Back to Journals » Infection and Drug Resistance » Volume 12
Whole-genome sequence analysis of multidrug-resistant uropathogenic strains of Escherichia coli from Mexico
Authors Paniagua-Contreras GL ![]() , Monroy-Pérez E
, Monroy-Pérez E ![]() , Díaz-Velásquez CE, Uribe-García A, Labastida A, Peñaloza-Figueroa F, Domínguez-Trejo P, García LR
, Díaz-Velásquez CE, Uribe-García A, Labastida A, Peñaloza-Figueroa F, Domínguez-Trejo P, García LR ![]() , Vaca-Paniagua F, Vaca S
, Vaca-Paniagua F, Vaca S ![]()
Received 31 January 2019
Accepted for publication 17 June 2019
Published 1 August 2019 Volume 2019:12 Pages 2363—2377
DOI https://doi.org/10.2147/IDR.S203661
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Joachim Wink
GL Paniagua-Contreras,1 E Monroy-Pérez,1 CE Díaz-Velásquez,2 A Uribe-García,1 A Labastida,3 F Peñaloza-Figueroa,3 P Domínguez-Trejo,4 LR García,4 F Vaca-Paniagua,2,5,6 S Vaca1
1Facultad de Estudios Superiores Iztacala, Universidad Nacional Autónoma de México, Tlalnepantla, México; 2Laboratorio Nacional en Salud: Diagnóstico Molecular y Efecto Ambiental en Enfermedades Crónico-
Degenerativas, FES-Iztacala, UNAM, Tlalnepantla, Edo. de México, México; 3OMICS Analysis, Ciudad de México, México; 4Instituto Mexicano del Seguro Social, México City, México; 5Subdirección de Investigación Básica, Instituto Nacional de Cancerología, México City, México; 6Unidad de Biomedicina, Facultad de Estudios Superiores Iztacala, Universidad Nacional Autónoma de México, Tlalnepantla, México
Background: Escherichia coli is the main bacterium associated with urinary tract infections (UTIs), including cystitis and pyelonephritis. Uropathogenic E. coli (UPEC) harbors numerous genes that encode diverse virulence factors contributing to its pathogenicity. The treatment of UTIs has become complicated due to the natural selection of E. coli strains that are multiresistant to several groups of antibiotics regularly used in clinical settings such as hospitals. Genomic reports of the global composition and distribution of the antibiotic resistance and virulence genes of these pathogenic strains are lacking in the Mexican population.
Purpose and methods: The aim of this study was to globally characterize the genomes of a group of UPEC strains by massive parallel sequencing to determine the prevalence and distribution of virulence and antibiotic resistance genes associated with different serotypes and phylogenetic groups.
Results: The strains exhibited 138-197 virulence genes and 29 antibiotic resistance genes related to antibiotics that are commonly used in clinical practice.
Conclusions: These findings are relevant to the definition of new strategies for treating urinary tract infections in public hospitals and private practice. To further define the epidemiological distribution and composition of these virulence and antibiotic resistance genes, larger studies are needed.
Keywords: whole-genome sequencing, virulence genes, antibiotic resistance genes
Introduction
Escherichia coli is the main bacteria associated with urinary tract infections (UTIs),1 including cystitis and pyelonephritis.2 Uropathogenic E. coli (UPEC) harbors numerous genes that encode diverse virulence factors contributing to its pathogenicity. These virulence factors include adhesins, toxins and capsule, serum resistance and iron uptake systems, among others.3 E. coli strains are identified serologically by their superficial antigens O (lipopolysaccharide), H (flagellar), and K (capsular).4 Phylogenetic analyses group E. coli into eight main phylogenetic groups, seven belonging to E. coli sensu stricto (A, B1, B2, C, D, E and F) and one belonging to the cryptic clade I.5 The treatment of UTIs has become complicated due to the selection of E. coli strains that are resistant to several groups of antibiotics regularly used in clinical settings such as hospitals.6
The size of the genome of E. coli strains differs by approximately 1 Mb, ranging from 4.5 to 5.5 Mb.7 For example, the genome size of E. coli K-12 MG1655 is 4.64 Mb,8 while strain EHEC O157:H7 Sakai has a genome of 5.50 Mb,9 and the genome size of UPEC CFT073 is 5.23 Mb.10 These genomic differences are due principally to the insertion or deletion of large chromosomal regions that encode pathogenicity-associated islands, whose deletion significantly decreases the virulence of UPEC strains in a mouse infection model.11,12
A few reports from Mexico have focused on the molecular analysis of UPEC strains;13–15 however, work addressing the genomics of these pathogens is lacking. Therefore, the purpose of this study was to globally characterize the genomes of a group of UPEC strains to determine the prevalence and distribution of virulence genes and resistance to antibiotics associated with serotypes and phylogenetic groups.
Interestingly, we found between 138-197 virulence genes in each strain that drive pathogenicity and 29 antibiotic resistance genes overall. The serotypes and sequence types of the strains were mainly O25:H4-ST131 and O8:H9-ST423, belonging to the B2 and B1 phylogroups, respectively. This is the first genomic analysis of UPEC strains in Mexico and highlights the need to continue with the genotypic characterization of UPEC strains to understand genomic bacterial factors that are clinically relevant to urinary tract infections.
Materials and methods
Urinary tract E. coli strains
We used 24 UPEC strains isolated from 20 women and 4 men (age range 36–73 years) with community-acquired, nonrecurrent urinary tract infections who were attending a Family Medical Unit (UMA) belonging to the Mexican Institute of Social Security (IMSS) in the municipality of Tlalnepantla, Edo. de Mexico, from August to December 2013. The local ethics committee of the UMF approved the study, and the patients signed an informed consent letter to participate in the study. These strains were selected according to their high frequency of resistance to beta-lactam antibiotics (ampicillin, cephalotin and carbenicillin), fluoroquinolones (pefloxacin) and trimethoprim-sulfamethoxazole and their high frequency of virulence factors detected by PCR,16 such as fimH (type-1 fimbriae), iha (iron-regulated-gene homologue adhesin), usp (uropathogenic-specific protein), irp2 (iron-repressible protein) and kpsMT (K-antigen). Each strain was cultured in EMB (eosin methylene blue) and isolated in clonal colonies.
DNA extraction
Genomic bacterial DNA (gDNA) was extracted with the DNeasy Blood & Tissue Kit (Qiagen) following the manufacturer’s instructions. The DNA concentration was quantified by fluorometry with the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, USA), and the integrity and purity of the material were verified by agarose gel electrophoresis and spectrophotometry, respectively.
Library preparation
The gDNA extracted from each UPEC clone was used for library preparation with the Nextera XT Kit (Illumina) following the manufacturer’s instructions. Briefly, 1 ng of DNA was fragmented and amplified with Nextera XT barcodes in limited-cycle PCR (12 cycles). The PCR product was purified with Agencourt AMPure XP magnetic beads (Beckman Coulter). The libraries were diluted to 4.0 nM and then pooled. Library quality was evaluated by DNA quantification with Qubit and by Bioanalyzer (Agilent) profiling with a High-Sensitivity DNA Kit. The pooled barcoded libraries were diluted to 12 pM.
Whole-genome sequencing
Whole-genome sequencing was performed at the Laboratorio Nacional en Salud, FES-Iztacala, UNAM, in a MiSeq (Illumina) instrument with pair-end reads (V3 2×300). E. coli CFT073 (GenBank: AE014075.1)10,17 was used as reference genome 1, and Escherichia coli EC958 O25b:H4 ST131 (GenBank: HG941718.1 and HG941719.1)17,18 was used as reference genome 2.
Quality filtering, alignment and de novo assembly
FastQ files were depurated by trimming Illumina adaptors, bases below Q20, uncalled bases, and end-sequences below Q20 with cutadapt 1.4 (https://cutadapt.readthedocs.org/). The R package Rqc v1.10.2 was used to measure the Q score and A, T, G, and C contents. The quality-filtered reads were aligned to the E. coli CFT073 reference with Bowtie v2.3.3.1 (Ref.19) and sorted according to chromosomal coordinates with Picard Tools v2.15.0 (Broad Institute). The mapping qualities of all reads were collected with the Rsamtools package v1.28.0.20 Overlapping reads were merged into single reads with FLASH2 v2.2.00.21 To avoid failing to detect any genes that were not present in the reference E. coli strains, de novo genome assembly (without any guiding reference genome) was performed with SPAdes v3.11.1 (Ref.22) in SE (single-end) mode, with multiple assembly k-mers (21, 33, 55, 77, 99, 107, 117, 127) and with the forward reads (for the read pairs with no overlap) and FLASH2 merged reads as input.
To evaluate the quality of each of our genome assemblies, we used QUAST v4.5 to measure their total length and their N50 value.23 The obtained values were compared with the genome size of the E. coli model strains MG1655 and CFT073 and with the N50 values previously reported for Illumina paired-end libraries of E. coli MG1655 by the SPAdes developers (http://cab.spbu.ru/software/spades/).
As an additional quality test, we aligned the reads of each of our 24 UPEC libraries against the corresponding genome assemblies with Bowtie 2. The percentage of reads aligned to the genome was used as a measure of the completeness of the assembly. The complete results for the genome assembly quality tests can be found in Supplementary materials.
Gene identification, pangenome and synteny analyses
The genome assemblies were annotated with RAST (online version Nov 2017)24,25 and Prokka v 1.12.26 For both tools, the default settings were used. The COG triangles and the OrthoMCL algorithms included in the GET_HOMOLOGUES27,28 software package were used to identify orthologous gene families among the genes annotated by Prokka and RAST in the 24 genome assemblies. GET_HOMOLOGUES was also used to measure the number of orthologous gene families shared by different numbers of the 24 strains and, thus, to define the size of the core and pangenomes of these isolates. Finally, we used GET_HOMOLOGUES to determine whether the pangenome was open or closed by measuring the increase in the pangenome size while the genes of the 24 strains were added to the gene set randomly, one strain at a time.29
To study the synteny among our 24 strains and the genomes of the UPEC model strains CFT073 and EC958 ST-131, the orthologue cluster data generated by GET_HOMOLOGUES were transformed into the i-ADHoRe software input.30 The i-ADHoRe tool was then used to identify syntenic blocks of at least 5 contiguous genes with a maximum discrepancy of 1 gene (only one gene lost or added) between each pair of strains.
Serotype, MLST analysis and phylogenetic group assignation
The genome assemblies were uploaded to SeroTypeFinder v1.1 (Ref.31), which identifies E. coli serotypes from draft or complete genome assemblies. This online tool depends on a database of alleles of the O-antigen-related genes wzx, wzy, wzm, and wzt and the H-antigen-related genes fliC, flkA, fllA, flmA, and flnA. The assemblies were also subjected to MultiLocus Sequence Typing (MLST) with the online tool MLST v1.8 (Ref.32), which determined the sequence type (ST) for each of the 24 assembled genomes. Each ST corresponds to a specific combination of alleles of the adk, fumC, gyrB, icd, mdh, purA and recA genes. To determine the location of the 24 UPEC isolates within the known E. coli phylogroups (A, D, E, B1 or B2), we carried out alignment of the sequences of adk, fumC, gyrB, icd, mdh, purA and recA from the 24 assembled genomes and 17 E. coli model strains whose phylogroup is known. We constructed a phylogenetic tree from a MAFT 7.313 (Ref.33) nucleotide alignment with the maximum likelihood method (Figure S1). The phylogroups of the 24 isolates were determined from their location in the phylogeny.
Identification of acquired antimicrobial resistance genes
To identify antimicrobial resistance genes, the genome assemblies were uploaded to ResFinder v2.1.34 ResFinder identifies acquired antimicrobial resistance genes in totally or partially sequenced isolates of bacteria from a manually curated gene database. An identity threshold of 90% and a minimum coverage of 60% were used as filters for the ResFinder search. The search was performed for the 24 assembled genomes and two additional E. coli model strains: CFT073 and EC958 ST-131.
Identification of virulence genes
To identify the virulence genes of the 24 UPEC genomes, we first obtained the FASTA sequences of the genes included in the Virulence Factor Database (VFDB),35 consulted on December 7, 2107. This VFDB version included 30178 genes that encode 1796 different virulence (or virulence related) factors (VFs) in 30 different bacterial genera. We used Nucleotide-Nucleotide BLAST v2.7.1+ to align the complete set of VFDB genes with each of the 24 genome assemblies and the genomes of CFT073 and EC958 ST-131. Only the alignments that covered 100% of the VFDB query gene with a sequence identity ≥90% were retained. When several VFDB genes exhibited alignments to the same locus of an assembly, only the VFDB gene that successfully aligned with the greatest number of the 26 genomes was considered. Finally, the positions of the VFs found with the VFDB gene alignments were compared to the Prokka and RAST annotations to unify the feature names and identify those putative VF loci that were not detected by the genome annotators (Table S1). The 349 VFDB genes whose orthologues were identified in the 26 studied genomes were manually classified under several functional categories based on the gene descriptions annotated in the VFDB (Table S2).
Results
Resistance to antibiotics in UPEC strains
We first evaluated the antibiotic resistance phenotypes of the strains. In these assays, 100% of the E. coli strains analyzed (n=24) were resistant to cephalotin, ampicillin and carbenicillin (Table 1) and 79% (n=19) to pefloxacin, 75% (n=18) to cefotaxime, 71% (n=17) to trimethoprim-sulfamethoxazole, 54% (n=13) to gentamicin, 50% (n=12) to ceftriaxone, 37.5% (n=9) to netilmicin, 29% (n=7) to chloramphenicol, and 25% (n=6) to nitrofurantoin and amikacin.
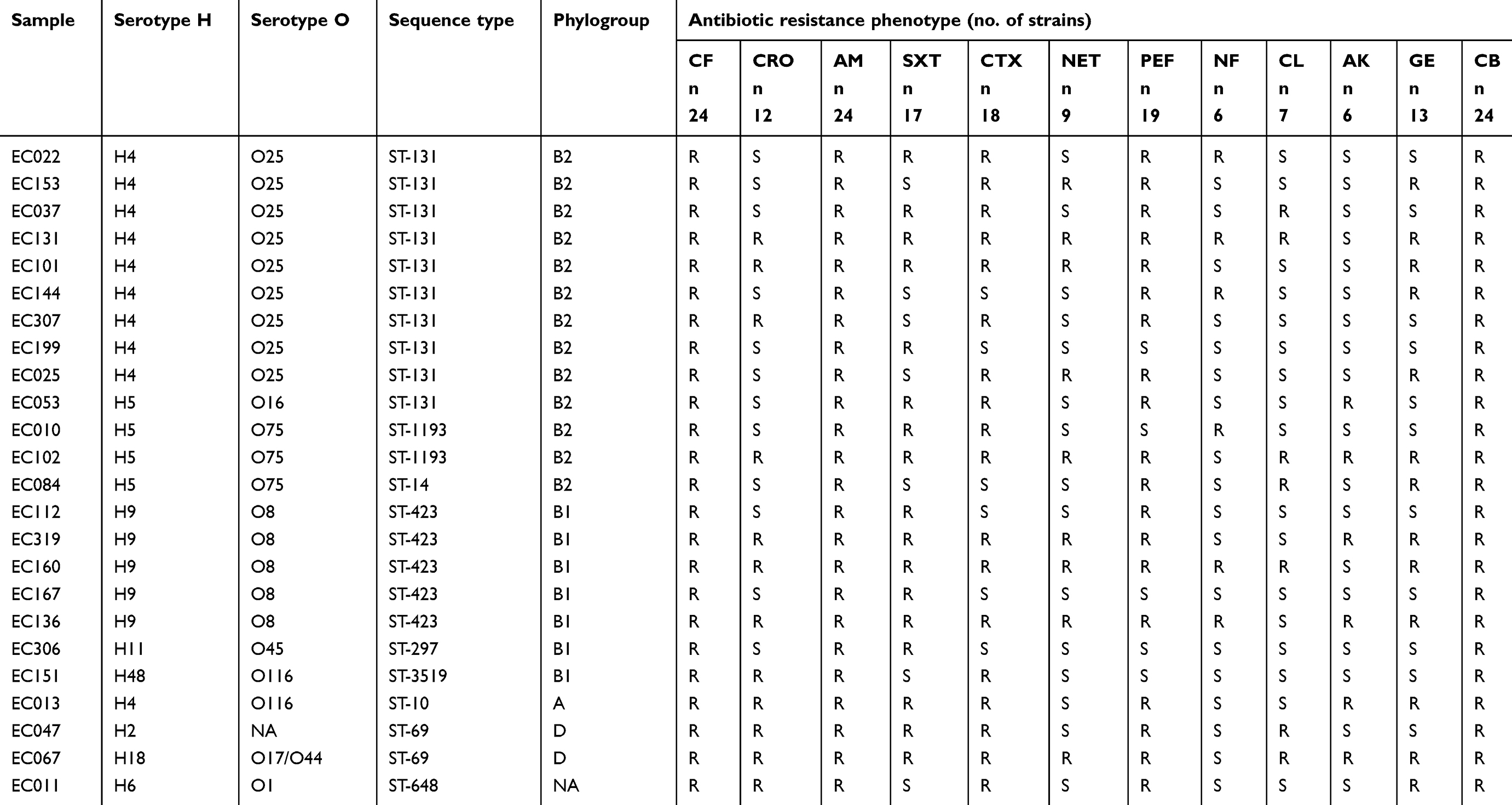 |
Table 1 Serotypes (O and H antigens), sequence types (STs), phylogenetic group assignations and antibiotic resistance phenotypes. The strains are grouped by their specific combination of serotype and ST |
Massive parallel sequencing metrics
To better cover the genomic diversity of UPEC, we performed sequencing considering their different patterns of virulence genes measured by PCR and a high frequency of antibiotic resistance to multiple agents, which is representative of the diversity of these infections.16 To measure the quality of the sequencing first, we evaluated the Phred score and the mapping parameters of the trimmed sequences. The per-base quality showed a typical Illumina pattern with an overall Q30 of >80%, and all libraries exhibited at least 25% of high-quality mapped reads and at least 100,000 reads. The assembly of all genomes presented a median N50 of 177,458 (SD: 51,745), and the average number of contigs was 123 (SD: 62), which suggests relatively low fragmentation in the assembly. As a second test to evaluate the quality and completeness of our genome assemblies, the forward reads of each library were aligned with Bowtie 2 against each of the whole-genome assemblies. The percentages of aligned reads were 97.76 (PE; SD: 1.45%) and 99.49 (SE; SD: 0.47), which indicated a high level of completeness of the assemblies.
Serotype, MLST analysis and phylogenetic group assignation
The most abundant serotype-ST combinations were O25:H4 (ST-131) and O8:H9 (ST-423), with 9 and 5 representatives among the 24 UPEC strains, respectively. The serotypes and the sequence types (ST) of the 24 studied strains are indicated in Table 1.
The 24 UPEC genome isolates belonged to 4 of the E. coli phylogroups (as determined from their position in the E. coli phylogeny; see Figure S1) and presented 10 specific serotype-ST combinations. Only the serotypes of strains EC047 and EC067 were ambiguous or undetermined. EC067 can be assigned to the O17 and O44 antigens due to antigen cross-reactions; in addition, SeroTypeFinder can erroneously report the O17 antigen instead of O44.31
Gene content comparison and pangenome composition
The mean number of protein-coding genes detected by Prokka in the 24 genome assemblies was 4,853 (range: 4,418–5,113), while RAST detected 5,064 (range: 4,574–5,368). In comparison, the UPEC model strain CFT073 and EC958 ST-131 genomes harbor 5,379 and 5,100 genes, respectively (Table 2).10,17,18
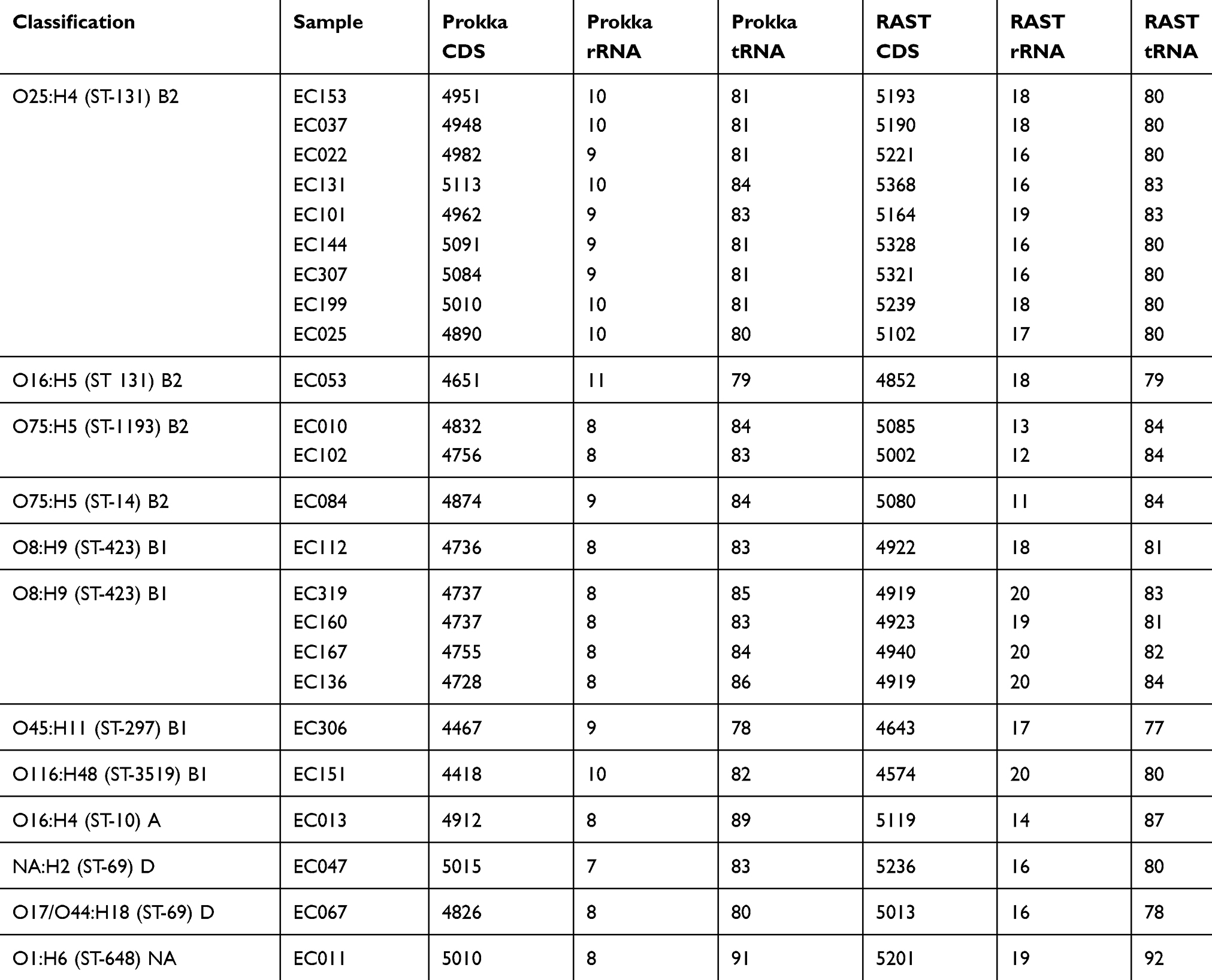 |
Table 2 Numbers and types of genes detected by Prokka and RAST |
Orthologous families were clustered in the following groups: (1) core (present in all samples), (2) soft core (present in at least 95% of the samples), (3) shell (present only in a few genomes) and (4) cloud (the remaining gene families). A total of 8,383 and 9,408 orthologous clusters were detected by GET_HOMOLOGUES in the Prokka and RAST annotations, respectively (Figure 1). The number of orthologous clusters present in all the strains was 3,136 for the Prokka annotations and 3,105 for the RAST annotations. Depending on the annotation method (either Prokka or RAST), an average of 4,853 or 5,064 genes were detected in the 24 UPEC strains, which approaches the number of genes in UPEC model strains CFT073 and EC958 (5,379 and 5,100). The difference in the numbers of genes detected by Prokka and RAST is due to accuracy differences between the annotation algorithms that are reported in the literature.26
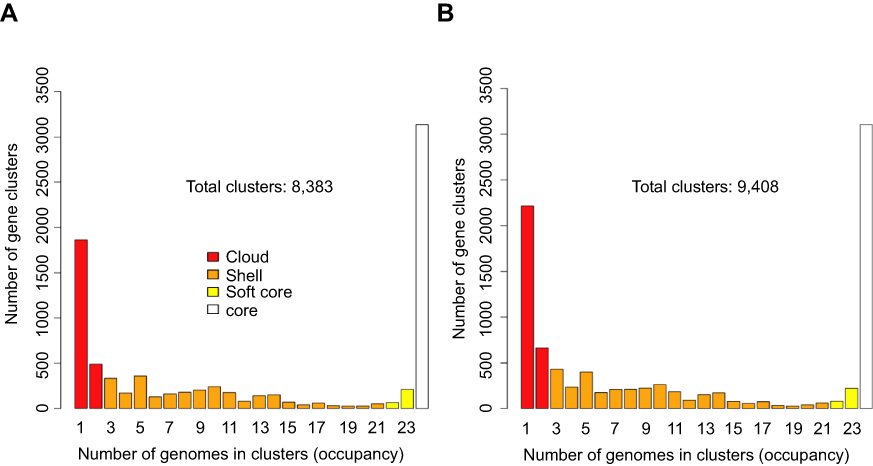 |
Figure 1 Occurrence of homologous gene families detected in the 24 UPEC strains. The number of homologous gene families ((y-axis) present in increasing numbers of strains (ranging from presence in at least 1 strain to all 24 strains) is shown. The numbers are displayed separately for the (A) Prokka and (B) RAST annotations. The genes of the cloud, shell, soft-core and core genome are indicated by color. |
The total number of orthologous gene families detected in the 24 UPEC strains (their pangenome) comprises between 8,383 genes and 9,408 orthologous gene families. Among these pangenome gene families, we identified a total of 307 virulence factors (Table S4).
After clustering homologous gene families, a distance tree based on the absence/presence of the genes was built with GET-HOMOLOGUES. This allowed us to group samples with a similar gene content. The clustering results of the 24 UPEC isolates calculated from the Prokka and the RAST annotations were very similar (Figure 2). This clustering strictly mirrors the distribution of the 24 UPEC isolates in different E. coli phylogroups and their grouping by serotype and ST.
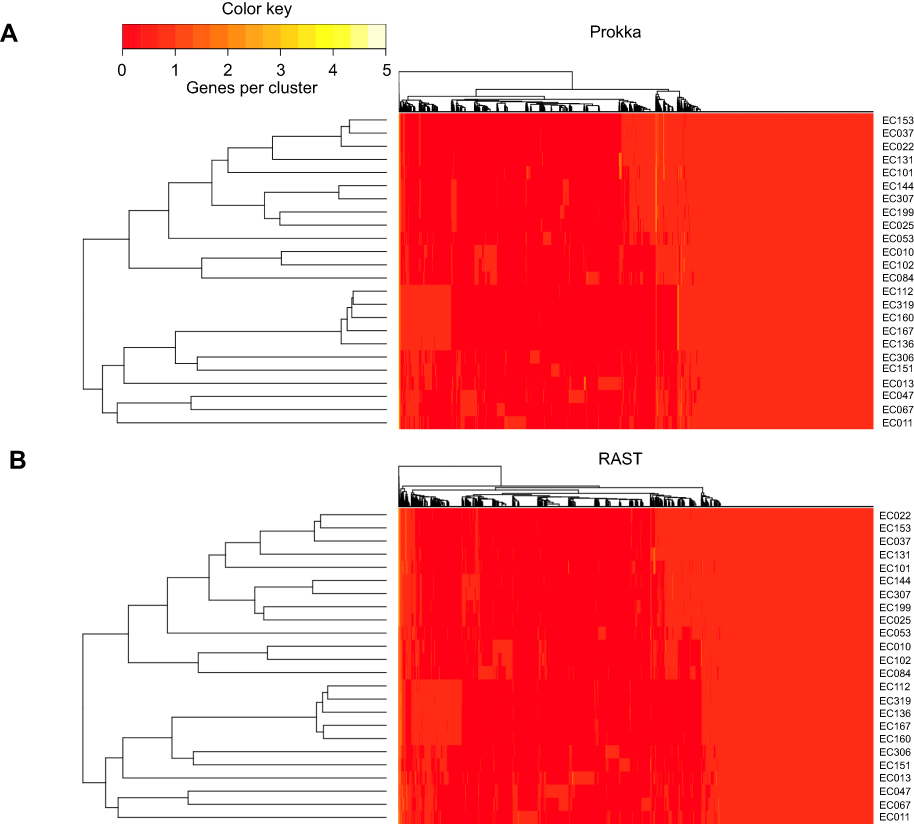 |
Figure 2 Hierarchical clustering of the strains by gene presence/absence with genes annotated by Prokka and RAST. The pangenome was used for clustering with (A) Prokka and (B) RAST. A distance tree was constructed based on the presence or absence of genes. Samples with a similar gene content clustered together. |
We also explored whether the pangenome of the 24 UPEC strains was open (ie, comprising an orthologous family set that would grow in number if more UPEC strains were included in the analysis). To this end, we followed the approach developed by Tettelin et al.29 Briefly, the increase in pangenome size was measured as the genes of the 24 strains and those of UPEC model strains CFT073 and EC958 ST-131 were added to the gene set randomly, one strain at a time. The generated data were used to estimate a mathematical function that describes the growth tendency of the pangenome (Figure S2). The pangenomes of the 24 UPEC strains showed a continuous growth tendency, such that we can predict that its size would reach ~10,000 or ~15,000 total genes if 39 or 97 total strains were included in the calculations.
Synteny conservation of the 24 strains and 2 UPEC model strains
We used i-ADHoRe software to identify the syntenic genomic regions between each pair of strains.30 The medium number of contiguous genes (present in the same contig) that belonged to these syntenic regions ranged from 42 to 281 for different strain comparisons. The assemblies with the highest N50, which tend to exhibit larger contigs, also present larger syntenic regions (represented in one or more fragments in other assemblies) (Supplementary materials).
For each pair (a, b) of strain assemblies, the average percentage (of the genes) of the contigs in a that presented the same synteny in one or more regions of b varied from 71.3 to 100%. The hierarchical clustering of the 24 UPEC and strains CFT073 and EC958 ST-131 according to this metric reproduced the distribution of the strains in different serotypes and E. coli phylogroups (Figure 3).
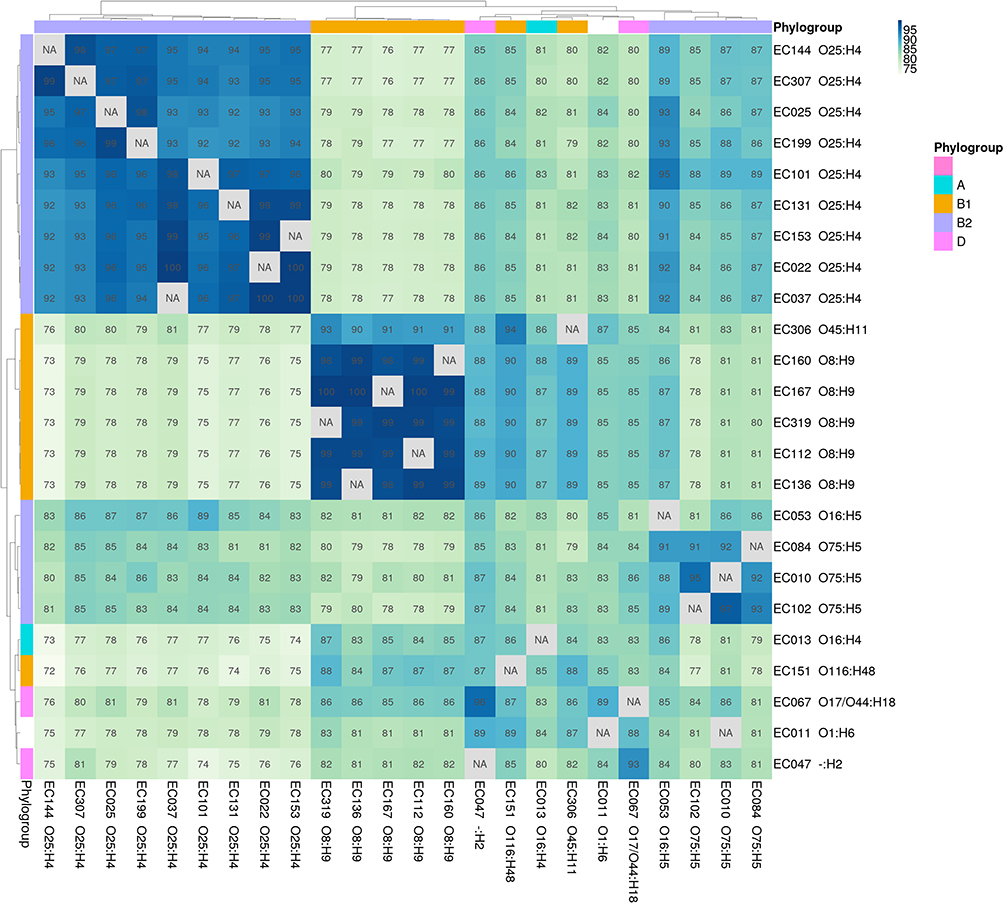 |
Figure 3 Average percentage of the contigs (of their genes) of each strain (column labels) that appears in one or more fragments with the same synteny in strain b (row labels). The values for all the comparisons between the 24 UPEC strains and the model strains CFT073 and EC958 ST-131 are shown. The strains were hierarchically clustered according to these values. The phylogroups and serotypes of the strains are indicated at the heatmap margins and in the row and column labels, respectively. |
The percentage of the total genes of a given strain that showed the same synteny in another strain ranged from 73.2 to 98.9%, but the values were higher (from 92 to 100%) when we took into account only the genes that were shared between each pair of strains. The clustering of the strains according to these statistics also reflected the phylogroup and serotype distribution (Supplementary materials).
Identification of acquired antimicrobial resistance genes
We used the ResFinder v2.1 online tool to identify antibiotic resistance genes in our 24 genome assemblies.34 There was a wide distribution of antibiotic resistance genes, which comprised 29 genes conferring resistance to aminoglycosides, fluoroquinolones, β-lactams, chloramphenicol, sulfonamides, tetracycline and trimethoprim. On average, each strain exhibited 6 antibiotic resistance genes, and the strain with the most exhibited 11. The genes with the highest prevalence among the strains were strA, strB, blaTEM1B and sul2 (Table 3). Overall, the antibiotic resistance genotypes showed concordance with the observed phenotypes (Table S3).
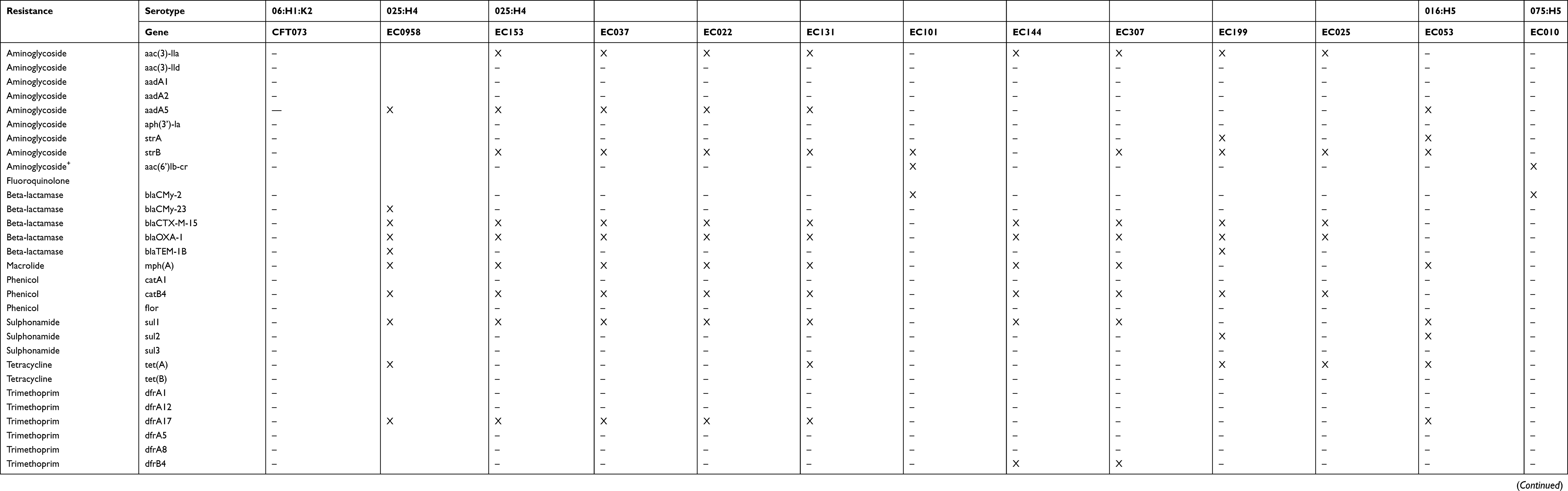 |
Table 3 Distribution of antibiotic resistance genes |
Detection of virulence genes
We identified virulence factor genes by performing Blast alignments of the complete set of genes from the Virulence Factor Database (VFDB) against our 24 genome assemblies. We detected between 138 and 197 virulence factor genes in the 24 genome assemblies; 90 of them were shared by at least 23 of the UPEC isolates (and by the reference strains CFT073 and EC958 ST-131) (Figure 4; Table S4). These conserved genes included the E. coli common pilus (ECP), curli and type I fimbriae genes; the che, fig, fli and mot genes related to motility and chemotaxis; the enterobactin siderophore genes and the ompA gene, among others. Adherence; iron uptake; motility and chemotaxis; and secretion systems were the most represented functional categories for the VFs in all strains. There was a characteristic functional category distribution of the genes dependent on the presence of different phylogroups, serotypes and STs. For instance, only the O25:H4 ST-131 and ST-14 isolates exhibited pap adherence genes. On the other hand, the yersiniabactin siderophore genes were found in all the isolates except for O8:1-H9 (ST-423), but the salmochelin siderophore genes occurred only in O8:H9 (ST-423) strains (Table S4).
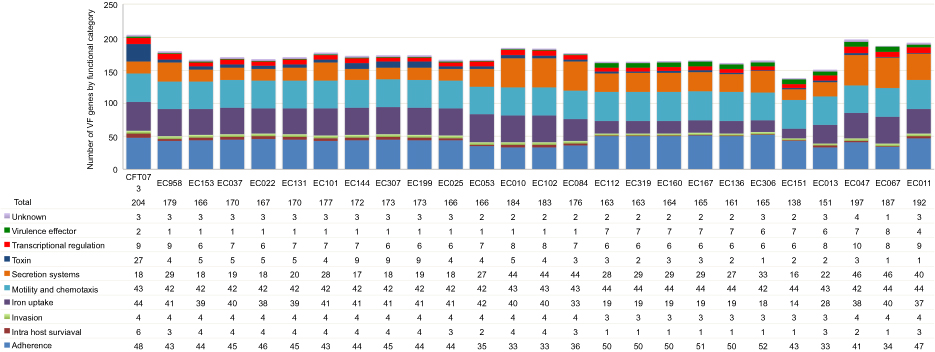 |
Figure 4 Number of virulence factor genes per functional category for the 24 assembled UPEC genomes. The strains are ordered by the phylogroup-ST. Each functional category is color coded. |
Discussion
In this work, we characterized the full genomes of 24 UPEC strains by massive parallel sequencing to investigate their contents of antibiotic resistance and virulence factor genes. These UPEC strains are of clinical relevance given that around the world, 150 million people develop urinary tract infections each year, and E. coli is their primary infectious agent.36 In Mexico, the incidence of UPEC infections is high, reaching 3,000 per 100,000 habitants in 2018.37 We previously characterized UPEC strains from Mexican patients via nongenomic approaches that are limited in terms of gene detection and identified the collective participation of a large repertoire of expressed virulence genes during in vitro infection; therefore, this work represents an advance of this development.16,38
Overall, these metrics are similar to those in published works and were considered valid for the purpose of this study.39,40
The size of our 24 UPEC pangenome is smaller than the E. coli species pangenome previously reported (~18,000 clusters of orthologues), which was calculated with a strain set of comparable size (20 isolates) that included commensal and pathogenic strains from different E. coli phylogroups.41 Although factors such as the completeness of the assemblies or the orthologous search method can contribute to the difference between the size of our UPEC pangenome and the reported E. coli pangenome, we expect that several genes that are characteristic of other E. coli lifestyles or phenotypes (for example, intestinal pathogens) are present in the species pangenome but absent in the UPEC pangenome.
The 24 UPEC-strain pangenome shows a highly pronounced growth tendency, such that when the genes of the 24 isolates (along with strains CFT073 and EC958 ST-131) are added to the gene set one strain at a time, the pangenome continues to grow without reaching a saturation point, and we can expect that it would continue to increase steeply if more UPEC strains were taken into account (Figure S2). We therefore conclude that the pangenome of these UPEC strains is open, reflecting the behavior previously reported for the E. coli species, whose members gain and lose genes frequently by horizontal gene transfer.42
The 24 UPEC-strain core genome (the group of gene families shared by all the strains) includes at least 3,136 gene families and is therefore larger than the E. coli species core genome (which includes ~2,000 orthologous gene families).41 This is explained by the fact that the UPEC strains share a group of genes related to their specific pathogenic lifestyle that may be absent in other members of the E. coli species. Among the genes shared by all the strains (or absent in just one strain), we found 90 virulence factors. These conserved genes include well-known UPEC virulence determinants, such as the E. coli common pilus (ECP), curli and type I fimbriae genes, as well as the che, fig, fli and mot genes, which are related to motility and chemotaxis.43
When the 24 UPEC strains were clustered according to their gene family content, the isolates from the same E. coli phylogroups and/or serotypes clearly grouped together. The same was true when the strains were clustered according to the number of syntenic contig regions that they shared (Figures 2 and 3). The highest levels of both shared synteny and gene content similarity were observed between strains of the same serotype. The values were lower when two strains of the same phylogroup with different serotypes were compared, and the lowest numbers were observed between strains of different phylogroups. Together, these results indicate that among these 24 UPEC strains, there is a distinct gene content for different serotypes or phylogroups and that these distinctive genes tend to share the same synteny. The percentage of the genes of a given strain that exhibit the same synteny in another strain is between 73 and 99% for different strain pairs, but it is much higher (between 92 and 100%) if only the genes shared by both strains are considered. This suggests that the gain or loss of groups of genes is the factor that contributes most to the differences in genomic structure between these 24 strains, rather than the rearrangement of their genes. This is in accordance with previous studies of the genome dynamics of E. coli, which have suggested that the members of the species share a common genomic backbone that presents conserved synteny (with very limited rearrangements) and is composed of genes of the core genome. The accessory genes, such as those related to pathogenicity, are inserted at prophages, pathogenicity islands (PAIs) or insertion sequences (IS) at different positions in the backbone.10,41
To better characterize the strains, we defined the serotype and sequence type in silico. All but one of the strains was successfully assigned. The phylotype was also assigned in silico. Then, we used the gene annotation data to hierarchically cluster the strains. The strains clustered in the same fashion as the distribution of phylogroups, serotypes and sequence types, which supports the validity of our genomic classification of the strains.
The O25:H4 (ST 131) strains have been extensively studied as a group of globally widespread, multiresistant UPEC clones. The most common ST-131 isolates belong to lineage C, which has the O25:H4 serotype and is best known for its fluoroquinone resistance (acquired through mutations in gyrA and parC) and for the spreading of the CTX-M-15 extended-spectrum beta lactamase gene.44,45 In most strains, 90 genes involved in the adherence, motility and chemotaxis, iron uptake and secretion systems (Table S4) were identified. In addition, 87.5% (n=21) of the genomes of the strains showed different antibiotic resistance genes, including genes related to aminoglycoside, fluoroquinolone, beta-lactamases, macrolide, phenicol, sulfonamide, tetracycline and trimethoprim resistance (Table 3), where the identified phenotype and genotype showed multidrug resistance (MDR) against β-lactams, sulfonamides and trimethoprim (Table S3). Recently, in a large study conducted in uropathogenic E. coli O25b-B2-ST131 (n=248), the most frequently identified virulence genes were kpsM2, sat, iucD, iutA, iha, fimA, fyuA, ompT, csgA and traT, and multidrug resistance to ceftazidime, cefotaxime, cefazolin, coamoxiclav, amoxicillin, erythromycin, tetracycline, tigecycline, gentamicin, and ciprofloxacin was also observed.46 In another study carried out in UPEC strains (n=167), the most frequent ST identified was ST131 (n=20), mainly corresponding to phylogenetic group B2, which also includes ST ST-1193, ST-14, ST-10 and ST-69, found in the present study (Table 1).47 In this study, group O25:H4-ST-131, belonging to phylogenetic group B2 of E. coli was the most prevalent group in the patients studied. Therefore, it is important to establish epidemiological monitoring programs in the region, focused specifically on controlling bacterial resistance to antimicrobials. A recent epidemiological study conducted in Mexico showed that strains of E. coli O25:H4-ST-131 isolated from cystitis and prostatitis patients were carriers of the fimH, papD, sfa, uspA, ipaH, hofB and hofC genes. The majority of the cystitis strains were resistant to trimethoprim-sulfamethoxazole, ticarcillin and ciprofloxacin, whereas most strains associated with prostatitis were resistant to tetracycline, azithromycin, doxycycline, ticarcillin and ciprofloxacin.14
The antibiotic resistance gene composition and distribution are among the major challenges in the clinical treatment of UPEC infections. We detected acquired antibiotic resistance genes in all the studied genomes except for CFT073. We found the CTX-M-15 gene related to extended-spectrum beta lactamase (characteristic of E. coli 025:H4 ST-131) in EC958 ST-131 and in eight of the nine 025:H4 ST-131 UPEC isolates studied here. The identification of the gyrA1AB and parCloAB alleles, responsible for the fluoroquinolone resistance of the ST-131 H25:H4 strains, would require a search for antibiotic resistance-conferring mutations in the corresponding genes.44,45 The 025:H4 ST-131 and 08:H9 ST-423 strains exhibit characteristic antibiotic resistance gene content patterns.
One limitation of this work is that we did not analyze the genetic context of the antibiotic resistance and virulence genes. However, the antibiotic resistance genes sul1 (sulfamethoxazole), dfrA17 (trimethoprim), and aadA5 (aminoglycosides) identified in the genomes of the UPEC strains (Table 3) have been found in the variable region gene cassette of class I integrons that can be mobilized by plasmids or transposons.48 Furthermore, several of the virulence genes detected in the analyzed UPEC genomes have been found in islands of pathogenicity (PAIs), such as PAI IV536 (yersiniabactin siderophore system), PAI I536 (α-hemolysin, CS12 fimbriae, and F17-like fimbrial adhesion) and PAI IIJ96 (α-hemolysin, Prs fimbriae, cytotoxic, necrotizing factor).49
The detection of strains belonging to distinct serogroups that are resistant to four or five unrelated families of antibiotics is of great concern. This high incidence of antibiotic resistance is probably related to several types of inappropriate use of antibiotics in Mexico.50 For example, antibiotics were sold without medical prescription in the drugstores in Mexico until 2010. Eighty-three percent of the strains are resistant to sulfonamides (sul1, sul2 or sul3), and most of them are resistant to both trimethoprim and sulfonamides, a combination of antibiotics that is commonly prescribed for treating UTIs in Mexico, whose continued use could be the cause of the selection of resistant strains. Most strains were resistant to 6–11 antibiotics, and only one was resistant to a single antibiotic, which is epidemiologically relevant given that there is no current knowledge of their prevalence in Mexico. Further studies involving a large number of isolated strains are needed to better establish the distribution and composition of antibiotic resistance genes and virulence factors in UPEC strains from Mexico.14
Conclusion
This is the first genomic analysis of UPEC strains to be carried out in Mexico. The characterized strains exhibited a phenotype and genotype of multidrug resistance and harbored a large number of virulence genes that are commonly used in clinical practice. Group O25:H4-ST-131, belonging to phylogenetic group B2 of E. coli, was the most prevalent in the patients. These findings are relevant to define new strategies for treating urinary tract infections in public hospitals and in private practice. To further define the epidemiological distribution and composition of the virulence and antibiotic resistance genes, larger studies are needed.
Author contributions
GLPC, FVP, and SV conceived and designed the study. PDT and LRG, collected samples and analyzed the clinical data. GLPC, EMP, AUG, and CEDV, cultured and isolated the bacterial strains and extracted DNA. CEDV prepared the libraries and performed the sequencing. AL, FPF, and FVP analyzed and interpreted the sequencing results. All authors contribuited to data analysis, manuscript drafting and revision, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yun KW, Kim HY, Park HK, Kim W, Lim IS. Virulence factors of uropathogenic Escherichia coli of urinary tract infections and asymptomatic bacteriuria in children. J Microbiol Immunol Infect. 2014;47:455–461. doi:10.1016/j.jmii.2013.07.010
2. Chiou YY, Chen MJ, Chiu NT, Lin CY, Tseng CC. Bacterial virulence factors are associated with occurrence of acute pyelonephritis but not renal scarring. J Urol. 2010;184:2098–2102. doi:10.1016/j.juro.2010.06.135
3. Momtaz H, Karimian A, Madani M, et al. Uropathogenic Escherichia coli in Iran: serogroup distributions, virulence factors and antimicrobial resistance properties. Ann Clin Microbiol Antimicrob. 2013;12:8. doi:10.1186/1476-0711-12-8
4. Li D, Liu B, Chen M, et al. A multiplex PCR method to detect 14 Escherichia coli serogroups associated with urinary tract infections. J Microbiol Methods. 2010;82:71–77. doi:10.1016/j.mimet.2010.04.008
5. Clermont O, Christenson JK, Denamur E, Gordon DM. The Clermont Escherichia coli phylo-typing method revisited: improvement of specificity and detection of new phylo-groups. Environ Microbiol Rep. 2013;5:58–65. doi:10.1111/1758-2229.12019
6. Ochoa SA, Cruz-Cordova A, Luna-Pineda VM, et al. Multidrug- and extensively drug-resistant uropathogenic Escherichia coli clinical strains: phylogenetic groups widely associated with integrons maintain high genetic diversity. Front Microbiol. 2016;7:2042. doi:10.3389/fmicb.2016.02042
7. Bergthorsson U, Ochman H. Distribution of chromosome length variation in natural isolates of Escherichia coli. Mol Biol Evol. 1998;15:6–16. doi:10.1093/oxfordjournals.molbev.a025847
8. Blattner FR, Plunkett G, Bloch CA, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi:10.1126/science.277.5331.1453
9. Hayashi T, Makino K, Ohnishi M, et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157: h7and genomic comparison with a laboratory strain K-12. DNA Res. 2001;8:11–22. doi:10.1093/dnares/8.1.11
10. Welch RA, Burland V, Plunkett G, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 2002;99:17020–17024. doi:10.1073/pnas.252529799
11. Rode CK, Melkerson-Watson LJ, Johnson AT, Bloch CA. Type-specific contributions to chromosome size differences in Escherichia coli. Infect Immun. 1999;67:230–236.
12. Brzuszkiewicz E, Bruggemann H, Liesegang H, et al. How to become a uropathogen: comparative genomic analysis of extraintestinal pathogenic Escherichia coli strains. Proc Natl Acad Sci U S A. 2006;103:12879–12884. doi:10.1073/pnas.0603038103
13. Molina-Lopez J, Aparicio-Ozores G, Ribas-Aparicio RM, et al. Drug resistance, serotypes, and phylogenetic groups among uropathogenic Escherichia coli including O25-ST131 in Mexico city. J Infect Dev Ctries. 2011;5:840–849.
14. Morales-Espinosa R, Hernandez-Castro R, Delgado G, et al. UPEC strain characterization isolated from Mexican patients with recurrent urinary infections. J Infect Dev Ctries. 2016;10:317–328. doi:10.3855/jidc.6652
15. Reyna-Flores F, Barrios H, Garza-Ramos U, et al. Molecular epidemiology of Escherichia coli O25b-ST131 isolates causing community-acquired UTIs in Mexico. Diagn Microbiol Infect Dis. 2013;76:396–398. doi:10.1016/j.diagmicrobio.2013.03.026
16. Paniagua-Contreras GL, Monroy-Perez E, Rodriguez-Moctezuma JR, Dominguez-Trejo P, Vaca-Paniagua F, Vaca S. Virulence factors, antibiotic resistance phenotypes and O-serogroups of Escherichia coli strains isolated from community-acquired urinary tract infection patients in Mexico. J Microbiol Immunol Infect. 2017;50:478–485. doi:10.1016/j.jmii.2015.08.005
17. Agarwala R, Barrett T, Beck J, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016;44:D7–D19. doi:10.1093/nar/gkv1290
18. Forde BM, Ben Zakour NL, Stanton-Cook M, et al. The complete genome sequence of Escherichia coli EC958: a high quality reference sequence for the globally disseminated multidrug resistant E. coli O25b: H4-ST131clone. PLoS One. 2014;9:e104400. doi:10.1371/journal.pone.0104400
19. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi:10.1038/nmeth.1923
20. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi:10.1093/bioinformatics/btp698
21. Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi:10.1093/bioinformatics/btr507
22. Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi:10.1089/cmb.2012.0021
23. Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi:10.1093/bioinformatics/btt086
24. Aziz RK, Bartels D, Best AA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi:10.1186/1471-2164-9-75
25. Overbeek R, Olson R, Pusch GD, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014;42:D206–D214. doi:10.1093/nar/gkt1226
26. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi:10.1093/bioinformatics/btu153
27. Contreras-Moreira B, Vinuesa P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol. 2013;79:7696–7701. doi:10.1128/AEM.02411-13
28. Vinuesa P, Contreras-Moreira B. Robust identification of orthologues and paralogues for microbial pan-genomics using GET_HOMOLOGUES: a case study of pIncA/C plasmids. Methods Mol Biol. 2015;1231:203–232. doi:10.1007/978-1-4939-1720-4_14
29. Tettelin H, Masignani V, Cieslewicz MJ, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A. 2005;102:13950–13955. doi:10.1073/pnas.0506758102
30. Proost S, Fostier J, De Witte D, et al. i-ADHoRe 3.0–fast and sensitive detection of genomic homology in extremely large data sets. Nucleic Acids Res. 2012;40:e11. doi:10.1093/nar/gkr955
31. Joensen KG, Tetzschner AM, Iguchi A, Aarestrup FM, Scheutz F. Rapid and easy in silico serotyping of escherichia coli isolates by use of whole-genome sequencing data. J Clin Microbiol. 2015;53:2410–2426. doi:10.1128/JCM.00008-15
32. Larsen MV, Cosentino S, Rasmussen S, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50:1355–1361. doi:10.1128/JCM.06094-11
33. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi:10.1093/molbev/mst010
34. Zankari E, Hasman H, Cosentino S, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–2644. doi:10.1093/jac/dks261
35. Chen L, Zheng D, Liu B, Yang J, Jin Q. VFDB 2016: hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 2016;44:D694–7. doi:10.1093/nar/gkv1239
36. Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol. 2015;13:269–284. doi:10.1038/nrmicro3432
37. Salud SD. Sistema Nacional de Vigilancia Epidemiológica. Panorama Epidemiológico de las ingecciones de vias urinarias en México 2003-2008. Epidemiología. 2009;51:1–28.
38. Paniagua-Contreras GL, Hernandez-Jaimes T, Monroy-Perez E, et al. Comprehensive expression analysis of pathogenicity genes in uropathogenic Escherichia coli strains. Microb Pathog. 2017;103:1–7. doi:10.1016/j.micpath.2016.12.008
39. Bradnam KR, Fass JN, Alexandrov A, et al. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. Gigascience. 2013;2:10. doi:10.1186/2047-217X-2-10
40. Salzberg SL, Phillippy AM, Zimin A, et al. GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2012;22:557–567. doi:10.1101/gr.131383.111
41. Touchon M, Hoede C, Tenaillon O, et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 2009;5:e1000344. doi:10.1371/journal.pgen.1000344
42. Zhang Y, Lin K. A phylogenomic analysis of Escherichia coli/Shigella group: implications of genomic features associated with pathogenicity and ecological adaptation. BMC Evol Biol. 2012;12:174. doi:10.1186/1471-2148-12-174
43. Terlizzi ME, Gribaudo G, Maffei ME. UroPathogenic Escherichia coli (UPEC) infections: virulence factors, bladder responses, antibiotic, and non-antibiotic antimicrobial strategies. Front Microbiol. 2017;8:1566. doi:10.3389/fmicb.2017.01566
44. Pitout JD, DeVinney R. Escherichia coli ST131: a multidrug-resistant clone primed for global domination. F1000Res. 2017;6. doi:10.12688/f1000research.10493.2
45. Schembri MA, Zakour NL, Phan MD, Forde BM, Stanton-Cook M, Beatson SA. Molecular characterization of the multidrug resistant Escherichia coli ST131 clone. Pathogens. 2015;4:422–430. doi:10.3390/pathogens4030422
46. Mostafavi SKS, Najar-Peerayeh S, Mobarez AM, Parizi MK. Characterization of uropathogenic E. coli O25b-B2-ST131, O15: K52:H1,and CGA: neutrophils apoptosis, serum bactericidal assay, biofilm formation, and virulence typing. J Cell Physiol. 2019. doi:10.1002/jcp.28459
47. Matsukawa M, Igarashi M, Watanabe H, et al. Epidemiology and genotypic characterisation of dissemination patterns of uropathogenic Escherichia coli in a community. Epidemiol Infect. 2019;147:e148. doi:10.1017/S0950268819000426
48. Vinue L, Saenz Y, Somalo S, et al. Prevalence and diversity of integrons and associated resistance genes in faecal Escherichia coli isolates of healthy humans in Spain. J Antimicrob Chemother. 2008;62:934–937. doi:10.1093/jac/dkn331
49. Ostblom A, Adlerberth I, Wold AE, Nowrouzian FL. Pathogenicity island markers, virulence determinants malX and usp, and the capacity of Escherichia coli to persist in infants’ commensal microbiotas. Appl Environ Microbiol. 2011;77:2303–2308. doi:10.1128/AEM.02405-10
50. Dreser A, Wirtz VJ, Corbett KK, Echaniz G. [Antibiotic use in Mexico: review of problems and policies]. Salud Publica Mex. 2008;50(Suppl 4):S480–7.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.