprotein.34 XKB decreased mitochondrial superoxide, mitochondrial fragmentation, expression of GTPase dynamin-related protein 1, and the mitochondrial membrane potential in PC12 cells.35,36 These data provided preliminary evidence for the validation of predicted molecular targets of XKB using a bioinformatic approach.
XKB interacts with human CYP3A4 and rat Cyp3a2 by hydrogen bond formation
Following prediction and validation of the molecular targets, we examined the possible interactions between XKB, midazolam (a CYP3A4 probe substrate), ketoconazole (a CYP3A4 inhibitor), and probucol (an antihyperlipidemic drug) with a human CYP3A4 and rat Cyp3a2 homology model using the Discovery Studio program 3.1. As shown in Table 5, the CDOCKER interaction energy ranged from 25 to 46 kcal/mol between CYP3A4 and XKB, midazolam, ketoconazole, and probucol. XKB formed a hydrogen bond with Gly481 at the active site of human CYP3A4 (PDB ID 4K9W; Figure 2A). Both midazolam and ketoconazole bound to the active site of human CYP3A4 via π-π stacking with Phe108 (Figure 2B) and Arg105 (Figure 2C), respectively. However, probucol did not show hydrogen bond formation, π-π stacking, or charge interaction with human CYP3A4 (Figure 2D).

|
Table 5 Molecular interactions between XKB, MDZ, KTZ, and probucol and the human CYP3A4 and rat Cyp3a2 homology model |
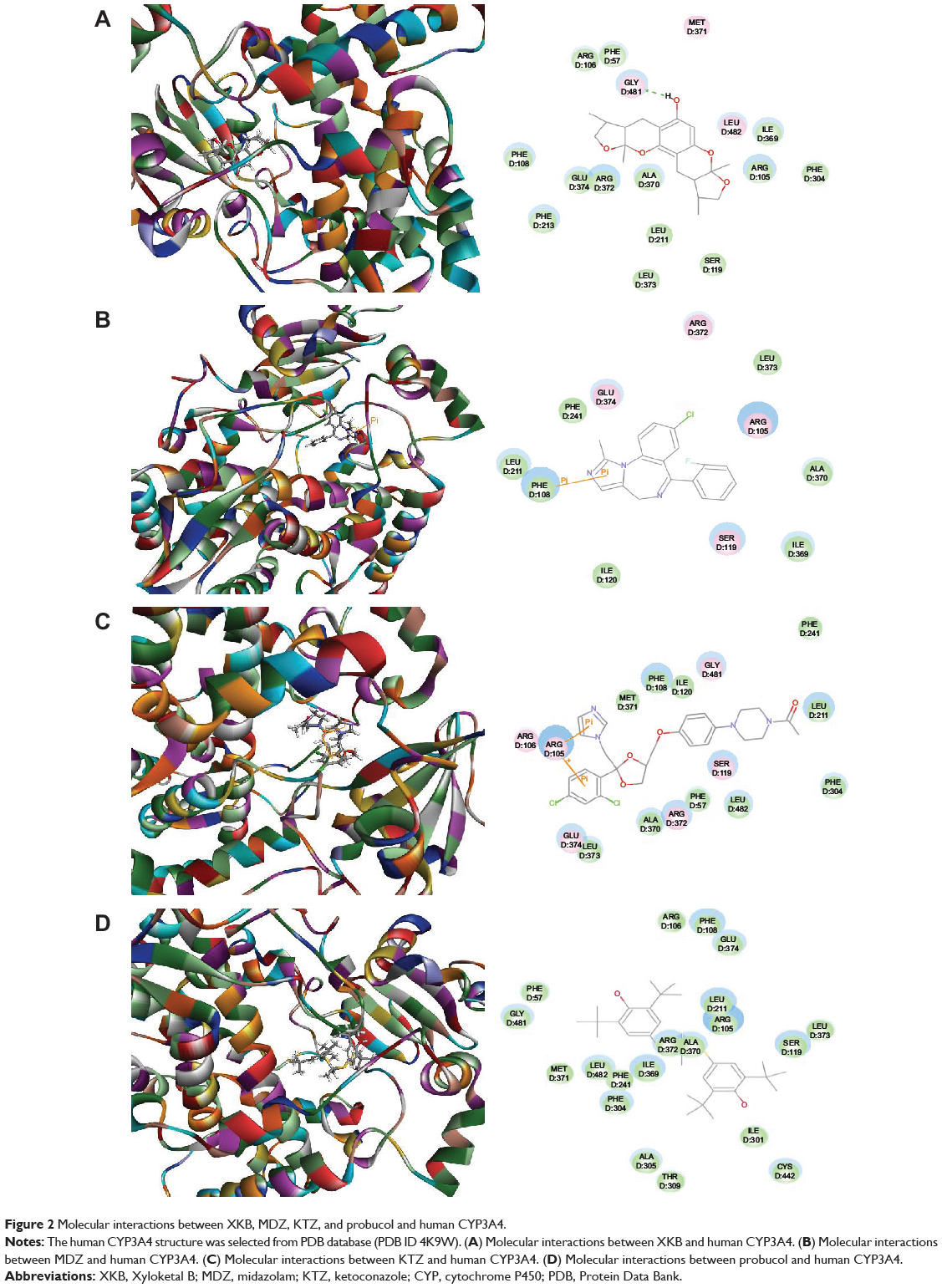
|
Figure 2 Molecular interactions between XKB, MDZ, KTZ, and probucol and human CYP3A4. |
Further, we built a homology model of rat Cyp3a2 based on the crystal structure of human CYP3A4 (PDB ID 4K9W). The sequence similarity and identity between the human CYP3A4 and rat Cyp3a2 homology model was 83.3% and 68.4%, respectively. The CDOCKER interaction energy ranged from 23 to 34 kcal/mol (Table 5). The docking results showed that XKB bound to the active site of rat Cyp3a2 homology model by hydrogen bond formation with Ala482 (Figure 3A). Midazolam was readily docked into the active site of the rat Cyp3a2 homology model by π-π stacking with Phe305 (Figure 3B). Ketoconazole bound with Arg105 by hydrogen bond formation at the active site of the rat Cyp3a2 homology model (Figure 3C). However, there was no hydrogen bond formation, π-π stacking, or charge interaction between probucol and the rat Cyp3a2 homology model (Figure 3D). The compound-CYP complexes with the highest CDOCKER interaction energy were selected, and two-dimensional and three-dimensional images were taken (Figures 2 and 3).

|
Figure 3 Molecular interactions between XKB, MDZ, KTZ, and probucol and the rat Cyp3a2 homology model. |
Taken together, the results showed that XKB could bind to the active sites of human CYP3A4 and rat Cyp3a2 via hydrogen bond formation and/or π-π stacking. XKB may serve as a substrate and/or inhibitor of these two enzymes.
Predicted ADMET properties of XKB, midazolam, ketoconazole, and probucol
The predicted ADMET properties of XKB, midazolam, ketoconazole, and probucol are shown in Table 6. The aqueous solubility value (log [molar solubility]) of XKB was between −6.0 and −4.0. XKB showed a moderate ability to penetrate the blood–brain barrier and a good intestinal absorption profile. All properties and optimal prediction space components of XKB were within the expected ranges with regard to the CYP2D6 ligand, hepatotoxicity, and plasma protein binding level. Midazolam and ketoconazole showed similar ADMET profiles to XKB, except for hepatotoxicity. ADMET Predictor showed that the optimal prediction space PC25 values for midazolam and optimal prediction space PC30 values for ketoconazole were out of range, suggesting that both compounds might be hepatotoxic (Table 6). Probucol had very low aqueous solubility and very low intestinal absorption, which are in agreement with the data reported for this drug (Table 6). The logP value for probucol is 10.91,38 and its oral bioavailability is only about 10%, but can be improved using nanotechnology.39–41
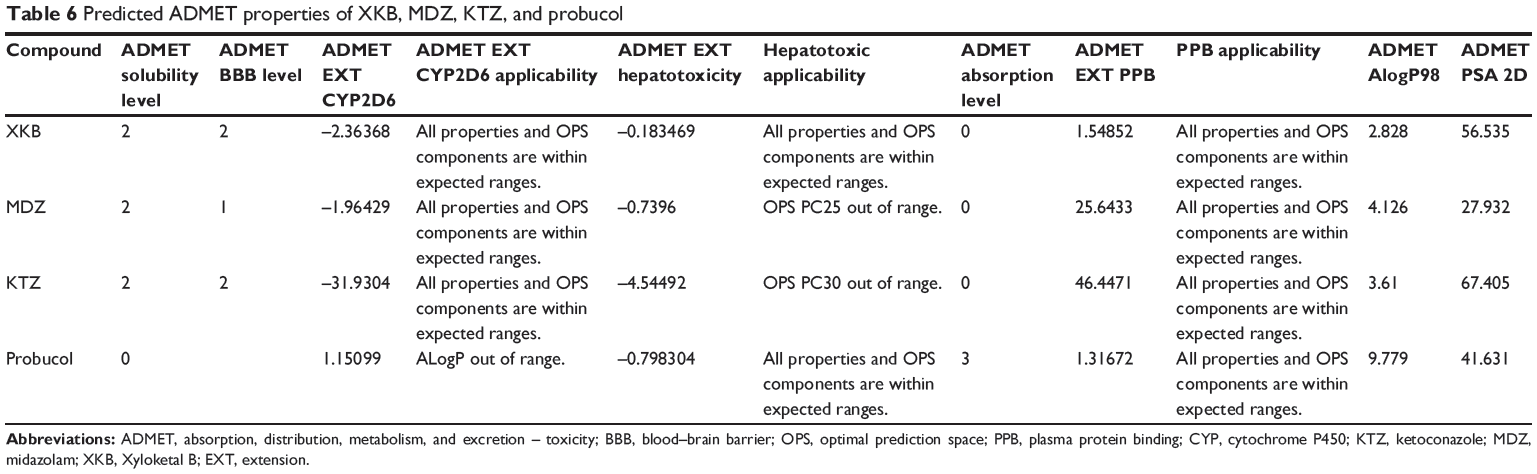
|
Table 6 Predicted ADMET properties of XKB, MDZ, KTZ, and probucol |
Validation of HPLC method for determination of midazolam and 1′-OH-MDZ in rat plasma
Validation of the HPLC method was performed prior to determination of the plasma PKs of midazolam and 1′-OH-MDZ in rats. This included an examination of specificity, linearity, lower limit of quantification, precision, accuracy, recovery, and stability.
Specificity
Figure 4 shows the representative chromatogram of blank rat plasma and rat plasma spiked with midazolam, 1′-OH-MDZ, and diazepam. The results indicate that midazolam, 1′-OH-MDZ, and diazepam were separated completely under the assay conditions and no endogenous interfering peaks were observed at their retention times.
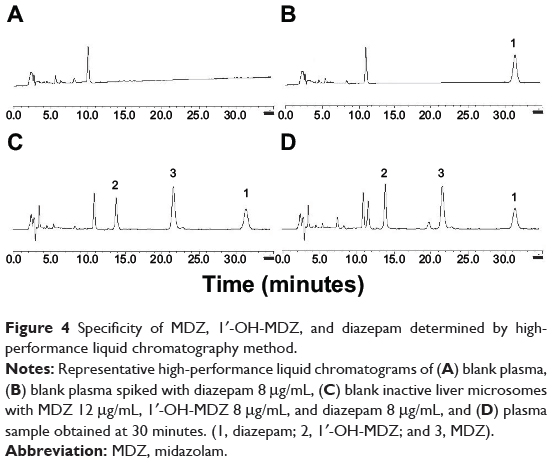
|
Figure 4 Specificity of MDZ, 1′-OH-MDZ, and diazepam determined by high-performance liquid chromatography method. |
Linearity and lower limit of quantification
The calibration curves for midazolam and 1′-OH-MDZ in rat plasma were linear in the concentration range from 0.0375 to 4.8 μg/mL and from 0.025 to 3.2 μg/mL, respectively. The regression equation of the calibration curve (obtained from seven points) of midazolam was y =1.6443x −0.0181 with a correlation coefficient of 0.9999. For 1′-OH-MDZ, the regression equation of the calibration curve was y =1.6619x −0.046 with a correlation coefficient of 0.9995, where y was the peak area ratio of midazolam and 1′-OH-MDZ over diazepam, and x was the plasma concentration of midazolam and 1′-OH-MDZ. The lower limit of quantification was established by determining the concentrations of five spiked calibration standards in rat plasma and found to be 0.0375 μg/mL and 0.025 μg/mL for midazolam and 1′-OH-MDZ, respectively. Taken together, the results show good linearity, indicating that this method can be used to measure plasma samples.
Precision and accuracy
The precision and accuracy of the method were determined using quality control samples at three concentrations of midazolam (0.075, 0.30, and 4.80 μg/mL) and 1′-OH-MDZ (0.05, 0.40, 3.20 μg/mL). The precision of the assay was expressed by the relative standard deviation of the mean value from the nominal concentration. Accuracy was expressed as the percentage of the mean value calculated from the nominal concentration. The results are shown in Table 7. These values were within the acceptable range, indicating that the developed method was reproducible, accurate, and reliable for quantitative determination of midazolam and 1′-OH-MDZ in rat plasma.
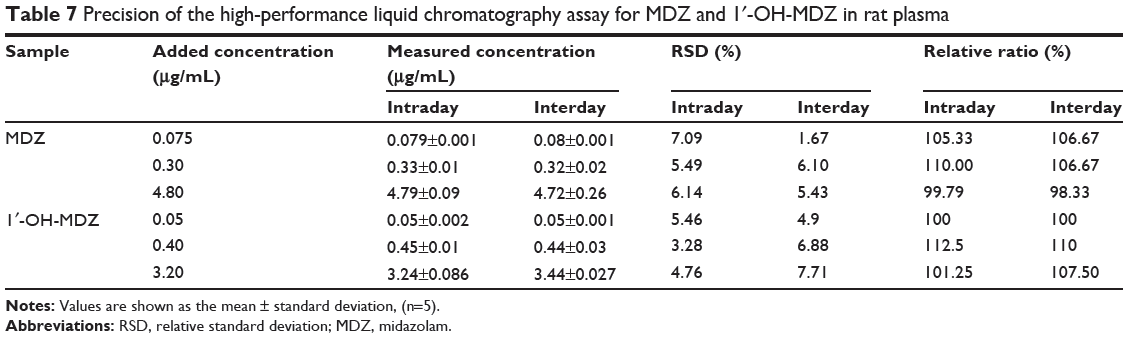
|
Table 7 Precision of the high-performance liquid chromatography assay for MDZ and 1′-OH-MDZ in rat plasma |
Recovery
The mean extraction recovery of midazolam at the three concentrations of 0.075, 0.30, and 4.80 μg/mL was 93.06%±3.55%, 94.00%±4.90%, and 82.32%±3.34%, respectively (n=5). The mean extraction recovery of 1′-OH-MDZ at the three concentrations of 0.05, 0.40, and 3.20 μg/mL was 88.995%±3.22%, 85.81%±2.11%, and 85.23%±3.93%, respectively (n=5). Recovery of diazepam from rat plasma was 90.18%±4.52% at the concentration of 0.40 μg/mL.
Stability
Stability tests were performed at low, medium, and high concentrations of the quality control samples and the results are shown in Table 8. Stock solutions of midazolam and 1′-OH-MDZ were stable for at least 30 days when stored at −20°C. Further, the concentrations of midazolam and 1′-OH-MDZ did not change significantly after exposure of the samples at room temperature for 2 and 12 hours or three freeze–thaw cycles. The results indicate that midazolam and 1′-OH-MDZ are stable enough to be analyzed using this method within a predetermined timeframe.
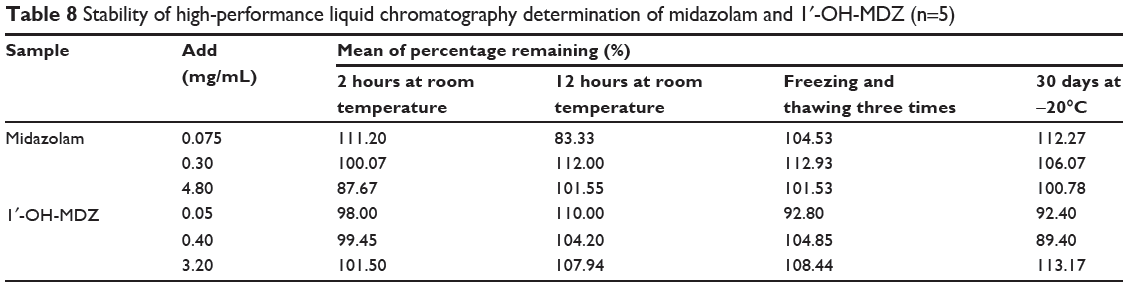
|
Table 8 Stability of high-performance liquid chromatography determination of midazolam and 1′-OH-MDZ (n=5) |
XKB alters plasma PKs of midazolam and 1′-OH-MDZ in rats
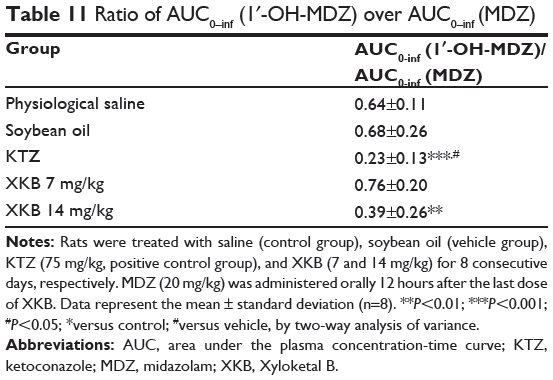
Given that we had observed potential interactions between XKB and rat Cyp3a2, we then examined the effect of XKB on Cyp3a-mediated drug metabolism. Successive oral administration of XKB was scheduled using midazolam as a probe drug. Midazolam is a well-known probe substrate for human CYP3A4/5, but it is unclear if it is an appropriate probe for determining rat Cyp3a activity in vivo. The major metabolite of midazolam in humans is 1′-OH-MDZ.42 The mean plasma profiles of midazolam and 1′-OH-MDZ after 8 consecutive days of treatment with XKB are shown for each group in Figures 5 and 6. The associated PK parameters are listed in Tables 9–11. The results indicate there were significant changes in the PK parameters of midazolam and 1′-OH-MDZ in rats treated with XKB for 8 days. In comparison with vehicle-treated rats, treatment with 14 mg/kg XKB increased the Cmax, AUC0–t, AUC0–inf, and AUMC of midazolam by 2.9-fold, 2.7-fold, 2.6-fold, and 2.0-fold, respectively (P<0.05). In contrast, the CL/F and V/F of midazolam were decreased by 63.0% and 84.0% in rats treated with 14 mg/kg XKB, respectively, compared with rats receiving vehicle only. The ka, ke, t1/2ka, t1/2ke, Tmax, and mean residence time for midazolam remained unchanged after rats were given 14 mg/kg XKB orally for 8 days. Our data also show that oral administration of XKB at 7 mg/kg significantly changed the AUC0–t of midazolam, with a 1.4-fold increase. For 1′-OH-MDZ, oral administration of XKB 7 mg/kg significantly increased AUC0–t, AUC0–inf, and AUMC by 1.5-fold, 1.6-fold, and 1.9-fold, respectively, when compared with the control rats treated with vehicle only. Similarly, there was a 1.4-fold, 1.5-fold, and 2-fold increase in AUC0–t, AUC0–inf, and AUMC, respectively, in rats treated with XKB 14 mg/kg for 8 days when compared with control rats. Compared with vehicle-treated rats, the ratio of plasma AUC for 1′-OH-MDZ over that of midazolam was significantly decreased by 42.6% in rats treated orally with XKB 14 mg/kg. However, treatment of XKB at either dose did not change the CL/F and V/F of midazolam or 1′-OH-MDZ in rats.
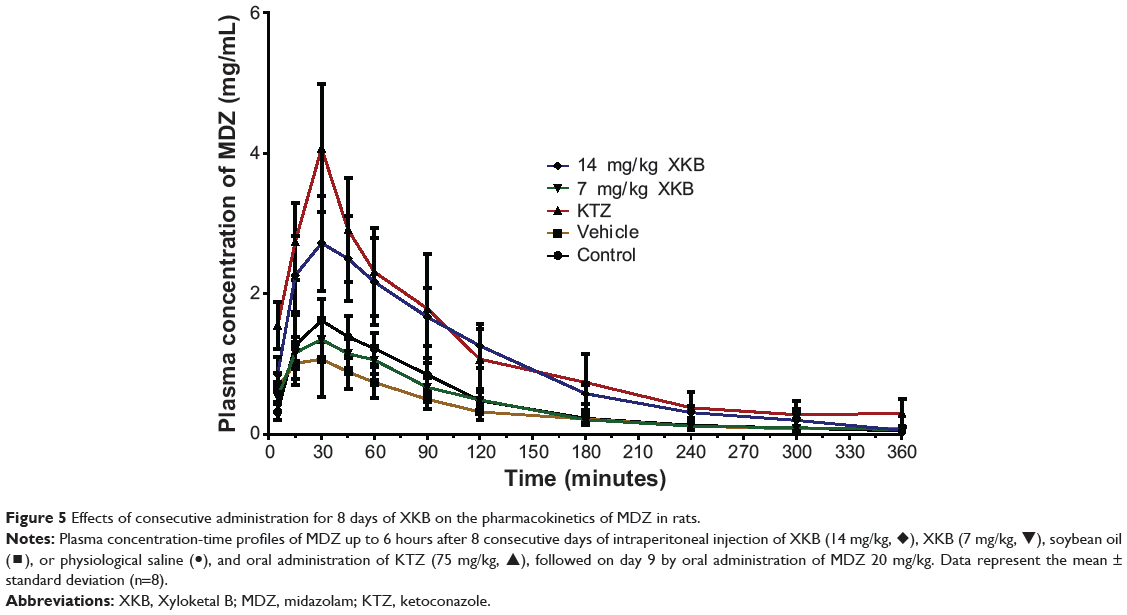
|
Figure 5 Effects of consecutive administration for 8 days of XKB on the pharmacokinetics of MDZ in rats. |
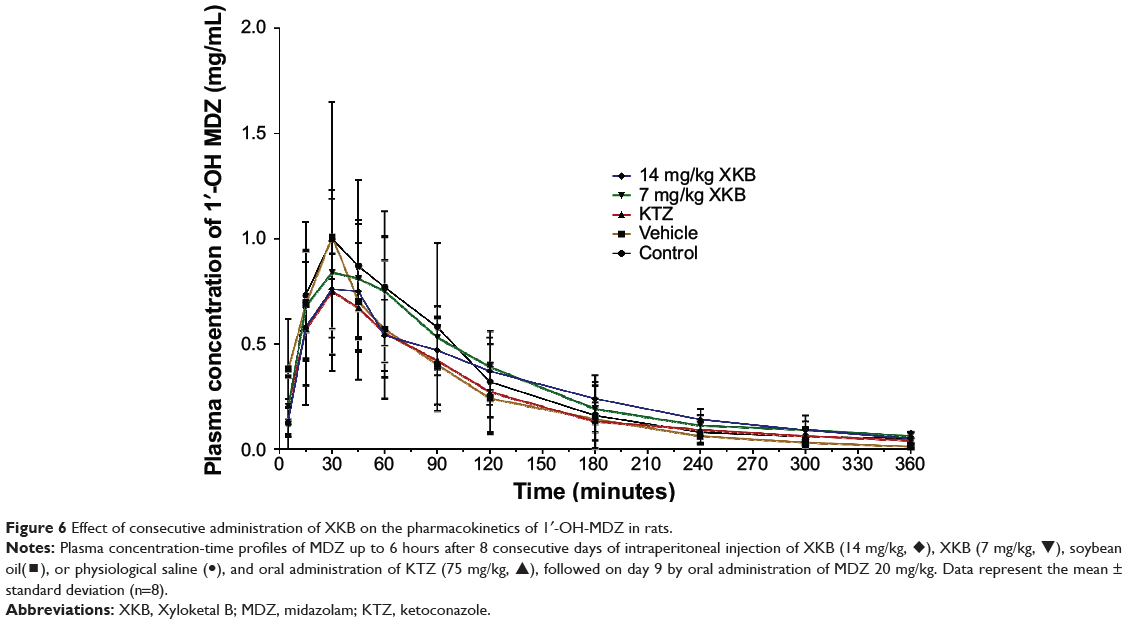
|
Figure 6 Effect of consecutive administration of XKB on the pharmacokinetics of 1′-OH-MDZ in rats. |
|
Table 9 Pharmacokinetic parameters for midazolam in Sprague-Dawley rats |

|
Table 10 Pharmacokinetic parameters of 1′-OH-MDZ in Sprague-Dawley rats |
|
Table 11 Ratio of AUC0–inf (1′-OH-MDZ) over AUC0–inf (MDZ) |
In addition, compared with normal saline or soybean oil, a single dose of ketoconazole 75 mg/kg significantly increased the Cmax, AUC0–t, and AUC0–inf of midazolam by 3.5-fold, 3.6-fold, and 3.6-fold, respectively, and decreased the V/F and CL/F of midazolam by 75.2% and 73.7%, respectively. Further, compared with vehicle-treated rats, the ratio of the plasma AUC of 1′-OH-MDZ over that of midazolam was significantly decreased by 66.2% in rats treated orally with ketoconazole 75 mg/kg.
Collectively, the above results show that XKB 7 or 14 mg/kg significantly reduced the metabolism of midazolam in rats without marked dose dependence, probably due to the decreased activity of Cyp3a. Since the total clearance and distribution of midazolam and its metabolite were not altered by XKB, the possibility of modulation of other pathways by XKB cannot be excluded.
Oral administration of XKB for 8 days inhibits activity and expression of rat hepatic Cyp3a
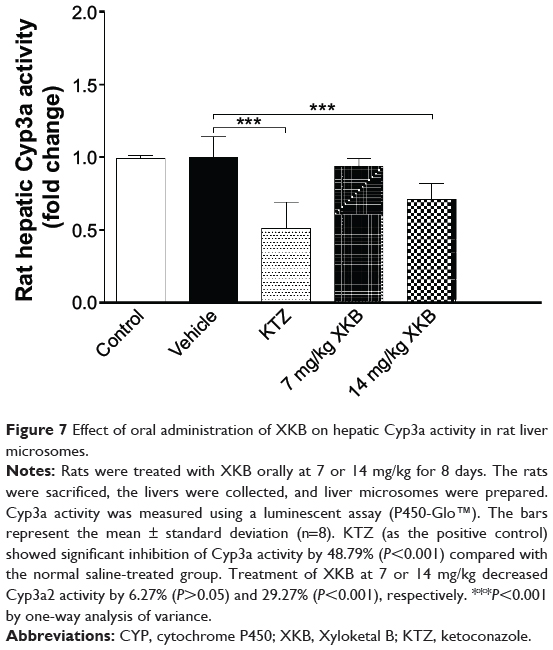
Following observation of the reduced metabolism of the probe substrate, hepatic Cyp3a activity was measured using a luminescent assay after the rats were dosed with XKB for 8 days. As shown in Figure 7, the activity of rat hepatic Cyp3a was significantly inhibited by XKB. In comparison with the vehicle group (soybean oil), there was a 6.27% (P>0.05) and 29.27% (P<0.001) decline in the activity of Cyp3a when rats were treated with 7 or 14 mg/kg XKB, respectively. Ketoconazole also showed significantly inhibited Cyp3a activity, with a 48.79% reduction (P<0.001) compared with the saline-treated group.
|
Figure 7 Effect of oral administration of XKB on hepatic Cyp3a activity in rat liver microsomes. |
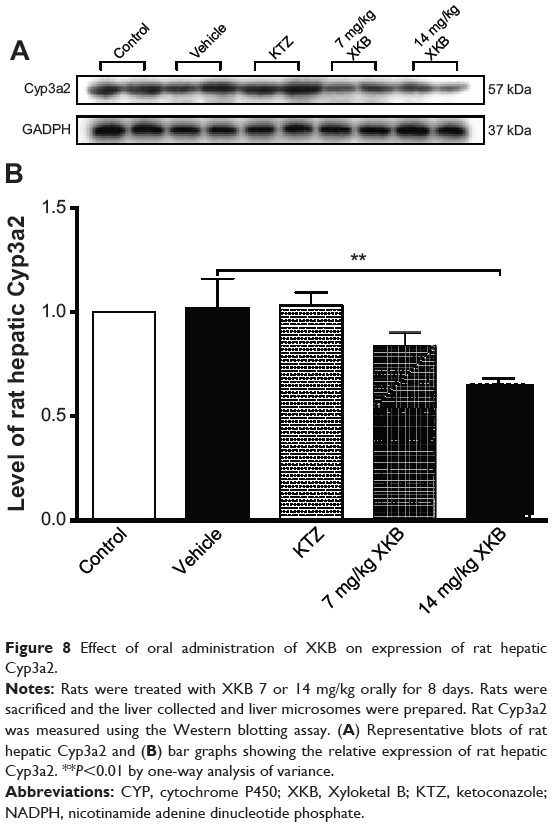
To examine further the regulatory effect of XKB on Cyp3a levels, Cyp3a2 expression was determined by Western blotting assay. As shown in Figure 8, administration of XKB for 8 days significantly inhibited expression of Cyp3a2 in the rat liver. Compared with the vehicle group (soybean oil), there was a 19.58% and 28.97% (P<0.001) decrease in expression of Cyp3a2 when rats were treated with 7 or 14 mg/kg XKB, respectively. However, administration of ketoconazole did not change Cyp3a2 expression in the rat liver. Taken together, XKB has a potent inhibitory effect on Cyp3a2 expression and activity in the rat liver. The mechanisms for the downregulating effect of XKB on hepatic Cyp3a2 in rats are unknown.
|
Figure 8 Effect of oral administration of XKB on expression of rat hepatic Cyp3a2. |
Discussion
XKB is a potent antioxidant with great potential for the treatment of CVD but its molecular targets and effects on drug-metabolizing enzymes remain unknown.37 Human CYP3A is a major CYP subfamily that metabolizes over 60% of endogenous and exogenous materials.6 Modulation of the activity and expression of CYP3A would cause substantial changes in drug metabolism in the clinical setting. In the present study, we predicted over 324 molecular targets and 61 related signaling pathways regulated by XKB. We found that XKB interacted with the rat hepatic Cyp3a2 homology model via hydrogen bond formation. Further, we found that XKB increased the AUC0–t and Cmax of midazolam, and inhibited the activity and expression level of Cyp3a in rats treated with XKB for 8 days. Our findings indicate that the beneficial effects of XKB can be attributed to regulation of a network of molecular targets and signaling pathways and that XKB can regulate Cyp3a-mediated drug metabolism by suppressing the activity and expression of Cyp3a in vivo.
Bioinformatic approaches have become a valuable way of predicting the interactome of a chemical molecule, so we used a DDI-CPI tool to predict the potential targets and employed DAVID to analyze the molecular targets and related signaling pathways regulated by XKB. Our findings show that XKB can modulate a number of functional proteins and related signaling pathways. These proteins and signaling pathways have important roles in the regulation of redox homeostasis, cell proliferation, apoptosis, energy metabolism, metabolism of xenobiotics, lipid and carbohydrate metabolism, and the inflammatory response. Interestingly, XKB shared a number of molecular targets with probucol, which was initially developed as an antihyperlipidemic drug for the treatment of coronary artery diseases but was removed from the USA market in 1995 after it was found to prolong the QT interval.33,43 The overlapped molecular targets suggest that XKB can regulate the same targets and signaling pathways as probucol. Probucol has been reported to lower cholesterol levels in the blood by increasing the rate of low-density lipoprotein catabolism, inhibiting cholesterol synthesis, and delaying cholesterol absorption.44 It is also a potent antioxidant that suppresses the oxidation of cholesterol in low-density lipoproteins. The cholesterol efflux regulatory protein ABCA1 is considered its main target.45 As a natural marine compound, XKB may be a promising agent for the treatment of CVD when its potential cardiovascular side effects can be minimized or avoided.
The DDI-CPI approach provides a rapid and inexpensive strategy to predict potential targets, identify drug repositioning potential, and evaluate and determine adverse drug reactions of a chemical/drug via molecular docking of small compound across human proteome,23,24,27,46,47 although this web-based program has several limitations that may affect the accuracy of the outcome.24 For example, DDI-CPI is able to fully reveal potential targets due to the lack of a human protein structure and the assessment stringency.24
XKB has shown potent antioxidative, anti-inflammatory, and anticancer effects in preclinical studies.17–18 This is supported by our prediction that XKB can target multiple pathways associated with oxidative stress, redox homoeostasis, cell proliferation, and apoptosis.
In order to characterize the role of XKB in the regulation of CYPs, we next performed a homology modeling experiment to examine the interactions between XKB and rat Cyp3a2. The rat Cyp3a2 homology model was built based on human CYP3A4 (PDB ID 4K9W). Our data show that the rat Cyp3a2 homology model shared 83.3% sequence similarity and 68.4% identity with human CYP3A4. It has been reported that rat Cyp3a2 exhibits a 73% homology amino acid sequence to human CYP3A4.48 This difference may affect the interaction between XKB and rat Cyp3a2. Indeed, our findings show that XKB interacted with the rat Cyp3a2 homology model via hydrogen bond formation at Ala482 located in the active site of the enzyme. However, there was no interaction between XKB and human CYP3A4 Ala482 located in the active site, but Gly481. Collectively, our modeling study shows that XKB can act as a substrate and/or inhibitor for both rat Cyp3a2 and human CYP3A4. However, when we extrapolate these results to humans, we should bear in mind that Cyp3a2 and CYP3A4 share a certain degree of sequence similarity identity, so functional validation is always needed.
The ADMET Predictor showed that XKB had moderate water solubility and oral absorption and that probucol had very low water solubility and oral absorption, which is consistent with previously reported data.38–41 The predicted ADMET profiles show that XKB might be a better oral agent than probucol for drug development.
Midazolam is a probe substrate for CYP3A with sedative, amnesic, anxiolytic, muscle relaxant, and anticonvulsant properties in humans. It has been widely used to examine the degree of drug interaction and inhibitory effect of xenobiotics on the activity and function of CYP3A4/5 via evaluation of urinary and/or plasma levels of midazolam and its main metabolite 1′-OH-MDZ.49,50 In our study, plasma concentrations of midazolam and 1′-OH-MDZ were determined using a validated HPLC method with a high specificity, linearity, precision, accuracy, and recovery rate for midazolam and 1′-OH-MDZ, and the tested samples showed high stability. This indicates that the HPLC method is reliable for determining the plasma concentration of midazolam and 1′-OH-MDZ. Our present findings show that oral administration of XKB 14 kg/mg for 8 days significantly elevated the AUC0–t and Cmax of midazolam, but decreased its CL/F. XKB also showed an inhibitory effect on the metabolism of midazolam similar to that seen with ketoconazole in vivo. However, there were not remarkable changes in the ka, ke, t1/2ka, t1/2ke, and V/F of midazolam and 1′-OH-MDZ in rats treated with XKB. Collectively, the data strongly suggest that XKB decreased Cyp3a-mediated metabolism and clearance of midazolam in rats. Notably, it has been reported that midazolam has inducing effects on expression of Cyp3a1 and Cyp2b in rat hepatocytes,51 which in turn may affect its own metabolism and that of other Cyp3a substrate drugs.
Further, we found that oral administration of XKB for 8 days significantly reduced the expression and activity of hepatic Cyp3a via unknown mechanisms, but probably due to post-transcriptional and/or post-translational modulation of the target gene/protein. It has been reported that protein degradation is an important regulator of CYP turnover at the post-translational level.52 In our study, we predicted that XKB might target ubiquitin carboxyl-terminal hydrolase 7 and 14 (USP7 and USP14), which play important roles in protein degradation. XKB might regulate ubiquitin-mediated protein degradation to downregulate the content of rat hepatic Cyp3a2. In addition, epigenetic mechanisms cannot be excluded. For example, microRNAs are the major determinants of gene expression at the post-transcriptional level, suppressing gene expression by mRNA cleavage or translational inhibition.53,54 Moreover, it has been reported that the expression of rat hepatic Cyp3a2 is predisposed to regulation by nuclear transcription factors or nuclear receptors,55–57 which may also be involved in the regulatory effect of XKB on expression of rat Cyp3a2.
These data have important implications in drug development when XKB is used as a lead compound. For example, XKB may have profound therapeutic effects due to interactions with its potential multiple targets; however, a multitargeted agent may also result in side effects due to unwanted binding with toxicity-associated targets (eg, hERG). The potential interactions between XKB and CYP3A4/Cyp3a2 may lead to unfavorable drug interactions. In addition, the short elimination half-life of XKB may negatively affect its concentration at the site of action and compromise the therapeutic effect in clinical practice. Therefore, these findings provide clues for further XKB-based drug development and clinical use, including structural modification or nanotechnology-based formulations to minimize or avoid its potential side effects and prolong its elimination half-life to achieve therapeutic concentrations at the site of action.
In summary, we found that the medicinal effects of XKB could be ascribed to modulation of the networks of functional proteins and the related signaling pathways, and that XKB significantly inhibited the Cyp3a-mediated metabolism of midazolam in rats in vivo via reduction of the activity and expression of hepatic Cyp3a. Our findings suggest that XKB may behave as a CYP3A4/Cyp3a2 substrate and/or inhibitor, probably altering the PK profile of coadministered drugs metabolized by CYP3A4/Cyp3a2 in the body. The molecular targets and modulating effects of XKB on human CYPs need to be further examined and functionally validated in the future.
Acknowledgment
The authors appreciate the financial support received from the Startup Fund of the College of Pharmacy, University of South Florida, Tampa, FL, USA, National Natural Science Foundation of China (grant 81202454), Major Scientific and Technological Special Project of Administration of Ocean and Fisheries of Guangdong Province (A201301C08), National Natural Science Foundation of China (21172271), and the Natural Science Foundation of Guangdong Province, People’s Republic of China (S2011020001231). Dr Zhi-Wei Zhou holds a postdoctoral scholarship from the College of Pharmacy, University of South Florida. The authors also thank Jun Xu, a postdoctoral researcher, for excellent technical assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Ghosh N, Ghosh R, Mandal V, Mandal SC. Recent advances in herbal medicine for treatment of liver diseases. Pharm Biol. 2011;49(9):9709–9788. | ||
Haeckel R. Verification, validation and evaluation of analytical procedures in laboratory medicine. Clin Chem Lab Med. 2004;42(1):111–112. | ||
Mishra BB, Tiwari VK. Natural products: an evolving role in future drug discovery. Eur J Med Chem. 2011;46(10):4769–4807. | ||
International Conference on Harmonisation. Validation of analytical procedures: text and methodology, 2005. Available from: http://www.ich.org/products/guidelines/quality/quality-single/article/validation-of-analytical-procedures-text-and-methodology.html. Accessed October 1, 2014. | ||
Wijnen PA, Op den Buijsch RA, Drent M, et al. Review article: the prevalence and clinical relevance of cytochrome P450 polymorphisms. Aliment Pharmacol Ther. 2007;26 Suppl 2:211–219. | ||
Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105(6):2253–2277. | ||
Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 2008;9(4):310–322. | ||
Zhou SF, Xue CC, Yu XQ, Li C, Wang G. Clinically important drug interactions potentially involving mechanism-based inhibition of cytochrome P450 3A4 and the role of therapeutic drug monitoring. Ther Drug Monit. 2007;29(6):687–710. | ||
Nagata K, Ogino M, Shimada M, Miyata M, Gonzalez FJ, Yamazoe Y. Structure and expression of the rat CYP3A1 gene: isolation of the gene (P450/6βB) and characterization of the recombinant protein. Arch Biochem Biophys. 1999;362(2):242–253. | ||
Dai D, Bai R, Hodgson E, Rose RL. Cloning, sequencing, heterologous expression, and characterization of murine cytochrome P450 3a25*(Cyp3a25), a testosterone 6β-hydroxylase. J Biochem Mol Toxicol. 2001;15(2):90–99. | ||
World Health Organization. Global status report on noncommunicable diseases 2010. Geneva, Switzerland: World Health Organization; 2011. Available from: http://www.who.int/nmh/publications/ncd_report_full_en.pdf. Accessed September 26, 2014. | ||
World Health Organization. Global atlas on cardiovascular disease prevention and control. Geneva, Switzerland: World Health Organization; 2011. Available from: http://whqlibdoc.who.int/publications/2011/9789241564373_eng.pdf. Accessed September 26, 2014. | ||
Hamilton CA, Miller WH, Al-Benna S, et al. Strategies to reduce oxidative stress in cardiovascular disease. Clin Sci (Lond). 2004;106(3):219–234. | ||
Sachidanandam K, Fagan SC, Ergul A. Oxidative stress and cardiovascular disease: antioxidants and unresolved issues. Cardiovasc Drug Rev. 2005;23(2):115–132. | ||
Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75(3):311–335. | ||
Kuete V, Efferth T. Cameroonian medicinal plants: pharmacology and derived natural products. Front Pharmacol. 2010;1:123. | ||
Blunt JW, Copp BR, Keyzers RA, Munro MH, Prinsep MR. Marine natural products. Nat Prod Rep. 2012;29(2):144. | ||
Jha RK, Zi-Rong X. Biomedical compounds from marine organisms. Mar Drugs. 2004;2(3):123–146. | ||
Handa K, Nakagome I, Yamaotsu N, Gouda H, Hirono S. Three-dimensional quantitative structure-activity relationship analysis of inhibitors of human and rat cytochrome P4503A enzymes. Drug Metab Pharmacokinet. 2013;28(4):345–355. | ||
Pettigrew JD, Wilson PD. Synthesis of xyloketal A, B, C, D, and G analogues. J Org Chem. 2006;71(4):1620–1625. | ||
Wu XY, Liu XH, Lin YC, et al. Xyloketal F: a strong L-calcium channel blocker from the mangrove fungus Xylaria sp.(# 2508) from the South China Sea coast. Eur J Org Chem. 2005;2005(19):4061–4064. | ||
Zhang W, Liu Y, Yang H, et al. A validated high-performance liquid chromatographic method with diode-array detection for the estimation of xyloketal B in rat plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;885–886:24–29. | ||
Luo H, Chen J, Shi L, et al. DRAR-CPI: a server for identifying drug repositioning potential and adverse drug reactions via the chemical-protein interactome. Nucleic Acids Res. 2011;39 (Web Server issue):W492–W498. | ||
Luo H, Zhang P, Huang H, et al. DDI-CPI, a server that predicts drug-drug interactions through implementing the chemical-protein interactome. Nucleic Acids Res. 2014;42(Web Server issue):W46–W52. | ||
Wang R, Fang X, Lu Y, Wang S. The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J Med Chem. 2004;47(12):2977–2980. | ||
Yang L, Luo H, Chen J, Xing Q, He L. SePreSA: a server for the prediction of populations susceptible to serious adverse drug reactions implementing the methodology of a chemical-protein interactome. Nucleic Acids Res. 2009;37(Web Server issue):W406–W412. | ||
Yang L, Chen J, Shi L, Hudock MP, Wang K, He L. Identifying unexpected therapeutic targets via chemical-protein interactome. PLoS One. 2010;5(3):e9568. | ||
Wang ZX, Sun J, Howell CE, et al. Prediction of the likelihood of drug interactions with kinase inhibitors based on in vitro and computational studies. Fundam Clin Pharmacol. 2014;28(5):551–582. | ||
Nagata K, Buppodom P, Matsunaga T, et al. Purification and characterization of seven distinct forms of liver microsomal cytochrome P-450 from untreated and inducer-treated male Wistar rats. J Biochem. 1985;97(6):1755–1766. | ||
Li WL, Xin HW, Su MW. Inhibitory effects of continuous ingestion of Schisandrin A on CYP3A in the rat. Basic Clin Pharmacol Toxicol. 2012;110(2):187–192. | ||
Zhang Y, Huo M, Zhou J, Xie S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99(3):306–314. | ||
Zhou ZW, Zhou SF. Application of mechanism-based CYP inhibition for predicting drug-drug interactions. Expert Opin Drug Metab Toxicol. 2009;5(6):579–605. | ||
Yamamoto A. A uniqe antilipidemic drug-probucol. J Atheroscler Thromb. 2008;15(6):304–305. | ||
Chen WL, Qian Y, Meng WF, et al. A novel marine compound xyloketal B protects against oxidized LDL-induced cell injury in vitro. Biochem Pharmacol. 2009;78(8):941–950. | ||
Zhao J, Li L, Ling C, et al. Marine compound Xyloketal B protects PC12 cells against OGD-induced cell damage. Brain Res. 2009;1302:240–247. | ||
Lu XL, Yao XL, Liu Z, et al. Protective effects of xyloketal B against MPP+-induced neurotoxicity in Caenorhabditis elegans and PC12 cells. Brain Res. 2010;1332:110–119. | ||
Li ZX, Chen JW, Yuan F, et al. Xyloketal B exhibits its antioxidant activity through induction of HO-1 in vascular endothelial cells and zebrafish. Mar Drugs. 2013;11(2):504–522. | ||
Gershkovich P, Hoffman A. Uptake of lipophilic drugs by plasma derived isolated chylomicrons: linear correlation with intestinal lymphatic bioavailability. Eur J Pharm Sci. 2005;26(5):394–404. | ||
Nishino Y, Kubota A, Kanazawa T, Takashima Y, Ozeki T, Okada H. Improved intestinal absorption of a poorly water-soluble oral drug using mannitol microparticles containing a nanosolid drug dispersion. J Pharm Sci. 2012;101(11):4191–4200. | ||
Sha X, Wu J, Chen Y, Fang X. Self-microemulsifying drug-delivery system for improved oral bioavailability of probucol: preparation and evaluation. Int J Nanomedicine. 2012;7:705–712. | ||
Zhang Z, Huang J, Jiang S, et al. A high-drug-loading self-assembled nanoemulsion enhances the oral absorption of probucol in rats. J Pharm Sci. 2013;102(4):1301–1306. | ||
Krupka E, Venisse N, Lafay C, et al. Probe of CYP3A by a single-point blood measurement after oral administration of midazolam in healthy elderly volunteers. Eur J Clin Pharmacol. 2006;62(8):653–659. | ||
Nogawa H, Kawai T, Yajima M, Miura M, Ogawa T, Murakami K. Effects of probucol, a typical hERG expression inhibitor, on in vivo QT interval prolongation in conscious dogs. Eur J Pharmacol. 2013;720(1–3):29–37. | ||
Miida T, Seino U, Miyazaki O, et al. Probucol markedly reduces HDL phospholipids and elevated prebeta1-HDL without delayed conversion into alpha-migrating HDL: putative role of angiopoietin-like protein 3 in probucol-induced HDL remodeling. Atherosclerosis. 2008;200(2):329–335. | ||
Favari E, Zanotti I, Zimetti F, Ronda N, Bernini F, Rothblat GH. Probucol inhibits ABCA1-mediated cellular lipid efflux. Arterioscler Thromb Vasc Biol. 2004;24(12):2345–2350. | ||
Yang L, Wang KJ, Wang LS, et al. Chemical-protein interactome and its application in off-target identification. Interdiscip Sci. 2011;3(1):22–30. | ||
Yang L, Wang K, Chen J, et al. Exploring off-targets and off-systems for adverse drug reactions via chemical-protein interactome- clozapine-induced agranulocytosis as a case study. PLoS Comput Biol. 2011;7(3):e1002016. | ||
Soucek P, Gut I. Cytochromes P-450 in rats: structures, functions, properties and relevant human forms. Xenobiotica. 1992;22(1):83–103. | ||
Shin KH, Choi MH, Lim KS, Yu KS, Jang IJ, Cho JY. Evaluation of endogenous metabolic markers of hepatic CYP3A activity using metabolic profiling and midazolam clearance. Clin Pharmacol Ther. 2013;94(5):601–609. | ||
Yamano K, Yamamoto K, Katashima M, et al. Prediction of midazolam-CYP3A inhibitors interaction in the human liver from in vivo/in vitro absorption, distribution, and metabolism data. Drug Metab Dispos. 2001;29(4 Pt 1):443–452. | ||
Hoen PA, Bijsterbosch MK, van Berkel TJ, Vermeulen NP, Commandeur JN. Midazolam is a phenobarbital-like cytochrome p450 inducer in rats. J Pharmacol Exp Ther. 2001;299(3):921–927. | ||
Yang J, Liao M, Shou M, et al. Cytochrome p450 turnover: regulation of synthesis and degradation, methods for determining rates, and implications for the prediction of drug interactions. Curr Drug Metab. 2008;9(5):384–394. | ||
Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8(2):93–103. | ||
Hobert O. Gene regulation by transcription factors and microRNAs. Science. 2008;319(5871):1785–1786. | ||
Khan AA, Chow EC, van Loenen-Weemaes AM, Porte RJ, Pang KS, Groothuis GM. Comparison of effects of VDR versus PXR, FXR and GR ligands on the regulation of CYP3A isozymes in rat and human intestine and liver. Eur J Pharm Sci. 2009;37(2):115–125. | ||
Ni S, Wang X, Wang J, Zeng S, Zhao Z. Expression of CYP3A23/1, CYP3A2, PXR, CAR and HNF4alpha in large-for-gestational-age neonatal rats. Pharmazie. 2009;64(4):252–257. | ||
De Martin S, Gabbia D, Albertin G, et al. Differential effect of liver cirrhosis on the PXR-mediated induction of cytochromes P450 3A1 and 3A2 in the rat. Drug Metab Dispos. 2014;42(10):1617–1626. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.