")
Back to Journals » International Journal of General Medicine » Volume 16
Whole-Exome Sequencing Reveals Mutational Signature of Hypertrophic Cardiomyopathy
Authors Wang XQ , Yuan F, Yu BR
Received 19 June 2023
Accepted for publication 21 September 2023
Published 11 October 2023 Volume 2023:16 Pages 4617—4628
DOI https://doi.org/10.2147/IJGM.S422598
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Yuriy Sirenko
Xi-Qin Wang,1 Fang Yuan,2 Bao-Rui Yu2
1Department of Internal Medicine, Yuhua Yunfang Integrated Traditional Chinese and Western Medicine Clinic, Shijiazhuang, Hebei, 050023, People’s Republic of China; 2Department of Cardiovascular Medicine, Fuwai Central China Cardiovascular Hospital, Zhengzhou, Henan, 450000, People’s Republic of China
Correspondence: Xi-Qin Wang, Department of Internal Medicine, Yuhua Yunfang Integrated Traditional Chinese and Western Medicine Clinic, NO. 59 Ta’nan Road, Shijiazhuang, Hebei, 050023, People’s Republic of China, Tel +86-13522595882, Email [email protected] Fang Yuan, Department of Cardiovascular Medicine, Fuwai Central China Cardiovascular Hospital, NO. 1 Fuwai Avenue, Zhengzhou, Henan, 450000, People’s Republic of China, Tel +86-13653821840, Email [email protected]
Background: Hypertrophic cardiomyopathy (HCM) is an extremely insidious and lethal disease caused by genetic variation. It has been studied for nearly 70 years since its discovery, but its cause of the disease remains a mystery. This study is aimed to explore the genetic pathogenesis of HCM in order to provide new insight for the diagnosis and treatment of HCM.
Methods: Patients with HCM at 4 hospitals from January 1, 2020, to December 31, 2021, were collected. Peripheral blood of these patients was collected for whole exome sequencing. Moreover, data on the HCM transcriptome were analyzed in the GEO database.
Results: Totally, 14 patients were enrolled, and 6 single-nucleotide variation (SNV) mutant genes represented by MUC12 were observed. Most of the gene mutations in HCM patients were synonymous and non-synonymous, and the types of base mutations were mainly C > T and G > A. Copy number variants (CNVs) predominantly occurred on chromosome 1 in HCM patients. Furthermore, we found that the only ATP2A2 gene was differentially expressed in 3 groups of transcriptome data in GEO database, and the presence of ATP2A2 mutation in 10 samples was observed in this study.
Conclusion: In summary, 7 mutated genes represented by MUC12 and ATP2A2 were found in this study, which may provide novel insights into the pathogenic mechanism of HCM.
Keywords: hypertrophic cardiomyopathy, whole exome sequencing, single-nucleotide variation, copy number variants, gene mutation
Introduction
Hypertrophic cardiomyopathy (HCM), characterized by asymmetric hypertrophy of the ventricular wall, is a condition where the heart becomes thickened without a distinct inducement.1,2 Epidemiological investigation shows that the estimated prevalence rate of HCM in the general population is 1:500.3,4 The clinical manifestations vary greatly, with no symptoms and mild symptoms, even dyspnea, heart failure and even sudden cardiac death.5,6 Unfortunately, early diagnosis of HCM is usually difficult, because many patients have almost no symptoms, and their condition is quite serious at the time of onset.2,5,6 So far, the pathogenesis of HCM is still an unsolved problem, and the danger of HCM has attracted widespread attention.
Hypertrophic cardiomyopathy is usually regarded as a monogenic disease with autosomal dominant inheritance, which is often characterized by familial aggregation, called familial HCM, and 20–30% is sporadic HCM.7 Previous studies reported that HCM was mainly caused by gene mutations encoding sarcomere proteins, including MYH7, MYBPC3, TNNI2, TNNI3, TPM1, ACTC1, MYL2 and MYL3, among which MYH7 and MYBPC3 were the most common.8 In recent years, some pathogenic mutations of non-sarcomere genes were also found.9 In addition, studies have shown that especially for sarcomere-negative HCM, the heritability of single nucleotide polymorphism (SNP) indicates a strong polygenic effect, and destyrosine tubulin can be used as a therapeutic target for sarcomere mutation-induced HCM.10 However, because the clinical phenotype of sporadic HCM is heterogeneous, and the family history and incomplete penetrance are more complicated, it is necessary to further study the genetic background of HCM, which is helpful to understand the pathogenesis of HCM more comprehensively.
With the development of high-throughput sequencing technology, whole exon sequencing (WES) and other technologies are gradually widely used in the field of genetics.11 WES uses the target sequence capture technology to capture all exon DNA of the genome and then high-throughput sequencing, which can not only quickly find the pathogenic genes of rare genetic diseases but also be used for common diseases caused by multiple genes, greatly shortening the detection period and making the diagnosis and treatment of diseases enter the era of precision.12,13 In addition, WES can accurately detect about 98% point mutations and most copy number variants, CNVs in the coverage area, which provides a new and relatively comprehensive detection method for finding pathogenic genes of diseases.
In this study, we applied WES to sporadic HCM patients, trying to find new pathogenic genes and mutations. At the same time, in order to investigate the mutant genes of HCM patients more generally, we further analyzed the transcriptome data in the public database in order to have a more comprehensive understanding of the pathogenesis of HCM.
Materials and Methods
Patients
Patients with HCM at the First Affiliated Hospital of Anhui Medical University, the First Affiliated Hospital of University of Science and Technology of China, the Second Affiliated Hospital of Hebei Medical University, and Henan Provincial People’s Hospital and Fuwai Central China Cardiovascular Hospital from January 1, 2020, to December 31, 2021, were, respectively, collected. The clinical indications and peripheral blood were collected from the patients with the informed and consent of the HCM patients. This study has been performed in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Yuhua Yunfang Integrated Traditional Chinese and Western Medicine Clinic.
Only one case per family was included in the present analysis. Clinic evaluations: Patients with HCM were diagnosed based on medical history, physical examination, electrocardiogram, and echocardiogram showing maximum left ventricular wall thickness (MLVWT) ≥13 mm in at least one myocardial segment, or MLVWT exceeding two SDs corrected for age, size and gender, in the absence of other diseases that could explain the hypertrophy.14
Whole Exome Sequencing
DNA was extracted with 10 mL peripheral blood using the DNeasy Blood & Tissue Kit (DP348) and stored at −20 °C until use. The DNA content and quality were assessed using Nanodrop and agarose gel electrophoresis, respectively. Subsequently, the genomic DNA was fragmented randomly and purified. The Roche SeqCap EZ MedExome Kit was employed for whole exome capture, followed by quality control enrichment. The captured fragments were then utilized for genomic library preparation using the AxyPrep Mag PCR Kit. The double-sided high flux PE150 sequencing was conducted using the IIIumina HiSeq X Ten sequencer, resulting in an average sequencing depth of 100X and over 95% of the region achieving coverage of at least 20X.
QC of Raw Sequencing Data
To ensure the accuracy of subsequent bioinformatics analysis, raw sequencing data was filtered to obtain high-quality sequencing data to ensure the smooth progress of subsequent analysis. Raw reads were quality controlled with FastQC and SeqPrep.15,16 Detailed methods were as follows: (1) adapter sequences in reads were removed; (2) Bases (quality values <20) were trimmed; (3) reads with an N ratio of more than 10% were removed; (4) Sequences (<20 bp in length) were discarded.
The Pipeline of WES Analysis
Briefly, the remaining good data was performed alignment by using BWA-MEM with default parameters.17 Repeated fragments (the main source for PCR amplification) that have a great impact on the discovery of genomic mutations were removed. Picard was used to remove marked repeats. Then, we used the BaseRecalibrator module and RealignerTargetCreator module in GATK for local indel-realignment and base recalibration.18 Significantly, Strelka software was selected to determine the single-nucleotide variations (SNVs) and indels.19
Rare Mutation Analysis
The pathogenicity of mutations was compared and analyzed in Clinvar database and ACMG Genetic Variation Classification Standard and Guide. Among them, “pathogenic”, “possibly pathogenic”, “unclear meaning” and mutations that do not exist in healthy people were identified as pathogenic mutations. The frequency of mutation in population was determined by searching the databases of DBSNP (http://www.ncbi.nlm.nih.gov/snp) and 1000 Genomes (http://browser.1000genomes.org).
To identify pathogenic variants associated with HCM, we prioritized rare SNV and Indel variants. Detailed processes were as follows: 1) mutation sites with mutation frequency ≥0.01 in 1000genome or ExAC database were screened out; 2) synonymous mutations and variant sites located in introns were re-screened out; 3) predicted benign/probably benign mutations in ClinVar, InterVar, SIFT and Polyphen databases were filtered. In order to reduce the false-positive rate of copy number variations (CNVs), fragments with length ≤300 bp were filtered out, and then screened by annotation information (CNV prediction quality score >40).
Variant Annotation Analysis
Variation sites were annotated using ANNOVAR and Tapes software, including gene structure annotation, genome feature annotation, non-synonymous mutation deleteriousness prediction, known variant database annotation, and variant-related gene function annotation.20,21 RefSeq and GENCODE were utilized to annotate the gene structure of variant sites (such as mRNA, non-coding RNA, small RNA, and microRNA) and genomic features of variant sites including CG islands, karyotypes, genome repeats, transcription factor binding sites, etc.
Differential Expression Analysis
To further study the molecular mechanisms of HCM, we searched the GEO database for transcriptomic data of HCM, including GSE89714, GSE13305422 and GSE160997.23 Limma (R package) was used to perform differential expression analysis of transcriptomic data between normal vs HCM patients. Threshold was false discovery rate (FDR) <0.05.24
Function Enrichment and Protein–Protein Interaction (PPI) Analysis
The clusterProfiler was used to perform function enrichment analysis, including gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) analysis.25 PPI analysis was performed by using STRING. Significantly enriched GO and KEGG terms were identified under the screening criteria of FDR < 0.05.
Statistical Analysis
The statistical significance of GO enrichment and pathway enrichment were analyzed by using the hypergeometric distribution and false discovery rate was calculated to correct the P‑value using Bonferroni correction, with P≤0.05 considered to indicate a statistically significant difference. For comparison of two groups using the NOI‑seq method, a gene was declared as a DEG with probability is ≥0.8 and fold change ≥2. Data analysis uses R version 3.0.1 with the addition of the ggplot2 package.
Results
SNV Characteristics of HCM
To investigate the pathology of HCM disease, we performed WES of 14 HCM patients. Clinical information for all HCM patients is provided in Supplementary Table S1. Sequencing data information is provided in Supplementary Table S2. Aiming to analyze rare mutations in HCM, we used rigorously selection pipeline. Finally, we obtained the SNVs and indels of HCM patients. The results showed that each patient had more than 1000 SNVs or indels, which involved mutations in more than 800 genes (Table 1). Moreover, annotation results of the variant regions indicated that most of the mutations were synonymous (45.5%) or non-synonymous (43.7%) (Figure 1A). Mutation annotation for each HCM patient also showed similar results (Figure 1B). Mutant base type analysis showed that the most significant base mutation was C > G and followed was G > A (Figure 1C). Notably, we obtained the top 30 genes involving in mutation of HCM patients. The results showed that most mutations were associated with MUC3A (Figure 1D).
 |
Table 1 Variant Gene and Locus Statistics |
 |
Figure 1 Characteristics of SNV in HCM patients. (A), Statistics of exon region variation type. Different colors represent different mutation types. (B), Mutation signatures of different mutation types in different samples. (C), Top 15 mutation base type statistics. (D), Top 30 variant genes. |
Identification of Shared Mutation Genes of HCM
To search for the causative gene of HCM, we searched for the shared mutated genes in these 14 patients and obtained 13 shared genes, including MUC3A, LILRB3, FCGBP, CGREF1, GOLGA6L7, NBPF10, CNTNAP3B, FRG2C, GPRIN2, MUC2, MUC12, PRAMEF18 and PRAMEF22. Moreover, after discarding the synonymous mutant genes in the shared mutant genes, a total of 6 non-synonymous mutant genes were obtained, including FCGBP, GOLGA6L7, GPRIN2, MUC2, MUC12 and MUC3A. GO and KEGG pathway enrichment analysis of these genes was performed. The GO analysis showed that these 6 non-synonymous mutant genes were associated with O-glycan processing, positive regulation of innate immune response, immune response-activating cell surface receptor signaling pathway, etc. (Figure 2A). KEGG pathway enrichment analysis showed that these 6 non-synonymous mutant genes were participated in amoebiasis and gastric cancer (Figure 2B). Interestingly, the results of PPI network indicated that these 6 non-synonymous mutant genes were able to form an interaction network and work together. Among them, MUC12 was a key gene in this interaction network (Figure 2C).
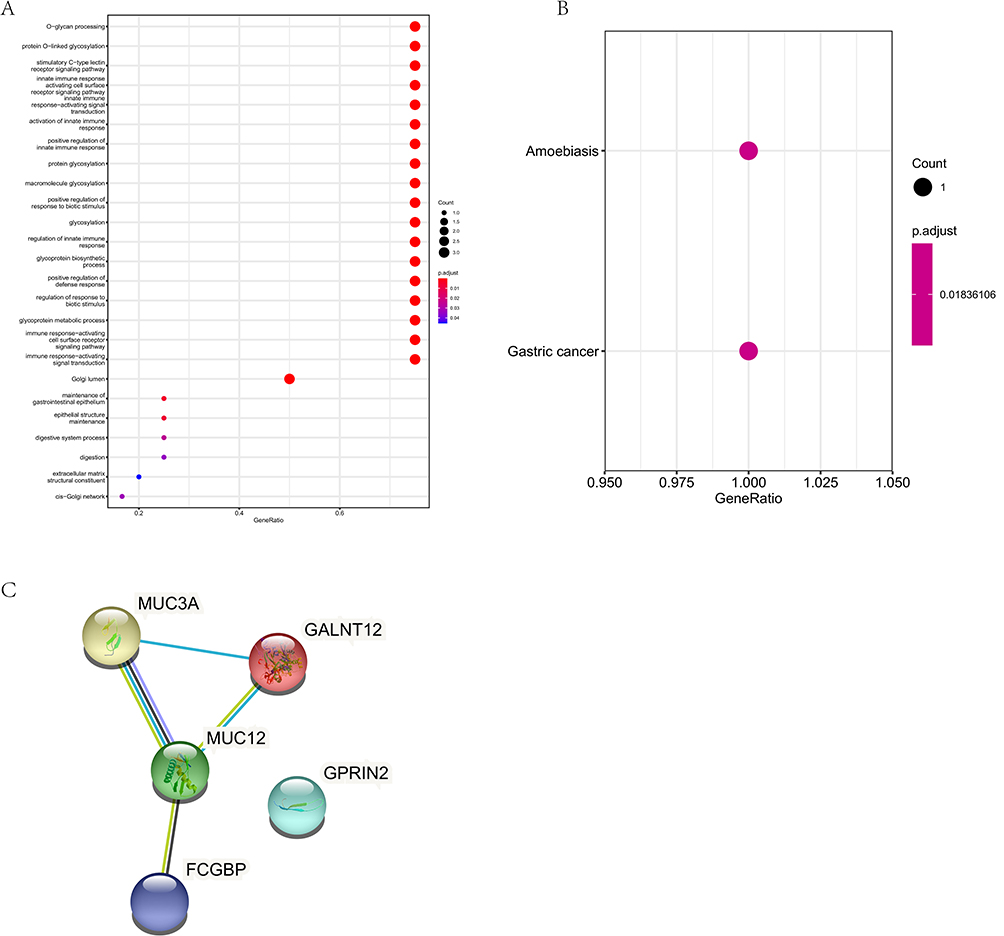 |
Figure 2 SNV-related variant gene functional enrichment analysis. (A), The GO analysis of SNV-related variant genes. Different color represent the FDR value. (B), The KEGG pathway analysis of SNV-related variant genes. Different color represent the FDR value. (C), The PPI analysis of SNV-related variant genes. |
Mutation Characteristics of Shared Mutation Genes of HCM
The results of the mutation status of the shared mutated genes in the patients showed that the number of mutations in all patients was more than 100 times, including up to 150 times in one patient (Table 2). Further, the analysis results of the mutation sites of the shared mutant genes showed that the GPRIN2 mutation was the most complex, with various non-synonymous mutations and non-frameshift insertion (Table 3).
 |
Table 2 The Number of Variants in Genes Shared by HCM Patients |
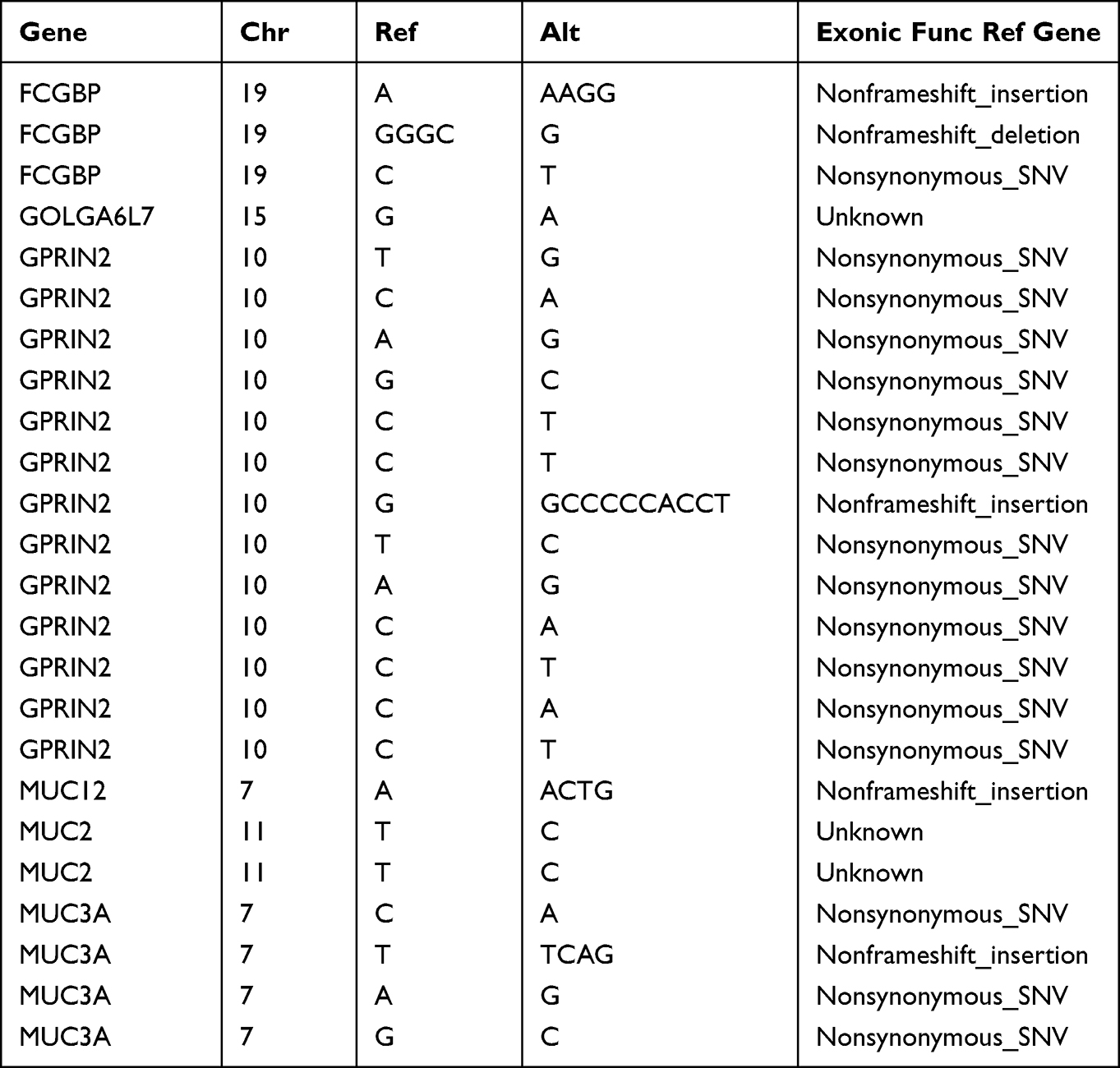 |
Table 3 The Common Variant Loci of Shared Genes in HCM Patients |
CNV Characteristics of HCM
To reduce the false positive rate of CNV in 14 patients, strict quality control conditions were used. CNV characteristics of each patient are shown in Table 4. More than 1000 CNVs could be detected in each patient, and the highest patient detected 3085 CNVs (Figure 3A). CNV statistics of each chromosome showed that chromosome 1 had the most CNVs and chromosome 20 had the least CNVs (Figure 3B). Furthermore, a comparative analysis of CNV in 14 HCM patients showed that a total of 5 CNV regions were obtained that could be detected in all patients (Table 5). Significantly, we obtained 11 CNV-shared genes that fall entirely within the CNV region.
 |
Table 4 Statistics of Variant CNV Regions in HCM Patients |
 |
Table 5 Gene Statistics in Shared CNV Regions |
 |
Figure 3 Characteristics of CNV variants in HCM patients. (A), CNV variants signature of each HCM patient. (B), Statistics of CNV variants characteristics for each chromosome. |
Function Enrichment of CNV-Shared Genes
The GO analysis showed that these 11 CNV-shared genes were associated with post-Golgi vesicle-mediated transport, protein localization to the plasma membrane, protein localization to the cell periphery, etc. (Figure 4A). KEGG pathway enrichment analysis showed that these 11 CNV-shared genes were participated in SNARE interactions in vesicular transport (Figure 4B). The results of PPI network showed that CNV-shared genes did not functionally interact, so far (Figure 4C).
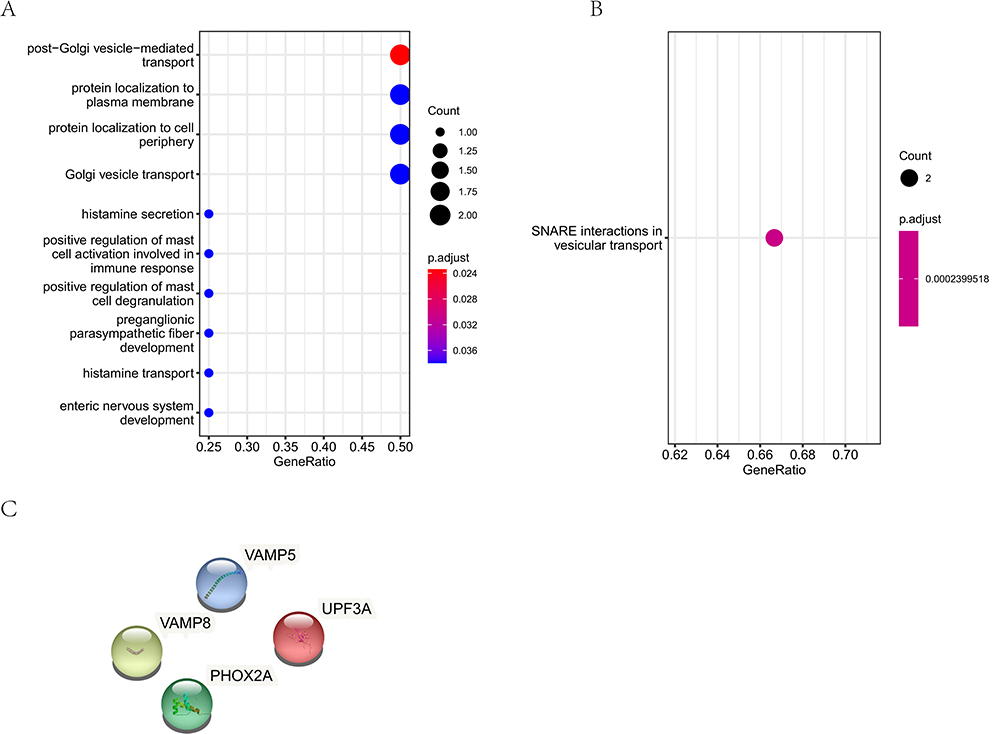 |
Figure 4 CNV-related variant gene functional enrichment analysis. (A), The GO analysis of CNV-related variant genes. (B), The KEGG pathway analysis of CNV-related variant genes. (C), The PPI analysis of CNV-related variant genes. |
Transcriptomic Analysis of HCM
To further dissect the pathogenesis of HCM, we obtained transcriptomic data of HCM patients from the GEO database. First, we analyzed the differential gene expression profiles of the HCM transcriptomic data separately. The results showed that compared with normal people, the transcriptional expression of HCM patients did not change drastically. For GSE89714, we obtained 413 differentially expressed genes (DEGs). For GSE133054, we gained 49 DEGs and 51 DEGs was got by GSE160997. Expression trends of transcriptomic data are shown in the heat map (Figure 5A). GO analysis of GSE89714 showed that the DEGs were involved in tricarboxylic acid cycle, oxidative phosphorylation, etc. (Figure 5B). KEGG pathway analysis of GSE8917 showed that the DEGs were involved in oxidative phosphorylation, propanoate metabolism, etc. (Figure 5C). GO analysis of GSE133054 showed that the DEGs were also associated with tricarboxylic acid cycle (Figure 5D). KEGG pathway analysis of GSE133054 showed that the DEGs were involved in citrate cycle (TCA cycle), 2-Oxocarboxylic acid metabolism, etc. (Figure 5E). For GSE133054, the GO analysis showed that the DEGs were related to cotranslational protein targeting to membrane, tricarboxylic acid cycle, etc. (Figure 5F). The KEGG pathway analysis showed that citrate cycle (TCA cycle) pathway was also enriched (Figure 5G). Significantly, we found 23 shared GO terms (Supplementary Table S3) and 11 common KEGG signaling pathways (Supplementary Table S4) in all HCM transcriptomic data. Moreover, we found that the only ATP2A2 gene was differentially expressed in 3 groups of transcriptome data in GEO database, and the presence of ATP2A2 mutation in 10 samples was observed in this study.
 |
Figure 5 Transcriptomic data analysis of HCM patients. (A), Heat map of DEGs in HCM patients. From left to right are GSE89714, GSE133054 and GSE160997. (B), The GO analysis of DEGs of GSE89714. (C), The KEGG pathway analysis of DEGs of GSE89714. (D), The GO analysis of DEGs of GSE133054. (E), The KEGG pathway analysis of DEGs of GSE133054. (F), The GO analysis of DEGs of GSE160997. (G), The KEGG pathway analysis of DEGs of GSE160997. |
Discussion
HCM is a condition in which the heart muscle is abnormally thickened or enlarged. The thickened heart muscle often makes it difficult for the heart to pump blood. It is often difficult to detect as patients have few obvious clinical symptoms prior to onset.3 In 1958, Donald Teare firstly described the feature of HCM.26 With nearly 70 years of research, although many features and causes of HCM have been discovered, the pathogenesis of HCM is still a cloud of mystery. Prior to the studies indicated here, the gene variant of HCM has not been well elaborated. By using WES and transcriptomic data, the transcription and mutation characteristics of HCM were highlighted.
WES is a widely used second-generation sequencing (NGS) method involving the sequencing of genome protein coding regions. Importantly, this method plays an important role in studying diseases caused by genetic variation.13,27 At present, HCM is considered as a disease that is the genetic heterogeneity disease caused by the mutation of myocardial protein coding genes (usually MYH7 and MYBPC3).15,28,29 At present, it has been reported that three members of the same HCM family have the mutation of MYH7 p.R671C. They found that although they carry the same mutant gene, different family members have different sex and clinical phenotype.28 In this study, we found that only 4 out of 14 patients had the mutation of MYH7, which is one of the most common mutation pathogenic genes of HCM, accounting for about 20–30% of the incidence of HCM, which is consistent with the results of this study. MYH7 gene contains 40 exons, and its encoded B-MHC constitutes the main structure of myosin, which is divided into three regions: head (S1 segment), neck (S1 segment) and tail (LMM).16 Rare mutations of ACTC1, MYL2, MYL3 were also detected in HCM patients,17 which often led to malignant outcomes including sudden cardiac death.18 Previous literature reported that TNNI2 and TNNI3 genes are also one of the main pathogenic genes of HCM, and carrying TNNT2 or TNNl3 gene mutation is a high-risk factor for sudden death.19,20 Previous studies have pointed out that the detection effect of WES may not be so ideal.21 In fact, there have been previous reports that mutations in TNNT2, TNNI3 and TPM1 lead to HCM,29 but the coverage of WES is apparently isolated for TNNI3 and PLN.21
Significantly, we found that 6 non-synonymous mutant genes in all HCM patients, including FCGBP, GOLGA6L7, GPRIN2, MUC2, MUC12 and MUC3A. Meanwhile, we found that the only ATP2A2 gene was differentially expressed in 3 groups of transcriptome data in GEO database, and the presence of ATP2A2 mutation in 10 samples was observed in the CNV mutation detection part of this study. Bioinformatics analysis found that MUC12 may be the gene most associated with HCM pathogenesis. MUC12, correlated with cell growth, is up-regulated in human cardiac myocytes.22 It is reported that MUC12 is associated with left ventricular noncompaction.23 CNTNAP3B may be involved in cardiac fibroblastogenesis.24 As far as the author knows, the relationship between other genes and HCM is almost rare. Transcriptome data analysis results suggest that ATP2A2 may be pathogenic to HCM. Previous reports have demonstrated that ATP2A2 and MYH7 gene promoters induce epigenetic switches in heart failure under stress overload.25 A previous study reported that in obstructive HCM, ATP2A2/ZTP2A1 played a key role.30 From completely asymptomatic to outflow tract obstruction, diastolic dysfunction, progressive heart failure, various tachyarrhythmias and sudden cardiac death, the abnormal individualization of clinical phenotype of HCM patients leads to difficulties in clinical decision-making and prognosis evaluation. In addition to significant genetic heterogeneity, the heterogeneity of clinical phenotype is also obvious among different HCMs, and the complex genotype-clinical phenotype relationship also brings challenges to the clinical diagnosis and genetic susceptibility assessment of HCM. This may also be one of the reasons for the inconsistent results obtained in this study.
Our study has some limitations. Firstly, the clinical sample size was small. Second, the prognosis of these clinical samples could not be obtained. Thirdly, further studies to confirm the role of these mutant genes in HCM through in vivo and in vitro experiments are needed.
Conclusions
In summary, 6 mutated genes represented by MUC12 in HCM patients and the only ATP2A2 expressed in all 3 groups of transcriptome data in GEO database were found in this study, which may provide novel insights into the pathogenic mechanism of HCM.
Data Sharing Statement
The datasets generated and/or analyzed during the current study are available in the BioProject repository, [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE234820].
Ethics Statement
This study was reviewed and approved by the Ethics Committee of Yuhua Yunfang Integrated Traditional Chinese and Western Medicine Clinic. All patients have signed the informed consent.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare no competing interests in this work.
References
1. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121:749–770. doi:10.1161/CIRCRESAHA.117.311059
2. McKenna WJ, Sen-Chowdhry S. From Teare to the present day: a fifty year odyssey in hypertrophic cardiomyopathy, a paradigm for the logic of the discovery process. Rev Esp Cardiol. 2008;61:1239–1244. doi:10.1016/S0300-8932(08)75730-0
3. Kuusisto J. Genetics of hypertrophic cardiomyopathy: what is the next step? Heart. 2020;106:1291–1292. doi:10.1136/heartjnl-2020-317043
4. Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–255. doi:10.1016/S0140-6736(12)60397-3
5. Ušaj M, Moretto L, Månsson A. Critical evaluation of current hypotheses for the pathogenesis of hypertrophic cardiomyopathy. Int J Mol Sci. 2022;23(4):2195. doi:10.3390/ijms23042195
6. Maron BJ, Rowin EJ, Maron MS. Global burden of hypertrophic cardiomyopathy. JACC Heart Fail. 2018;6:376–378. doi:10.1016/j.jchf.2018.03.004
7. Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines. Circulation. 2011;124:2761–2796. doi:10.1161/CIR.0b013e318223e230
8. Marsiglia JD, Pereira AC. Hypertrophic cardiomyopathy: how do mutations lead to disease? Arq Bras Cardiol. 2014;102:295–304. doi:10.5935/abc.20140022
9. Sedaghat-Hamedani F, Kayvanpour E, Tugrul OF, et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: a meta-analysis on 7675 individuals. Clin Res Cardiol. 2018;107:30–41. doi:10.1007/s00392-017-1155-5
10. Schuldt M, Pei J, Harakalova M, et al. Proteomic and functional studies reveal detyrosinated tubulin as treatment target in sarcomere mutation-induced hypertrophic cardiomyopathy. Circ Heart Fail. 2021;14:e007022. doi:10.1161/CIRCHEARTFAILURE.120.007022
11. Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet. 2014;59:5–15. doi:10.1038/jhg.2013.114
12. Fujimoto A, Totoki Y, Abe T, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–764. doi:10.1038/ng.2291
13. Ploski R, Pollak A, Müller S, et al. Does p.Q247X in TRIM63 cause human hypertrophic cardiomyopathy? Circ Res. 2014;114:e2–5. doi:10.1161/CIRCRESAHA.114.302662
14. Dubourg O, Isnard R, Hagège A, et al. Doppler echocardiography in familial hypertrophic cardiomyopathy: the French Cooperative Study. Echocardiography. 1995;12:235–241. doi:10.1111/j.1540-8175.1995.tb00544.x
15. Seeger T, Shrestha R, Lam CK, et al. A premature termination codon mutation in MYBPC3 causes hypertrophic cardiomyopathy via chronic activation of nonsense-mediated decay. Circulation. 2019;139:799–811. doi:10.1161/CIRCULATIONAHA.118.034624
16. Robert-Paganin J, Auguin D, Houdusse A. Hypertrophic cardiomyopathy disease results from disparate impairments of cardiac myosin function and auto-inhibition. Nat Commun. 2018;9:4019. doi:10.1038/s41467-018-06191-4
17. Kaski JP, Syrris P, Esteban MT, et al. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:436–441. doi:10.1161/CIRCGENETICS.108.821314
18. Bos JM, Will ML, Gersh BJ, Kruisselbrink TM, Ommen SR, Ackerman MJ. Characterization of a phenotype-based genetic test prediction score for unrelated patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2014;89:727–737. doi:10.1016/j.mayocp.2014.01.025
19. Mogensen J, Hey T, Lambrecht S. A systematic review of phenotypic features associated with cardiac troponin I mutations in hereditary cardiomyopathies. Can J Cardiol. 2015;31:1377–1385. doi:10.1016/j.cjca.2015.06.015
20. Mogensen J, Murphy RT, Kubo T, et al. Frequency and clinical expression of cardiac troponin I mutations in 748 consecutive families with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:2315–2325. doi:10.1016/j.jacc.2004.05.088
21. Mak TSH, Lee YK, Tang CS. Coverage and diagnostic yield of whole exome sequencing for the evaluation of cases with dilated and hypertrophic cardiomyopathy. Sci Rep. 2018;8:10846. doi:10.1038/s41598-018-29263-3
22. Ruan L, Yang Y, Huang Y, Ding L, Zhang C, Wu X. Functional prediction of miR-3144-5p in human cardiac myocytes based on transcriptome sequencing and bioinformatics. Medicine. 2017;96(32):e7539. doi:10.1097/MD.0000000000007539
23. Zhou Y, Qian Z, Yang J, et al. Whole exome sequencing identifies novel candidate mutations in a Chinese family with left ventricular noncompaction. Mol Med Rep. 2018;17:7325–7330. doi:10.3892/mmr.2018.8777
24. Masuda S, Matsuura K, Shimizu T. Inhibition of LYPD1 is critical for endothelial network formation in bioengineered tissue with human cardiac fibroblasts. Biomaterials. 2018;166:109–121. doi:10.1016/j.biomaterials.2018.03.002
25. Angrisano T, Schiattarella GG, Keller S, et al. Epigenetic switch at atp2a2 and myh7 gene promoters in pressure overload-induced heart failure. PLoS One. 2014;9:e106024. doi:10.1371/journal.pone.0106024
26. Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20:1–8. doi:10.1136/hrt.20.1.1
27. Belkadi A, Bolze A, Itan Y, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci USA. 2015;112:5473–5478. doi:10.1073/pnas.1418631112
28. Yu W, Huang MM, Zhang GH, Wang W, Chen CJ, Cheng JD. Whole-exome sequencing reveals MYH7 p.R671C mutation in three different phenotypes of familial hypertrophic cardiomyopathy. Exp Ther Med. 2021;22:1002. doi:10.3892/etm.2021.10434
29. Millat G, Bouvagnet P, Chevalier P, et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur J Med Genet. 2010;53:261–267. doi:10.1016/j.ejmg.2010.07.007
30. Lombardi M, Lazzeroni D, Pisano A, et al. Mitochondrial energetics and Ca2(+)-activated ATPase in obstructive hypertrophic cardiomyopathy. J Clin Med. 2020;9:1799. doi:10.3390/jcm9061799
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.