")
Back to Journals » Infection and Drug Resistance » Volume 16
Whole Genomic Analysis Revealed High Genetic Diversity and Drug-Resistant Characteristics of Mycobacterium tuberculosis in Guangxi, China
Authors Liang D, Song Z, Liang X, Qin H, Huang L, Ye J, Lan R, Luo D, Zhao Y, Lin M
Received 27 March 2023
Accepted for publication 21 June 2023
Published 3 August 2023 Volume 2023:16 Pages 5021—5031
DOI https://doi.org/10.2147/IDR.S410828
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Dabin Liang,1,2,* Zexuan Song,3,* Xiaoyan Liang,1,2,* Huifang Qin,1,2 Liwen Huang,1,2 Jing Ye,1,2 Rushu Lan,4 Dan Luo,5 Yanlin Zhao,3 Mei Lin1,2
1Guangxi Zhuang Autonomous Region Center for Disease Control and Prevention, Nanning, Guangxi, People’s Republic of China; 2Guangxi Key Laboratory of Major Infectious Disease Prevention and Control and Biosafety Emergency Response, Nanning, Guangxi, People’s Republic of China; 3National Tuberculosis Reference Laboratory, Chinese Center for Disease Control and Prevention, Beijing, People’s Republic of China; 4Department of Clinical Laboratory, Jiangbin Hospital of Guangxi Zhuang Autonomous Region, Nanning, Guangxi, People’s Republic of China; 5School of Public Health and Management, Guangxi University of Chinese Medicine, Nanning, Guangxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yanlin Zhao, National Tuberculosis Reference Laboratory, Chinese Center for Disease Control and Prevention, No. 155 Chang Bai Road, Changping District, Beijing, 102206, People’s Republic of China, Email [email protected] Mei Lin, Guangxi Zhuang Autonomous Region Center for Disease Prevention and Control, Nanning, 530023, People’s Republic of China, Email [email protected]
Background: Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is a major public health issue in China. Nevertheless, the prevalence and drug resistance characteristics of isolates vary in different regions and provinces. In this study, we investigated the population structure, transmission dynamics and drug-resistant profiles of Mtb in Guangxi, located on the border of China.
Methods: From February 2016 to April 2017, 462 clinical M. tuberculosis isolates were selected from 5 locations in Guangxi. Drug-susceptibility testing was performed using 6 common anti-tuberculosis drugs. The genotypic drug resistance and transmission dynamics were analyzed by the whole genome sequence.
Results: Our data showed that the Mtb in Guangxi has high genetic diversity including Lineage 1 to Lineage 4, and mostly belong to Lineage 2 and Lineage 4. Novelty, 9.6% of Lineage 2 isolates were proto-Beijing genotype (L2.1), which is rare in China. About 12.6% of isolates were phylogenetically clustered and formed into 28 transmission clusters. We observed that the isolates with the high resistant rate of isoniazid (INH, 21.2%), followed by rifampicin (RIF, 13.2%), and 6.7%, 12.1%, 6.7% and 1.9% isolates were resistant to ethambutol (EMB), streptomycin (SM), ofloxacin (OFL) and kanamycin (KAN), respectively. Among these, 6.5% and 3.3% of isolates belong to MDR-TB and Pre-XDR, respectively, with a high drug-resistant burden. Genetic analysis identified the most frequently encountered mutations of INH, RIF, EMB, SM, OFL and KAN were katG_Ser315Thr (62.2%), rpoB_Ser450Leu (42.6%), embB_Met306Vol (45.2%), rpsL_Lys43Arg (53.6%), gyrA_Asp94Gly (29.0%) and rrs_A1401G (66.7%), respectively. Additionally, we discovered that isolates from border cities are more likely to be drug-resistant than isolates from non-border cities.
Conclusion: Our findings provide a deep analysis of the genomic population characteristics and drug-resistant of M. tuberculosis in Guangxi, which could contribute to developing effective TB prevention and control strategies.
Keywords: tuberculosis, Mycobacterium tuberculosis, drug-resistance, genetic diversity, whole genome sequencing
Introduction
Tuberculosis (TB), caused by etiological Mycobacterium tuberculosis (Mtb), remains a public health worldwide and the leading cause of death from a single infectious agent until the COVID-19 pandemic.1 Previous studies revealed that M. tuberculosis originated in Africa and spread with human migration worldwide, it developed to a contemporary phylogeny consisting of nine recognized lineages (L1–L9) with vary in their distribution between countries and continents.2 In China, Lineage 2 (L2) and Lineage 4 (L4) are the most widely distributed throughout the country, whereas Lineage 1 (L1) and Lineage 3 (L3) are most prevalent in Southeast and Northwest, respectively.3
To date, the emergence of drug-resistant tuberculosis poses a great challenge for TB control and prevention. The WHO Global Tuberculosis Report of 2022 estimated that the burden of drug-resistant TB has increased between 2020 and 2021, with 450000 (95% UI: 399 000–501 000) new cases of rifampicin resistant in 2021.4 The treatment success rate for rifampicin-resistant TB is typically in the range of 50–75%.1 It is urgent to learn about the drug-resistant profile of the M. tuberculosis isolates in order to reduce the risk of infection and transmission. Whole-genome sequencing (WGS) is a useful tool for predicting the susceptibility to anti-TB drugs in clinical specimen.5 In some countries, WGS of M. tuberculosis is increasingly used in routine care settings for species identification, drug resistance profile determination and transmission investigation.6–8 Traditionally, a great number of single nucleotide polymorphisms (SNPs) mutation in some genes have been identified as associated with drug resistance.9
Although TB control has made noticeable progress over the past years, China remains a high burden country worldwide.1 Meanwhile, the prevalent genotype and drug resistance of the Mtb isolates show a great difference among diverse regions. Guangxi, located on the border of China and borders Vietnam, is one of the high TB-burden regions in South China. Previous studies have reported the epidemiology of Mtb in hot and cold spots of Guangxi,10,11 but the detailed lineage distribution evolution and drug-resistant profile of Mtb are limited. In this study, we analyzed the genome of M. tuberculosis isolates and the clinical system from pulmonary TB patients in Guangxi, which can provide insights into the genetic diversity and drug-resistant profile of M. tuberculosis isolates.
Methods
Study Setting and Sample Enrollment
In this retrospective cross-sectional investigation, five locations (Guigang, Fangchenggang, Baise, Chongzuo, and Guilin) in Guangxi were selected as study sites. Geographically, these locations are dispersed over the east, south, west, and north of Guangxi. To be specific, Baise, Chongzuo, and Fangchenggang bordering Vietnam represent the border monitoring points, and Guilin and Guigang belong to non-border monitoring points (Figure 1).
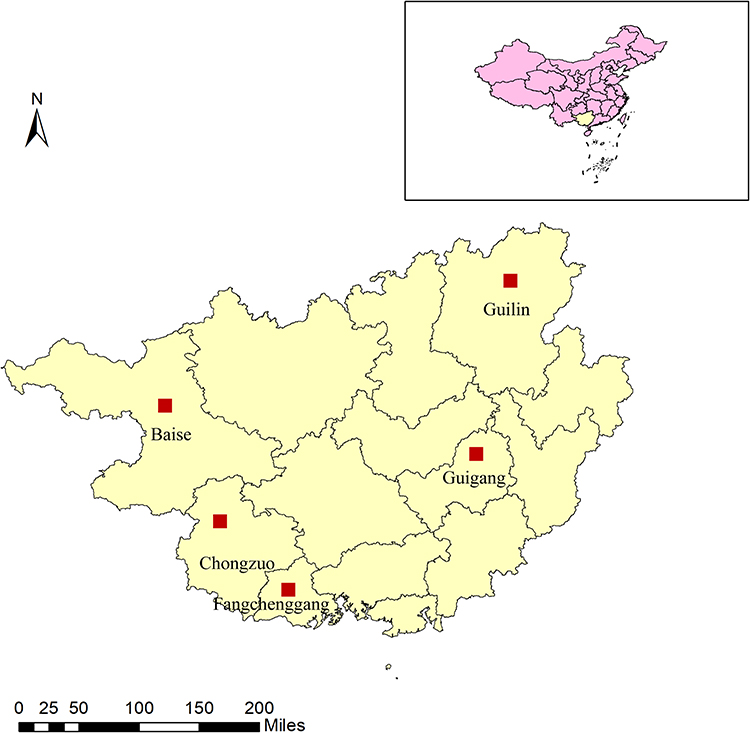 |
Figure 1 Distribution of the study site in Guangxi, China. The five cities located in border and non-border of Guangxi, which belongs to the southwest region of China. |
During February 2016 and August 2017, we collected 1638 Mtb strains isolated from pulmonary tuberculosis patients with sputum smears positive, who attended local designated hospitals. Then, 600 isolates were randomly selected for drug sensitivity testing and whole genome sequencing. Due to issues like unsuccessful re-culture, contamination, unsuccessful genome extraction, and failure to match the epidemiologic information, a total of 462 clinical isolates were ultimately included in the in-depth study and further analysis (Figure S1 and Table S1). The epidemiologic investigations were collected from the patient after written informed consent. The study received ethical approval, which complies with the Declaration of Helsinki, from the Ethics Committee of the Guangxi Center for Disease Control and Prevention (IRB00001594).
Drug-Susceptibility Testing
Drug-susceptibility testing (DST) was performed by conventional proportion methods as the described previously,12 with the following drug concentration: isoniazid (0.2 μg/mL), rifampin (40 μg/mL), ethambutol (2 μg/mL), streptomycin (4 μg/mL), kanamycin (30 μg/mL), ofloxacin (2 μg/mL). The isolate was regarded as resistant if the proportion of growth on the medium containing drug was 1% of that observed in drug-free medium. The reference isolate H37Rv (ATCC 27294) as a quality control.
DNA Extraction and Sequencing
All the M. tuberculosis isolates were cultured on Lowenstein Jensen media and genome DNA was extracted following the protocol of the cetyltrimethylammonium bromide (CTAB) method.13 The whole genome sequence was performed on purified DNA using Illumina HiSeq 2500 platform. All the whole genome sequencing operations were completed by Annoroad Gene Technology company (Beijing, China).
Bioinformatic Analysis
The WGS data analysis was carried out using procedures that were similar to those reported previously. The paired-end reads were examined using FastQC (v0.11.9) and trimmed using Trimmomatic (v0.39).14 The qualified reads were mapped against the H37Rv genome (NC_000962.3) by BWA-MEM algorithm (v0.7.16). Samtools (v1.13) and bcftools (v1.13) were used to variant calling from the sorted mapped sequences. The variant filtration was performed based on the following criteria: mapping quality greater than 30 and minimum read depth of 10. SnpEff (v4.3) was used to annotate the filter variant SNPs. And the isolate mutation conferring resistance to six anti-tuberculosis drugs was identified in TBProfiler.15
Phylogenetic Analysis
Phylogenetic analysis was performed by snippy pipeline (v4.3.6) for alignment of the core SNPs (https://github.com/tseemann/snippy). SNPs’ positions within PE/PPE genes or other repetitive regions with low mapping scores were removed. Then, the recombinant region of the alignment was filtered with Gubbins. The maximum-likelihood phylogenetic tree based on the core SNPs was construct by RAxML, with 1000 bootstrap iterations and the GTR+G model. The lineage of the M. tuberculosis isolates was identified using fast-lineage-caller v1.0 (https://github.com/farhat-lab/fast-lineage-caller). And we used Coll et al and Shitikov et al SNP schemes to define the sub-lineages and genotype of lineage 2, respectively.16,17 The genomic cluster was defined as strains with a genetic distance of 12 SNPs or less, suggesting they were the result of recent transmission.18 The genomic pairwise distance was calculated using the program snp-dists (https://github.com/tseemann/snp-dists).
Statistical Analysis
All statistical analysis was used with SPSS (v18.0). The Chi-square test or Fisher’s exact test was performed for this study data. P < 0.05 was considered statistically significant.
Results
Demographic Characteristics of the Patients
Among the enrolled patients, 78.6% (363/462) were male, and the age of 41.8% of patients belong to 45–65 years. The majority of patients are living in rural (83.5%) and being Han (68.0%). And 34.9% and 40.5% of the patients have primary education and junior education, respectively. In addition, most patients do not have diabetes (77.7%) or hepatitis (82.7%). We also investigated the clinical symptoms of the patients. Most patients have the symptoms of cough (98.5%) and expectoration (92.4%), and various percentages of patients also have symptoms like hemoptysis (11.7%), chest pain (8%), fever (9.9%), weak (17.5%), night sweat (10.2%), and other conditions (6.7%). Other detailed information is shown in Table 1.
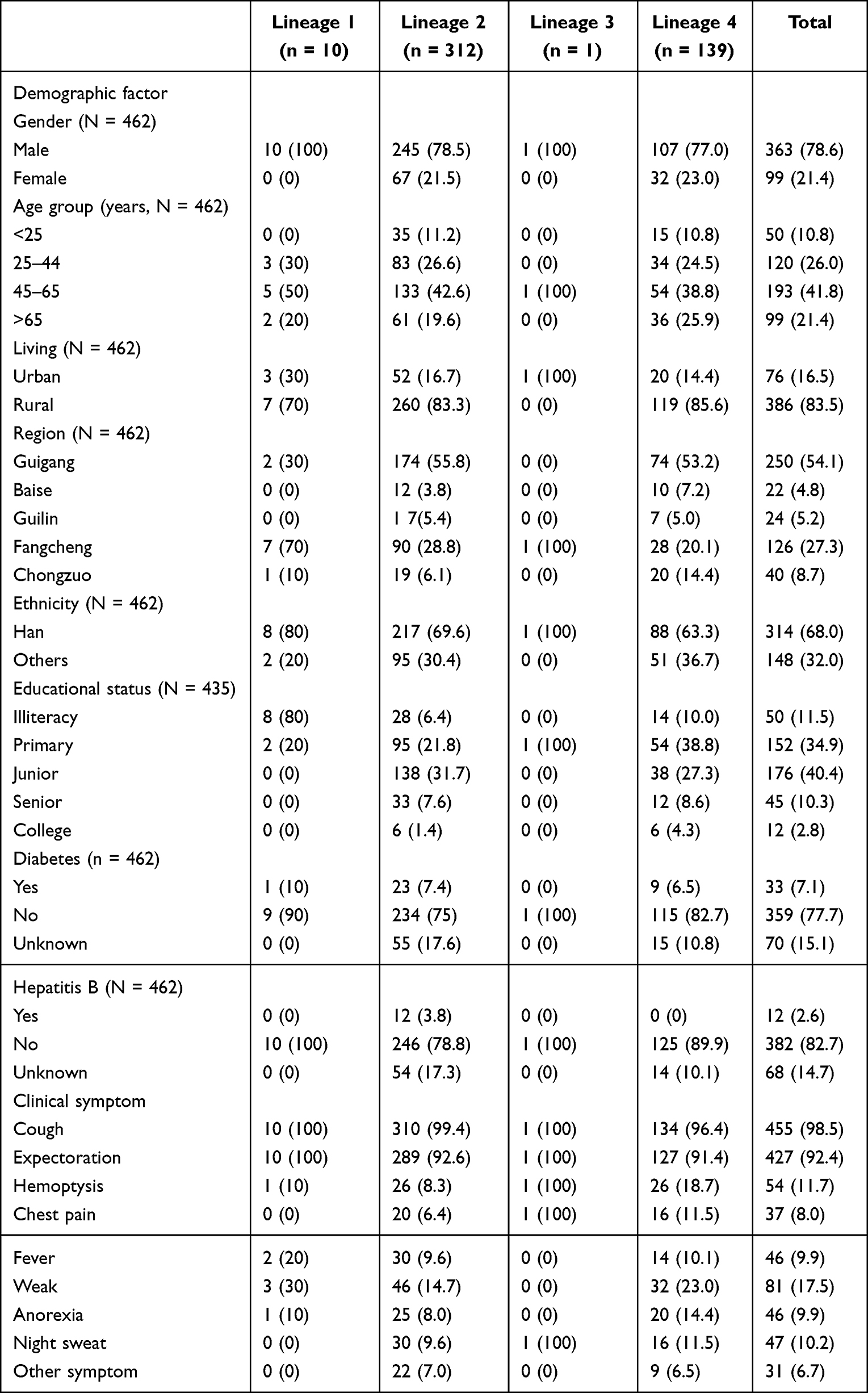 |
Table 1 The Demographic Information and Clinical Characteristics of the Mtb Infection Patients |
Population Structure of M. tuberculosis
The population structure of Mtb isolates in Guangxi was investigated (Figure 2). The maximum likelihood phylogenetic tree and the barcode-SNP–based genotyping show that majority of isolates (58.8%, 272/462) belong to lineage 2.2.1 (L2.2.1), and 2.2% (10/462) isolates to lineage 2.2.2 (L2.2.2), which are the subgroup of Beijing lineage (L2). Unexpectedly, 6.5% (30/462) belong to the proto-Beijing sub-lineage (L2.1), which is not found in most areas of China and other regions of the world.3,19 Within lineage 4 (L4), 48.2% (67/139) isolates belong to lineage 4.4, followed by lineage 4.5 (28.1%, 39/139) and lineage 4.2 (23.0%, 32/139), only one isolate is lineage 4.1. In addition, 2.2% (10/462) isolates belong to lineage 1 (L1) and one isolate belongs to lineage 3 (L3) in this study.
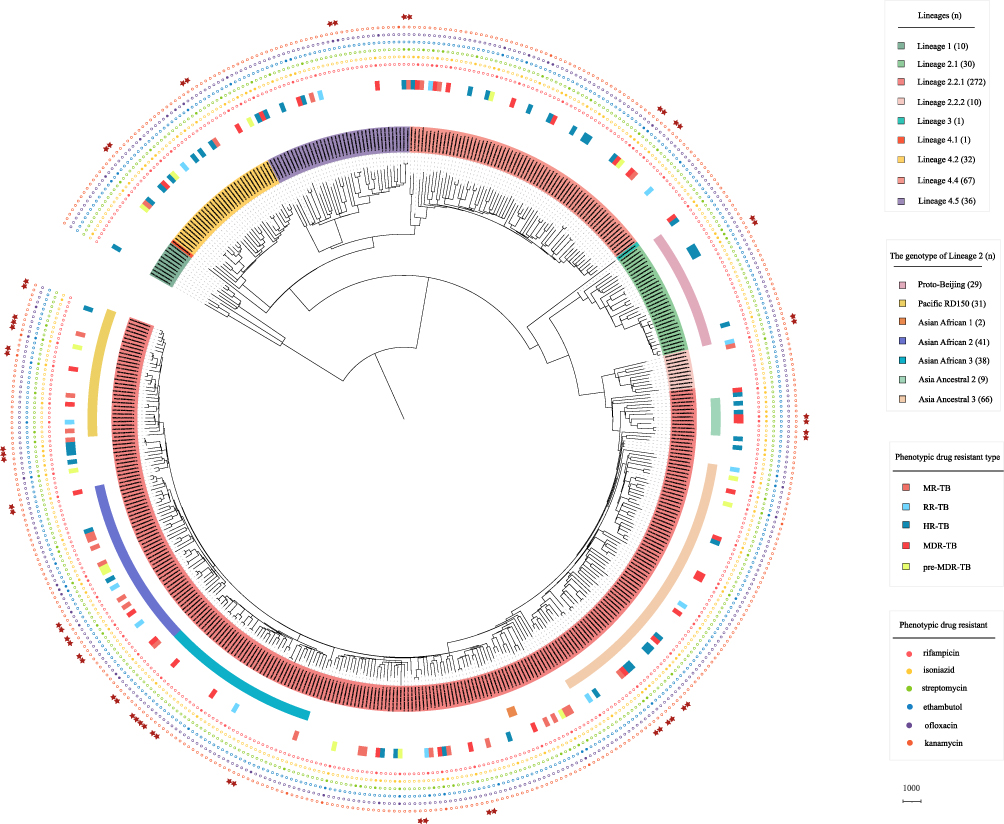 |
Figure 2 Phylogenetic tree of the 462 Mtb isolates in this study. The maximum likelihood phylogenetic tree was constructed by Raxml, with 1000 bootstraps replicates. The sub-lineages, drug resistant type and drug resistant profile of the isolates are shown on the tree (from inner to outer circles), according to the color legend shown on the right. The red stars indicate the cluster isolates. |
Meanwhile, we further classified the isolates of lineage 2 according to Shitikov’s schemes,17 which divided the L2 isolates into 11 genotypes based on genetic markers. The results showed that 69.2% (216/312) of L2 isolates were divided into seven genotypes in our study (Figure 2). Specifically, 21.2% (66/312) of L2 is Asia Ancestral 3, followed by Asian African 2 (13.1%, 41/312), Asian African 3 (12.2%, 38/312), Pacific RD150 (9.9%, 31/312) and Proto-Beijing (9.3%, 29/312). And small amounts of isolates belong to Asia Ancestral 2 (2.9%, 9/312) and Asian African 1 (0.6%, 2/312). However, there are some L2 isolates that do not belong to these genotypes and more detailed classification may be needed to identify them. Overall, our results indicate that Mtb isolates in Guangxi with high genetic diversity, and majority of TB cases were caused by L2 and L4 isolates.
The Transmission Dynamic Analysis
The cluster analysis employing a threshold of a maximum distance of 12 SNPs grouped 58 (12.6%) of the 462 isolates into 28 clusters, ranging in size from two to three isolates (Figure 3). The genetic differences between isolates in each cluster ranged from 1 to 11 SNPs; 16 pairs differed by less than 8 SNPs and 12 pairs by 10 to 11 SNPs. The isolates from different cities are interspersed in the phylogenetic tree, but the clustering rate in border areas is higher than that in non-border areas (P < 0.01). Among all the cluster isolates, 25 pairs (89.3%) isolate from the same city and only 3 pairs (10.7%) isolate from separate cities (Figure 3). We also noted that there was a statistical difference in the clustering rate between the Beijing genotype and non-Beijing genotype (P < 0.05).
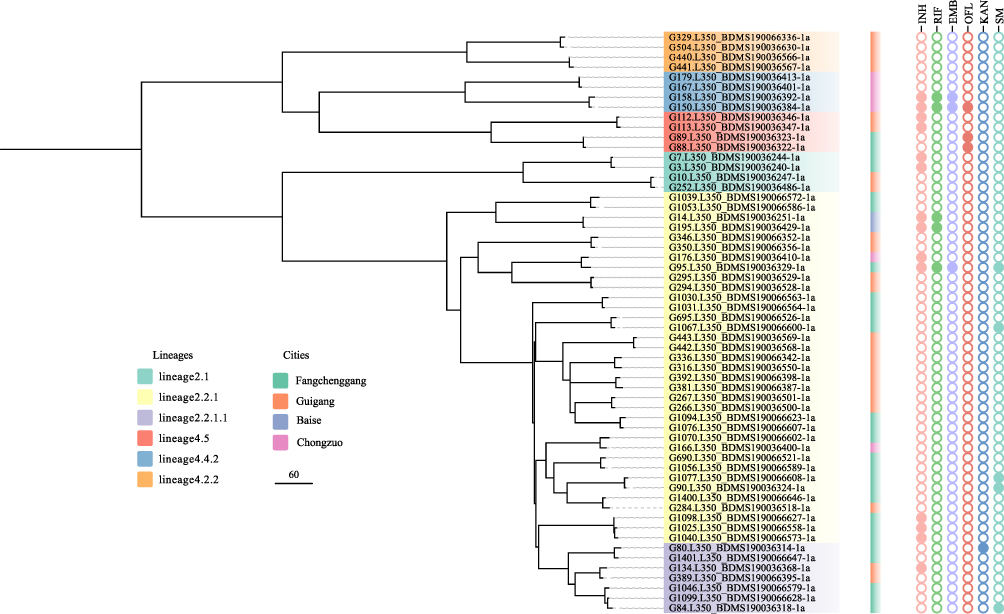 |
Figure 3 Phylogenetic tree of the 58 Mtb isolates with 28 clusters in this study. The lineages, cities and genotypic drug resistance of isolates are shown on the tree according to the color legend. |
Phenotypic Drug-Resistant Profile
Among all the isolates in our study, 21.1% (98/462) were resistant to isoniazid, 13.2 (61/462) to rifampicin, 6.7% (31/462), 12.1% (56/462), 6.7% (31/462) and 1.9% (9/462) isolates were resistant to ethambutol, streptomycin, ofloxacin and kanamycin, respectively (Figure 4A). According to the latest definition of drug-resistant TB,20 1.3% (6/462) isolates are RR-TB (rifampicin-resistant tuberculosis), 7.1% (33/462) isolates are MDR-TB (multidrug-resistant tuberculosis) and 3.0% (14/462) are Pre-XDR (pre-extensively drug-resistant), whereas 7.1% (33/462) and 11.0% (51/462) isolates belong to MR-TB (mono-resistant tuberculosis) and Hr-TB (isoniazid-resistant tuberculosis), respectively (Figure 4B). These results showed that high phenotypic resistance proportions of the Mtb isolates in Guangxi, especially for the isoniazid resistance.
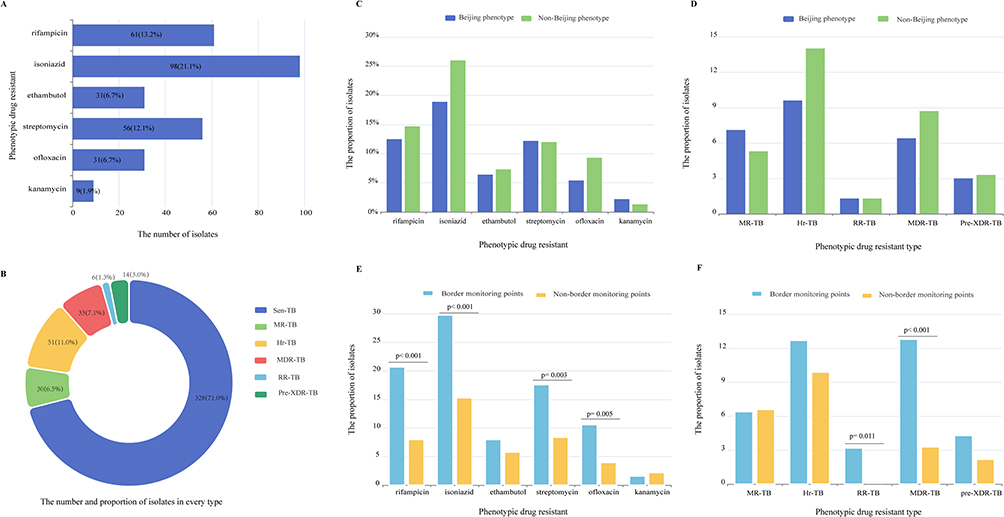 |
Figure 4 The drug-resistant profiles of the isolates in this study. (A and B) The phenotypic drug resistance and different phenotypic drug type of the isolates in this study. (C and D) Comparison of drug-resistant profiles between Beijing phenotype and non-Beijing phenotype. (E and F) Comparison of drug-resistant profiles between Border monitoring points and non-border monitoring points. |
Unlike the previous report showed that high prevalence of XDR-TB in proto-Beijing (L2.1),19 we found that only 13.3% (4/30) isolates belong to Hr-TB and one isolate is MR-TB in L2.1 of this study (Figure 2). By analyzing, the prevalence of drug resistance of non-Beijing genotype was higher than that of Beijing genotype strains, especially for isoniazid resistance (Figure 4C), but there was no significant difference between these two genotypes, including the drug types (Figure 4D). In addition, our data showed that the isolates of border monitoring points are more likely to be resistant to rifampicin, isoniazid, streptomycin and ofloxacin resistant than those of no-border monitoring points (P < 0.01) (Figure 4E). RR-TB and MDR-TB isolates are more likely to be present in border monitoring points (p < 0.05) (Figure 4F).
Drug-Resistance Related Gene Mutations
The drug resistance profiles based on the whole genome sequence were assessed. Of the 61 phenotypic rifampicin resistant isolates, the most prevalent mutation was Ser450 Leu (37.7%, 23/61), followed by His445Asn (9.8%, 6/61) and His445Tyr (6.5%, 4/61). Combination mutations were detected in 7 (11.5%, 7/61) isolates, and two of these isolates also carried mutations in the rpoC gene (Table S2). Eighty-five (18.3%, 85/462) isolates detected mutations related to isoniazid resistance genes including katG, ahpC, fabG1and inhA (Table S3). Of these mutations, KatG Ser315Thr (62.2%, 61/98) was the most common mutation, followed by fabG1 −15C>T (10.2%, 10/98). And eight isolates (8.2%, 8/98) carried combined mutations. Thirty-one isolates predicted ethambutol resistance were mainly related to the mutation embB Met306Val (45.2%, 14/31) and Met306 Ile (22.6%, 7/31) (Table S4). Mostly, phenotypically streptomycin resistant isolates were detected with Lys43Arg (53.57%, 30/56) mutation in rpsL gene (Table S5). In addition, 54.8% (17/31) phenotypically fluoroquinolone-resistant isolates were identified with mutations in the gyrA gene like Asp94Gly (29.0%, 9/31) and Ala90Val (22.6%, 7/31), but no isolates have mutation in gyrB gene (Table S6). Among kanamycin-resistant isolates, 66.7% of isolates (6/9) have 1401a>g mutation in rrs gene (Table S7).
Discussion
Tuberculosis remains a major public health problem in China, and the population characteristics and drug resistance profiles in different regions showed a great difference.3,21,22 In this study, we provide the population characteristics, transmission dynamics and drug resistance characteristics of Mtb in Guangxi, China. Our results show an initial picture of the Mtb population structure in Guangxi, which has high genetic diversity including lineage 1 to lineage 4. Our data show that 67.5% and 30.1% of Mtb isolates belonged to lineage 2 and Lineage 4, respectively, which is in line with the previous report that lineage 2 and lineage 4 are most prevalent and distributed throughout in China.3,21,23,24 Although most isolates belonged to Beijing lineage (L2), 9.6% of them are proto-Beijing genotype (L2.1), which is not common in other regions of China and is a significant character of the Mtb isolates in Guangxi.3 Previous studies have shown that L2.1 isolates are also present in parts of Vietnam and Thailand,19,25 especially in Thailand where L2.1 has a great propensity to transmit. Overall, the sub-lineage L2.1 is geographically restricted and need more widespread surveillance in other areas.
Drug resistance was formerly assumed to be related to the Beijing genotype,26 but a recent study from China showed that the Beijing genotype is less associated with drug resistance.27 Conversely, the non-Beijing genotype was more likely to be resistant to isoniazid, rifampicin and ofloxacin in this study, which showed the findings have been inconsistent because of different geographic settings and isolates populations. This question requires more research, particularly from the standpoint of molecular evolution. Our data showed that the isolates belonging to L2.1 exhibit low drug resistance, which is inconsistent with the recent report that proto-Beijing genotype (L2.1) isolates have a great propensity to be XDR-TB in Thailand.19 Here, we also found that the drug-resistance in border regions was higher than in non-border regions, which suggests that we should pay more attention to generating efficient tuberculosis prevention and control strategies in border regions.
Our data show a high rate of isoniazid resistance among the Mtb isolates in Guangxi, and the prevalence of Hr-TB is significantly higher than the prevalence of rifampicin resistance, which includes MDR-TB. Importantly, the rate of Hr-TB is higher than the previous report in China (5.7%)28 and the global prevalence of Hr-TB (7.4% in new TB patients and 11.4% in retreated TB patients).29 According to a meta-analysis, patients with INH-resistant TB who received first-line HRZE therapy had high rates of unfavorable outcomes, such as failure, relapse, and acquired multidrug resistance (11%, 10%, and 8%, respectively).29 To date, isoniazid is one of the most powerful drugs for the treatment of TB, but the procedures are not standardized and effective enough for the diagnosis and promptly modified treatment for Hr-TB in some areas, which emphasizes the urgent necessity to prioritize its rapid detection and effective therapy to curb the future epidemics of MDR-TB.
Concerning the molecular mechanism of resistance to anti-TB drugs, the associated gene mutations were screened by the whole genome sequence. In concordance with previous studies, the rifampicin resistance-determining region (RRDR) of rpoB and katG 315 codon mutations clearly predominated in the assessment of rifampicin- and isoniazid-resistant mutations.30–32 And we also found that there are some combinations of mutations like secondary mutations in rpoC and non-RRDR of rpoB, which may have a compensatory function of the isolates.33 The study showed that EMB resistance was associated with embCAB mutations and embB was the most frequently mutated gene.34 Previous studies show that SM resistance has been shown to be associated with rpsL, rrs, and gid genes, of which rpsL mutations were predominant,35 and this study showed 64.3% of isolates have mutation in rpsL. Mutation in gyrA and gyrB have been known to be related to ofloxacin, the current study has found 77.4% of the isolates have mutation in gyrA, but no isolates in gyrB. This is consistent with the report that gyrA mutations are more common than gyrB mutations.32 More importantly, some Mtb strains with phenotypical resistance against anti-TB drugs (eg, rifampicin, isoniazid, ofloxacin, etc.) had no detectable gene mutations in our study, suggesting that there may be other genomic variants, such as insertions and deletions,36 which need further clarification. In addition, alternative mechanisms, such as drug efflux pump and decreased cell wall permeability to drugs, may also be related to drug resistance in MTBC.37,38
Our study also found that 12.6% (58/462) of the pulmonary TB cases had genomic-clustered isolates in Guangxi during a period of fewer than two years. The clustering rate was similar to Luodian (13.1%) in a one-year study,21 lower than Shenzhen (15.2%) in five years study.28 The clustering rate in the current study may have been underestimated because molecular epidemiological studies of tuberculosis typically utilize a sampling period of two years or longer. Thus, further long-term investigation is required to comprehensively characterize TB transmission. Of note, most isolates within a cluster were collected from the same county in our study, indicating that the recent transmission predominantly occurred in the local area. Interestingly, some isolates of clusters showed different drug-resistant types, suggesting the progression and accumulation of mutations linked to drug resistance.39 But some of them were isolated in the same year, this result could be partially explained by the fact that not all cases from the possible cluster were enrolled in this study, and some index cases with primary resistance may have been overlooked in the chosen population.39
There are several limitations in the current study. First, the proportion of recent transmission events might be underestimated since our collection of isolates is a period of less than two years. Second, the isolates were only selected from five sites, which may not be representative of the overall situation of the Guangxi area. Thus, the generalizability of the results obtained in this study may be limited. Third, the clinical information collected in this study was relatively limited, with only basic clinical symptoms, thus the correlation between clinical symptoms and genomic variants was not analyzed. In addition, due to the retrospective nature of this study, treatment outcome was not collected, which prevented us from exploring the correlation between drug-resistance profile and clinical outcomes.
Conclusion
In summary, we obtained the diversity of Mtb isolates and the trend of drug resistance, and get a better understanding of the epidemic characteristic of TB in Guangxi, China. We found that the genomic of Mtb isolates is high diversity including four major lineages (L1-L4), and it has rare proto-Beijing isolates (L2.1). The genomic clusters contained 58 isolates, with the clustering rate of 12.6%. In addition, our data show the high rate of isoniazid resistance among the Mtb isolates in Guangxi, and the prevalence of Hr-TB is significantly higher than the prevalence of rifampicin resistance, including MDR-TB, which emphasizes the urgent necessity to prioritize its rapid detection and effective therapy for Hr-TB and to curb the future epidemics of MDR-TB. The isolates in border monitoring points are more likely to be drug resistant than no-border monitoring points, which needs to be noted. Overall, this work provided a deep analysis of the genomic population characteristics and drug resistance of Mtb in Guangxi, which could contribute to developing effective TB prevention and control strategies.
Data Sharing Statement
The accession numbers of sequenced genomes from this study were shown in Table S1.
Funding
This work was supported by grants from Guangxi Medical and health key discipline construction project and National Natural Science Foundation of China (81560549).
Disclosure
The authors declare that they have no competing interests.
References
1. World Health Organization. Global tuberculosis report 2021. Geneva: World Health Organization; 2021.
2. Freschi L, Vargas R, Husain A, et al. Population structure, biogeography and transmissibility of Mycobacterium tuberculosis. Nat Commun. 2021;12(1):6099. doi:10.1038/s41467-021-26248-1
3. Liu Q, Ma A, Wei L, et al. China’s tuberculosis epidemic stems from historical expansion of four strains of Mycobacterium tuberculosis. Nat Ecol Evol. 2018;2(12):1982–1992. doi:10.1038/s41559-018-0680-6
4. World Health Organization. Global tuberculosis report 2022. Geneva; 2022.
5. Allix-Béguec C, Arandjelovic I, Bi L, et al. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N Engl J Med. 2018;379(15):1403–1415. doi:10.1056/NEJMoa1800474
6. Pankhurst LJ, Del Ojo Elias C, Votintseva AA, et al. Rapid, comprehensive, and affordable mycobacterial diagnosis with whole-genome sequencing: a prospective study. Lancet Respir Med. 2016;4(1):49–58. doi:10.1016/s2213-2600(15)00466-x
7. Rivière E, Heupink TH, Ismail N, et al. Capacity building for whole genome sequencing of Mycobacterium tuberculosis and bioinformatics in high TB burden countries. Brief Bioinform. 2021;22(4). doi:10.1093/bib/bbaa246
8. Tagliani E, Anthony R, Kohl TA, et al. Use of a whole genome sequencing-based approach for Mycobacterium tuberculosis surveillance in Europe in 2017–2019: an ECDC pilot study. Eur Respir J. 2021;57(1). doi:10.1183/13993003.02272-2020
9. Papaventsis D, Casali N, Kontsevaya I, Drobniewski F, Cirillo DM, Nikolayevskyy V. Whole genome sequencing of Mycobacterium tuberculosis for detection of drug resistance: a systematic review. Clin Microbiol Infect. 2017;23(2):61–68. doi:10.1016/j.cmi.2016.09.008
10. Lin D, Cui Z, Chongsuvivatwong V, et al. The geno-spatio analysis of Mycobacterium tuberculosis complex in hot and cold spots of Guangxi, China. BMC Infect Dis. 2020;20(1):462. doi:10.1186/s12879-020-05189-y
11. Lin D, Wang J, Cui Z, Ou J, Huang L, Wang Y. A genome epidemiological study of mycobacterium tuberculosis in subpopulations with high and low incidence rate in Guangxi, South China. BMC Infect Dis. 2021;21(1):840. doi:10.1186/s12879-021-06385-0
12. Rastogi N, Goh KS, David HL. Drug susceptibility testing in tuberculosis: a comparison of the proportion methods using Lowenstein-Jensen, Middlebrook 7H10 and 7H11 agar media and a radiometric method. Res Microbiol. 1989;140(6):405–417. doi:10.1016/0923-2508(89)90016-8
13. Larsen MH, Biermann K, Tandberg S, Hsu T, Jacobs WR. Genetic manipulation of Mycobacterium tuberculosis. Curr Protoc Microbiol. 2007. doi:10.1002/9780471729259.mc10a02s6
14. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi:10.1093/bioinformatics/btu170
15. Phelan JE, O’Sullivan DM, Machado D, et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 2019;11(1):41. doi:10.1186/s13073-019-0650-x
16. Coll F, McNerney R, Guerra-Assunção JA, et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat Commun. 2014;5:4812. doi:10.1038/ncomms5812
17. Shitikov E, Kolchenko S, Mokrousov I, et al. Evolutionary pathway analysis and unified classification of East Asian lineage of Mycobacterium tuberculosis. Sci Rep. 2017;7(1):9227. doi:10.1038/s41598-017-10018-5
18. Meehan CJ, Moris P, Kohl TA, et al. The relationship between transmission time and clustering methods in Mycobacterium tuberculosis epidemiology. EBioMedicine. 2018;37:410–416. doi:10.1016/j.ebiom.2018.10.013
19. Srilohasin P, Prammananan T, Faksri K, et al. Genomic evidence supporting the clonal expansion of extensively drug-resistant tuberculosis bacteria belonging to a rare proto-Beijing genotype. Emerg Microbes Infect. 2020;9(1):2632–2641. doi:10.1080/22221751.2020.1852891
20. World Health Organization. WHO consolidated guidelines on drug-resistant tuberculosis treatment; 2019.
21. Liu M, Xu P, Liao X, et al. Molecular epidemiology and drug-resistance of tuberculosis in Luodian revealed by whole genome sequencing. Infect Genet Evol. 2021;93:104979. doi:10.1016/j.meegid.2021.104979
22. Li Q, Wang Y, Li Y, et al. Characterisation of drug resistance-associated mutations among clinical multidrug-resistant Mycobacterium tuberculosis isolates from Hebei Province, China. J Glob Antimicrob Resist. 2019;18:168–176. doi:10.1016/j.jgar.2019.03.012
23. Jiang Q, Liu Q, Ji L, et al. Citywide transmission of multidrug-resistant tuberculosis under China’s rapid urbanization: a retrospective population-based genomic spatial epidemiological study. Clin Infect Dis. 2020;71(1):142–151. doi:10.1093/cid/ciz790
24. Liu Q, Liu H, Shi L, et al. Local adaptation of Mycobacterium tuberculosis on the Tibetan Plateau. Proc Natl Acad Sci U S A. 2021;118(17). doi:10.1073/pnas.2017831118
25. Holt KE, McAdam P, Thai PVK, et al. Frequent transmission of the Mycobacterium tuberculosis Beijing lineage and positive selection for the EsxW Beijing variant in Vietnam. Nat Genet. 2018;50(6):849–856. doi:10.1038/s41588-018-0117-9
26. Cox HS, Kubica T, Doshetov D, Kebede Y, Rüsch-Gerdess S, Niemann S. The Beijing genotype and drug resistant tuberculosis in the Aral Sea region of Central Asia. Respir Res. 2005;6(1):134. doi:10.1186/1465-9921-6-134
27. Zhao LL, Li MC, Liu HC, et al. Beijing genotype of Mycobacterium tuberculosis is less associated with drug resistance in south China. Int J Antimicrob Agents. 2019;54(6):766–770. doi:10.1016/j.ijantimicag.2019.08.005
28. Yang C, Shen X, Peng Y, et al. Transmission of Mycobacterium tuberculosis in China: a population-based molecular epidemiologic study. Clin Infect Dis. 2015;61(2):219–227. doi:10.1093/cid/civ255
29. Dean AS, Zignol M, Cabibbe AM, et al. Prevalence and genetic profiles of isoniazid resistance in tuberculosis patients: a multicountry analysis of cross-sectional data. PLoS Med. 2020;17(1):e1003008. doi:10.1371/journal.pmed.1003008
30. Dookie N, Rambaran S, Padayatchi N, Mahomed S, Naidoo K. Evolution of drug resistance in Mycobacterium tuberculosis: a review on the molecular determinants of resistance and implications for personalized care. J Antimicrob Chemother. 2018;73(5):1138–1151. doi:10.1093/jac/dkx506
31. Shea J, Halse TA, Kohlerschmidt D, et al. Low-level rifampin resistance and rpoB mutations in mycobacterium tuberculosis: an analysis of whole-genome sequencing and drug susceptibility test data in New York. J Clin Microbiol. 2021;59(4). doi:10.1128/JCM.01885-20
32. He W, Tan Y, Liu C, et al. Drug-resistant characteristics, genetic diversity, and transmission dynamics of rifampicin-resistant Mycobacterium tuberculosis in Hunan, China, revealed by whole-genome sequencing. Microbiol Spectr. 2022;10(1):e0154321. doi:10.1128/spectrum.01543-21
33. Ma P, Luo T, Ge L, et al. Compensatory effects of M. tuberculosis rpoB mutations outside the rifampicin resistance-determining region. Emerg Microbes Infect. 2021;10(1):743–752. doi:10.1080/22221751.2021.1908096
34. Safi H, Fleischmann RD, Peterson SN, Jones MB, Jarrahi B, Alland D. Allelic exchange and mutant selection demonstrate that common clinical embCAB gene mutations only modestly increase resistance to ethambutol in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2010;54(1):103–108. doi:10.1128/aac.01288-09
35. Wang Y, Li Q, Gao H, et al. The roles of rpsL, rrs, and gidB mutations in predicting streptomycin-resistant drugs used on clinical Mycobacterium tuberculosis isolates from Hebei Province, China. Int J Clin Exp Pathol. 2019;12(7):2713–2721.
36. Gomes LC, Campino S, Marinho CRF, Clark TG, Phelan JE. Whole genome sequencing reveals large deletions and other loss of function mutations in Mycobacterium tuberculosis drug resistance genes. Microb Genom. 2021;7(12). doi:10.1099/mgen.0.000724
37. Vaziri F, Kohl TA, Ghajavand H, et al. Genetic diversity of multi- and extensively drug-resistant Mycobacterium tuberculosis isolates in the capital of Iran, revealed by whole-genome sequencing. J Clin Microbiol. 2019;57(1). doi:10.1128/jcm.01477-18
38. Louw GE, Warren RM, Gey van Pittius NC, McEvoy CR, Van Helden PD, Victor TC. A balancing act: efflux/influx in mycobacterial drug resistance. Antimicrob Agents Chemother. 2009;53(8):3181–3189. doi:10.1128/aac.01577-08
39. Nonghanphithak D, Chaiprasert A, Smithtikarn S, et al. Clusters of drug-resistant mycobacterium tuberculosis detected by whole-genome sequence analysis of nationwide sample, Thailand, 2014–2017. Emerg Infect Dis. 2021;27(3):813–822. doi:10.3201/eid2703.204364
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.