Back to Journals » Journal of Inflammation Research » Volume 15
Whole Exome Sequencing Revealed Variants That Predict Pulmonary Artery Involvement in Patients with Takayasu Arteritis
Authors Liu L ![]() , Chen J, Li J
, Chen J, Li J ![]() , Yang Y, Zeng X, Tian X
, Yang Y, Zeng X, Tian X
Received 13 June 2022
Accepted for publication 6 August 2022
Published 24 August 2022 Volume 2022:15 Pages 4817—4831
DOI https://doi.org/10.2147/JIR.S377402
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Lingyu Liu,1 Jing Chen,2 Jing Li,1 Yunjiao Yang,1 Xiaofeng Zeng,1 Xinping Tian1
1Department of Rheumatology and Clinical Immunology, Chinese Academy of Medical Sciences & Peking Union Medical College, National Clinical Research Center for Dermatologic and Immunologic Diseases (NCRC-DID), Ministry of Science & Technology, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital (PUMCH), Key Laboratory of Rheumatology and Clinical Immunology, Ministry of Education, Beijing, People’s Republic of China; 2Department of Rheumatology, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, People’s Republic of China
Correspondence: Xiaofeng Zeng; Xinping Tian, Department of Rheumatology and Clinical Immunology, Chinese Academy of Medical Sciences & Peking Union Medical College, National Clinical Research Center for Dermatologic and Immunologic Diseases (NCRC-DID), Ministry of Science & Technology, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital (PUMCH), Key Laboratory of Rheumatology and Clinical Immunology, Ministry of Education, Beijing, People’s Republic of China, Tel +86 10 69158795, Email [email protected]; [email protected]
Purpose: To conduct the first whole exome sequencing (WES) on Takayasu arteritis (TAK) to identify common and rare variants responsible for disease susceptibility.
Patients and Methods: A total of 200 patients and 1675 healthy controls from China were recruited for this study. Site-based association analysis for common variants and gene-based burden analysis for rare variants were conducted. A weighted genetic risk score (wGRS) was calculated for each patient with TAK based on the independent risk alleles identified in the association analyses. The ability of the patient wGRS to discriminate between different phenotypes was evaluated.
Results: In the site-based analysis, the top association signal was CCHCR1 (rs1265067, p = 8.27 × 10− 12, OR = 2.41), a proxy for HLA-B*52:01. HLA-DQB1 (rs9273902), HLA-DQB2 (rs34109750), and a haplotype block in the human leukocyte antigen (HLA) class III region (represented by rs3130618) also exhibited significant associations independently. In addition, four novel non-HLA susceptibility loci were identified: PRRT4, TLL2, LRP1B, and DLGAP2. Twelve independently associated single nucleotide polymorphisms were used to calculate the wGRS. TAK patients with a higher wGRS were found to have an increased risk of pulmonary artery involvement compared with those with a lower wGRS (p = 5.76 × 10− 7, OR = 13.92). The wGRS algorithm showed good predictive capability for pulmonary artery involvement in TAK (sensitivity, 92.1%; specificity, 59.9%). In the gene-based analysis, risk genes that reached exome-wide significance were not identified.
Conclusion: This WES study on TAK supports a previously reported association within the HLA region. Moreover, novel susceptibility loci were identified outside the HLA region. These risk alleles showed potential associations with pulmonary artery involvement in TAK. However, additional studies are warranted to verify our findings.
Keywords: Takayasu arteritis, whole exome sequencing, pulmonary artery, human leukocyte antigen
Introduction
Takayasu arteritis (TAK) is a rare chronic inflammatory disease that mainly affects the medium and large arteries, including the aorta and its major branches, as well as the coronary and pulmonary arteries. Vascular inflammation leads to wall thickening, stenosis, fibrosis, dilation, and progressive occlusion, resulting in severe ischemic complications.1,2 TAK is thought to be the result of an interplay between genetic and environmental factors.3 However, the etiology of TAK remains largely unknown. Epidemiological research has shown that TAK occurs more frequently in females than in males, with a female to male ratio of 5–12:1, and typically affects patients under the age of 40. Despite its worldwide distribution, TAK has been predominantly reported in East Asia, India, Turkey, and Mexico.4 Ethnic differences in prevalence suggest that there are diverse genetic bases underlying the predisposition to TAK in various populations.
In recent decades, genome-wide association studies (GWAS) have achieved substantial advances in identifying common genetic variants associated with TAK. The association with the human leukocyte antigen (HLA) region, especially the HLA-B*52 allele, remains the strongest genetic signal in TAK among various large-scale GWAS.5–9 Furthermore, GWAS have revealed the association of a few non-HLA susceptibility loci with TAK, including RPS9/LILRB3, LILRA3, FCGR2A/3A, IL6, and IL12B,5–7 several of which encode immune response regulators, mediators of humoral immunity, and proinflammatory cytokines, which may directly participate in the pathophysiological processes of TAK. However, most of the risk loci identified in GWAS reside in non-coding regions and do not affect the amino acid sequence. Furthermore, GWAS using single nucleotide polymorphism (SNP) arrays are designed to capture only common and low-frequency variations and are, thus, not sensitive enough to discover rare variants. The contribution of rare coding variants, which are more likely to influence protein structure and function, has not yet been fully explored. Whole exome sequencing (WES) can theoretically cover all SNPs in the coding regions of the genome, thus overcoming the limitations of GWAS. Hence, WES has become a powerful and effective approach for identifying coding variants, especially rare deleterious coding variants. To date, WES has succeeded in discovering pathogenic variations in complex diseases.10–12
In this study, we aimed to identify both common and rare variants responsible for TAK susceptibility in a cohort of Chinese patients with TAK using WES.
Methods
Patient Selection
A total of 200 patients with TAK aged 16–65 years were recruited from the Peking Union Medical College Hospital (PUMCH), a national referral center. All the patients were Han Chinese and had no first- or second-degree relatives with TAK. TAK was diagnosed according to the 1990 American College of Rheumatology classification criteria.13 Data on sex, age at onset, and vascular involvement were available for all patients with TAK. The control group comprised 1675 sex-matched healthy subjects who were randomly selected from the Novo-Zhonghua project (a high-depth sequencing project providing in-house, Chinese population-specific exome sequencing data). All controls were Han Chinese. This study was approved by the institutional review board of PUMCH (JS-2038). Written informed consent for DNA analysis was obtained from each participant and/or their guardian at the time of sample collection, and the study was conducted in accordance with the Declaration of Helsinki. A flow diagram illustrating the study design is shown in Figure 1.
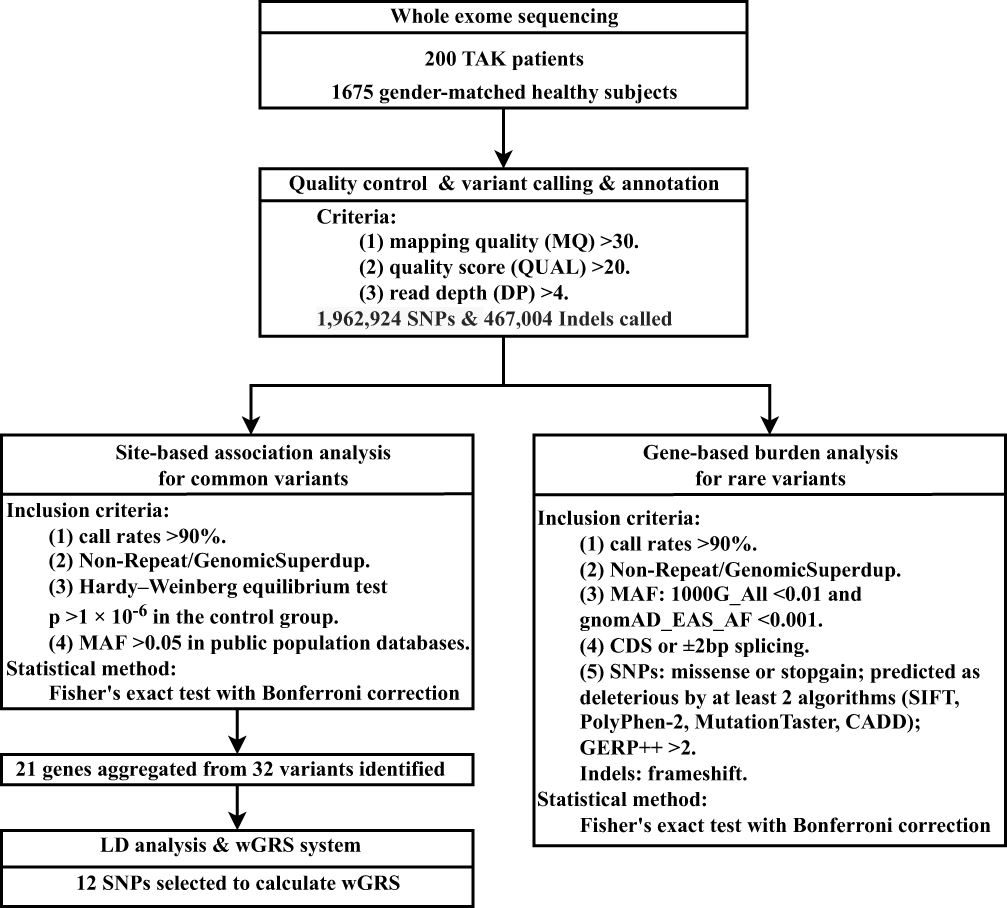 |
Figure 1 The basic workflow chart of the study. Abbreviations: TAK, Takayasu arteritis; SNP, single-nucleotide polymorphism; Indel, insertion/deletion; MAF, minor allele frequency; LD, linkage disequilibrium; wGRS, weighted Genetic Risk Score; 1000G, 1000 Genomes Project; gnomAD, Genome Aggregation Database; EAS, East Asian; AF, allele frequency; CDS, coding sequence. |
DNA Extraction
Genomic DNA was extracted from the peripheral blood of each subject using the Magnetic Universal Genomic DNA Kit (TIANGEN, Beijing, China) and sonicated into fragments of 180–280 bp for the subsequent construction of DNA libraries.
Whole Exome Sequencing and Data Processing
The Agilent SureSelect Human All ExonV6 Kit (Agilent Technologies, Santa Clara, CA, USA) was used for exome capture, following the manufacturer’s instructions. Paired-end DNA sequencing (2 × 150 bp) was performed on an Illumina Novaseq 6000 platform (Illumina Inc., San Diego, CA, USA) to provide a mean coverage of over 100×. After converting the sequencing-derived original image files into raw sequenced reads (raw data), the following quality control criteria were applied to obtain clean reads (clean data): (1) removing paired reads containing adapters (>10 nucleotides mapped to the adapter sequences, allowing ≤10% mismatches), (2) removing paired reads with an uncertain nucleotide (N) ratio of >10% in one of the paired reads, and (3) removing paired reads containing >50% low-quality (Phred quality <5) nucleotides in either read. Then, the clean data were aligned to the human reference genome (GRCh37/hg19) using the Burrows-Wheeler Aligner tool.14 The resultant BAM files were sorted and indexed using SAMtools.15 Reads mapped to multiple locations were marked as duplicates using the Sambamba tools.16 Subsequently, BAM files were subjected to variant calling to identify single nucleotide variants and small insertions/deletions (indels) for each subject using SAMtools and BCFtools.17 The raw variations were further filtered using the following inclusion criteria: (1) Root-Mean-Square mapping quality (MQ) >30, (2) variant quality score (QUAL) >20, and (3) read depth (DP) >4. Finally, all filtered variants of each subject were merged using VCFtools and functionally annotated using ANNOVAR.18 Minor allele frequency (MAF) was obtained from the 1000 Genomes Project (1000G)19 and Genome Aggregation Database (gnomAD). Variant deleteriousness was predicted using SIFT,20 Polyphen-2,21 MutationTaster,22 and CADD23 algorithms. To reduce the influence of population stratification, principal component analysis (PCA) of the sequenced common variants with a MAF of >0.05 was performed to identify the population genetic structure of the cases and controls.
Common SNP Association Analysis
Association analyses were performed to explore the effects of the genetic variants on TAK susceptibility. For common variants (MAF >0.05), site-based association analysis was performed, in which every variant was individually tested for association with TAK. Allele frequencies between the case and control groups were compared using the Fisher’s exact test. P-values and odds ratios (ORs) were calculated for each SNP. For quality control, only SNPs meeting the following conditions were included in the analysis: SNP and sample call rates >90%, SNPs not in the repeat region, SNPs that passed the Hardy-Weinberg equilibrium test in controls (p >1 × 10−6), and MAF >0.05. SNPs with unknown frequencies in public population databases were excluded. Accounting for multiple hypothesis testing, a P-value of <5 × 10−7 was considered statistically significant after Bonferroni correction. Enrichment analysis was conducted for the resultant candidate genes using the WebGestalt online server (WEB-based Gene Set Analysis Toolkit, http://www.webgestalt.org/). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were used for the analysis. False discovery rate (FDR)-corrected P-values of <0.05 (FDR <0.05) were considered significant.
Weighted Genetic Risk Score (wGRS) System
To further investigate the correlation between the genotypes and clinical phenotypes of patients with TAK, we applied the wGRS system described by De Jager et al24 to evaluate the cumulative effects of significant common SNPs. Considering that potential linkage disequilibrium (LD) might exist among these SNPs and that the wGRS might be dominated by SNPs in the LD block, an LD analysis for these disease-associated significant common SNPs was performed using the R package “LDheatmap.” Only one representative SNP from each LD block with the lowest P-value was selected to calculate the wGRS. The wGRS for each patient with TAK was calculated by summing the products of the weight of each SNP and the number of corresponding risk alleles (0, 1, or 2), according to the following formula:
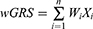
where i is the SNP, n is the number of SNPs included (12 in this study), Wi is the weight of the SNP, defined as the natural logarithm of the odds ratio of that allele, ln(OR), and Xi is the number of the corresponding effect alleles. The wGRS of different phenotypic subgroups were compared using the Mann–Whitney U-test in R. Patients with TAK were then divided into three groups based on the distribution of the wGRS, including group 1 (wGRS < bottom tertile), group 2 (bottom tertile ≤ wGRS < top tertile), and group 3 (wGRS ≥ top tertile). The presence or absence of the phenotype was compared between the groups using the Fisher’s exact test, with group 1 as the reference. To assess the discriminatory and predictive capability of the wGRS, receiver operating characteristic (ROC) curve analysis was performed. The 95% confidence intervals for each discrimination threshold and the areas under the ROC curve (AUC) were calculated. The optimal cut-off value was determined, and the corresponding sensitivity and specificity were measured.
Rare Variant Burden Analysis
Rare deleterious variants are more likely than common variants to affect protein function and have a larger effect on the disease. However, rare variants offer limited power in single-variant association tests. Therefore, to improve the statistical power, a gene-based burden analysis was conducted, which enabled the evaluation of the cumulative effects of rare variants within a gene. Variants were filtered to retain rare and likely damaging variants as follows: 1) SNP and sample call rates >90%, 2) single nucleotide variants that were limited to missense, nonsense, and splicing sites; indels that were limited to frameshift and splicing sites, 3) variants that were not located in the genomic repeat region, 4) variants that were predicted as “damaging” or “probably damaging” by at least two of the four prediction algorithms including SIFT, PolyPhen-2, MutationTaster, and CADD, 5) single nucleotide variants with conservation scores >2 assessed by GERP++, and 6) variants with a MAF of <1% in 1000G and 0.1% in gnomAD East Asian or variants with undetermined allele frequencies. For each gene, the number of alleles from rare deleterious variants was counted. The combined gene-level allele frequencies were compared between cases and controls using a two-tailed Fisher’s exact test. FDR was also calculated using the Benjamini-Hochberg and Benjamini-Yekutieli procedures. The significance level was defined as 4.83×10−6 (P-value of 0.05 divided by the 10,353 tested genes).
Statistical Analysis
Results are reported as percentages, median (range), and mean ± standard deviation, where appropriate. Genotype/allele frequencies and qualitative variables were compared using the Fisher’s exact test. The Mann–Whitney U-test was used to analyze non-normally distributed quantitative variables. All statistical tests were two-tailed and a P-value of <0.05 was considered statistically significant. Data analysis and plot generation were conducted using the R software (version 4.0.5).
Results
Study Population Description and WES
In this study, whole exome sequencing of 200 patients with TAK and 1675 sex-matched controls was performed to investigate variants associated with TAK in China. The demographic and clinical features of the TAK cohort are described in Table 1. Briefly, the affected individuals were mainly female (88.0%) with a median age at onset of 25 years (range, 11–62 years). The majority of the patients (91.5%) were no more than 40 years old at the time of presentation. Numano type V (50.5%, 101/200) was the most prevalent angiographic pattern observed in our cohort. In addition, we focused on special artery involvement (defined as wall thickening, stenosis, occlusion, or aneurysm), including pulmonary (19.0%, 38/200) and coronary (9.5%, 19/200) artery involvement. WES data were generated with a mean coverage of 125.88× (100.09–228.94×) on the target exome regions for each sample, and 99.2% of exomes had a sequencing depth >10×. On average, 159,002 SNPs and 22,011 indels were detected per sample. After applying variant quality control measures, 1,962,924 SNPs and 467,004 short indels were used for further analyses. These variants were annotated to genes using ANNOVAR.
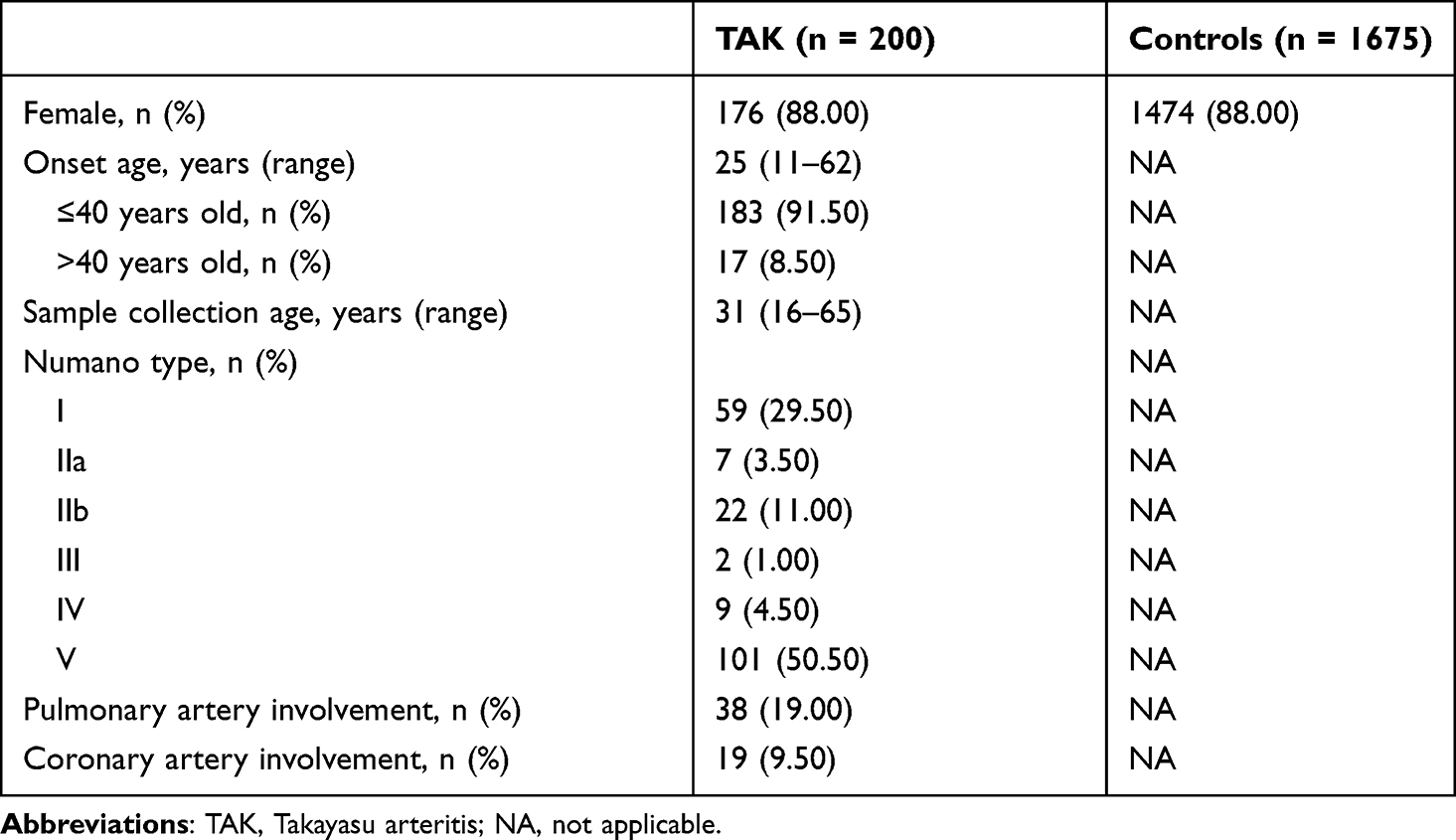 |
Table 1 Demographic and Clinical Characteristics of TAK Patients and Healthy Controls |
Risk Loci Associated with TAK in GWAS
For the site-based association test, all variants that passed the quality control measures and had a MAF of at least 0.05 were individually tested for associations with TAK. In total, 103,516 SNPs were included in the final analysis. First, PCA was performed to detect population stratification. The results confirmed the homogenous population structure between all cases and controls, as both groups were clustered together and were close to the subjects of Chinese ancestry in 1000G (Figure 2). Then, a single-variant association test was conducted using the Fisher’s exact test. The -log10 P-value of each variant was plotted against its chromosomal position in a Manhattan plot (Figure 3).
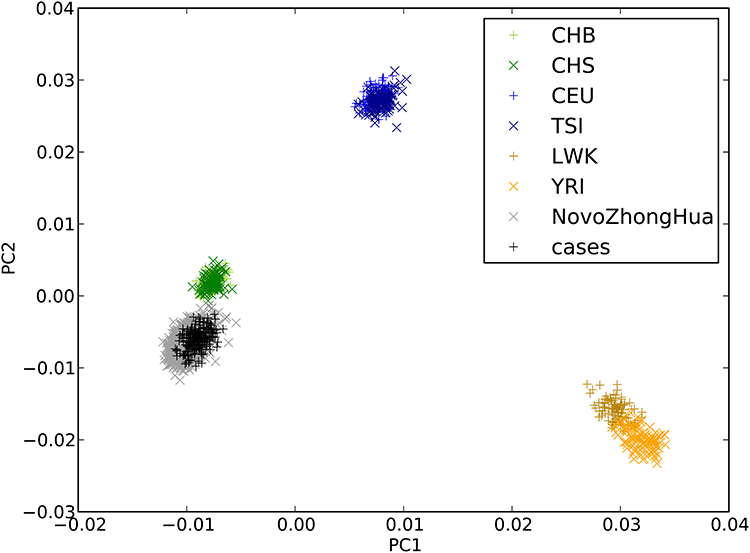 |
Figure 2 Principal component analysis (PCA) for ancestry of TAK patients and controls. PCA was performed to evaluate population stratification by projecting exome-sequencing data of samples onto 1000 Genomes samples. TAK patients were shown as “+” in black, and controls were shown as “×” in black. Cases and controls were clustered together and close to Han Chinese (in green), indicating that there was no excess population stratification. Abbreviations: CHB, Han Chinese in Beijing, China; CHS, Southern Han Chinese; CEU, Utah residents with Northern and Western European ancestry; TSI, Toscani in Italia; LWK= Luhya in Webuye, Kenya; YRI= Yoruba in Ibadan, Nigeria. |
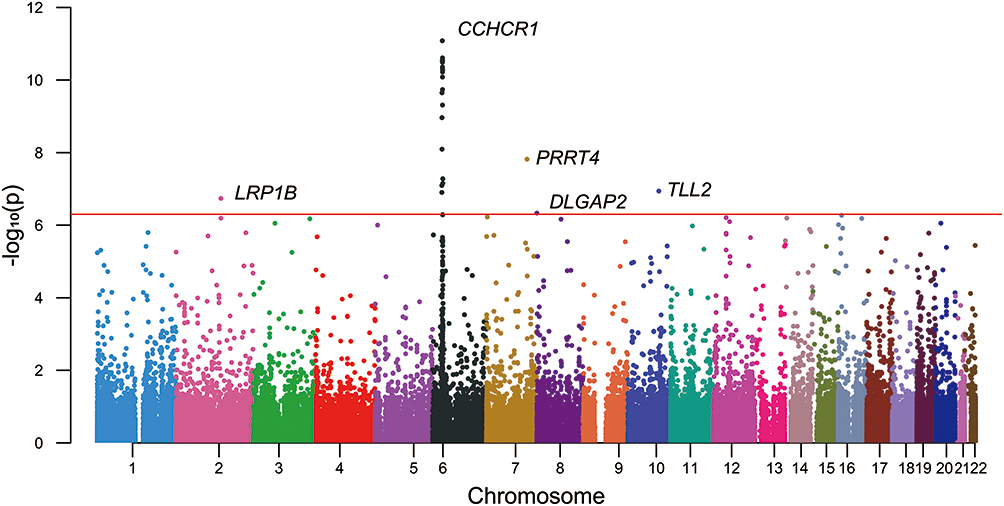 |
Figure 3 Manhattan plot from the single-variant association test for common SNPs. The X-axis represents chromosome position and the Y-axis shows the -log10 of p-values. The horizontal red line indicates the threshold value for significance. Twenty-eight SNPs located in the Human leukocyte antigen (HLA) locus on chromosome 6 indicated a strong association with TAK, with an SNP in CCHCR1 expressing the strongest signal. Outside the HLA region, SNPs in LRP1B, PRRT4, DLGAP2, and TLL2 also achieved genome-wide significance. |
A total of 32 variants approached genome-wide significance (p <5 × 10−7) with an OR of >1, as summarized in Table 2. As expected, a peak association signal within the HLA loci was observed among patients with TAK. Specifically, among the 28 significantly associated SNPs in the HLA region, SNP rs1265067 in CCHCR1 showed the strongest association with a P-value of 8.27 × 10−12, whereas rs3130627 in PRRC2A showed the highest OR of 2.61. Interestingly, four SNPs (rs1265067, rs1576, rs2517985, and rs1265078) in CCHCR1 formed a haplotype block (see below), and the previously reported TAK-associated SNP rs9263739 was also located in this haplotype block.6 Moreover, rs9263739 was considered as a proxy for HLA-B*52:01 because of its high LD.6 In addition, a significant independent association with HLA-DQB1 (rs9273902), which is also a previously reported candidate gene,5 was observed. Overall, our results were partly consistent with those of previous GWAS, which confirmed the genetic background of our TAK cohort.
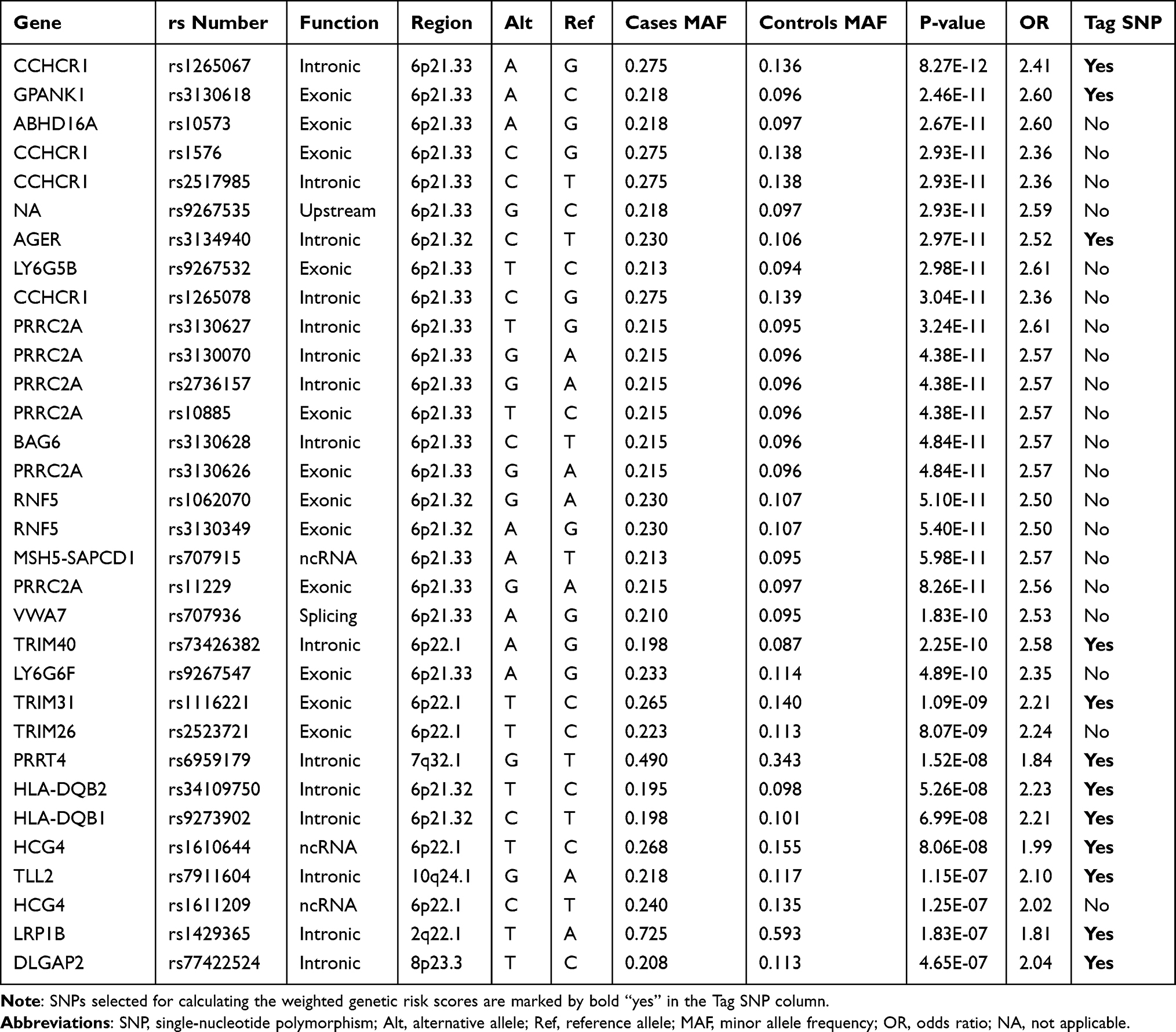 |
Table 2 Common SNPs That Were Significantly Associated with Takayasu Arteritis from the Site-Based Association Analysis Results |
Notably, half of the identified risk-associated SNPs on chromosome 6 (14 out of 28) were located in the HLA class III region (6p21.33) and were in high LD (see below). Outside the HLA region, we also identified four novel SNPs with P values achieving a genome-wide significance threshold, including introns in PRRT4 (p = 1.52 × 10−8; OR = 1.84), TLL2 (p = 1.15 × 10−7; OR = 2.10), LRP1B (p = 1.83 × 10−7; OR = 1.81), and DLGAP2 (p = 4.65 × 10−7; OR = 2.04).
Next, to investigate the related biological pathways, functional enrichment analyses of 21 genes aggregated from the 32 identified significant variants were performed. Four genes (HLA-DQB1, HLA-DQB2, TRIM26, and TRIM31) were found to be involved in the interferon-gamma (IFN-γ)-mediated signaling pathway (GO:0060333, FDR <0.05), which played a critical role in TAK pathogenesis. The other annotated GO terms did not reach the FDR-corrected significance level. No significant variants were enriched in the KEGG pathway analysis.
The wGRS Predicted Pulmonary Artery Involvement in TAK
The prevalent hypothesis to explain the contribution of common genetic variants to complex diseases is that the risk comes from the cumulative burden of the alleles identified in GWAS. Therefore, the wGRS system proposed by De Jager et al24 was used to explore the overall effects of these identified candidate loci from the single-variant analysis on clinical phenotypes. Data on onset age, Numano type, and the presence of coronary and pulmonary artery involvement were used as grouping variables. Before calculating the wGRS for each patient, an LD analysis was carried out to exclude SNPs in high LD on chromosome 6. As shown in Figure 4, the SNPs were highly linked, and there were five haplotype blocks. Only the SNPs with the lowest P-value in each block were retained for analysis. Finally, 12 representative tag SNPs were selected to calculate the wGRS (see Table 2 Tag SNP column and method section). Then, the wGRS among different phenotype subgroups were compared, and the results showed that there was a significant difference between the wGRS of patients with and without pulmonary artery involvement (p = 1.28×10−8). No statistical difference was found in the wGRS among patients with different age at onset, Numano type, and coronary artery involvement (Table 3). Furthermore, the ability of the wGRS to predict the occurrence of pulmonary artery involvement in TAK was investigated. Patients were categorized into the following three subgroups according to the tertile points of the wGRS: group 1 (wGRS <2.87), group 2 (2.87≤wGRS<6.75), and group 3 (wGRS ≥6.75). As shown in Table 4, patients in group 3 had a significantly higher risk of pulmonary artery involvement than those in group 1 (p = 5.76 × 10−7, 95% CI = 3.90–76.30). The OR was approximately 13.92, which suggested that the risk of pulmonary artery involvement was estimated to increase nearly 14-fold for patients in group 3 compared with that of patients in group 1. The sensitivity and specificity of the wGRS in predicting the occurrence of pulmonary artery involvement in group 3 versus group 1 were 90.0% and 61.2%, respectively. To further assess the performance of the wGRS in discriminating patients with TAK who had pulmonary artery involvement from those who had not, an ROC curve was constructed (Figure 5). The results revealed the good discriminatory capability of the wGRS, with an AUC of 0.797. The optimal cut-off value of the wGRS was 4.9, and the corresponding sensitivity and specificity were 92.1% and 59.9%, respectively.
 |
Table 3 Weighted Genetic Risk Score (wGRS) Between Different Phenotype Subgroups |
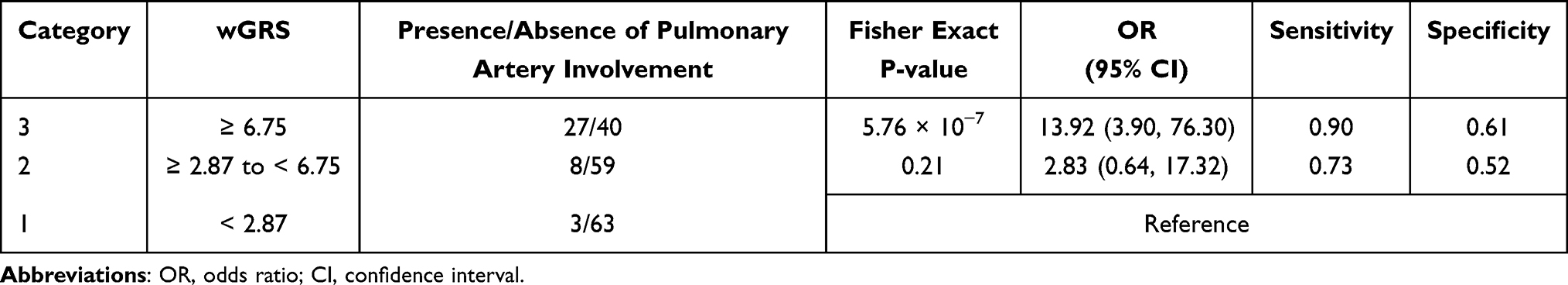 |
Table 4 Weighted Genetic Risk Score (wGRS) Analysis Among 3 Patient Subgroups |
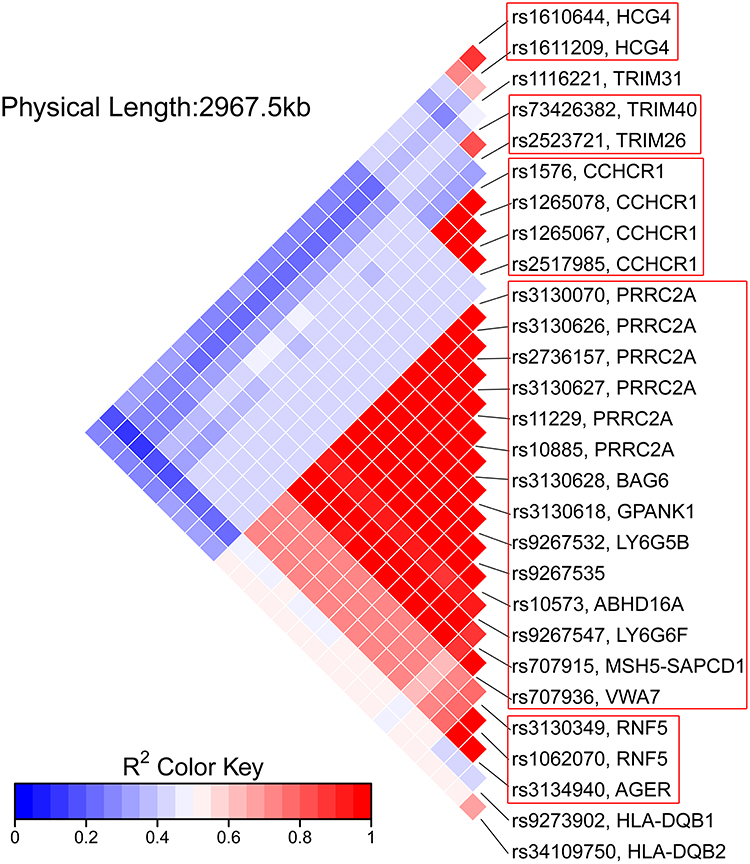 |
Figure 4 Linkage disequilibrium (LD) map among significant SNPs on chromosome 6. The color within each diamond represents the pairwise correlation (R2) between SNPs defined by the upper right and the lower right sides of the diamond. A darker red color represents a stronger LD. Five haplotype blocks are observed (outlined in red boxes). Abbreviation: SNP, single nucleotide polymorphism. |
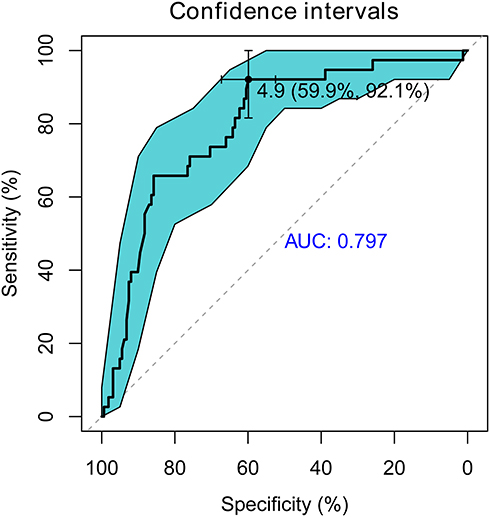 |
Figure 5 Receiver operating characteristic (ROC) curve analysis of weighted genetic risk score (wGRS). The y-axis represents sensitivity and the x-axis represents specificity. DeLong method was used to calculate the confidence intervals of ROC. The best predictive value of wGRS (4.9) and the corresponding specificity and sensitivity (59.9% and 92.1%, respectively) were shown. |
Gene-Based Burden Analysis
One of the key advantages of WES is its ability to discover the rare disease-causing variants in rare diseases. For this purpose, common variants were filtered out using a strict frequency filter threshold (MAF <1% in 1000G and <0.1% in gnomAD East Asians, or with uncertain allele frequencies) because such low frequencies could better capture rare coding variants with large OR. Among the 45,920 variants (located in 10,353 genes), 15 genes nominally reached P-values below 0.001. However, none of these genes approached the Bonferroni-corrected P-value threshold (P <4.83 × 10−6) or FDR corrected q-value threshold (q <0.05). The quantile-quantile (Q-Q) plot for the gene-based burden tests, which reveals the similarity between the observed and expected significance values, is displayed in Figure 6. Furthermore, we sought to identify rare, deleterious variants that were present in patients with TAK and absent in controls. A total of 14 such genes were found (data not shown). However, the maximum number of risk alleles of these genes in patients with TAK was only two, and no homozygous variant was found in patients with TAK. Therefore, we did not further focus on them. A list of the 15 genes that yielded the strongest associations in the gene-based burden analyses between patients with TAK and controls is shown in Table 5. Of these, EXOC4 encodes a component of the exocyst complex, which plays a role in mediating the migration of primary human microvascular endothelial cells in response to vascular endothelial growth factor-A.25 KCNB1 encodes a member of the voltage-gated potassium channel found in artery smooth muscle cells, and its level of expression was found to influence vasoconstriction.26 Endothelial loss of thioredoxin reductase 2, a key enzyme in mitochondrial redox control encoded by TXNRD2, leads to a proinflammatory vascular phenotype in mice.27 A deficiency of 5’-ectonucleotidase encoded by NT5E has been reported to result in arterial calcification.28 These genes appear to be potentially associated with vascular diseases.
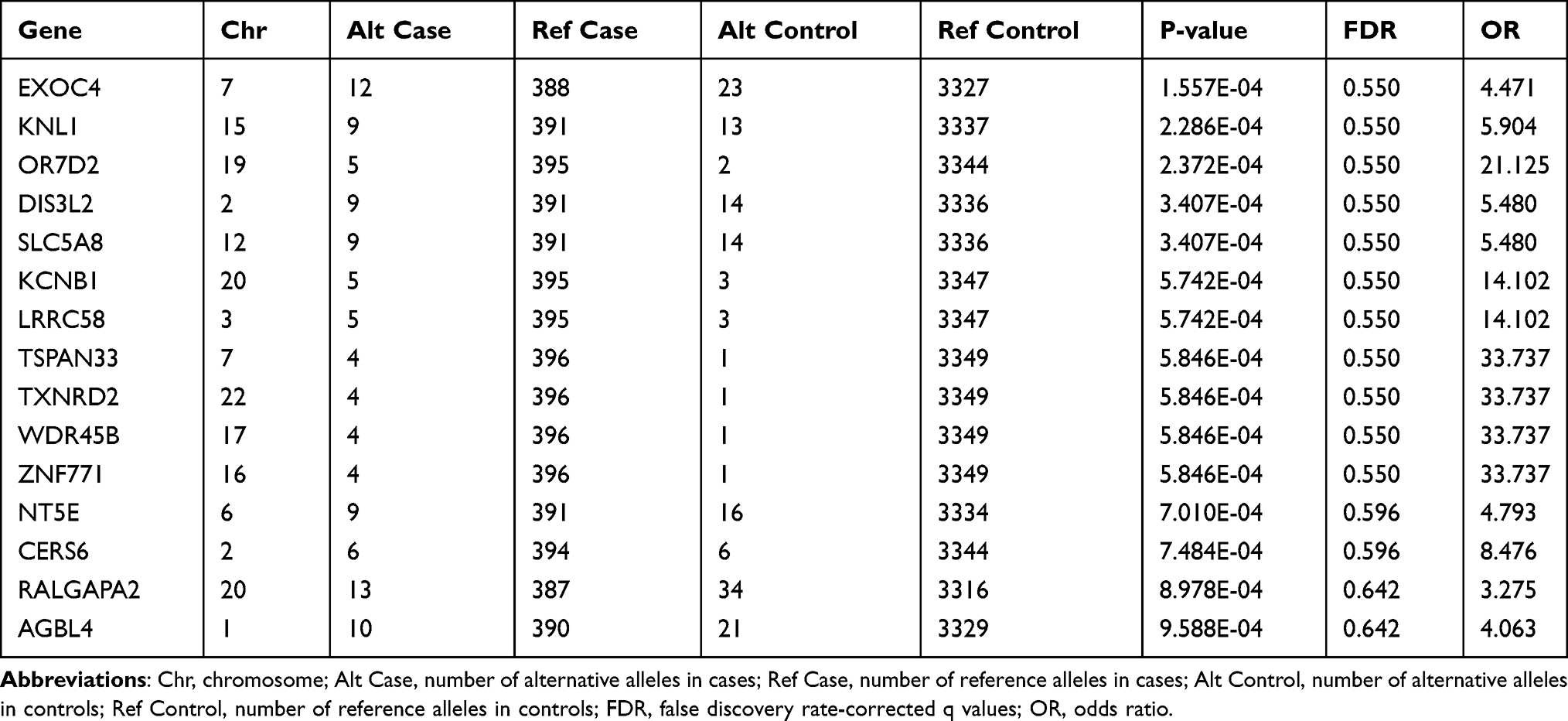 |
Table 5 The Top 15 Associated Genes from Gene-Based Burden Analysis |
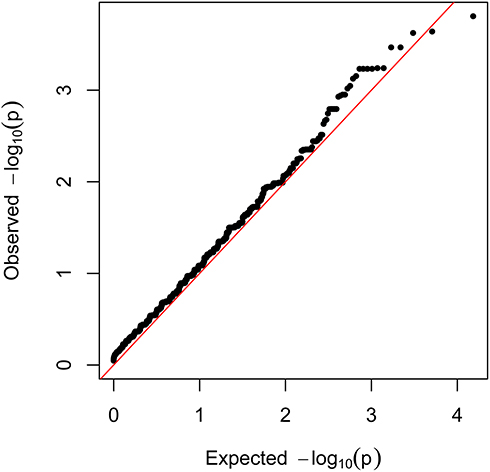 |
Figure 6 Quantile-quantile plot from gene-based burden analysis. The observed -log10 P values were plotted. The red line represents the expected line under the null distribution. |
Discussion
In the present study, WES and case-control association analyses of common and rare variants were carried out to investigate the causal variants associated with TAK in China. To the best of our knowledge, this is the first WES study on TAK. In line with the findings from multiple previous GWAS of TAK, the strongest association signals resided in the HLA region.5–9 Specifically, the top association in the HLA loci was rs1265067 in CCHCR1, which lies in a haploblock in high LD with HLA-B*52:01. The genetic link between HLA-B*52 and TAK has been repeatedly verified in several ethnicities.3 Therefore, we speculated that the most significant association signal of CCHCR1 in our study was also attributed to HLA-B*52:01. In addition, two independent variants of the HLA class II region (rs9273902 in HLA-DQB1 and rs34109750 in HLA-DQB2) showed significant associations. In a GWAS of Turkish and North American populations, a similar association was observed in the HLA-DQB1/HLA-DRB1 locus, and its independent genetic susceptibility to TAK was confirmed.5 Therefore, our results confirm a common genetic background among different ancestries.
Furthermore, several independent signals in the HLA region were identified. Of these, AGER (rs3134940) encodes a receptor for advanced glycosylation end products (RAGE),29 which participates in the RAGE/Toll-like receptor 4 (TLR4)/STAT1 signaling pathway.30 Interestingly, the activation of TLR4 has been shown to cause transmural panarteritis.31 However, Kabeerdoss et al32 found that RAGE mRNA expression in peripheral blood mononuclear cells (PBMC) was reduced in patients with TAK compared to that in healthy controls. Considering that TLR4 is highly expressed in aortic tissues of patients with TAK,33 the expression of RAGE in PBMC may not necessarily reflect the real conditions in the tissues. In addition, RAGE expression was upregulated in human pulmonary arterial smooth muscle cells under hypoxic conditions, thus inducing the development of pulmonary artery hypertension (PAH).34 Moreover, RAGE can activate endotheliocytes in the arterial wall and cause endothelial dysfunction.35 Taken together, AGER has been implicated in vascular inflammatory processes. However, given that rs3134940 was in an intron, its exact impact on the gene needs further validation.
Notably, half of the significant SNPs identified on chromosome 6 reside within the HLA class III region and are closer to the HLA class I region, where many immune function-related genes are located, including MHC I chain-related protein A (MICA) and B (MICB). Remarkably, these significant SNPs in the HLA class III region showed independent association from the HLA class I region represented by variants in CCHCR1. Similarly, a GWAS confirmed the independent susceptibility of the HLA-B/MICA locus tagged with rs12524487 from the classical HLA allele HLA-B*5201/HLA-Cw*1202.5 MICA encodes a natural ligand for the immune receptor NKG2D, an activating molecule for natural killer cells, CD8+ T cells, and gamma delta T cells. Abundant infiltration of NKG2D-expressing immune cells, as well as enhanced expression of MICA, was observed in the aortic tissues of patients with TAK, suggesting their possible roles in disease pathogenesis.36
Significant GO enrichment was found in the IFN-γ-mediated signaling pathway, which is the central link in the pathogenesis of TAK. Upon antigen presentation by dendritic cells, CD4+ Th1 cells secrete IFN-γ and promote granuloma formation.37 Four genes (HLA-DQB1, HLA-DQB2, TRIM26, and TRIM31) were enriched in the IFN-γ-mediated signaling pathway. IFN-γ is known to stimulate the expression of MHC class II molecules and the TRIM protein family,38 which are jointly involved in the IFN-γ response.
Outside the HLA region, the identified novel susceptibility loci of PRRT4, TLL2, and DLGAP2 encode proteins that have not been previously linked to any related diseases. However, LRP1B encodes a member of the low-density lipoprotein (LDL) receptor family, which has been extensively studied in atherosclerosis.39 Although several members of the LDL receptor family have been discovered to play crucial roles in PAH,40 LRP1B has not yet been directly linked with PAH. However, the role of LRP1B has been studied in vascular smooth muscle cells, and the results showed that LRP1B interacts with platelet-derived growth factor receptor-β, leading to a decrease in ERK1/2 signaling and inhibits cell migration.40 In addition, the LRP1B genetic polymorphism is correlated with the occurrence of coronary artery aneurysms in Kawasaki disease, which is an acute and systemic vasculitis.41 These findings suggest the potential involvement of LRP1B in vascular inflammation; however, its functional effects remain to be clarified.
An interesting finding of our study was that these identified association signals across the single-variant association tests could predict the involvement of the pulmonary artery in TAK. The clinical value of a single disease-predisposing variant is less informative, resulting in difficulties in converting genetic data into clinical applications. Therefore, wGRS analysis was employed as it aggregates the ORs from multiple genetic risk variants, thus enabling the evaluation of their cumulative effects and improving the predictive accuracy of clinical phenotypes.24
Cardiovascular events are major complications and causes of mortality in patients with TAK.42 A susceptibility locus in IL12B was significantly associated with the incidence of aortic regurgitation and hypertension in TAK.43 Kitamura et al44 reported that HLA-B52 positive patients with TAK often develop aortic regurgitation, pulmonary infarction, and ischemic heart disease. These findings suggest that these clinical phenotypes can be partly explained by the genetic background. The present study found that the wGRS was significantly higher in patients with pulmonary artery involvement than in those without. Specifically, patients with a higher genetic burden (wGRS in the upper tertile) had a nearly 14-fold increased risk of pulmonary artery involvement compared to those with a lower genetic burden (wGRS in the lower tertile), which calls for more aggressive monitoring in such at-risk patients. Moreover, the ROC analysis confirmed the good discriminatory capability of this predictive model, which has important clinical implications. As TAK patients with PAH have severely compromised hemodynamics and poor prognosis,45 early recognition may improve outcomes. Overall, these results provide evidence for the synergistic effects of susceptibility variants on the TAK phenotype.
WES has the unique advantage of detecting deleterious rare variants in coding regions in order to explain the missing heritability that common variants fail to explain. We adopted several approaches to improve the statistical power including enrolling a homogeneous population with the same race and sex ratio and without population stratification, using a relatively large sample size, applying stringent filtering criteria to better capture rare damaging variants with large OR, and performing a gene-based burden analysis to enrich the mutations. Nevertheless, we failed to identify rare deleterious variants that reached the significance threshold. This can be attributed to the following reasons. First, the sample size was still not large enough to provide sufficient statistical power for identifying novel candidate genes among unrelated patients with TAK. Larger sample sizes are required for future studies. Secondly, there was a lack of samples with extreme illness phenotypes. Patient selection was only based on the fulfillment of the diagnostic criteria. Third, there was a lack of samples from pedigrees. However, to date, familial cases of TAK have only been sporadically reported46 and we have not observed familial TAK cases in the Chinese population.
This study had some limitations. First, the novel susceptible loci identified in site-based association tests have not been validated in functional experiments. Further studies are necessary to elucidate their specific impacts on the disease. Another drawback is that the performance of the wGRS algorithm was not replicated in an independent TAK cohort. However, the low prevalence of TAK makes it difficult to collect sufficient additional DNA samples for validation. Multicenter studies are needed to confirm our findings.
Conclusion
This first WES study of TAK reinforced the association of previously reported loci in the HLA region as robust susceptibility loci shared across ethnicities. In addition, four common SNPs outside the HLA region were discovered as novel susceptibility loci, but no TAK-associated rare variants were identified. Furthermore, a genetic risk scoring system based on common risk variants could predict the occurrence of pulmonary artery involvement in patients with TAK.
Abbreviations
1000G, 1000 Genomes Project; AUC, areas under the ROC curve; FDR, false discovery rate; gnomAD, Genome Aggregation Database; GO, Gene Ontology; GWAS, genome-wide association studies; HLA, human leukocyte antigen; IFN-γ, interferon-gamma; Indels, insertions/deletions; KEGG, Kyoto Encyclopedia of Genes and Genomes; LD, linkage disequilibrium; LDL, low-density lipoprotein; MAF, minor allele frequency; MICA, MHC I chain-related protein A; OR, odds ratio; PAH, pulmonary artery hypertension; PCA, principal component analysis; RAGE, receptor for the advanced glycosylation end product; ROC, receiver operating characteristic; SNP, single-nucleotide polymorphism; TAK, Takayasu arteritis; WES, whole exome sequencing; wGRS, weighted genetic risk score.
Data Sharing Statement
The raw data supporting the findings of this study are available from the corresponding author on reasonable request.
Acknowledgments
We thank the bioinformatics teams for their technical support and assistance in the analysis of sequencing data.
Funding
This study was supported by the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-005).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kobayashi Y, Numano F. 3. Takayasu arteritis. Intern Med. 2002;41(1):44–46. doi:10.2169/internalmedicine.41.44
2. Kerr GS, Hallahan CW, Giordano J, et al. Takayasu arteritis. Ann Intern Med. 1994;120(11):919–929. doi:10.7326/0003-4819-120-11-199406010-00004
3. Renauer P, Sawalha AH. The genetics of Takayasu arteritis. Presse Med. 2017;46(7–8 Pt 2):e179–e187. doi:10.1016/j.lpm.2016.11.031
4. Montufar-Robles I, Soto ME, Jimenez-Morales S, et al. Polymorphisms in TNFAIP3, but not in STAT4, BANK1, BLK, and TNFSF4, are associated with susceptibility to Takayasu arteritis. Cell Immunol. 2021;365:104375. doi:10.1016/j.cellimm.2021.104375
5. Saruhan-Direskeneli G, Hughes T, Aksu K, et al. Identification of multiple genetic susceptibility loci in Takayasu arteritis. Am J Hum Genet. 2013;93(2):298–305. doi:10.1016/j.ajhg.2013.05.026
6. Terao C, Yoshifuji H, Kimura A, et al. Two susceptibility loci to Takayasu arteritis reveal a synergistic role of the IL12B and HLA-B regions in a Japanese population. Am J Hum Genet. 2013;93(2):289–297. doi:10.1016/j.ajhg.2013.05.024
7. Renauer PA, Saruhan-Direskeneli G, Coit P, et al. Identification of susceptibility loci in IL6, RPS9/LILRB3, and an intergenic locus on chromosome 21q22 in Takayasu arteritis in a Genome-Wide Association Study. Arthritis Rheumatol. 2015;67(5):1361–1368. doi:10.1002/art.39035
8. Terao C, Yoshifuji H, Matsumura T, et al. Genetic determinants and an epistasis of LILRA3 and HLA-B*52 in Takayasu arteritis. Proc Natl Acad Sci U S A. 2018;115(51):13045–13050. doi:10.1073/pnas.1808850115
9. Ortiz-Fernandez L, Saruhan-Direskeneli G, Alibaz-Oner F, et al. Identification of susceptibility loci for Takayasu arteritis through a large multi-ancestral genome-wide association study. Am J Hum Genet. 2021;108(1):84–99. doi:10.1016/j.ajhg.2020.11.014
10. Do R, Stitziel NO, Won HH, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518(7537):102–106. doi:10.1038/nature13917
11. Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371(1):22–31.
12. Tang H, Jin X, Li Y, et al. A large-scale screen for coding variants predisposing to psoriasis. Nat Genet. 2014;46(1):45–50. doi:10.1038/ng.2827
13. Arend WP, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990;33(8):1129–1134. doi:10.1002/art.1780330811
14. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
15. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi:10.1093/bioinformatics/btp352
16. Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31(12):2032–2034. doi:10.1093/bioinformatics/btv098
17. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–2993. doi:10.1093/bioinformatics/btr509
18. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi:10.1093/nar/gkq603
19. Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi:10.1038/nature15393
20. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1081. doi:10.1038/nprot.2009.86
21. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi:10.1038/nmeth0410-248
22. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–576. doi:10.1038/nmeth0810-575
23. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi:10.1038/ng.2892
24. De Jager PL, Chibnik LB, Cui J, et al. Integration of genetic risk factors into a clinical algorithm for multiple sclerosis susceptibility: a weighted genetic risk score. Lancet Neurol. 2009;8(12):1111–1119. doi:10.1016/S1474-4422(09)70275-3
25. Barkefors I, Fuchs PF, Heldin J, et al. Exocyst complex component 3-like 2 (EXOC3L2) associates with the exocyst complex and mediates directional migration of endothelial cells. J Biol Chem. 2011;286(27):24189–24199. doi:10.1074/jbc.M110.212209
26. O’Dwyer SC, Palacio S, Matsumoto C, et al. Kv2.1 channels play opposing roles in regulating membrane potential, Ca(2+) channel function, and myogenic tone in arterial smooth muscle. Proc Natl Acad Sci U S A. 2020;117(7):3858–3866. doi:10.1073/pnas.1917879117
27. Kirsch J, Schneider H, Pagel JI, et al. Endothelial dysfunction, and A prothrombotic, proinflammatory phenotype is caused by loss of mitochondrial thioredoxin reductase in endothelium. Arterioscler Thromb Vasc Biol. 2016;36(9):1891–1899. doi:10.1161/ATVBAHA.116.307843
28. Markello TC, Pak LK, St Hilaire C, et al. Vascular pathology of medial arterial calcifications in NT5E deficiency: implications for the role of adenosine in pseudoxanthoma elasticum. Mol Genet Metab. 2011;103(1):44–50. doi:10.1016/j.ymgme.2011.01.018
29. Serveaux-Dancer M, Jabaudon M, Creveaux I, et al. Pathological implications of receptor for Advanced Glycation End-Product (AGER) gene polymorphism. Dis Markers. 2019;2019:2067353. doi:10.1155/2019/2067353
30. Liu Z, Ma Y, Cui Q, et al. Toll-like receptor 4 plays a key role in advanced glycation end products-induced M1 macrophage polarization. Biochem Biophys Res Commun. 2020;531(4):602–608. doi:10.1016/j.bbrc.2020.08.014
31. Deng J, Ma-Krupa W, Gewirtz AT, et al. Toll-like receptors 4 and 5 induce distinct types of vasculitis. Circ Res. 2009;104(4):488–495. doi:10.1161/CIRCRESAHA.108.185777
32. Kabeerdoss J, Thomas M, Goel R, et al. High expression of S100 calgranulin genes in peripheral blood mononuclear cells from patients with Takayasu arteritis. Cytokine. 2019;114:61–66. doi:10.1016/j.cyto.2018.11.033
33. Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific Toll-like receptor profiles in human medium and large arteries. Circulation. 2008;118(12):1276–1284. doi:10.1161/CIRCULATIONAHA.108.789172
34. Jia D, He Y, Zhu Q, et al. RAGE-mediated extracellular matrix proteins accumulation exacerbates HySu-induced pulmonary hypertension. Cardiovasc Res. 2017;113(6):586–597. doi:10.1093/cvr/cvx051
35. Porembskaya O, Toropova Y, Tomson V, et al. Pulmonary artery thrombosis: a diagnosis that strives for its independence. Int J Mol Sci. 2020;21(14):5086. doi:10.3390/ijms21145086
36. Seko Y, Sugishita K, Sato O, et al. Expression of costimulatory molecules (4-1BBL and Fas) and major histocompatibility class I chain-related A (MICA) in aortic tissue with Takayasu’s arteritis. J Vasc Res. 2004;41(1):84–90. doi:10.1159/000076437
37. Watanabe R, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Cellular signaling pathways in medium and large vessel vasculitis. Front Immunol. 2020;11:587089. doi:10.3389/fimmu.2020.587089
38. Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi:10.1146/annurev.immunol.15.1.749
39. Mineo C. Lipoprotein receptor signalling in atherosclerosis. Cardiovasc Res. 2020;116(7):1254–1274. doi:10.1093/cvr/cvz338
40. Calvier L, Herz J, Hansmann G. Interplay of low-density lipoprotein receptors, LRPs, and lipoproteins in pulmonary hypertension. JACC Basic Transl Sci. 2022;7(2):164–180. doi:10.1016/j.jacbts.2021.09.011
41. Lin YJ, Liu X, Chang JS, et al. Coronary artery aneurysms occurrence risk analysis between Kawasaki disease and LRP1B gene in Taiwanese children. Biomedicine. 2014;4(2):10. doi:10.7603/s40681-014-0010-5
42. Park SJ, Kim HJ, Park H, et al. Incidence, prevalence, mortality and causes of death in Takayasu Arteritis in Korea - A nationwide, population-based study. Int J Cardiol. 2017;235:100–104. doi:10.1016/j.ijcard.2017.02.086
43. Kadoba K, Watanabe R, Iwasaki T, et al. A susceptibility locus in the IL12B but not LILRA3 region is associated with vascular damage in Takayasu arteritis. Sci Rep. 2021;11(1):13667. doi:10.1038/s41598-021-93213-9
44. Kitamura H, Kobayashi Y, Kimura A, Numano F. Association of clinical manifestations with HLA-B alleles in Takayasu arteritis. Int J Cardiol. 1998;66(Suppl 1):S121–S126. doi:10.1016/S0167-5273(98)00159-4
45. Jiang X, Zhu YJ, Zhou YP, et al. Clinical features and survival in Takayasu’s arteritis-associated pulmonary hypertension: a nationwide study. Eur Heart J. 2021;42(42):4298–4305. doi:10.1093/eurheartj/ehab599
46. Morishita KA, Rosendahl K, Brogan PA. Familial Takayasu arteritis - a pediatric case and a review of the literature. Pediatr Rheumatol Online J. 2011;9:6. doi:10.1186/1546-0096-9-6
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.